

Abstract
Objective
The 24‐week equivalent efficacy and comparable safety results of the biosimilar SB5 and reference adalimumab (ADA) from the phase III randomized study in patients with moderate‐to‐severe rheumatoid arthritis (RA) have been reported previously. We undertook this transition study to evaluate patients who switched from ADA to SB5 or who continued to receive SB5 or ADA up to 52 weeks.
Methods
In this phase III study, patients were initially randomized 1:1 to receive SB5 or ADA (40 mg subcutaneously every other week). At 24 weeks, patients receiving ADA were rerandomized 1:1 to continue with ADA (ADA/ADA group) or to switch to SB5 (ADA/SB5 group) up to week 52; patients receiving SB5 continued with SB5 for 52 weeks (SB5 group). Efficacy, safety, and immunogenicity were evaluated up to 52 weeks.
Results
The full analysis set population consisted of 542 patients (269 in the SB5 group, 273 in the ADA overall group [patients who were randomized to receive ADA at week 0], 125 in the ADA/SB5 group, and 129 in the ADA/ADA group). The percentages of patients meeting the American College of Rheumatology 20%, 50%, or 70% improvement criteria (achieving an ACR20, ACR50, or ACR70 response) at week 24 were maintained after the transition from ADA to SB5, and these response rates were comparable across treatment groups throughout the study. ACR20 response rates ranged from 73.4% to 78.8% at week 52. Radiographic progression was minimal and comparable across treatment groups. The safety profile and the incidence of antidrug antibodies were comparable across treatment groups after transition.
Conclusion
SB5 was well tolerated over 1 year in patients with RA, with efficacy, safety, and immunogenicity comparable to those of ADA. Switching from ADA to SB5 had no treatment‐emergent issues such as increased adverse events, increased immunogenicity, or loss of efficacy.
Biologic disease‐modifying antirheumatic drugs (bDMARDs), including tumor necrosis factor (TNF) inhibitors such as adalimumab (ADA), have been used successfully to treat patients with rheumatoid arthritis (RA) 1. According to the updated 2016 European League Against Rheumatism (EULAR) recommendations, addition of a bDMARD should be considered in patients who do not achieve the treatment target and have poor prognostic factors 2. In the absence of poor prognostic factors, other conventional synthetic DMARDs (csDMARDs) may be tried. The American College of Rheumatology (ACR) similarly recommends the step‐up therapy; however, either bDMARDs or csDMARDs may be used regardless of prognostic factors 3. Treatment with bDMARDs is often associated with high costs 4, 5, and the advent of biosimilars provides the potential to reduce costs and increase patient access to such therapies 4, 6. Several biosimilars targeting TNF have been approved recently for use in the US and Europe, and the use of these biosimilars is recommended by EULAR 2.
ADA is a recombinant human IgG1 specific for TNF that is approved for the treatment of RA as well as several other inflammatory conditions 7. SB5 (Imraldi; Samsung Bioepis) was developed as an ADA biosimilar and has an identical amino acid sequence and physicochemical and in vitro functional properties similar to those of reference ADA 8. The European Commission granted a marketing authorization for SB5 in August 2017. Pharmacokinetic (PK) equivalence and comparable safety for SB5 and ADA were demonstrated in a phase I study in healthy individuals 8. In a phase III randomized study in patients with moderate‐to‐severe RA, equivalent efficacy was demonstrated for SB5 and ADA, as seen in percentages of patients meeting the ACR 20% improvement criteria (achieving an ACR20 response) 9 (72.4% and 72.2%, respectively) and additional efficacy end points up to 24 weeks; SB5 was well tolerated, with PK, safety, and immunogenicity profiles comparable to those of ADA 10.
Growing numbers of biosimilars for various biologic agents have been approved or are in various stages of clinical development; however, there are limited clinical and real‐world data regarding the effects of switching from reference biologic agents to biosimilars 11. An important clinical consideration for the use of biosimilars is whether switching from reference product might result in loss of efficacy or increased immunogenicity or other safety concerns. Data derived from appropriately designed switching clinical trials and real‐world experience can help fill this information gap and provide useful evidence in clinical decision‐making 11.
As mentioned above, the 24‐week results of the phase III clinical study evaluating SB5 and ADA demonstrated comparable ACR20 response rates, PK, safety, and immunogenicity in patients with moderate‐to‐severe RA 10. The objective of the current 52‐week transition study was to evaluate the safety, immunogenicity, and efficacy of continuing SB5 treatment versus switching from ADA to SB5 (as will occur in clinical practice) versus continuing ADA treatment.
Patients and Methods
Methods have been described previously in detail 10 and are briefly summarized herein.
Patient inclusion and exclusion criteria. The study included patients ages 18–75 years with moderate‐to‐severe RA treated with methotrexate (MTX) for ≥6 months and receiving a stable MTX dosage of 10–25 mg/week for ≥4 weeks before screening. Patients had active disease (≥6 swollen joints, ≥6 tender joints, and either an erythrocyte sedimentation rate [ESR] ≥28 mm/hour or a serum C‐reactive protein [CRP] level ≥1.0 mg/dl). Pertinent exclusions included previous treatment with biologic agents and active or latent tuberculosis infection at screening 10.
Study design. This 52‐week, phase III, randomized, double‐blind, parallel‐group study was conducted at 51 sites in 7 countries. Patients were initially randomized 1:1 to receive SB5 or ADA (40 mg subcutaneously every other week). Patients receiving ADA were randomized again 1:1 at week 24 to continue with ADA or to switch to SB5 up to week 52; patients receiving SB5 were also randomized for purposes of blinding and continued with SB5 for the 52 weeks of the study (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40444/abstract).
The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with International Conference on Harmonisation Guidelines for Good Clinical Practice. Each study center's independent ethics committee or institutional review board reviewed and approved the protocol and study. All patients provided written informed consent before study entry.
Study assessments. The primary efficacy end point of the study, the ACR20 response rate at week 24, has been reported previously 10. Efficacy assessments up to week 52 included the ACR20, ACR50, and ACR70 response rates and the ACR index of improvement in RA response rate 12, the Disease Activity Score in 28 joints 13 using the ESR (DAS28‐ESR), and the EULAR response (good, moderate, or no response) 14. Post hoc analyses were performed on the Simplified Disease Activity Index (SDAI) 15, the Clinical Disease Activity Index (CDAI) 16, the remission rate based on the DAS28‐ESR (score of <2.6), SDAI (score of ≤3.3), and CDAI (score of ≤2.8), and the proportion of patients with low disease activity based on the DAS28‐ESR (score of ≤3.2), SDAI (score of 3.3–11.0), and CDAI (score of 2.8–10.0). A Boolean‐based remission was determined based on a swollen joint count of ≤1, a tender joint count of ≤1, a CRP level of ≤10 mg/liter, and a visual analog scale score of ≤10 mm on the patient's global assessment of disease activity.
Radiographs of the hands and feet were obtained at baseline and at week 52 and were evaluated centrally by 2 independent qualified readers who were blinded with regard to patient identity, treatment, and assessment time point. When the score change was within the top 5% of cases with the highest score differences between readers, the radiographs required consensus review by the primary readers. The mean joint erosion score and joint space narrowing score of the 2 readers were used to calculate the modified Sharp/van der Heijde score (SHS) 17.
Treatment‐emergent adverse events (AEs) (graded as mild, moderate, or severe), serious AEs (SAEs), vital sign abnormalities, and clinical laboratory abnormalities were monitored as part of safety assessments. Immunogenicity assessments included monitoring for the development of antidrug antibodies and neutralizing antibodies.
Immunogenicity was analyzed as emergent (positive for ≥1 antidrug antibody after transition among patients with negative 24‐week overall antidrug antibody results), boosted (increased titer of antidrug antibodies at any time compared with the highest titer up to week 24), and total (seroconverted to positive for antidrug antibodies or boosted their preexisting antidrug antibody titer during the transition period). Antidrug antibodies were detected using electrochemiluminescence bridging (Meso Scale Discovery), employing an SB5 single‐tagged immunoassay. Subgroup analyses based on the antidrug antibody status of the patients were performed in the treatment groups following transition at 24 weeks up to 52 weeks for the various efficacy parameters.
Statistical analysis. Sample size determination was described previously 10. Efficacy assessments were performed on the full analysis set, which comprised all randomized patients (intent‐to‐treat principle). The following treatment groups were compared: patients who were randomized to receive SB5 at week 0 (SB5 group), patients who were randomized to receive ADA at week 0 (ADA overall group), and patients in the ADA group who were rerandomized at week 24 to continue ADA (ADA/ADA group) or to switch to SB5 (ADA/SB5 group). Results are presented with no imputation for missing data. The safety population included all patients who received ≥1 dose of study drug after rerandomization at week 24. Safety results were compared for the SB5/SB5 group (patients in the SB5 group who were rerandomized to continue SB5), ADA/SB5 group, and ADA/ADA group. All efficacy and safety results were summarized descriptively by treatment group. Statistical analyses were performed using SAS software, version 9.2 or higher (SAS Institute).
Results
Patients. The study was initiated on May 12, 2014 and completed on October 19, 2015. A total of 544 patients were randomized to the SB5 (n = 271) or ADA overall (n = 273) groups; 254 patients (93.7%) in the SB5 group and 254 (93.0%) in the ADA overall group completed the 24‐week study 10. At week 24, the 254 patients in the SB5 group continued to receive SB5 (SB5/SB5 group); patients in the ADA overall group were randomized to transition to SB5 (ADA/SB5 group [n = 125]) or to continue ADA (ADA/ADA group [n = 129]). The full analysis set population consisted of 542 patients (269 in the SB5 group, 273 in the ADA overall group, 125 in the ADA/SB5 group, and 129 in the ADA/ADA group). Patient disposition for the 52‐week study is summarized in Figure 1.
Figure 1.
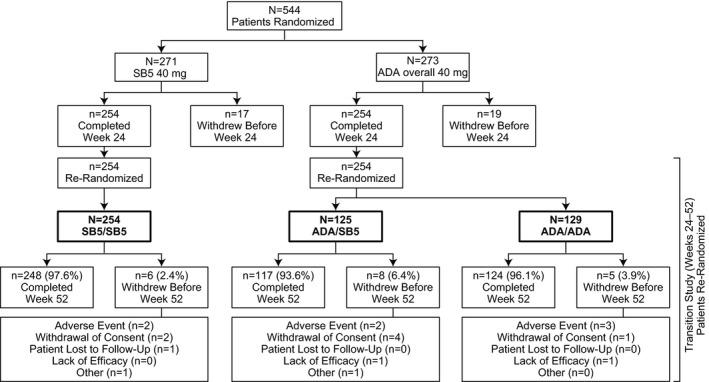
Patient disposition summary. The percentages of patients who completed or discontinued are based on the numbers of patients who were rerandomized at week 24. The SB5 group included patients who were randomized to receive the biosimilar SB5 at week 0. The adalimumab (ADA) overall group included patients who were randomized to receive reference ADA at week 0. The SB5/SB5 group included patients receiving SB5 who were rerandomized to continue SB5. The ADA/SB5 group included patients receiving ADA who were rerandomized to switch to SB5. The ADA/ADA group included patients receiving ADA who were rerandomized to continue ADA.
Patient demographics and baseline characteristics at study start (week 0) and disease activity at weeks 0 and 24 (rerandomization time point) are shown in Table 1. Overall, baseline characteristics and disease activity were well balanced across treatment groups.
Table 1.
Patient demographics at baseline and disease activity at baseline and week 24a
| SB5 group (n = 271) | ADA overall group (n = 273) | ADA/SB5 group (n = 125) | ADA/ADA group (n = 129) | |||||
|---|---|---|---|---|---|---|---|---|
| Demographics at baseline | ||||||||
| Age, years | 49.8 ± 12.7 | 52.5 ± 11.9 | 51.7 ± 11.3 | 52.8 ± 12.3 | ||||
| Sex, no. (%) | ||||||||
| Female | 217 (80.1) | 224 (82.1) | 105 (84.0) | 103 (79.8) | ||||
| Male | 54 (19.9) | 49 (17.9) | 20 (16.0) | 26 (20.2) | ||||
| BMI, kg/m2 | 26.2 ± 4.8 | 27.0 ± 5.1 | 27.2 ± 5.3 | 26.9 ± 5.0 | ||||
| Disease duration, years | 5.4 ± 4.4 | 5.5 ± 4.3 | 5.3 ± 4.1 | 5.6 ± 4.5 | ||||
| MTX dose, mg/week | 15.1 ± 4.6 | 15.4 ± 4.4 | 15.4 ± 4.5 | 15.2 ± 4.4 | ||||
| Duration of MTX use, months | 39.5 ± 38.4 | 37.8 ± 34.9 | 38.3 ± 33.8 | 39.5 ± 37.2 | ||||
| RF positive, no. (%) | 203 (74.9) | 185 (67.8) | 80 (64.0) | 94 (72.9) | ||||
| Disease activity at baseline | ||||||||
| Swollen joint count | 15.8 ± 8.0 | 15.5 ± 7.5 | 14.5 ± 6.4 | 16.3 ± 8.3 | ||||
| Tender joint count | 23.9 ± 11.7 | 24.1 ± 10.8 | 23.8 ± 10.5 | 24.3 ± 11.3 | ||||
| HAQ DI score | 1.3 ± 0.6 | 1.4 ± 0.6 | 1.4 ± 0.6 | 1.4 ± 0.7 | ||||
| DAS28‐ESR | 6.5 ± 0.7 | 6.5 ± 0.71 | 6.5 ± 0.6 | 6.4 ± 0.8 | ||||
| CRP, mg/liter | 11.5 ± 19.0 | 12.6 ± 19.0 | 13.0 ± 20.8 | 11.9 ± 15.7 | ||||
| ESR, mm/hour | 39.6 ± 13.3 | 39.6 ± 13.9 | 40.5 ± 14.3 | 39.3 ± 14.0 | ||||
| Physician's global assessment, 0–100‐mm VAS | 59.8 ± 16.9 | 60.6 ± 15.4 | 60.6 ± 14.7 | 61.0 ± 16.1 | ||||
| Patient's global assessment, 0–100‐mm VAS | 58.5 ± 20.3 | 59.4 ± 18.7 | 59.1 ± 18.0 | 59.9 ± 19.6 | ||||
| Patient's assessment of pain, 0–100‐mm VAS | 59.2 ± 20.7 | 60.8 ± 19.7 | 61.0 ± 19.6 | 60.6 ± 20.0 | ||||
| ACR‐N | NA | NA | NA | NA | ||||
| Disease activity at week 24 | ||||||||
| Swollen joint count | 3.2 ± 4.5 | 3.3 ± 4.9 | 3.3 ± 5.0 | 3.3 ± 4.7 | ||||
| Tender joint count | 7.4 ± 7.4 | 8.4 ± 9.6 | 8.1 ± 9.1 | 8.4 ± 10.0 | ||||
| HAQ DI score | 0.8 ± 0.6 | 0.9 ± 0.6 | 0.9 ± 0.6 | 0.9 ± 0.6 | ||||
| DAS28‐ESR | 3.7 ± 1.2 | 3.8 ± 1.4 | 3.7 ± 1.4 | 3.8 ± 1.3 | ||||
| CRP, mg/liter | 5.6 ± 10.3 | 5.8 ± 8.6 | 6.1 ± 9.9 | 5.3 ± 6.7 | ||||
| ESR, mm/hour | 18.9 ± 14.8 | 19.0 ± 14.2 | 18.6 ± 12.8 | 19.6 ± 15.5 | ||||
| Physician's global assessment, 0–100‐mm VAS | 23.6 ± 16.4 | 25.0 ± 17.8 | 25.0 ± 18.0 | 24.0 ± 17.1 | ||||
| Patient's global assessment, 0–100‐mm VAS | 33.7 ± 21.7 | 35.1 ± 22.0 | 36.7 ± 22.4 | 33.1 ± 21.4 | ||||
| Patient's assessment of pain, 0–100‐mm VAS | 35.6 ± 21.8 | 37.0 ± 23.3 | 38.6 ± 23.4 | 34.7 ± 22.9 | ||||
| ACR‐N | 40.2 ± 28.7 | 39.6 ± 29.2 | 38.4 ± 29.6 | 41.5 ± 28.8 | ||||
Except where indicated otherwise, values are the mean ± SD. SB5 group = patients randomized to receive the biosimilar SB5 at week 0; adalimumab (ADA) overall group = patients randomized to receive reference ADA at week 0; ADA/SB5 group = patients receiving ADA rerandomized to switch to SB5; ADA/ADA group = patients receiving ADA rerandomized to continue ADA; BMI = body mass index; MTX = methotrexate; RF = rheumatoid factor; HAQ DI = Health Assessment Questionnaire disability index; DAS28‐ESR = Disease Activity Score in 28 joints using the erythrocyte sedimentation rate; CRP = C‐reactive protein; VAS = visual analog scale; ACR‐N = American College of Rheumatology index of improvement in rheumatoid arthritis; NA = not applicable.
Efficacy assessments. The ACR response rates were not affected after transition from ADA to SB5 at week 24, and results for the ADA/SB5 and ADA/ADA groups were comparable up to week 52 (Figure 2). Efficacy was also comparable between the SB5 and ADA/ADA groups throughout the study. Efficacy was maintained from week 24 up to week 52 across treatment groups. Analysis of the DAS28, SDAI, and CDAI showed comparable trends across all treatment groups during the study; results from 24 weeks to 52 weeks (full analysis set) are shown in Supplementary Figure 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40444/abstract. Other efficacy end points (e.g., Boolean‐based remission; low disease activity based on the DAS28, SDAI, and CDAI) were also comparable at week 52 among treatment groups (Table 2).
Figure 2.
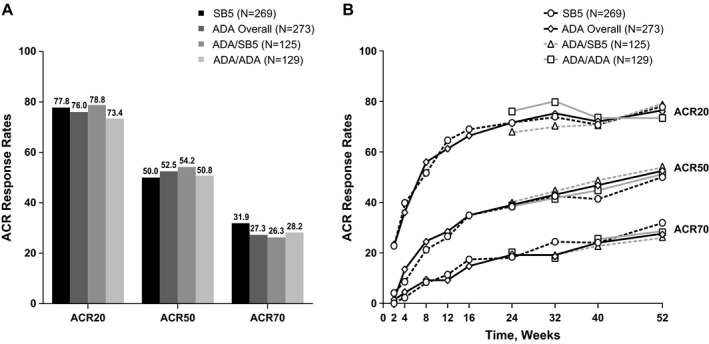
Percentages of patients meeting the American College of Rheumatology 20%, 50%, or 70% improvement criteria (achieving an ACR20, ACR50, or ACR70 response) in the full analysis set population. A, ACR response rates at 52 weeks in the different treatment groups. B, ACR response rates over the 52‐week period in the different treatment groups. See Figure 1 for other definitions.
Table 2.
Efficacy end points at week 52 in the full analysis seta
| SB5 group (n = 269) | ADA overall group (n = 273) | ADA/SB5 group (n = 125) | ADA/ADA group (n = 129) | |
|---|---|---|---|---|
| ACR response rate, % | ||||
| ACR20 | 77.8 | 76.0 | 78.8 | 73.4 |
| ACR50 | 50.0 | 52.5 | 54.2 | 50.8 |
| ACR70 | 31.9 | 27.3 | 26.3 | 28.2 |
| DAS28‐ESR | ||||
| Mean change from baseline | −3.05 | −2.97 | −3.02 | −2.92 |
| LDA, no./total no. (%)b | 118/247 (47.8) | 112/242 (46.3) | 55/118 (46.6) | 57/124 (46.0) |
| Remission, no./total no. (%)b | 75/247 (30.4) | 70/242 (28.9) | 34/118 (28.8) | 36/124 (29.0) |
| SDAI | ||||
| Mean change from baseline | −29.0 | −28.0 | −28.2 | −27.8 |
| LDA, no./total no. (%)b | 88/247 (35.6) | 95/242 (39.3) | 46/118 (39.0) | 49/124 (39.5) |
| Remission, no./total no. (%)b | 55/247 (22.3) | 46/242 (19.0) | 23/118 (19.5) | 23/124 (18.5) |
| CDAI | ||||
| Mean change from baseline | −28.5 | −27.4 | −27.50 | −27.3 |
| LDA, no./total no. (%)b | 84/248 (33.9) | 91/242 (37.6) | 45/118 (38.1) | 46/124 (37.1) |
| Remission, no./total no. (%)b | 52/248 (21.0) | 47/242 (19.4) | 23/118 (19.5) | 24/124 (19.4) |
| Boolean‐based remission, no./total no. (%)b, c | 35/247 (14.2) | 31/242 (12.8) | 17/118 (14.4) | 14/124 (11.3) |
| Radiographic results | ||||
| Change from baseline in joint erosion score, mean ± SD | 0.1 ± 1.6 | 0.2 ± 1.3 | 0.2 ± 1.4 | 0.2 ± 1.2 |
| Change from baseline in joint space narrowing score, mean ± SD | 0.1 ± 1.3 | 0.2 ± 1.8 | 0.1 ± 1.5 | 0.3 ± 1.9 |
| Change from baseline in SHS, mean ± SD | 0.2 ± 2.5 | 0.4 ± 2.6 | 0.3 ± 2.7 | 0.5 ± 2.4 |
| Proportion of patients with change from baseline in SHS >0, no./total no. (%)b | 48/241 (19.9) | 57/238 (23.9) | 22/114 (19.3) | 35/124 (28.2) |
ACR20 = American College of Rheumatology 20% improvement criteria; LDA = low disease activity; SDAI = Simplified Disease Activity Index; CDAI = Clinical Disease Activity Index; SHS = modified Sharp/van der Heijde score (see Table 1 for other definitions).
Number of patients with available data at each time point.
Determined based on 28–swollen joint count ≤1, 28–tender joint count ≤1, CRP level ≤10 mg/liter, and VAS score ≤10 mm on patient's global assessment.
Radiographic results were comparable across all treatment groups, as seen in the change from baseline at week 52 in joint erosion score, joint space narrowing score, SHS, and the proportion of patients with change from baseline in the SHS >0 (Table 2). Mean ± SD changes in SHS were 0.2 ± 2.5 in the SB5 group, 0.4 ± 2.6 in the ADA overall group, 0.3 ± 2.7 in the ADA/SB5 group, and 0.5 ± 2.4 in the ADA/ADA group. Radiographic progression was minimal, as seen in the cumulative probability plot of change from baseline in SHS at week 52, and was comparable for the different treatment groups (Figure 3).
Figure 3.
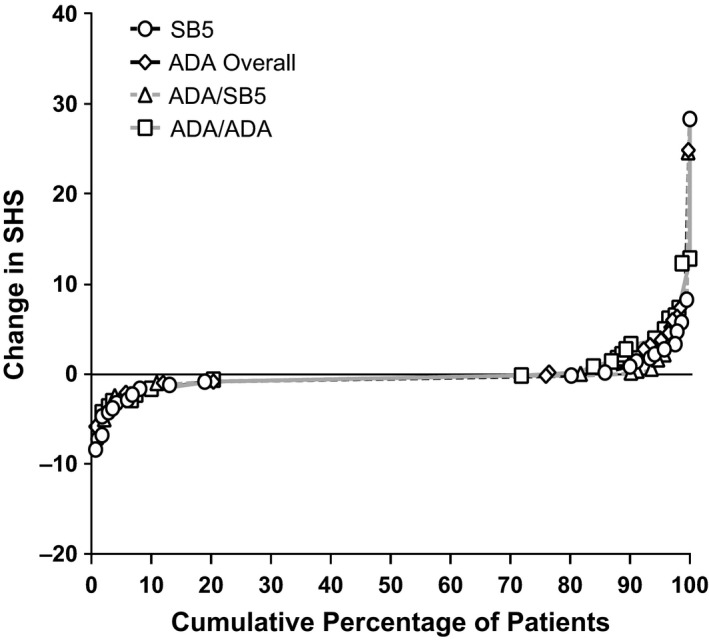
Cumulative probability change from baseline in modified Sharp/van der Heijde score (SHS) at week 52. Data are based on patients for whom radiographic assessment results were available at each visit. See Figure 1 for other definitions.
Subgroup analyses of ACR response rate, EULAR response, and proportion of patients with low disease activity and remission (according to the DAS28, SDAI, and CDAI) showed a trend toward decreased efficacy in antidrug antibody–positive patients (n = 84) compared with antidrug antibody–negative patients (n = 423) in all treatment groups (see Supplementary Table 1, http://onlinelibrary.wiley.com/doi/10.1002/art.40444/abstract). Mean DAS28, SDAI, and CDAI values tended to improve in antidrug antibody–negative patients and tended to worsen in antidrug antibody–positive patients across all treatment groups (24–52 weeks); the magnitude of the changes was comparable in all treatment groups (see Supplementary Figure 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40444/abstract).
Safety. Safety was comparable across treatment groups after week 24. Treatment‐emergent AEs that occurred after week 24 in the SB5/SB5, ADA/SB5, and ADA/ADA groups are summarized in Table 3. There were no cases of active tuberculosis in any treatment group and no deaths during the transition period. Malignancies were reported in 1 patient each in the SB5/SB5 group (small cell lung cancer), ADA/SB5 group (glioblastoma multiforme), and ADA/ADA group (seminoma). Injection‐site reactions were reported in 2 patients in the ADA/ADA group only. The proportion of patients with any treatment‐emergent AEs was comparable among the SB5/SB5 (32.3%), ADA/SB5 (37.6%), and ADA/ADA (33.1%) groups. Similar results were seen in the antidrug antibody–positive and antidrug antibody–negative subgroups within each treatment group (data not shown).
Table 3.
Summary of safety profile after transitiona
| SB5/SB5 group (n = 254) | ADA/SB5 group (n = 125) | ADA/ADA group (n = 127) | |
|---|---|---|---|
| Patients with ≥1 treatment‐emergent AE | 82 (32.3) | 47 (37.6) | 42 (33.1) |
| Treatment‐emergent AEs reported in ≥2% of patients | |||
| Nasopharyngitis | 11 (4.3) | 4 (3.2) | 3 (2.4) |
| Latent tuberculosis | 8 (3.1) | 1 (0.8) | 7 (5.5) |
| Spinal pain | 5 (2.0) | 3 (2.4) | 2 (1.6) |
| Bronchitis | 5 (2.0) | 2 (1.6) | 2 (1.6) |
| Urinary tract infection | 5 (2.0) | 3 (2.4) | 0 (0.0) |
| Rheumatoid arthritis | 4 (1.6) | 3 (2.4) | 4 (3.1) |
| Upper respiratory tract infection | 4 (1.6) | 5 (4.0) | 0 (0.0) |
| Increased ALT levelb | 3 (1.2) | 1 (0.8) | 3 (2.4) |
| Headache | 2 (0.8) | 3 (2.4) | 4 (3.1) |
| Positive Mycobacterium tuberculosis complex test resultc | 2 (0.8) | 4 (3.2) | 1 (0.8) |
| Any serious treatment‐emergent AE | 6 (2.4) | 4 (3.2) | 4 (3.1) |
| Treatment‐emergent AEs leading to study drug discontinuation | 1 (0.4) | 2 (1.6) | 3 (2.4) |
| Serious infection | 0 (0.0) | 2 (1.6) | 0 (0.0) |
| Active tuberculosis | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection‐site reactiond | 0 (0.0) | 0 (0.0) | 2 (1.6) |
| Malignancye | 1 (0.4) | 1 (0.8) | 1 (0.8) |
| Death | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Values are the number (%) of patients. SB5/SB5 group = patients in the SB5 group rerandomized to continue the biosimilar SB5; AE = adverse event; ALT = alanine aminotransferase (see Table 1 for other definitions).
Based on investigator's discretion.
Treatment‐emergent AE led to study drug discontinuation in 1 patient in the ADA/SB5 group and 1 patient in the ADA/ADA group.
Numbers based on the high‐level group term of administration site reaction.
Small cell lung cancer in SB5/SB5 group; glioblastoma multiforme in ADA/SB5 group; seminoma in ADA/ADA group.
Immunogenicity. The incidence of overall antidrug antibodies (emergent and boosted) was comparable across treatment groups after transition at week 24 (40 of 254 patients [15.7%] in the SB5/SB5 group, 21 of 125 patients [16.8%] in the ADA/SB5 group, and 23 of 126 patients [18.3%] in the ADA/ADA group). Emergent antidrug antibodies occurred in 9 of 160 patients (5.6%) in the SB5/SB5 group, 5 of 80 patients (6.3%) in the ADA/SB5 group, and 11 of 87 patients (12.6%) in the ADA/ADA group. Boosted responses were reported in 31 of 94 patients (33.0%) in the SB5/SB5 group, 16 of 45 patients (35.6%) in the ADA/SB5 group, and 12 of 39 patients (30.8%) in the ADA/ADA group.
Discussion
Previous results from this phase III randomized study showed equivalent efficacy for SB5 and ADA at week 24, and SB5 was well tolerated with PK, safety, and immunogenicity profiles comparable to those of ADA 10. In this 52‐week transition study, patients in the ADA group were rerandomized at week 24 to either continue with ADA or switch to SB5 up to week 52, and patients receiving SB5 continued with SB5 up to week 52. This study design allowed for the comparison of efficacy and safety of SB5 in patients who switched from ADA to SB5 with efficacy and safety in those who continued to receive SB5 or ADA for the entire 52‐week period.
The ACR response rates were similar across all treatment groups, both at week 52 and over the course of the study. Importantly, comparison of the ADA/SB5 and ADA/ADA treatment groups showed that efficacy was maintained after transition from ADA to SB5. In addition, there was minimal radiographic progression in all treatment groups over the course of 52 weeks. The ACR20, ACR50, and ACR70 response rates achieved in this study were similar to those described previously for ADA 18, 19, 20, 21, and the radiographic results were also consistent with historical data for ADA 19.
Analysis of treatment‐emergent AEs after transition showed that SB5 was well tolerated after switching from ADA, with comparable safety profiles across treatment groups after transition. This transition study was designed to assess ~100 patients per group so that an increase in the frequency of injection‐site reactions to ≥1% could be detected. No injection‐site reactions occurred in the ADA/SB5 group, and 2 patients in the ADA/ADA group experienced injection‐site reactions. Rates of SAEs and treatment‐emergent AEs leading to study drug discontinuation were low and similar across treatment groups, ranging from 2.4% to 3.2% of patients and from 0.4% to 2.4% of patients, respectively. No potential cases of anaphylaxis were identified based on retrospective analysis using related AEs (e.g., pruritus, flushing, dyspnea, hypotonia, syncope, incontinence, vomiting) and blood pressure (systolic blood pressure <90 mm Hg or >30% decrease from baseline) as defined by the National Institute of Allergy and Infectious Diseases/Food Allergy and Anaphylaxis Network criteria 22. There were no cases of active tuberculosis or deaths. Thus, based on the safety results from the 24‐week study 10 and the safety results presented herein, SB5 is well tolerated up to 1 year of treatment with no significant safety issues.
The incidence of antidrug antibodies after transition was comparable across treatment groups, and the occurrence of newly emergent antidrug antibodies in patients who switched from ADA to SB5 (ADA/SB5 group) was not higher than that of patients in the ADA/ADA group. The formation of antibodies to ADA may reduce efficacy because of increased clearance 7, 21, 23. Therefore, similar to previous findings 21, 23, it was not surprising that ACR response rates were decreased in patients who were antidrug antibody positive compared with those who were antidrug antibody negative. Consistent with historical results, there was no apparent correlation between the development of antidrug antibodies and AEs, including injection‐site reactions 7, 24.
Several biosimilars of ADA and other bDMARDs have either been marketed or are in clinical development 11, 25. One of the key clinical considerations when prescribing biosimilars are the effects of switching patients from a reference product to a biosimilar 11. Importantly, biosimilars are expected to have the same clinical effects as the reference product, and efficacy and safety should not be compromised when switching from a reference product to a biosimilar 26. To test for potential changes in safety and efficacy following transition from reference biologic agents to biosimilars, some studies have incorporated a transition design 25. Several of these switch studies use an open‐label extension design in which all patients receive the biosimilar product following a double‐blind randomized trial 25. For example, the transition studies evaluating the infliximab biosimilar CT‐P13 for RA (Program Evaluating the Autoimmune Disease Investigational Drug cT‐p13 in RA Patients) or ankylosing spondylitis (Program Evaluating the Autoimmune Disease Investigational Drug cT‐p13 in AS Patients) and those for the etanercept biosimilar SB4 were open‐label, single‐arm extension studies 11, 27, 28, 29. The ADA biosimilar ABP 501 has a similar ongoing single‐arm, open‐label extension study to evaluate efficacy and safety during transition from ADA to the biosimilar 11, 30. In other studies, patients are rerandomized after a blinded treatment phase to transition to the biosimilar from the reference product, such as the study with the infliximab biosimilar SB2 and the switching study of CT‐P13 in Norway (the NOR‐SWITCH trial) 11, 31. Such switching studies will provide valuable data that will be useful in clinical decision‐making for transition from reference biologic agents to biosimilars.
One of the strengths of the transitional design of this 52‐week study is that it allows for direct comparison between maintenance groups (the SB5/SB5 and ADA/ADA groups) and the transition group (the ADA/SB5 group) and shows that switching does not have negative effects in terms of reduced efficacy or increased AEs or immunogenicity. A limitation of this transition study was that it was not designed for statistical comparisons of equivalence; however, the results from this study provide valuable data on switching from a reference product to a biosimilar.
In conclusion, SB5 was well tolerated over 1 year in patients with RA, with efficacy, safety, and immunogenicity comparable to those of ADA. Switching from ADA to SB5 had no treatment‐emergent issues such as increased AEs, increased immunogenicity, or loss of efficacy.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Weinblatt had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Weinblatt, Baek, Ghil.
Acquisition of data
Baranauskaite, Dokoupilova, Zielinska, Jaworski, Racewicz, Pileckyte, Jedrychowicz‐Rosiak.
Analysis and interpretation of data
Weinblatt, Baek, Ghil.
Role of the study sponsor
Samsung Bioepis funded the study and facilitated the study design, provided writing assistance for the manuscript, and reviewed and approved the manuscript prior to submission. The authors independently collected the data, interpreted the results, and had the final decision to submit the manuscript for publication. Samsung Bioepis also funded medical writing assistance provided by Beena John, PhD (C4 MedSolutions, Yardley, PA; a CHC Group company). Publication of this article was not contingent upon approval by Samsung Bioepis.
Supporting information
Acknowledgments
The authors thank the patients who were involved in the study, the study personnel who made this work possible, and the study investigators in the following countries: Bosnia and Herzegovina (S. Sokolovic, M. Mekic, N. Prodanovic, B. Gajic, E. Karaselimovic‐Dzambasovic, B. Pojskic); Bulgaria (A. Toncheva, P. Dimitar, L. Rodina, M. Geneva‐Popova, I. Staykov, R. Stoilov); Czech Republic (L. Podrazilova, Z. Mosterova, G. Simkova, J. Kopackova, Z. Stejfova, J. Vencovsky, Z. Urbanova, L. Janska, D. Galatíkova); Lithuania (S. Stropuviene, I. Sniuoliene); Poland (J. Niebrzydowski, K. Sitek‐Ziolkowska, M. Rell‐Bakalarska, R. Kolasa, S. Daniluk, B. Sliwowska, M. Bartosik‐Twardowska, J. Brzezicki, M. Konieczny, S. Jeka); Republic of Korea (J. Choe, S. Bae, Y. Kang); Ukraine (L. Prystupa, Z. Vyacheslav, I. Gasanov, R. Yatsyshyn, D. Rekalov, O. Iaremenko, M. Stanislavchuk, V. Tseluyko). Medical writing assistance was provided by Beena John, PhD, from C4 MedSolutions (Yardley, PA), a CHC Group company, and was funded by Samsung Bioepis.
ClinicalTrials.gov identifier: NCT02167139.
Supported by Samsung Bioepis.
Dr. Weinblatt has received consulting fees and/or honoraria from AbbVie, Amgen, Novartis, Roche, GlaxoSmithKline, Merck, Samsung Bioepis, Crescendo Bioscience, and AstraZeneca (less than $10,000 each) and from Bristol‐Myers Squibb, Eli Lilly and Company, Pfizer, and UCB (more than $10,000 each) and research support from Amgen, Bristol‐Myers Squibb, Crescendo Bioscience, Sanofi, and UCB. Dr. Baranauskaite has received consulting fees (less than $10,000) and research support from Samsung Bioepis. Drs. Dokoupilova, Zielinska, Jaworski, Racewicz, Pileckyte, and Jedrychowicz‐Rosiak have received research support from Samsung Bioepis. Drs. Baek and Ghil own stock or stock options in Samsung Bioepis.
References
- 1. McInnes IB, O'Dell JR. State‐of‐the‐art: rheumatoid arthritis. Ann Rheum Dis 2010;69:1898–906. [DOI] [PubMed] [Google Scholar]
- 2. Smolen JS, Landewe R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Ann Rheum Dis 2017;76:960–77. [DOI] [PubMed] [Google Scholar]
- 3. Singh JA, Saag KG, Bridges SL Jr, Akl EA, Bannuru RR, Sullivan MC, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 2016;68:1–25. [DOI] [PubMed] [Google Scholar]
- 4. Dorner T, Strand V, Castaneda‐Hernandez G, Ferraccioli G, Isaacs JD, Kvien TK, et al. The role of biosimilars in the treatment of rheumatic diseases. Ann Rheum Dis 2013;72:322–8. [DOI] [PubMed] [Google Scholar]
- 5. Graudal N, Hubeck‐Graudal T, Faurschou M, Baslund B, Jürgens G. Combination therapy with and without tumor necrosis factor inhibitors in rheumatoid arthritis: a meta‐analysis of randomized trials. Arthritis Care Res (Hoboken) 2015;67:1487–95. [DOI] [PubMed] [Google Scholar]
- 6. Hirsch BR, Lyman GH. Biosimilars: a cure to the U.S. health care cost conundrum? Blood Rev 2014;28:263–8. [DOI] [PubMed] [Google Scholar]
- 7. Humira (adalimumab) medication guide. North Chicago: AbbVie; 2016. [Google Scholar]
- 8. Shin D, Lee Y, Kim H, Kornicke T, Fuhr R. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther 2017;42:672–8. [DOI] [PubMed] [Google Scholar]
- 9. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 10. Weinblatt ME, Baranauskaite A, Niebrzydowski J, Dokoupilova E, Zielinska A, Jaworski J, et al. Phase III randomized study of SB5, an adalimumab biosimilar, versus reference adalimumab in patients with moderate‐to‐severe rheumatoid arthritis. Arthritis Rheumatol 2018;70:40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moots R, Azevedo V, Coindreau JL, Dorner T, Mahgoub E, Mysler E, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep 2017;19:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schiff M, Weaver A, Keystone E, Moreland L, Spencer‐Green G. Comparison of ACR response, numeric ACR, and ACR AUC as measures of clinical improvement in RA clinical trials [abstract]. Arthritis Rheum 1999;42 Suppl:S81. [Google Scholar]
- 13. Prevoo ML, van ‘t Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty‐eight–joint counts: development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum 1995;38:44–8. [DOI] [PubMed] [Google Scholar]
- 14. Van Gestel AM, Prevoo ML, van ‘t Hof MA, van Rijswijk MH, van de Putte LB, van Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis: comparison with the preliminary American College of Rheumatology and the World Health Organization/International League Against Rheumatism criteria. Arthritis Rheum 1996;39:34–40. [DOI] [PubMed] [Google Scholar]
- 15. Smolen JS, Breedveld FC, Schiff MH, Kalden JR, Emery P, Eberl G, et al. A Simplified Disease Activity Index for rheumatoid arthritis for use in clinical practice. Rheumatology (Oxford) 2003;42:244–57. [DOI] [PubMed] [Google Scholar]
- 16. Aletaha D, Nell VP, Stamm T, Uffmann M, Pflugbeil S, Machold K, et al. Acute phase reactants add little to composite disease activity indices for rheumatoid arthritis: validation of a clinical activity score. Arthritis Res Ther 2005;7:R796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van der Heijde DM, van Riel PL, Gribnau FW, Nuver‐Zwart IH, van de Putte LB. Effects of hydroxychloroquine and sulphasalazine on progression of joint damage in rheumatoid arthritis. Lancet 1989;1:1036–8. [DOI] [PubMed] [Google Scholar]
- 18. Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double‐blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26–37. [DOI] [PubMed] [Google Scholar]
- 19. Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti–tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo‐controlled, 52‐week trial. Arthritis Rheum 2004;50:1400–11. [DOI] [PubMed] [Google Scholar]
- 20. Weinblatt ME, Keystone EC, Furst DE, Kavanaugh AF, Chartash EK, Segurado OG. Long term efficacy and safety of adalimumab plus methotrexate in patients with rheumatoid arthritis: ARMADA 4 year extended study. Ann Rheum Dis 2006;65:753–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti–tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003;48:35–45. [DOI] [PubMed] [Google Scholar]
- 22. Sampson HA, Munoz‐Furlong A, Campbell RL, Adkinson NF Jr, Bock SA, Branum A, et al. Second symposium on the definition and management of anaphylaxis: summary report–second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med 2006;47:373–80. [DOI] [PubMed] [Google Scholar]
- 23. Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long‐term follow‐up. JAMA 2011;305:1460–8. [DOI] [PubMed] [Google Scholar]
- 24. Van de Putte LB, Atkins C, Malaise M, Sany J, Russell AS, van Riel PL, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis 2004;63:508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open 2016;2:e000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. 2015. URL: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf.
- 27. Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT‐P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT‐P13 and continuing CT‐P13 in the PLANETRA extension study. Ann Rheum Dis 2017;76:355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez‐Urena S, et al. Efficacy and safety of switching from reference infliximab to CT‐P13 compared with maintenance of CT‐P13 in ankylosing spondylitis: 102‐week data from the PLANETAS extension study. Ann Rheum Dis 2017;76:346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emery P, Vencovsky J, Sylwestrzak A, Leszczynski P, Porawska W, Stasiuk B, et al. Long‐term efficacy and safety in patients with rheumatoid arthritis continuing on SB4 or switching from reference etanercept to SB4. Ann Rheum Dis 2017. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen S, Genovese MC, Choy E, Perez‐Ruiz F, Matsumoto A, Pavelka K, et al. Efficacy and safety of the biosimilar ABP 501 compared with adalimumab in patients with moderate to severe rheumatoid arthritis: a randomised, double‐blind, phase III equivalence study. Ann Rheum Dis 2017;76:1679–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jorgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT‐P13 compared with maintained treatment with originator infliximab (NOR‐SWITCH): a 52‐week, randomised, double‐blind, non‐inferiority trial. Lancet 2017;389:2304–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials