

Abstract
Selective anion extraction is useful for the recovery and purification of valuable chemicals, and in the removal of pollutants from the environment. Here we report that FeII 4L4 cage 1 is able to extract an equimolar amount of ReO4 −, a high‐value anion and a nonradioactive surrogate of TcO4 −, from water into nitromethane. Importantly, the extraction was efficiently performed even in the presence of 10 other common anions in water, highlighting the high selectivity of 1 for ReO4 −. The extracted guest could be released into water as the cage disassembled in ethyl acetate, and then 1 could be recycled by switching the solvent to acetonitrile. The versatile solubility of the cage also enabled complete extraction of ReO4 − (as the tetrabutylammonium salt) from an organic phase into water by using the sulfate salt of 1 as the extractant.
Keywords: anion receptor, coordination cage, liquid–liquid extraction, self-assembly, supramolecular chemistry
Rhenium is among the rarest elements in the Earth's crust,1 but it is a key ingredient for modern industry. It is used as catalyst for petroleum refining,2 in the high‐melting superalloys of jet engines,3 and in new superhard materials,4 to cite only three examples. The limited supply and great demand lead to a high cost, generating an economic incentive for new means to extract, separate, and recycle rhenium as perrhenate (ReO4 −).5
Because of its similar structure and almost identical charge density, perrhenate is also used as a nonradioactive surrogate for pertechnetate (99TcO4 −),6 which is an important radiopharmaceutical and one of the most problematic radioactive ions in nuclear waste.7 Significant advances have been made in designing sorbent materials for removing ReO4 −/TcO4 − from aqueous solution by liquid–solid extraction.7, 8 These solid materials take up anionic targets from water via anion exchange. An attractive alternative to such sorbents is the use of supramolecular receptors as liquid‐phase extractants,9 although only a few such ReO4 −/TcO4 − receptors have been reported.10 Compared to solid‐state anion exchange materials, supramolecular extractants functioning through molecular recognition offer the potential for better selectivity toward target anions. Their flexibility in solution may provide a better size and shape match in order to optimize specific interactions between receptors and substrates.7 Such receptors can thus help address the major challenge in supramolecular chemistry of anion recognition in water.11
Most supramolecular anion extractants have been robust covalent receptors12 as opposed to coordination cages.13 Such extractants must be stable in the presence of both water and organic solvents,14 properties that are easier to engineer for covalent systems. Nevertheless, compared to the synthesis of covalent cages, the preparation of self‐assembled coordination capsules usually involves less synthetic complexity. The dynamic nature of coordination bonds15 may also enable guest release and subsequent recycling of the extractant.16
We recently reported the water‐soluble sulfate salt of azaphosphatrane‐based FeII 4L4 tetrahedron 1 (Figure 1), which can adaptively encapsulate different anions via hydrogen bonding and electrostatic interactions in water.17 Herein, we develop 1 as an efficient and selective extractant, capable of extracting ReO4 − in either direction between organic and aqueous phases. We also establish a simple solvent‐switching procedure that allows 1 to be disassembled, releasing its anionic cargo and allowing it to be recycled.
Figure 1.
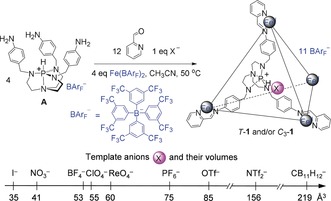
Subcomponent self‐assembly of 1 around 1 equiv of template anion.
Non‐coordinating tetrakis(3,5‐bis(trifluoromethyl)phenyl)borate (BArF −) was selected as the counter‐anion for 1 in this work based on its lipophilicity and bulk (Figure 1). The lipophilic nature of BArF − renders 1 soluble in water‐immiscible organic solvents such as nitromethane. BArF − is larger (968 Å3)18 than the cavity volume of 1 at its most expansive (253 Å3; see below), precluding competition with any of the anions discussed below.
The BArF − salt of subcomponent A (Figure 1) was obtained by anion metathesis (Supporting Information section 2.1). As was observed in water,17 the reaction of A (4 equiv) with Fe(BArF)2 (4 equiv) and 2‐formylpyridine (12 equiv) in acetonitrile failed to give the expected cage complex 1⋅[BArF]12, which required an internal template anion (listed in Figure 1) for its formation.
In acetonitrile, template anions with volumes below 53 Å3 gave rise to both a C 3‐symmetric isomer (C 3‐1, with one azaphosphatrane +P‐H group oriented away from the inner cavity and the other three pointed inward) and a T‐symmetric isomer (T‐1, containing four inwardly‐directed +P‐H groups) (Figure S1), whereas larger anionic templates, having volumes ≥55 Å3, resulted in the formation of T‐1 exclusively (Figure S2), as was observed in water.17 The initially obtained mixture of isomers in the former case is kinetically metastable and gradual interconversion between cage isomers was observed. Energy barriers of conversion in CD3CN at 323 K were determined to be similar to the values previously obtained in water at 298 K17 (Figures S3–S6).
We then tested the stability of the cage, as Tf2N−⊂ 1⋅[BArF]11 (Tf=CF3SO2), in ethyl acetate and nitromethane, both of which are water‐immiscible organic solvents suitable for liquid–liquid extraction experiments. Circa 65 % of 1 was observed to disassemble at a concentration of 1.5 mm in EtOAc after 4 h (Figure S7), with complete disassembly occurring at more dilute concentrations. In contrast, the cage was stable without any decomposition in CD3NO2 for at least two weeks at room temperature (Figure S9). We infer that the more polar solvent nitromethane offers a greater degree of stabilization to highly cationic 1 than does less polar ethyl acetate.19 Nitromethane was thus chosen as the organic solvent for liquid–liquid extractions.
Interestingly, cage reassembly was observed after evaporation of EtOAc and redissolution of 1 in CD3CN, indicating a reversible process (Figure S8). This phenomenon provides an original means of guest release and extractant recovery, as explored further below.
Through competitive guest exchange, we were able to gauge the relative binding affinities of different anions in CD3NO2. The following hierarchy was observed: CB11H12 − > ReO4 − > TfO− > PF6 − > ClO4 − > Tf2N− > BF4 − > I− > NO3 − (Figures S10–S17, Table S1). This ordering differs from the one observed in water: PF6 − > ReO4 − > TfO− > ClO4 − > CB11H12 − > Tf2N− > BF4 − > I− > NO3 −,17 especially as regards the binding affinity of CB11H12 −. To accommodate this largest anion, the cage framework must expand; we infer that this larger conformation in water is unfavorable because it involves greater exposure of hydrophobic surface to water. In both solvents, ReO4 − binds more strongly than other common anions, indicating potential for its selective extraction.
We obtained single crystals of 1 encapsulating the two most strongly bound anions in nitromethane, CB11H12 − and ReO4 −. X‐ray diffraction analyses20 (Figure 2) showed a T‐symmetric framework for both structures. The structures demonstrate the flexibility of the cage skeleton, allowing adaptation to guests of different sizes. Calculated cavity volumes of 157 Å3 and 253 Å3 were obtained for the ReO4 − (volume 60 Å3) and CB11H12 − (volume 219 Å3) complexes, respectively (Figure S18). Cavity expansion occurs through outward motion of the azaphosphatrane faces, resulting in a more open surface having pores of ca. 2.5 Å in CB11H12 −⊂1, compared to ca. 1.2 Å in ReO4 −⊂1.
Figure 2.
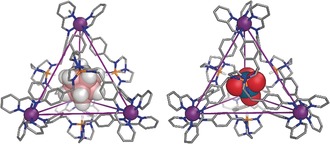
X‐ray crystal structures of CB11H12 −⊂1 (left) and ReO4 −⊂1 (right). Disorder, unbound counterions, non‐P‐bound hydrogen atoms, and solvents are omitted for clarity.
Since Tf2N− is the most weakly bound among anions capable of templating T‐1 exclusively, extraction of ReO4 − was initially investigated using Tf2N−⊂1⋅[BArF]11 as the extractant. After mixing 0.8 mm Tf2N−⊂1⋅[BArF]11 in CD3NO2 with 0.8 mm NaReO4 in D2O for 7 h, no further uptake of ReO4 − by 1 was observed. 1H NMR spectroscopy of the CD3NO2 phase revealed that 60 % of the ReO4 − from the aqueous phase had been extracted as ReO4 −⊂1⋅[BArF]11, with the remainder of 1 binding Tf2N− (Figure S19). After displacement by the extracted ReO4 −, free Tf2N− thus transferred from CD3NO2 to D2O as the sodium salt.
We investigated the effect of the counterions of the Tf2N− template by adding TBANTf2 (TBA=tetra‐n‐butylammonium), KNTf2, or LiNTf2 during the self‐assembly, but no cation effect on the efficiency of ReO4 − extraction was observed (Figure S20). Similarly, increasing the concentrations of Tf2N−⊂1⋅[BArF]11 in CD3NO2 and NaReO4 in D2O to 1.3 mm (Figure S20f) did not impact extraction efficiency.
The extraction of TfO− (using NaOTf) from water under identical liquid–liquid conditions was also successful but with a lower efficiency (43 %, Figure S21). Control experiments confirmed that without the cage, NaOTf did not transfer to the CD3NO2 phase (Figure S22).
In order to improve the extraction efficiency, we sought a more weakly bound template anion that avoided the complexity of generating a mixture of cage diastereomers. Such an anion was found to be n‐butyltrifluoroborate (nBuBF3 −). We found nBuBF3 − to be able to template T‐1 exclusively (Figures S23–S28), and the resultant nBuBF3 −⊂1⋅[BArF]11 to be stable in CD3NO2 for weeks. Moreover, 1 equiv of Tf2N− in CD3NO2 almost completely displaced the encapsulated nBuBF3 − (Figure S29), marking nBuBF3 − as the weaker binder.
When the extractant nBuBF3 −⊂1⋅[BArF]11 in CD3NO2 was mixed with an equimolar amount of NaReO4 in D2O, only ReO4 −⊂1⋅[BArF]11 was observed after extraction, indicating complete removal of ReO4 − from water (Figures 3 c and S30). Complete extraction of TfO− from aqueous NaOTf was also achieved by using nBuBF3 −⊂1⋅[BArF]11 (Figure S31).
Figure 3.
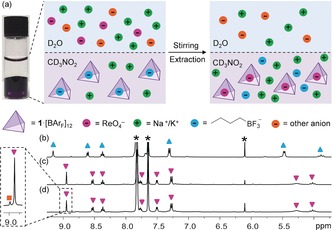
a) Selective liquid–liquid extraction of ReO4 − in the presence of other anions. Conditions: 0.8 mm nBuBF3 −⊂1⋅[BArF]11 in CD3NO2; 0.8 mm in D2O of each of NaReO4, NaF, NaCl, NaBr, NaI, Na2SO4, KClO4, KNO3, NaBF4, NaH2PO4, and NaOAc; 7 hours stirring at RT; b–d) Partial 1H NMR spectra of (b) the CD3NO2 phase before extraction, showing only the presence of nBuBF3 −⊂1⋅[BArF]11 (blue ▴); c) the CD3NO2 phase after extraction in the absence of competing anions, showing only the presence of ReO4 −⊂1⋅[BArF]11 (pink ▾); (d) the CD3NO2 phase after extraction in the presence of competing anions, showing the presence of 97 % ReO4 −⊂1⋅[BArF]11 (pink ▾) and 3 % ClO4 −⊂1⋅[BArF]11 (orange ▪). The peaks of BArF − and the trimethoxybenzene standard are denoted by asterisks.
Encouraged by these results, we evaluated the selectivity of 1 toward ReO4 − in the presence of 10 other different anions simultaneously in water: F−, Cl−, Br−, I−, SO4 2−, ClO4 −, NO3 −, BF4 −, H2PO4 −, and AcO− (1 equiv to ReO4 − in each case). The extraction efficiency for ReO4 − by nBuBF3 −⊂ 1⋅[BArF]11 in the presence of this anion library was 97 %, with ClO4 − comprising the other 3 % extracted (Figure 3).
We also developed a strategy to release and separate the extracted guest and recover the cage extractant by exploiting the instability of 1 in less polar solvents. As shown in Figure 4, after extraction, the nitromethane layer was separated and the solvent evaporated. The isolated cage was then redissolved in degassed EtOAc. As described above, the cage disassembled in this solvent. The extracted guest transferred to the water phase as KReO4, pairing with K+ from nBuBF3K, allowing its removal as the phases were separated. Regeneration of nBuBF3 −⊂1⋅[BArF]11, which could be reused for further extraction experiments, was realized by evaporating the ethyl acetate and adding acetonitrile, along with nBuBF3K (Figure S32).
Figure 4.
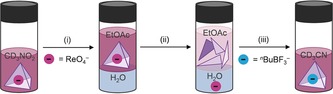
Illustration of cage extractant recycling: (i) After evaporation of CD3NO2, ReO4 −⊂1⋅[BArF]11 was redissolved in degassed EtOAc; degassed H2O was then added. (ii) After stirring for 4 h, the cage disassembled and ReO4 − was released, transferring to the H2O phase. (iii) After separation and evaporation of the EtOAc layer, addition of CD3CN and nBuBF3 − resulted in regeneration of the extractant nBuBF3 −⊂1⋅[BArF]11.
Interestingly, due to the versatile solubility of 1, either ReO4 − or TfO− could also be extracted from an organic phase into water, in the opposite direction to what was described above. In this case, Tf2N−⊂1⋅[SO4]5.5 as extractant completely removed either ReO4 − or TfO− from CD3NO2 into D2O (Figures 5 and S33). Control experiments showed that without the cage, TBAReO4 and TBAOTf did not transfer to D2O (Figures S34).
Figure 5.
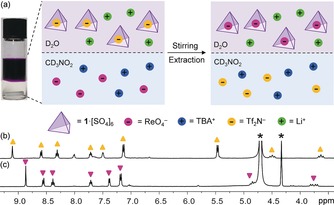
a) Illustration of the liquid–liquid extraction of ReO4 − from an organic phase into water. Conditions: 0.8 mm Tf2N−⊂1⋅[SO4]5.5 in D2O; 0.8 mm TBAReO4 in CD3NO2; 3 hours stirring. b,c) Partial 1H NMR spectra of (b) the D2O phase before extraction, showing only Tf2N−⊂1⋅[SO4]5.5 (yellow ▴), and (c) the D2O phase after extraction, showing only ReO4 −⊂1⋅[SO4]5.5 (pink ▾). HDO and CHD2NO2 peaks are represented by asterisks.
In summary, we have demonstrated for the first time the feasibility of using a coordination cage for biphasic extraction. By employing BArF − as counter‐anion and nBuBF3 − as template, nBuBF3 −⊂1⋅[BArF]11 was capable of completely extracting ReO4 − from water into nitromethane. An efficiency of 97 % was achieved even in the presence of 10 competing anions. A novel strategy for extractant regeneration was developed by taking advantage of the differential stability of 1 across solvents. Moreover, due to the versatile solubility of 1 when paired with different counter‐anions, complete extraction of ReO4 − (TBAReO4) from an organic phase into water could also be accomplished by using Tf2N−⊂1⋅[SO4]5.5. The selective extraction properties of the cage toward perrhenate suggest great potential for recycling rhenium compounds, purification of chemicals, and for pertechnetate removal from water. Concepts developed in this study may also be generalized to enable the purification of other species using different coordination cages.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the UK Engineering and Physical Sciences Research Council (EPSRC EP/M008258/1). The authors thank the Department of Chemistry NMR facility, University of Cambridge for performing some NMR experiments, and the EPSRC UK National Mass Spectrometry Facility at Swansea University for carrying out high resolution mass spectrometry. D.Z. acknowledges the Herchel Smith Research Fellowship from the University of Cambridge. We thank Diamond Light Source (UK) for synchrotron beamtime on I19 (MT15768).
D. Zhang, T. K. Ronson, J. Mosquera, A. Martinez, J. R. Nitschke, Angew. Chem. Int. Ed. 2018, 57, 3717.
References
- 1. Scerri E., Nat. Chem. 2010, 2, 598. [DOI] [PubMed] [Google Scholar]
- 2. D'Ippolito S. A., Vera C. R., Epron F., Samoila P., Especel C., Marécot P., Gutierrez L. B., Pieck C. L., Ind. Eng. Chem. Res. 2009, 48, 3771–3778. [Google Scholar]
- 3. Tian S., Su Y., Qian B., Yu X., Liang F., Li A., Mater. Des. 2012, 37, 236–242. [Google Scholar]
- 4. Kaner R. B., Gilman J. J., Tolbert S. H., Science 2005, 308, 1268–1269. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Lan X., Liang S., Song Y., Hydrometallurgy 2006, 82, 133–136; [Google Scholar]
- 5b. Ghorbanzadeh Mashkani S., Tajer Mohammad Ghazvini P., Agha Aligol D., Bioresour. Technol. 2009, 100, 603–608. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Shen J., Chai W., Wang K., Zhang F., ACS Appl. Mater. Interfaces 2017, 9, 22440–22448; [DOI] [PubMed] [Google Scholar]
- 6b. Banerjee D., Elsaidi S. K., Aguila B., Li B., Kim D., Schweiger M. J., Kruger A. A., Doonan C. J., Ma S., Thallapally P. K., Chem. Eur. J. 2016, 22, 17581–17584; [DOI] [PubMed] [Google Scholar]
- 6c. Banerjee D., Xu W., Nie Z., Johnson L. E., Coghlan C., Sushko M. L., Kim D., Schweiger M. J., Kruger A. A., Doonan C. J., Thallapally P. K., Inorg. Chem. 2016, 55, 8241–8243. [DOI] [PubMed] [Google Scholar]
- 7. Banerjee D., Kim D., Schweiger M. J., Kruger A. A., Thallapally P. K., Chem. Soc. Rev. 2016, 45, 2724–2739. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Bonnesen P. V., Brown G. M., Alexandratos S. D., Bavoux L. B., Presley D. J., Patel V., Ober R., Moyer B. A., Environ. Sci. Technol. 2000, 34, 3761–3766; [Google Scholar]
- 8b. Wang S., Alekseev E. V., Diwu J., Casey W. H., Phillips B. L., Depmeier W., Albrecht-Schmitt T. E., Angew. Chem. Int. Ed. 2010, 49, 1057–1060; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1075–1078; [Google Scholar]
- 8c. Zhu L., Sheng D., Xu C., Dai X., Silver M. A., Li J., Li P., Wang Y., Wang Y., Chen L., Xiao C., Chen J., Zhou R., Zhang C., Farha O. K., Chai Z., Albrecht-Schmitt T. E., Wang S., J. Am. Chem. Soc. 2017, 139, 14873–14876; [DOI] [PubMed] [Google Scholar]
- 8d. Zhu L., Xiao C., Dai X., Li J., Gui D., Sheng D., Chen L., Zhou R., Chai Z., Albrecht-Schmitt T. E., Wang S., Environ. Sci. Technol. Lett. 2017, 4, 316–322; [Google Scholar]
- 8e. Fei H., Bresler M. R., Oliver S. R., J. Am. Chem. Soc. 2011, 133, 11110–11113. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Holman K. T., Halihan M. M., Steed J. W., Jurisson S. S., Atwood J. L., J. Am. Chem. Soc. 1995, 117, 7848–7849; [Google Scholar]
- 9b. Gawenis J. A., Holman K. T., Atwood J. L., Jurisson S. S., Inorg. Chem. 2002, 41, 6028–6031; [DOI] [PubMed] [Google Scholar]
- 9c. Beer P. D., Hopkins P. K., McKinney J. D., Chem. Commun. 1999, 1253–1254; [Google Scholar]
- 9d. Farrell D., Gloe K., Gloe K., Goretzki G., McKee V., Nelson J., Nieuwenhuyzen M., Pal I., Stephan H., Town R. M., Wichmann K., Dalton Trans. 2003, 1961–1968; [Google Scholar]
- 9e. Wilson A. M., Bailey P. J., Tasker P. A., Turkington J. R., Grant R. A., Love J. B., Chem. Soc. Rev. 2014, 43, 123–134; [DOI] [PubMed] [Google Scholar]
- 9f. Izatt R. M., Clark G. A., Christensen J. J., Sep. Sci. Technol. 1986, 21, 865–872. [Google Scholar]
- 10.
- 10a. Katayev E. A., Kolesnikov G. V., Sessler J. L., Chem. Soc. Rev. 2009, 38, 1572–1586; [DOI] [PubMed] [Google Scholar]
- 10b. Alberto R., Bergamaschi G., Braband H., Fox T., Amendola V., Angew. Chem. Int. Ed. 2012, 51, 9772–9776; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9910–9914; [Google Scholar]
- 10c. Amendola V., Bergamaschi G., Boiocchi M., Alberto R., Braband H., Z. Naturforsch. B 2014, 5, 1820–1826; [Google Scholar]
- 10d. Custelcean R., Chem. Commun. 2013, 49, 2173–2182. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Busschaert N., Caltagirone C., Van Rossom W., Gale P. A., Chem. Rev. 2015, 115, 8038–8155; [DOI] [PubMed] [Google Scholar]
- 11b. Ballester P., Chem. Soc. Rev. 2010, 39, 3810–3830; [DOI] [PubMed] [Google Scholar]
- 11c. Zhou Y., Zhang J. F., Yoon J., Chem. Rev. 2014, 114, 5511–5571; [DOI] [PubMed] [Google Scholar]
- 11d. Sokkalingam P., Shraberg J., Rick S. W., Gibb B. C., J. Am. Chem. Soc. 2016, 138, 48–51; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Langton M. J., Serpell C. J., Beer P. D., Angew. Chem. Int. Ed. 2016, 55, 4629–4629; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4705–4705; [Google Scholar]
- 11f. Kang S. O., Llinares J. M., Day V. W., Bowman-James K., Chem. Soc. Rev. 2010, 39, 3980–4003; [DOI] [PubMed] [Google Scholar]
- 11g. Jo J., Olasz A., Chen C. H., Lee D., J. Am. Chem. Soc. 2013, 135, 3620–3632; [DOI] [PubMed] [Google Scholar]
- 11h. Kubik S., Chem. Soc. Rev. 2010, 39, 3648–3663; [DOI] [PubMed] [Google Scholar]
- 11i. Wang Q. Q., Day V. W., Bowman-James K., Angew. Chem. Int. Ed. 2012, 51, 2119–2123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2161–2165; [Google Scholar]
- 11j. Fabbrizzi L., Poggi A., Chem. Soc. Rev. 2013, 42, 1681–1699; [DOI] [PubMed] [Google Scholar]
- 11k. Lisbjerg M., Valkenier H., Jessen B. M., Al-Kerdi H., Davis A. P., Pittelkow M., J. Am. Chem. Soc. 2015, 137, 4948–4951. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. He Q., Peters G. M., Lynch V. M., Sessler J. L., Angew. Chem. Int. Ed. 2017, 56, 13396–13400; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13581–13585; [Google Scholar]
- 12b. Sisson A. L., Clare J. P., Davis A. P., Chem. Commun. 2005, 5263–5265; [DOI] [PubMed] [Google Scholar]
- 12c. Kim S. K., Vargas-Zuniga G. I., Hay B. P., Young N. J., Delmau L. H., Masselin C., Lee C. H., Kim J. S., Lynch V. M., Moyer B. A., Sessler J. L., J. Am. Chem. Soc. 2012, 134, 1782–1792; [DOI] [PubMed] [Google Scholar]
- 12d. Jia C., Wu B., Li S., Huang X., Zhao Q., Li Q. S., Yang X. J., Angew. Chem. Int. Ed. 2011, 50, 486–490; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 506–510; [Google Scholar]
- 12e. Kharel S., Joshi H., Bierschenk S., Stollenz M., Taher D., Bhuvanesh N., Gladysz J. A., J. Am. Chem. Soc. 2017, 139, 2172–2175; [DOI] [PubMed] [Google Scholar]
- 12f. Chi X., Peters G. M., Hammel F., Brockman C., Sessler J. L., J. Am. Chem. Soc. 2017, 139, 9124–9127. [DOI] [PubMed] [Google Scholar]
- 13. Wenzel M., Knapp Q. W., Plieger P. G., Chem. Commun. 2011, 47, 499–501. [DOI] [PubMed] [Google Scholar]
- 14. Percástegui E. G., Mosquera J., Nitschke J. R., Angew. Chem. Int. Ed. 2017, 56, 9136–9140; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9264–9268. [Google Scholar]
- 15.
- 15a. Zhou X.-P., Wu Y., Li D., J. Am. Chem. Soc. 2013, 135, 16062–16065; [DOI] [PubMed] [Google Scholar]
- 15b. Yang L., Tan X., Wang Z., Zhang X., Chem. Rev. 2015, 115, 7196–7239; [DOI] [PubMed] [Google Scholar]
- 15c. Wang W., Wang Y. X., Yang H. B., Chem. Soc. Rev. 2016, 45, 2656–2693; [DOI] [PubMed] [Google Scholar]
- 15d. Krieg E., Bastings M. M. C., Besenius P., Rybtchinski B., Chem. Rev. 2016, 116, 2414–2477; [DOI] [PubMed] [Google Scholar]
- 15e. Ray D., Foy J. T., Hughes R. P., Aprahamian I., Nat. Chem. 2012, 4, 757–762; [DOI] [PubMed] [Google Scholar]
- 15f. Erbas-Cakmak S., Fielden S. D. P., Karaca U., Leigh D. A., McTernan C. T., Tetlow D. J., Wilson M. R., Science 2017, 358, 340–343; [DOI] [PubMed] [Google Scholar]
- 15g. He Z., Jiang W., Schalley C. A., Chem. Soc. Rev. 2015, 44, 779–789; [DOI] [PubMed] [Google Scholar]
- 15h. Barry D. E., Caffrey D. F., Gunnlaugsson T., Chem. Soc. Rev. 2016, 45, 3244–3274; [DOI] [PubMed] [Google Scholar]
- 15i. Mauro M., Aliprandi A., Septiadi D., Kehr N. S., De Cola L., Chem. Soc. Rev. 2014, 43, 4144–4166; [DOI] [PubMed] [Google Scholar]
- 15j. Wang S., Sawada T., Ohara K., Yamaguchi K., Fujita M., Angew. Chem. Int. Ed. 2016, 55, 2063–2066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2103–2106; [Google Scholar]
- 15k. Wei P., Yan X., Huang F., Chem. Soc. Rev. 2015, 44, 815–832; [DOI] [PubMed] [Google Scholar]
- 15l. Custelcean R., Bonnesen P. V., Duncan N. C., Zhang X., Watson L. A., Van Berkel G., Parson W. B., Hay B. P., J. Am. Chem. Soc. 2012, 134, 8525–8534. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Samanta S. K., Moncelet D., Briken V., Isaacs L., J. Am. Chem. Soc. 2016, 138, 14488–14496; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Han M., Michel R., He B., Chen Y. S., Stalke D., John M., Clever G. H., Angew. Chem. Int. Ed. 2013, 52, 1319–1323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1358–1362; [Google Scholar]
- 16c. Chan A. K., Lam W. H., Tanaka Y., Wong K. M., Yam V. W., Proc. Natl. Acad. Sci. USA 2015, 112, 690–695; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Kurihara K., Yazaki K., Akita M., Yoshizawa M., Angew. Chem. Int. Ed. 2017, 56, 11360–11364; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11518–11522; [Google Scholar]
- 16e. Cook T. R., Stang P. J., Chem. Rev. 2015, 115, 7001–7045. [DOI] [PubMed] [Google Scholar]
- 17. Zhang D., Ronson T. K., Mosquera J., Martinez A., Guy L., Nitschke J. R., J. Am. Chem. Soc. 2017, 139, 6574–6577. [DOI] [PubMed] [Google Scholar]
- 18. Cullinane J., Jolleys A., Mair F. S., Dalton Trans. 2013, 42, 11971–11975. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Smith W. L., J. Chem. Educ. 1977, 54, 228–229; [Google Scholar]
- 19b. Roseman M., Jencks W. P., J. Am. Chem. Soc. 1975, 97, 631–640. [Google Scholar]
- 20.CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/anie.201800459 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary