

Abstract
Malignant atrophic papulosis (MAP) is a rare type of obliterating vasculopathy that can present as pure cutaneous lesions or a systemic entity affecting multiple organs. Systemic disease, such as gastrointestinal or central nervous system involvement, may predispose the patients to poorer or even fatal outcomes. We present a 30‐year‐old female patient with systemic manifestation of MAP 10 days after delivery of a full‐term pregnancy who subsequently developed motor aphasia and intestinal perforation. The patient was administrated empirical treatment with an antiplatelet, anticoagulant, methylprednisolone sodium succinate and alprostadil. Antibiotics were administrated due to intestinal perforation and secondary sepsis. Despite all treatment, the patient died a week later. We summarized all the previous reports of MAP based on thorough review of previous published work. Overall, this is the first patient with MAP combined with motor aphasia and intestinal perforation and may provide insights for future studies on the treatment of this disease.
Keywords: case report, intestinal perforation, malignant atrophic papulosis, motor aphasia, review of published work
Introduction
Malignant atrophic papulosis (MAP), also known as Köhlmeier–Degos disease, was first described by Köhlmeier in 1941.1 MAP is a rare entity characterized by unique cutaneous lesions alone or combined with multiple lesions of other organs, most commonly the central nervous system (CNS) and gastrointestinal tract.2 The disease is frequently fatal because no specific treatment has been proven to be effective against it. The most frequent causes of death are cerebral infarction, intestinal perforation and secondary peritonitis.3 Hereby, we present a 30‐year‐old female patient with MAP presenting with typical skin lesions combined with gastrointestinal and CNS involvement, who subsequently developed gastrointestinal perforation and neurological motor aphasia.
Case Report
A 30‐year‐old woman of Han race was admitted to the Chinese People's Liberation Army General Hospital on 12 February 2017 due to recurrent acute abdominal pain and speech disorder after giving birth to a full‐term healthy female infant at her local hospital on 31 January without any obvious discomfort. Two days later, the patient started to have abdominal pain with nausea and vomiting, and got worse during treatment at the local hospital. The patient started to have neurological disorders from 5 February, including right side limb movement disorder and motor aphasia. She had widespread skin lesions with chronic atrophic and scarring papules over the trunk and extremities, sparing the palms, face and genital areas. Examination of the skin revealed white to pink lesions of 3–10 mm in diameter, which had evolved into painless scars with a central porcelain‐white atrophic center surrounded by an erythematous telangiectatic peripheral halo (Fig. 1).
Figure 1.
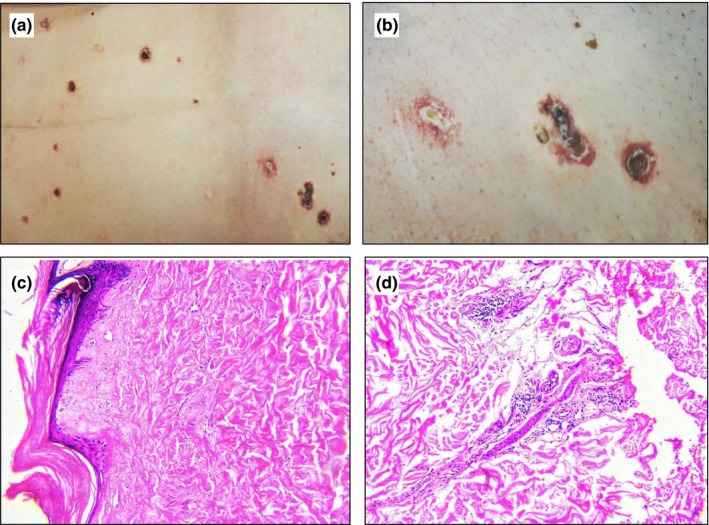
(a,b) Characteristic lesions of Degos disease with central porcelain‐white atrophy surrounding a rim of erythema on the trunk. (c,d) Histopathological sections showing absence of crests, atrophic epidermis, epidermal overlying orthokeratosis and a lymphocytic hyalinization of collagen infiltrate in the papillary dermis (hematoxylin–eosin, original magnification ×100).
This patient had no family history of the disease. Three years prior, she had noted some scattered erythematous papules on the thorax. No treatment was administrated then because she was found to be pregnant. The patient reported no clinical symptoms afterwards until the child was born.
After admission, various tests were performed. Cranial magnetic resonance imaging revealed tiny lesions in the left cerebral hemisphere suggesting ischemic small vessel disease, whereas the main branches of the cerebral arteries were normal (Fig. 2). Full blood cell count revealed increased white blood cell count (12.74 × 109/L) initially, which increased progressively to 24.61 × 109/L, with increased neutrophil proportion (95.4%) and reduced lymphocytes (0.033 × 109/L). It is worth noting that platelet count increased from 348 × 109/L to 577 × 109/L within 5 days. Other laboratory tests showed increased alkaline phosphatase (201.1 U/L), γ‐glutamyltransferase (149.2 U/L) and lactate dehydrogenase (256 U/L) levels, and decreased albumin (26.3 g/L) and serum calcium (1.83 mmol/L) levels. Elevated levels of C‐reactive protein (265 mg/L) and interleukin‐6 (1037 pg/L) and normal procalcitonin (0.15 ng/mL) was found during her hospitalization process. Immunoglobulin and complement profile levels were within normal ranges while coagulation test revealed prolonged plasma prothrombin time (15.5 s) and abnormal fibrinogen (7.08 g/L) and D‐dimer levels (10.35 μg/mL). There was no significant increase in rheumatoid factor and other rheumatoid immune markers and tumor markers.
Figure 2.
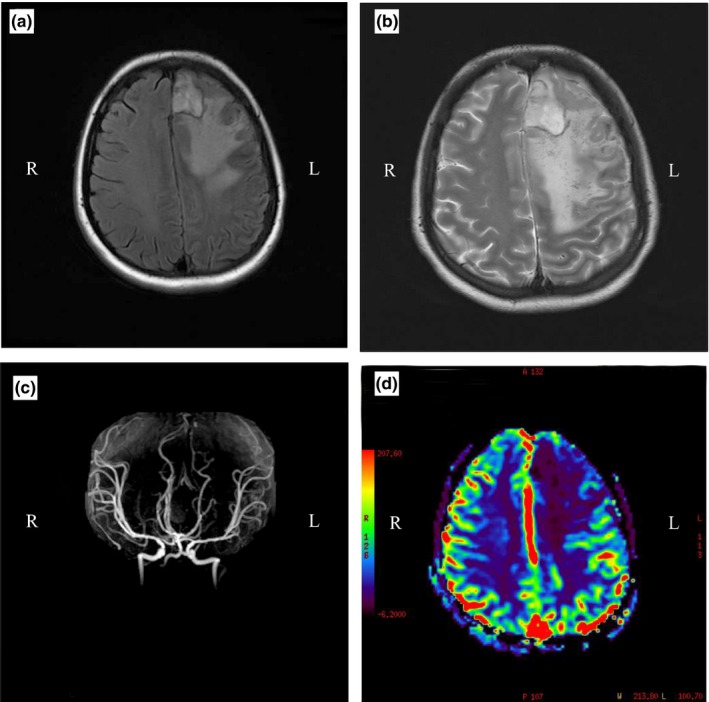
(a,b) Left side of the parietal lobes can be seen large patchy and patchy uneven long T1 and T2 signals, suggesting a large area of cerebral infarction with bleeding. (c) Magnetic resonance angiography shows that the main branches of cerebral arteries are normal. (d) Magnetic resonance perfusion shows a significant reduction in infarct perfusion.
Skin tissue biopsy suggested characteristics of malignant atrophic papulosis. The epidermis of overlying revealed hyperkeratosis, focal atrophy and absent acanthosis. Localized vacuolar degeneration was seen at the junction of the epidermis and the dermis, as well as significantly reduced vascellum and dermal appendages. Homogeneously hyperplastic collagen was found in the shallow dermal layer where a banded necrotic area was visible (Fig. 1).
Standard therapy for Degos disease has not been well established yet. The patient was administrated antiplatelet (100 mg q.d. bayaspirin) and anticoagulant therapy (2500 IU q.d. s.c. dalteparin sodium). Because Degos disease is pathologically a vasculitis, i.v. methylprednisolone sodium succinate (40 mg b.i.d. i.v.), alprostadil (10 μg q.d. i.v.) and i.v. immunoglobulin (400 mg/kg per day) were administrated. Antibiotics (1 g b.i.d. i.v. imipenem and cilastatin sodium, 0.4 g b.i.d. i.v. teicoplanin) were also administrated due to existing signs of infection. Unfortunately, despite the above‐mentioned treatment and supportive care, the patient continued to deteriorate and eventually died of intestinal necrosis and perforation.
Discussion
Malignant atrophic papulosis, also referred to as Degos disease, is a rare vasculopathy of unknown cause that mainly affects the skin, gastrointestinal tract, CNS and occasionally other organs. Less than 200 cases have been reported so far in the previous published work. The disease is usually categorized into two types, benign and malignant, according to the distinct prognoses between patients with pure cutaneous disease and MAP with systemic involvement.4 In the malignant type, prognosis is usually poorer and mortality is reported shortly after or within a few years of the onset of skin manifestations. The benign form is exclusively cutaneous and has a favorable outcome, although it may persist over years and can be lifelong.5
We searched works published between January 2000 and July 2017 using the key words “malignant atrophic papulosis” and “Degos disease” in the PubMed database. The search yielded 80 cases which were further analyzed for diagnostic procedures and disease outcomes (Table S1). The male : female ratio was 1:1.2 (37 males and 43 females). The average age at disease onset was 38.5 ± 19.0 years (95% confidence interval [CI], 32.1–44.8) in male patients and 37.9 ± 18.7 years (95% CI, 32.1–43.6; P > 0.05) in female patients. The overall mean age of onset was 38.1 ± 18.7 years (95% CI, 34.0–42.3). Fifty‐two of the 80 (65%) patients were identified to have the malignant systemic variant, whereas 28 of the 80 patients (35%) presented with only cutaneous signs throughout the follow‐up period. Among the 80 patients, 34 had a fatal outcome during follow up, four lost to follow up whose outcome was not available and 14 were stable throughout the follow up. The overall mortality was 42.5% (18 males [48.6%] and 16 females [37.2%], P > 0.05), and all dead patients had systemic involvement. The mortality in all patients with the systemic type was 65.3% (34/52), whereas no patient death was observed among the 28 patients with pure cutaneous involvement (P < 0.05). In patients with extracutaneous involvement (n = 43, excluding six patients whose information was not available and three patients with cutaneous involvement after systemic manifestations).6, 7, 8 Moreover, we found that in patients in the systemic category, the first systemic manifestation occurred at a median of 2.0 years (95% CI, 0–7.8) after disease onset. The gastrointestinal tract and CNS were involved in 43 of 52 (82.7%) and seven of 22 patients (42.3%), respectively, of whom 30 (69.8%) and 12 patients (54.5%) died during follow up. Our findings are consistent with previous published reports demonstrating the most frequent causes of death to be intestinal perforation, peritonitis and cerebral infarction.3 Several patients had involvement of other internal organs, such as the heart, eyes, lungs, bladder, pancreas, kidneys, spleen and vocal cords (Table 1).
Table 1.
Demographic data of patients with malignant atrophic papulosis from the current and previous studies
| Current review | Review by Burg et al.10 | |
|---|---|---|
| No. of patients | 80 | 106 |
| Male/female (ratio) | 37/43 (1:1.2) | 63/43 (1.47:1) |
| Age at diagnosis (years), median (min–max) | 37.9 ± 18.7 | 35 |
| Patients with systemic manifestations, n (%) | 52/80 (65) | 67/106 (63.2) |
| Multi‐organ involvement (≥3 organs), n (%) | 25/52 (48) | 33/67 (49) |
| Organs involved, n (%); fatal outcome, n (%) | ||
| GI tract | 43/52 (82.7), 30/43 (69.8) | 51/67 (76) |
| CNS | 22/52 (42.3), 12/22 (54.5) | 20/67 (30) |
| Lung | 7/52 (13.5), 6/7 (85.7) | 16/67 (24) |
| Eye | 5/52 (9.6), 4/5 (80) | 14/67 (21) |
| Heart | 7/52 (13.5), 2/7 (28.6) | NR |
| Kidney | 2/52 (3.8), 1/2 (50) | NR |
| Bladder | 2/52 (3.8), 0/2 (0) | NR |
| Pancreas | 1/52 (1.9), 1/1 (100) | NR |
| Spleen | 1/52 (1.9), 1/1 (100) | NR |
| Patients with cutaneous lesions only, n (%) | 28/80 (35) | 39/106 (36.8) |
| ANA positive | 4/28 (14.3) | NR |
| Coexisted with Behçet's disease | 1/28 (3.6) | NR |
| Mortality, n (%) | ||
| Total | 34/80 (42.5) | 51/106 (48.1) |
| Male | 18/37 (48.6) | 36/63 (57) |
| Female | 16/43 (37.2) | 15/43 (35) |
| Patients with systemic disease | 34/52 (65.3) | 50/67 (75) |
| Patients with skin lesions only | 0/28 (0) | 1/39 (3) |
ANA, antinuclear antibodies; CNS, central nervous system; GI, gastrointestinal; NR, not reported.
Unlike typical neurological manifestations of Degos disease,9 significant mass effect, compression of the lateral ventricle and sulci, and midline shift, which often appear in brain tumors, were seen in the patient with MAP we reported (Fig. 2). Our patient was stable throughout the pregnancy but unfortunately the lesion she had turned out to be of the malignant systemic form when she had just had her baby. Thus, pregnancy is possibly a risk factor to accelerate the development of this disease.
Medical treatment of MAP, of either cutaneous or systemic form, is currently obscure. Antiplatelet agents, anticoagulants, fibrinolytics, immunosuppressants, anti‐inflammatory drugs, vasodilator agents, chloroquine, monoclonal antibodies and plasma exchange have been tried, but not all were proven to be effective (Fig. S1). Compared with the previous review by Burg et al.,10 the differences in mortality can be attributed to the use of monoclonal antibodies such as eculizumab (n = 4) and the use of nicotine (n = 1). If these five cases were included in the treatment failure group, the total mortality would be 48.75%, and the mortality in patients with systemic disease would be 75%, which is comparable with the results in the previous review by Burg et al. Thus, monoclonal antibodies and nicotine may be effective in treating MAP.
In conclusion, MAP is a rare disease with potential lethal complications. The cause of MAP has yet to be discovered. Although white atrophic papules characterized in MAP can present in many other diseases, systemic MAP is pathognomonic and was associated with poor outcomes. Effective treatment modalities are to be identified for this potentially lethal disease.
Conflict of Interest
None declared.
Supporting information
Figure S1. Treatment for malignant systemic disease.
{kind=link}
Table S1. Case statistics of Degos disease in the period 2000–2017
References
- 1. Köhlmeier W. Multiple Hautnekrosen bei Thrombangiitis obliterans. Arch Dermatol Syph 1941; 181: 783–792. [Google Scholar]
- 2. Scheinfeld N. Malignant atrophic papulosis. Clin Exp Dermatol 2007; 32: 483–487. [DOI] [PubMed] [Google Scholar]
- 3. Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Kohlmeier‐Degos disease) ‐ a review. Orphanet J Rare Dis 2013; 8: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heymann WR. Degos disease: considerations for reclassification. J Am Acad Dermatol 2009; 61: 505–506. [DOI] [PubMed] [Google Scholar]
- 5. Theodoridis A, Konstantinidou A, Makrantonaki E, Zouboulis CC. Malignant and benign forms of atrophic papulosis (Kohlmeier‐Degos disease): systemic involvement determines the prognosis. Br J Dermatol 2014; 170: 110–115. [DOI] [PubMed] [Google Scholar]
- 6. Magro CM, Wang X, Garrett‐Bakelman F, Laurence J, Shapiro LS, DeSancho MT. The effects of Eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis 2013; 8: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karaoglu P, Topcu Y, Bayram E et al Severe neurologic involvement of Degos disease in a pediatric patient. J Child Neurol 2014; 29: 550–554. [DOI] [PubMed] [Google Scholar]
- 8. Gmuca S, Boos MD, Treece A et al Degos disease mimicking primary vasculitis of the CNS. Neurol Neuroimmunol Neuroinflamm 2016; 3: e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Amato C, Ferri R, Elia M et al Nervous system involvement in Degos disease. AJNR Am J Neuroradiol 2005; 26: 646–649. [PMC free article] [PubMed] [Google Scholar]
- 10. Burg G, Vieluf D, Stolz W, Landthaler M, Braun‐Falco O. [Malignant atrophic papulosis (Kohlmeier‐Degos disease)]. Hautarzt 1989; 40: 480–485. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Treatment for malignant systemic disease.
Table S1. Case statistics of Degos disease in the period 2000–2017