

Abstract
The use of electricity instead of stoichiometric amounts of oxidizers or reducing agents in synthesis is very appealing for economic and ecological reasons, and represents a major driving force for research efforts in this area. To use electron transfer at the electrode for a successful transformation in organic synthesis, the intermediate radical (cation/anion) has to be stabilized. Its combination with other approaches in organic chemistry or concepts of contemporary synthesis allows the establishment of powerful synthetic methods. The aim in the 21st Century will be to use as little fossil carbon as possible and, for this reason, the use of renewable sources is becoming increasingly important. The direct conversion of renewables, which have previously mainly been incinerated, is of increasing interest. This Review surveys many of the recent seminal important developments which will determine the future of this dynamic emerging field.
Keywords: electrolysis, flow electrochemistry, organocatalysis, renewable resources, synthetic methods
1. Introduction
The application of electrochemical methods to the synthesis of organic molecules has undergone a revival during the last few decades.1 In terms of the ecological footprint,2, 3, 4 the substitution of chemical redox reagents by electricity is an inevitable step towards green chemical processes.5 A variety of valuable synthetic pathways for electrochemical synthesis has been described in the previous review “Electrifying Organic Synthesis”.171 Besides the continuous development of new electrochemical reactions and the synthesis of complex organic molecules, significant progress has been made in the realization of electrochemical methods. These innovations include the merger of electrochemistry with conventional chemical ideas, such as organocatalysis and flow electrochemistry, as well as new procedures for controlling the selectivity of electrochemical transformations such as the “cation‐pool” method, the redox‐tag approach, and bio‐electrochemistry. In addition, the electrochemical conversion of renewables provides a sustainable alternative for the synthesis of valuable fine chemicals from current waste streams. All these innovative methods will help in the development of selective electrochemical transformations for value‐added organic products and help in the scale‐up for technical applications. A variety of these recent developments will be described in this Review.
All electrochemical methods are based on simple electron transfers from the electrode to the substrate or vice versa. In electroorganic syntheses, roughly three different scenarios for the electron transfer from the electrode to the substrate are feasible. The classical way is to use an inert electrode. In this case, the electroconversion occurs at the electrode surface and selectivity can be achieved by adjusting the appropriate electrode potential (Figure 1 a).
Figure 1.

Different operation modes of electrodes in electrosynthetic applications.
Since many molecular moieties, such as alcohols or double bonds, cannot be selectivity addressed in complex molecules, an electrocatalytic approach is required. This can be achieved either by an active electrode or by using a mediator. An active electrode has electrocatalytically active species on the surface, which can be considered as immobilized redox‐active reagents (Figure 1 b). In the best case, a compact and electrically conductive coating is formed, which is electrochemically regenerated in situ.6 Such active electrodes usually provide a unique reactivity. In this particular case, the electroconversion becomes less dominated by the applied potential since the redox‐active layer serves as a redox filter. In addition, the immobilization of the electrocatalyst simplifies the experimental set‐up. Operation in undivided cells and at constant current is commonly the case. Since the redox‐active component stays on the surface because of its low solubility, such electrodes are not consumed and can even be easily operated in simple flow cells. Although this concept is well‐established for anodes, it is rarely applied for electroreductions. Typical examples are nickel‐based anodes for oxidation reactions in alkaline media or fluorinations (e.g. Scheme 20).
Scheme 20.
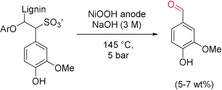
Electrochemical degradation of lignosulfonate at nickel anodes.89
The third option describes electroactive species that are soluble in the electrolyte (Figure 1 c). Here, the redox‐active species are mediators and can be considered as electrochemically regenerated reagents. Besides the unique reactivity, large over‐potentials can usually be avoided. Therefore, the electroconversion can be conducted at milder potentials compared to an electrolysis without a mediator.7 The disadvantage of this approach can be the generation of additional waste and costs.
In general, the application of novel organic synthesis concepts mostly relies on the use of inert electrodes, since the reactivity is not determined by a surface layer. Operation at constant current has several advantages: Firstly, the electronic periphery is of low cost, since simple power supplies (DC‐type) can be employed. Secondly, no reference electrode is required. Thirdly, the reaction scale‐up is more straightforward in a two‐electrode arrangement. Although many novel electrode materials have been developed and established in electroorganic synthesis, the workhorse is still the carbon electrode. Such carbon systems range from various types of graphite to glassy carbon. Besides the compact electrode material, porous versions are also employed. For more extreme electrochemical potentials, doped‐diamond seems to be the material of choice.8 Since electrodes do not remain intact forever and corrosion or fouling might occur, carbon electrodes will be the only sustainable solution without using up critical metals or resources.
2. Organocatalysis in Electrosynthesis
Organocatalysis gained significant attention in the late 1990s and evolved into a popular research field of organic chemistry,9 although there were some reports of using organic molecules as catalysts in the last century. The potential for avoiding chemical waste, saving costs, and facilitating experimental procedures awakened the interest of many research groups. The combination of the advantages of organocatalysis with the sustainability of electroorganic reactions yielded a collection of effective synthetic strategies. Ogawa and Boydston recently reviewed this unification in detail.10
Pyrrolidine derivatives are broadly used as catalysts in organocatalyzed reactions, in particular for asymmetric synthesis.11 Jang and co‐workers developed a method for the anodic organocatalyzed α‐oxyamination of aldehydes under constant current conditions by using pyrrolidine as a catalyst.12 Earlier reported chemical oxidants such as ceric ammonium nitrate (CAN) or Cp2FeBF4 were not required; thus, the electrochemical variation is an elegant version to conduct this reaction (Scheme 1).13
Scheme 1.
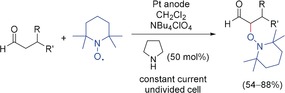
Electrochemical organocatalyzed α‐oxyamination of aldehydes.12
The reaction of different aldehydes with a variety of chiral secondary amines was tested for the applicability of this method in asymmetric synthesis. Only pyrrolidine‐based chiral catalysts led to a successful conversion with moderate enantiomeric excess, with l‐proline methyl ester hydrochloride providing the best results. Cyclic voltammetry and control experiments enabled the mechanism to be elucidated. The in situ formed enamine derivatives exhibited a lower oxidation potential than the corresponding aldehydes, and the radical cations generated were intercepted by the 2,2,6,6‐tetramethylpiperidinyloxyl (TEMPO) radical present as the coupling partner. The formation of TEMPO+ species by anodic oxidation could be excluded.
This approach was expanded to the electrochemical α‐functionalization of aldehydes.14 Different aliphatic aldehydes were coupled with xanthene (Scheme 2). The use of cycloheptatriene, instead of xanthene, as the coupling partner led to poor yields. Different organocatalysts were tested to improve the yield and stereoselectivity.
Scheme 2.

Electrochemical organocatalyzed α‐alkylation of aldehydes.14 Bn=benzyl.
Two plausible mechanisms were postulated: The first involved the formation of a xanthene cation and subsequent nucleophilic attack of the enamine. The other included the simultaneous oxidation of the enamine and xanthene, followed by a recombination of the radicals formed. The oxidation potentials of all the components were determined and control experiments were conducted. The addition of the radical inhibitor 2,6‐di‐tert‐butyl‐4‐methylphenol prevented the formation of product, thereby indicating the latter‐mentioned radical‐based mechanism was operative. The reported procedure was a more‐sustainable method for performing this reaction, as there was no need for photoredox catalysts or stoichiometric chemical oxidants, as used in previous studies.15
Another interesting contribution to this topic came from Jørgensen et al. Starting from aliphatic aldehydes and N‐tosylated 4‐aminophenols in the presence of a pyrrolidine‐based organocatalyst, electrochemical oxidation at a constant current led to meta‐substituted anilines in good yields and high enantiomeric excess (Scheme 3).16
Scheme 3.
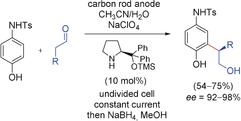
Electrochemical organocatalyzed synthesis of meta‐substituted anilines.16 TMS=trimethylsilyl, Ts=tosyl.
The electrochemical oxidation of the 4‐aminophenols at the anode induced an Umpolung of the aromatic system. The catalytically formed enamine underwent a Michael addition, and a subsequent condensation formed the dihydrobenzofuran. Reduction of the dihydrobenzofuran enabled access to meta‐substituted anilines.
Even before the wide interest in organocatalysis, Chiba et al. reported the first organocatalyzed anodic oxidation in 1982. The oxidation of aldehydes in methanol in the presence of sodium cyanide by using platinum electrodes and a constant current led to the corresponding methyl carboxylates, as shown in Scheme 4.17 Aromatic, non‐activated aldehydes were converted into methyl esters in good yields, whereas the oxidation of aliphatic substrates competed with side reactions such as aldol condensations.
Scheme 4.

Anodic oxidation of aldehydes in methanolic NaCN solution.17
Control experiments using different electrolytes highlighted the necessity of cyanide for the oxidation. The oxidation potential of benzaldehyde versus the saturated calomel electrode (SCE) in a lithium perchlorate/acetonitrile solution is about 2.3 V, whereas the oxidation in methanolic NaCN solution occurs at 1.7 V. Therefore, Chiba et al. assumed the electroactive intermediate was the formed cyanohydrin. The same mechanism was predicted in 1968 by Corey et al., who used an excess of manganese dioxide as the oxidant.18
N‐Heterocyclic carbenes (NHC) are also of significant interest for organocatalyzed reactions. Inspired by the oxidation of other electron‐rich alkenes, Boydston and co‐workers focused their studies on the oxidation of the Breslow intermediate formed by NHCs with aldehydes. The electrochemical oxidation of aldehydes in the presence of NHC and DBU led to esters.19 The low substrate concentrations necessitated the reaction being performed under potentiostatic control. The authors were able to convert a broad range of substrates, including aliphatic and aromatic aldehydes as well as sterically unhindered alcohols. The best results were obtained with a combination of graphite as the anode material and platinum as the cathode (Scheme 5, top). As previously mentioned, the effectiveness of the reaction was based on the lower oxidation potential of the intermediate species compared to the corresponding aldehyde. This principle is similar to the work of Chiba et al. with cyanohydrin intermediates. Besides esters, thioesters could also be successfully generated. It is noteworthy that the conversion did not work under the previously used conditions. The use of DBU as the base yielded about 58 % of the disulfide. Changing the base to DMAP and halving the base load to a 0.075 m concentration led to the ratio of thioester to disulfide being significantly improved. The method worked for aromatic and aliphatic aldehydes, and even bulky thiols could be used (Scheme 5, top).20 The predominant reason for these phenomena was the enhanced reactivity of thiols as nucleophiles.
Scheme 5.

NHC‐mediated electrochemical oxidation of aldehydes to esters, thioesters, or amides.19, 20, 22 DIPP=2,6‐diisopropylphenyl, DMAP=4‐(dimethylamino)pyridine, DMF=N,N‐dimethylformamide, DBU=1,8‐diazabicyclo[5.4.0]undec‐7‐ene, Mes=mesityl, PVDF=polyvinylidene difluoride, TBAB=tetrabutylammonium fluoride, Tf=triflyl.
Recently, Brown and co‐workers expanded the approach developed by Boydston and co‐workers by carrying out the electrochemical oxidation of the Breslow intermediate in a flow cell.21 Moreover, extension to amide formation was possible.22 Different aromatic aldehydes and aliphatic amines were tested. The method generally tolerated aromatic substitutions on the amidic alkyl chain. The application of a flow cell and the synthesis of amides underlined the versatility of the method and general applicability (Scheme 5, bottom).
As a consequence of the crucial role of NHCs such as thiamine pyrophosphate as enzymatic cofactors, their oxidation mechanism has been of interest to several research groups for many years.23 In 1990, Schlegel et al. used cyclic voltammetry and ESR spectroscopy to demonstrate a one‐electron oxidation via a thiazolium cation radical.24 The mechanistic studies were continued by Fukuzumi and co‐workers, who investigated the activated aldehydes of several substrates. For steric reasons, the radical intermediates resulting from single‐electron transfers were stable enough for detection by ESR spectroscopy.25 The first preparative electrochemical approach for the oxidation was reported by Diederich et al. in 1992. To investigate the biochemical process, they conducted the transformation with flavin as the mediator. They were able to produce methyl esters from aliphatic and aromatic aldehydes, whereby aromatic derivatives gave high yields and good faradaic efficiencies in methanol.26
The field of NHC‐catalyzed electrochemical transformations was expanded by Inesi and co‐workers, whose work was based on the electrochemical activation of an ionic liquid as an NHC catalyst. After electrolysis of the ionic liquid, the aldehyde was added. Catalyzed by the activated ionic liquid, the formation of the benzoin adduct of benzaldehyde was possible in good yields (Scheme 6, top).27 The substrate scope was later extended to enals, but instead of benzoin adducts, α,β‐saturated esters were formed in very good yields (Scheme 6, bottom).28 The major reason for this might be the electrolysis in the presence of the substrate. In this case, the mechanism proceeded analogously to the oxidation of the cyanohydrin or the Breslow intermediate. By implementation of an ionic liquid, Inesi and co‐workers added a further concept of sustainable chemistry.
Scheme 6.

In conclusion, the combination of organocatalysis and electrochemistry is highly valuable for the efficient oxidation of aldehydes to esters, amides, and thioesters; thus, this method could also be interesting for late‐stage functionalization of more complex molecules. Moreover, the large variety of examples for the carbon functionalization of enols proves that this method is broadly applicable and can be used in various fields of organic chemistry.
3. The “Cation‐Pool” Method
In 1999, Yoshida et al. presented the so‐called “cation‐pool” method.29 Since then, this method has evolved into a versatile and valuable tool. In electrochemistry and conventional methods, the combination of a cation with a nucleophile is a challenging task, due to the exothermic nature of this conversion. Nucleophiles often do not tolerate the conditions which are necessary for generation of the cation. In electrochemistry, a nucleophile can also be unstable under anodic conditions.
For this reason, the cation‐pool concept relies on the idea of separating the cation generation and the nucleophile addition spatially and in time (Scheme 7). Electrochemistry enables the mild and reagent‐free generation of cations without the necessity to remove any reagent waste afterwards. The key to this method is the enhanced lifetime and accumulation of the cations when the electrochemical oxidation is conducted at −78 °C. A nucleophile is subsequently added to afford the product.30
Scheme 7.
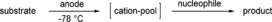
With this method, Yoshida and co‐workers were able to generate a broad scope of cations such as N‐acyliminium,29, 31 alkoxycarbenium,32 diarylcarbenium,33 glycosyl,34 silyl,35 iodine,36 alkoxysulfonium,37, 38 benzylaminosulfonium,39 arene,40 and thioarenium cations,41 as well as thionium cations.42 Depending on the cation and the added nucleophile, various products were accessible with a high selectivity.43 It is noteworthy that radical cations were sometimes formed. Then, at least two equivalents of cations were required for the generation of the final product.44 Another possibility was the reduction of a cation pool to the corresponding radical, thereby leading to dimerization.45 The generation of the cations was also possible in a mediated fashion if aryl disulfides served as the mediators.46 This indirect “cation‐pool” method overcame the problem of the relative low efficiency of the reaction with regards to cation generation. Moreover, these mediators could also serve as reaction partners, which underwent addition to the substrate, and the concept of flow chemistry could also be applied.47 Although the cation‐pool method is valuable, the stabilization of the cationic intermediates sometimes demands expensive electrolyte systems.39, 40
Scheme 8 summarizes the major reaction types and cations which are accessible with the cation‐pool method. Some features of the reaction will be described in more detail. In the last few years, Yoshida and co‐workers focussed on stabilization of the cation at elevated temperatures. The key for this was the incorporation of the mostly unstable cations in a stabilizing chemical structure. Alkoxysulfoniumions are important intermediates in chemical oxidations and represent a class of relatively stable cations. DMSO reacts with the initially formed oxidized species of, for example, halogens or diarylsulfides to generate alkoxysulfoniumions. Besides the accumulation of these ions and a subsequent quenching to generate ketones or alcohols, it could also be combined with halogenation (Scheme 9).37
Scheme 8.
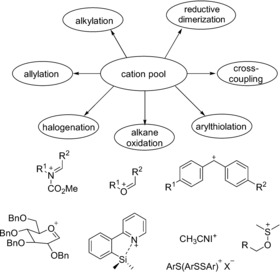
Potential reaction pathways and accumulated cations.
Scheme 9.
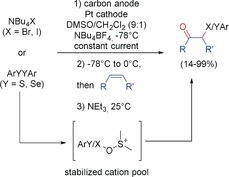
Stabilized “cation‐pool” method for an integrated reaction sequence.37 DMSO=dimethyl sulfoxide.
The stabilized “cation‐pool” method also allowed cross‐coupling between aromatic and benzylic C−H groups. However, the use of another cation‐stabilizing agent, such as diphenylsulfilimine, is necessary (Scheme 10).39 Amination reactions are also possible using this reagent.48
Scheme 10.
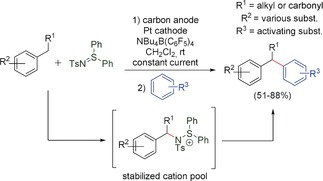
Stabilized “cation‐pool” for cross‐coupling between aromatic and benzylic groups.39
Recently, Yoshida and co‐workers accumulated a new type of cation by the indirect cation‐pool method. With aryldisulfides as mediators, thionium ions were generated and analyzed by NMR, UV/Vis, and IR spectroscopy. Several reaction pathways are possible, for example, cross‐coupling and homoallylation.42 Scheme 11 displays the conversion with a silyl ketenacetal as the nucleophile.
Scheme 11.
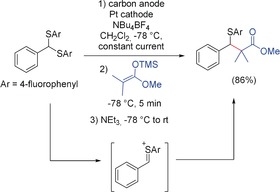
Accumulation and conversion of thionium ions.42
The “cation‐pool” method is of great synthetic value, since the conversion of relatively unstable intermediates is possible with “green” methods such as electrochemistry. Nevertheless, much preparative effort is necessary and scale‐up is difficult. The generation of low temperatures is energy‐consuming and the electrochemical set‐ups are rather sophisticated.
4. Bioelectrochemical Systems
The archetypal microbial bio‐electrochemical systems (BES) are microbial fuel cells (MFCs). In a microbially catalyzed process, organic or inorganic substances are oxidized and the produced electrons are transferred to the anode. Thus, the surplus of reducing equivalents in living systems is exploited. The chemical energy contained in wastewater streams is partly converted into electric energy by the catalytic (metabolic) activity of bacteria. If the cathodic process is the oxygen reduction reaction (ORR), the overall cell generates a surplus of energy. The cathodic process can be catalyzed microbially, enzymatically, or chemically (e.g. by the use of noble metals). However, the energy efficiency of such systems proved to be relatively low (e.g. 11 % for the bio‐electrochemical hydrogen production process reported by Kreysa et al.).49, 50, 51, 52, 53
This led to the development of microbial electrolysis cells (MECs), whereby the generated energy was directly used for the cathodic hydrogen evolution reaction (HER). As the potential delivered by the microbial anode was not sufficient for the HER, an additional voltage (0.2–0.7 V) had to be applied. However, this voltage is significantly lower than that required for the electrolysis of water (practically >1.6 V).49, 50, 51, 54 Furthermore, bio‐electrochemical systems were further developed by using the cathodic reaction for the synthesis of value‐added organic products through the reduction of CO2 or organic substrates (microbial electrosynthesis cell). This would be highly desirable, not only because of the production of valuable chemicals, but also because of the recycling of CO2. However, these processes are still at an early stage of development. Challenges arising on scale‐up, possible migration between the anodic and cathodic compartments, as well as the lifetime of the catalyst are just a few issues to mention.50, 51, 55, 56 Furthermore, reduction of CO2 leads to rather low‐value products such as methane, the biofuels ethanol and butanol, or carboxylates such as acetate or butyrate.51, 52, 55, 56, 57 Only a few examples of the production of high‐value compounds, such as 6‐bromo‐2‐tetralol (6‐bromo‐3,4‐dihydro‐2(1H)‐naphthol), have been reported. 6‐Bromo‐2‐tetralol, an intermediate in the synthesis of a potassium channel blocker, was obtained from 6‐bromo‐2‐tetralone in a biotransformation process by yeast cells of Trichosporon capitatum or its partially purified Br‐β‐tetralone reductase on a 10 mm scale.55, 58
Several redox‐enzymes require cofactors for their operation. These cofactors usually represent the reducing agents. The combination of high enantioselectivity with the sustainability of electroconversions is, thus, very appealing.59, 60, 61
The reduction of prochiral ketones to optically enriched alcohols is of particular interest. For this, NAD+ has to be cathodically regenerated to NADH. As a consequence of the phosphorylated nature of this cofactor, a direct reduction at the cathode is hampered. The use of cationic rhodium mediators can solve this challenge (Scheme 12).61, 62, 63 However, even the mediators of the second generation provided a very limited total turnover of 113 h−1.62 Alternative mediators not based on precious metals and exhibiting a longer performance/durability are highly desired. Thus, much more research appears to be necessary to develop attractive bio‐electrochemical syntheses.
Scheme 12.

Indirect electrochemical regeneration of the cofactor.62 bpy=2,2′‐bipyridine, Cp*=C5Me5.
5. Redox Tags in Electrochemical Synthesis
Single electron transfer (SET) processes initiate a large variety of reactions in organic electrochemistry. Such reactions lead to open‐shell reactivity, often with complementary outcomes compared to traditional polarity‐driven reactions.64 Ashby has reported the universality of SET mechanisms and attempted to replace conventional polar mechanisms.65 Here, SN2 reactions are often discussed as polar processes, whereas Grignard reactions have been explained by SET mechanisms. Typically, these SETs take place between the electrode and the substrate or in an intermolecular manner. Nevertheless, intramolecular SET processes are key to mechanistic investigations, facilitate reaction pathways which would be forbidden, and offer important synthetic routes such as [2+2] cycloadditions66, 67 or Diels–Alder reactions.68 Intramolecular SET processes are rather elusive compared to intermolecular types, as these processes are net redox‐neutral and cannot be simply regarded as oxidations or reductions. These processes can be understood in combination with associated bond formations and bond cleavages. Such intramolecular SET processes can be regulated by so‐called “redox tags”. Electrochemical investigations on the synthetic application of this concept have been mainly conducted by Chiba and co‐workers. The main electrolyte used for the reported electrochemical reactions is lithium perchlorate in nitromethane. As a consequence of the unique Lewis acidity of lithium cations and weakly or noncoordinated counterions, this mixture is known to accelerate and promote a variety of chemical transformations, for example, Diels–Alder reactions.69
5.1. SET‐Triggered Formal [2+2] Cycloaddition versus Olefin Metathesis
Chiba and co‐workers reported the anodic treatment of enol ethers in MeNO2/LiClO4 in the presence of terminal olefins to yield cyclobutane derivatives by formal [2+2] cycloaddition reactions.67 The premise for a successful cycloaddition was the presence of an electron‐rich benzene ring in one of the reaction partners. The proposed mechanism is shown in Scheme 13.70 The enol ether is oxidized to the corresponding radical cation. This reactive intermediate can be trapped by the terminal olefin, which is present in large excess. Crucial for a successful cycloaddition is the subsequent intramolecular reduction of the cyclobutyl radical cation by a SET from the methoxyphenyl ring to form a relatively long‐lived aromatic radical cation. This species is then capable of oxidizing the starting enol ether to start the radical cation chain mechanism. The electrocatalytic nature of this reaction is also apparent because complete conversion is already achieved after 0.5 F (F=Faraday constant). For this type of reaction, the aromatic ring acts first as an electron donor and subsequently as an electron acceptor and, therefore, is a so‐called redox tag. This redox tag has the essential role of regulating the intramolecular SET process and, therefore, makes the cycloaddition reaction possible. Chiba and co‐workers investigated the limitations for the electron density of the aromatic ring and found that electron‐rich aromatic moieties such as either mono‐ or dimethoxy or mono‐, di‐, and trimethylbenzenes work as redox tags. Extremely electron‐rich benzene moieties with three methoxy groups or electron‐releasing nitrogen or sulfur moieties are not capable of working as redox tags, because they are too readily oxidized. The lower the oxidation potential of the redox tag, the more efficient is the cycloaddition reaction, but its oxidation potential has to be higher than the oxidation potential of the enol ether. This strictly limits the scope for synthetic applications, but gave an interesting insight for mechanistic investigations.
Scheme 13.

Proposed mechanism for the [2+2] cycloaddition of enol ethers and terminal olefins. The oxidation potentials E Ox were measured versus the Ag/AgCl reference electrode.70
Carrying out the reaction with derivatives not containing any substitution at the phenyl ring also does not result in the desired [2+2] cycloaddition product. In fact, this reaction opened a new synthetic pathway, since the unsubstituted phenyl ring could not work as a redox tag. The reaction pathway is depicted in Scheme 14. First, oxidation takes places at the enol ether to form the radical cation. Trapping of this species by the olefin will result in the formation of the cyclobutyl radical cation. As a consequence of the ineffective intramolecular SET process, the final formation of the cyclobutane moiety cannot occur. Instead, the cyclobutyl radical cation is decomposed again. The decomposition can take place either to give the starting combination of the enol ether radical cation and the terminal olefin, or result in an olefin metathesis.71
Scheme 14.

Proposed mechanism for olefin metathesis as a result of the ineffective redox tag.71
The use of an olefin with a cyclohexyl ring instead of the phenyl ring enhanced the efficacy of the metathesis pathway (Scheme 15). Thus, interestingly, the investigation of the limits of redox tags led to access to electrochemically initiated olefin metathesis reactions.72
Scheme 15.
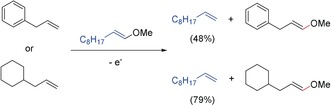
Comparison of olefins in electrocatalytic olefin metathesis.72
Boydston and co‐workers were able to use this concept to develop a metal‐free, photochemical ring‐opening metathesis polymerization.73 In general, electrochemical metathesis reactions provide a pathway to energy‐efficient, metal‐ and/or catalyst‐free metathesis reactions. Therefore, this route is a sustainable alternative to conventional metathesis reactions, when substrate combinations are suitable.
5.2. Anodic SET‐Triggered Diels–Alder Reactions
Compared to [2+2] cycloaddition reactions, SET‐triggered Diels–Alder reactions (or [4+2] cycloadditions between a diene and a dienophile) have a wider field of application in synthetic organic chemistry. SET processes are the most straightforward approaches to realize Umpolung and, therefore, achieve electronically mismatching Diels–Alder reactions. Chiba and co‐workers investigated electrochemical SET‐triggered Diels–Alder reactions.74 Comparison of this approach to [2+2] cycloadditions showed the mechanism is also based on a crucial intramolecular SET process. The first evidence for this is seen on comparing the reaction of trans‐anethole and 1‐propenylbenzene with isoprene (Scheme 16, top part). Whereas the SET‐triggered Diels–Alder reaction of trans‐anethole and isoprene gave excellent yields after application of just 0.1 F, no Diels–Alder reaction was observed with 1‐propenylbenzene.
Scheme 16.

Anodic SET‐triggered Diels–Alder reaction with trans‐anethole and 1‐propenylbenzene as well as the proposed mechanism reaction involving an intramolecular SET process. The oxidation potentials E Ox were measured versus the Ag/AgCl reference electrode.74 Q=amount of charge.
The proposed mechanism is depicted in the lower part of Scheme 16. Based on the oxidation potentials, initial oxidation takes place on trans‐anethole to form a radical cation. This intermediate is then trapped by the diene to form the cyclohexenyl ring. To form the desired reaction product, this radical cation has to be immediately reduced through an intramolecular SET process by the methoxyphenyl redox tag. The resulting aromatic radical cation will again initiate the radical chain reaction. Although the oxidation potential of 1‐propenylbenzene (1.51 V versus Ag/AgCl) was suitable to generate the corresponding radical cation, similar to trans‐anethole, no desired product was obtained. This is explained by the inefficiency of the unsubstituted phenyl ring to act as a redox tag. Therefore, the intramolecular SET from the redox tag is a crucial part of a successful SET‐triggered Diels–Alder reaction.
Similar to the observations for the [2+2] cycloaddition, further substitution with methoxy groups at the redox tag decreased the yield of the Diels–Alder reaction product. This was rationalized by the reduced reactivity of the initially formed radical cation by multiple methoxy groups. Chiba et al. showed the versatility of this method by carrying out further investigations on anodic SET‐triggered Diels–Alder reactions and the reaction mechanism.
6. Electroconversion of Renewables
In the past 70 years, naphtha and natural gas have become the primary feedstock for the industrial production of organic chemicals.75, 76 Nowadays, the use of fossil raw materials is questionable due to the increasing environmental awareness of society and the guiding principle of “sustainable development”, to which the chemical industry professes.77 Biomass seems to be a promising alternative as a renewable and sustainable source for fuels and chemicals.75 Moreover, organic electrosynthesis is a powerful and ecological method,4, 78 since the combination of organic electrosynthesis and the use of renewable biomass would be a sustainable and “green” approach for fuel and chemical production.
6.1. Carbon Dioxide as a Renewable Feedstock
A long‐standing challenge is closing the anthropogenic carbon cycle by recycling CO2 from various sources into feedstock materials for fuels and chemical manufacturing. The direct electrochemical reduction of CO2 seems to be a powerful ecological method and, therefore, an appropriate intermediate step towards a carbon‐free future. The challenge in the electrochemical conversion of the greenhouse gas is the selectivity and faradaic efficiency. Here, the electrode plays a crucial role (Scheme 17).79, 80, 81
Scheme 17.

Electrochemical reduction of carbon dioxide to various products.79, 80, 81
During the past few decades, the electrocatalytic and electrochemical reduction of CO2 at metal cathodes and related electrodes has been extensively investigated in aqueous and anhydrous (organic, ionic liquids or mixtures of them) media. The reduction products were found to depend strongly on the cathode material. Cu cathodes led to methane and ethylene as the major products, whereas Au, Ag, and Zn as the cathode led to the formation of CO. Formic acid was a major product in the reduction of CO2 at Pb and Hg cathodes.80, 82 It is noteworthy that only ethylene and, in particular, CO represent products of significantly added value. These products are in high demand and can be employed in conventional chemical plants for the generation of commodities.76
Recently, Koper and co‐workers published various methods for the direct electrochemical and electrocatalytical reduction of CO2. They used immobilized cobalt protoporphyrin on a pyrolytic graphite electrode and obtained CO as the major product of CO2 reduction. They enhanced the faradaic efficiency and the rate of CO formation significantly by designing a three‐dimensional porous hollow fiber Cu electrode. Besides Cu being used as an electrode material, it can also be used as an electrocatalyst. Thus, the electrocatalytic reduction of CO2 to methane and ethylene could be performed with a high turnover in aqueous media by using a Cu‐porphyrin complex. The use of PdxPt100−x/C nanoparticles as the electrocatalyst was reported for the selective production of formic acid with a high faradaic efficiency.83
Besides metal‐based electrodes for the direct electrochemical reduction of CO2, various carbon electrodes, such as graphite, glassy carbon, BDD, and carbon nanotubes, were also used.81, 84 Compared to other electrode materials, BDD electrodes have a wide potential window and high electrochemical stability. As a consequence of the high over‐potential for cathodic hydrogen evolution, BDD is well‐suited for the cathodic reduction of CO2. Encouraging results for the reduction of carbon dioxide to formaldehyde is reported in Scheme 18. Here, Einaga and co‐workers showed the highly selective electrochemical production of formaldehyde from CO2 and seawater at BDD cathodes. The seawater served as the electrolyte and source of protons and electrons (Scheme 18).85
Scheme 18.

Electrochemical reduction of carbon dioxide to formaldehyde at boron‐doped diamond anodes in seawater.85
In addition, carbon electrodes, in particular BDD, are very attractive for carboxylation processes by electrosynthesis with CO2. A drawback of electrochemical carboxylations is that sacrificial electrodes are required. However, the use of BDD cathodes can reduce the required amount of sacrificial anodes. Thus, the electrochemical conversion of methional into 2‐hydroxy‐4‐methylsulfanylbutyric acid (MHA, an important technical product used as an additive for animal food), as well as 1‐hydroxy‐3‐methylsulfanylpropanol was achieved at BDD cathodes in the presence of Mg sacrificial anodes (Scheme 19).86
Scheme 19.

Carboxylation of methional at BDD cathodes.86
6.2. Degradation of Lignin to Valuable Fine Chemicals
Besides cellulose and hemicellulose, lignin is one of the most abundant polymers in nature. It represents the major part of plant biomass. The polyphenolic structure of the biopolymer lignin qualified it as a potential sustainable and renewable feedstock for fuels and aromatic fine chemicals. Furthermore, about 50 million tons of lignin are produced every year as waste material by pulping industries. Kraft pulping is the predominant process used for the production of cellulose. As a consequence of the harsh reaction conditions, significant modifications in the native lignin take place. Technical Kraft lignin is characterized by inertness and degradation robustness, which complicates the selective degradation.87, 88
Lignosulfonate is a waste stream from the almost replaced sulfite pulping process. Utley and co‐workers carried out the electrooxidative cleavage of lignosulfate in alkaline media at Ni electrodes at 145 °C and 500 kPa. The major product was the aroma chemical vanillin in a yield of 5–7 wt % (Scheme 20). Moreover, a rationale for the mechanism was found on studying model lignin dimers. The system was also transferred to a flow reactor based on a filter press to allow continuous application.89, 90
As was mentioned before, Kraft lignin is the major waste product from the pulping industry. The electrochemical degradation of Kraft lignin to vanillin has been shown on Pt, Au, Ni, Cu, DSA‐O2 (DSA‐O2=dimensionally stable anode for oxygen evolution), and PbO2.91 The conversion and chemical yields depend mostly on the applied current density, while the nature of the electrode influences the reaction rate. The Waldvogel research group described the highly selective generation of the aroma chemical vanillin by anodic degradation of Kraft lignin at activated porous Ni/P‐foam electrodes under mild reaction conditions (Scheme 21). Furthermore, a combined electrochemical process with product isolation by adsorption on strong anionic exchange resins was established. This allowed isolation of vanillin without neutralization of the whole electrolyte or affecting the waste streams.92
Scheme 21.
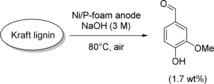
Highly selective electro‐depolymerization of Kraft lignin to vanillin on porous Ni/P‐foam electrodes.92
Stiefel et al. reported a controlled Kraft lignin depolymerization in an electrochemical reactor with an in situ nanoporous membrane.93 Another interesting method was reported by Tian et al. They described a novel approach which combined electrochemical and photochemical oxidation for the modification and degradation of Kraft lignin. In this approach, a Ta2O5‐IrO2 thin film was used as the electrocatalyst and TiO2 nanotubes arrays used as the photocatalyst.94 However, no yields for the degradation products were reported for either method.
Lignin prepared from different organic‐solvent‐based procedures are known as organosolv lignins. The anodic oxidation of organosolv lignin from spruce was reported in alkaline media at high temperatures. Lower temperatures could be used by adding nitrobenzene or 1,3‐dinitrobenzene as a co‐catalyst. The major degradation products were vanillic acid, 4‐hydroxybenzaldehyde, and vanillin, but no absolute yields were given.95 Crude enzymatically derived lignin was depolymerized by Zhu et al. The degradation was achieved through a combination of direct anodic oxidation at a RuO2/Ti mesh and oxidation by cathodic generated H2O2 at a graphite felt. The depolymerization was unselective and a broad spectrum of monomers and dimers were obtained.96 Aspen lignin was depolymerized at Pb/PbO2 electrodes in an alkaline electrolyte. This lignin was degraded by hydroxyl radicals and hydrogenated by alkaline water electrolysis to afford 4‐methylanisol and other products.97 Pb/PbO2 anodes were also used for the degradation of bamboo lignin in alkali solution. The main products consisted of vanillin, syringaldehyde, and p‐coumaric acid (Scheme 22).98 The Stephenson research group developed a selective one‐pot method for the oxidative β‐O‐ether bond cleavage of lignin‐type dimers and native‐like lignin. The method was a combination of electrocatalytic oxidation and photocatalytic fragmentation at ambient temperature.99
Scheme 22.

Products from the electrochemical depolymerization of bamboo lignin: vanillin (left), syringaldehyde (center), and p‐coumaric acid (right).98
Several anodic degradations of different lignin types have been reported, usually aiming to generate vanillin and related compounds (Schemes 20–22). Besides the mostly anodic cleavage of lignin, there are a few cathodic degradation methods. They are usually based on the electrocatalyzed reduction of lignin. Most of the methods were reviewed by Weckhuysen and co‐workers.88 However, electrochemistry can be used to obtain aromatic lignin degradation or extraction products. Here, the Moeller research group is notable. They reported the temperature‐ and pressure‐controlled solvolysis of sawdust to generate cinnamyl ether and/or aryl aldehyde products. Those electron‐rich aromatic compounds were used to synthesize a variety of more‐complex platform chemicals. Here, electrochemistry has been identified as a sustainable method to accomplish these transformations.100
6.3. Electrochemical Conversion of Sugars
Carbohydrates, such as C5 and C6 sugars, are extremely abundant in nature and can be used as renewable feedstock for fuels and chemicals. The selective oxidation of carbohydrates, in particular to uronic acids, has been intensively studied in the last few decades and continues to be an area of current interest. Schäfer et al. established a selective method for the direct anodic oxidation of carbohydrates to their corresponding uronic acids in moderate to excellent yields. The key step was the use of TEMPO as a redox mediator. In this way, methyl‐β‐d‐glucopyranoside was converted into the corresponding acid derivative.101 Besides the glycoside, various monosaccharides were anodically oxidized under these TEMPO‐mediated conditions. Furthermore, di‐, oligo‐, and polysaccharides were electrochemically converted into their corresponding uronic acid derivatives. In this way, unprotected d‐maltose was oxidized at the anomeric center to the corresponding triacid, without significant oxidation of the five secondary alcohol groups (Scheme 23).101, 102
Scheme 23.

Selective anodic TEMPO‐mediated oxidation of the primary hydroxy group of a glycoside (left), methyl‐l‐sorbopyranose (middle), and d‐maltose (right).
The direct anodic oxidation of saccharides at Cu or Au anodes continues to be of significant interest and was reviewed by Torto.103 Matsumoto et al. investigated the electrooxidation of glucose on Hg adatom‐modified Au eletrodes, while Park and co‐workers studied the same reaction on electrodes modified with Ag nanoparticles. Both groups used cyclic voltammetry as an electrochemical tool.104
Schröder and co‐workers reported the first one‐pot electrochemical deoxygenation of xylolactone to δ‐valerolactone. The process was realized by the partially separated selective oxidation with electrogenerated chlorine and cathodic reduction (Scheme 24).105
Scheme 24.

Simplified pathway of the electrochemical deoxygenation of xylolactone to δ‐valerolactone.105 DSA=dimensionally stable anode.
The most important electrochemical reduction of sugars is the conversion of glucose into sorbitol. The polyol sorbitol is used in foodstuffs as well as in cosmetic, medical, and industrial applications. For example, sorbitol is used as a feedstock for the synthesis of vitamin C. The electroreduction of glucose can be performed directly at a Pb cathode or indirectly by electrocatalytic hydrogenation at a Raney‐Ni cathode (Scheme 25).106 Other pentoses, such as ribose and xylose can also be electrochemically reduced at amalgamated Pb cathodes under galvanostatic conditions.107
Scheme 25.

Electrochemical reduction of glucose to sorbitol.106
Tessonnier and co‐workers reported recently an interesting route that combines biotechnology with electrochemical transformation to convert glucose into bio‐based unsaturated nylon‐6,6. In this approach, the monosaccharide was converted into muconic acid by fermentation. This resulting diene diacid was further electrochemically hydrogenated, without prior isolation, to 3‐hexenedioic acid in 94 % yield. The cathodic electrosynthesis promotes the generation of isolated double bonds. It is noteworthy that the fermentation broth is directly subjected to electroconversion. The cathodic hydrogenation was carried out under ambient conditions at a lead rod cathode at a constant potential of −1.5 V versus Ag/AgCl. Afterwards the unsaturated nylon was finally obtained by a polycondensation reaction of the 3‐hexenedioic acid with hexamethylenediamine (Scheme 26).108 The obtained polyamide can undergo subsequent modifications, such as cross‐linking.
Scheme 26.
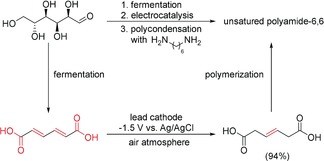
Combined method of bio‐ and electrochemical transformation for the conversion of glucose into bio‐based nylon‐6,6.108
Biologically produced muconic acid has emerged as a platform chemical for the synthesis of a wide range of bio‐based monomers. The conversion, selectivity, and current efficiency can be tuned by varying the nature of the metal cathode and the applied potential.109
Glycosylations are essential processes in the chemical synthesis of oligosaccharides. Electrochemical oxidation is a powerful method to activate glycosyl donors. For example, Nokami et al. developed a highly efficient electrochemical glycosylation reaction with Bu4NOTf as a supporting electrolyte. They also combined the electroglycosylation with a subsequent one‐pot cleavage of a fluorenylmethoxycarbonyl (Fmoc) protecting group. This one‐pot reaction can be used as a highly practical method for the synthesis of oligosaccharides (Scheme 27).110 A complete overview of electrochemical glycosylation was reported by Nokami et al.111
Scheme 27.

Electrochemical glycosylation and sequential one‐pot cleavage of the fluorenylmethoxycarbonyl (Fmoc) group.110 Bz=benzoyl.
The generation of furan derivatives from sugars is a classical conversion in chemistry. Furfural and 5‐hydroxymethylfurfural can be generated by the acid‐catalyzed dehydration of pentoses (C5) and hexoses (C6), respectively. The possibility of oxidation, dehydration, and hydrogenation of these furanic compounds makes them potential alternative commodity chemicals to fossil‐fuel‐based platform chemicals. Direct electrochemical or electrocatalytic conversion has emerged as a useful technology for both oxidation and hydrogenation processes. The electrochemically reduced products, such as methylfuran and dimethylfuran, can be used as biofuels, whereas electrooxidation leads to 2‐furancarboxylic acid and 2,5‐difurandicarboxylic acid, which are bulk chemicals for renewable biopolymers. The dicarboxylic acid has potential as a biogenic substitute for terephthalic acid (Scheme 28).112
Scheme 28.
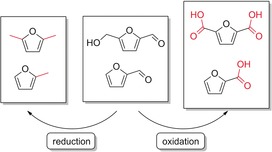
Electrochemical conversion of furanic compounds.112
An important technical process is the electrochemical dialkoxylation of furans.113 Breinbauer and co‐workers reported the indirect eletroorganic synthesis of 2,5‐dimethoxylated furanic compounds on a solid support. Here, Br2 was used as a mediator and the chemical transformation had a broad product scope.114 The anodic methoxylation of furans can be used for the synthesis of pyridoxine derivatives such as a vitamin B6 precursor (Scheme 29).115
Scheme 29.
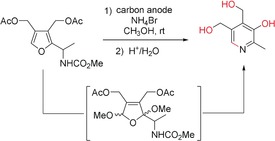
Electrochemical dialkoxylation of a furan derivative and synthesis of a pyridoxine derivative.115
6.4. Conversion and Modification of Fatty Acids
Oil and fat are high‐energy storage materials of biological organisms, which makes their usage very attractive as fuels and for chemical synthesis.116 Electrochemistry is a sustainable and powerful tool for the modification and conversion of fatty acids. The electroactive sites of fatty acids are the carboxy group, C−C double bonds, and activated C−H bonds. Carboxy groups can be decarboxylated at the anode to form radicals; this reaction is better known as Kolbe electrolysis. High current densities and hydrogen atoms in the α‐position facilitate the formation of radicals. Thus, the Kolbe electrolysis is a preferred method for the conversion of fatty acids.117, 118 The homocoupling of two identical fatty acids affords a symmetric dimer. For example, the dimerization of the ricinoleic acid derivative shown in Scheme 30.119
Scheme 30.

Homocoupling of a ricinoleic acid derivative by Kolbe electrolysis.119
Two different fatty acids with pK a values in the same range can be unsymmetrical cross‐coupled by co‐electrolyzation, with the intermediate radicals coupling statistically. For an increased formation of the cross‐coupling product, the less expensive acid is used in excess. The radicals generated from the electrochemical decarboxylation (Kolbe reaction) can react with double bonds (Scheme 31, top).117 Dimethyl pentadecandioate was electrogenerated from monomethyl dodecanedioate and monomethyl glutarate. The obtained diester can be further converted into rac‐muscone (Scheme 31, bottom).117, 119
Scheme 31.
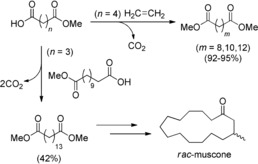
Top: Anodic addition of a Kolbe radical from monomethyladipate to ethylene. Bottom: Unsymmetrical electrochemical coupling of two fatty acids to the precursor to generate a muscone precursor.117, 119
Double bonds can also be directly oxidized at very positive potentials. For example, double bonds in vicinal dialkyl olefins are oxidized at potentials above +1.8 V versus SCE. A radical cation is probably formed that can react with a nucleophile or deprotonates to form an allylic radical. For example, methyl oleate can be oxidized in acetic acid at a Pt anode. Two isomeric diacetates were formed as major products after consumption of 4 F (Scheme 32).120
Scheme 32.
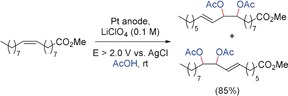
Anodic diacetoxylation of methyl oleate.120
The oxidation potential of conjugated dienes is lower than the oxidation potential of the isolated olefin system. For example, the anodic oxidation of the conjugated fatty diene shown in Scheme 33, which can be prepared from linoleic acid ester, takes places at 1.4 V versus SCE in AcOH/AcONa. The electrochemical conversion provides in good yields the 1,4‐diacetate, which has applications as a plasticizer.101
Scheme 33.

Electrochemical diacetoxylation of a doubly unsaturated fatty acid derived from linoleic acid ester.101
Activated allylic hydrogen bonds can be oxidized to the corresponding ketone by using substoichiometric amounts of TEMPO as a mediator. The use of the mediator enables a significantly lower potential to be applied. This TEMPO‐mediated anodic oxidation allows the trienone to be generated from methyl linolenoate (Scheme 34).121
Scheme 34.
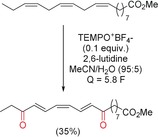
TEMPO‐mediated anodic oxidation of methyl linolenoate.121
Furthermore, electroorganic synthesis can be used for the production of biofuels from fatty acids. With good coulomb efficiencies, the electrochemical decarboxylation of fatty acids in methanolic and ethanolic solution leads to the formation of diesel‐like olefin/ether mixtures (Scheme 35). In addition, the electrochemical conversion of levulinic acid into octane has been reported.122
Scheme 35.

Anodic decarboxylation of oleic acid to diesel‐like compounds.122
The cathodic reduction of enones derived from fatty acids in DMF afforded the corresponding hydrodimers (Scheme 36).123
Scheme 36.
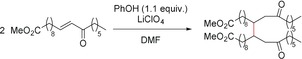
Hydrodimerization by cathodic reduction of a fatty acid enone.123
6.5. Amino Acids as Feed Stock for Nitrogen‐Containing Chemicals
The Baizer process is probably one of the best‐known electrochemical procedures and provides access to the important bulk chemical adiponitrile. This dinitrile is used for the production of the popular polymer nylon‐6.6.124 Remarkably, all the industrial routes for the synthesis of adiponitrile, including the Baizer process, are based on petrochemicals and require external nitrogen sources (i.e. NH3, NaCN, or HCN). Amino acids represent a renewable biomass, which could be used as a sustainable nitrogen source. The electrochemical synthesis of adiponitrile from glutamic acid has been reported. Glutamic acid, the most abundant non‐essential amino acid in plant proteins, was conventionally converted into the mono ester methylglutamate.125 After that, the anodic decarboxylation of the monomethyl glutamate to 3‐cyanopropanoic acid methyl ester was performed with NaBr as the supporting electrolyte and mediator. Adiponitrile was obtained from the 3‐cyanopropanoic acid methyl ester in a final one‐pot reaction with electrochemical substeps (Scheme 37).126
Scheme 37.
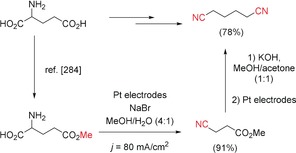
Sequential electrochemical synthesis of adiponitrile from glutamic acid.126
Another electrochemical conversion of an amino acid was demonstrated by De Vos and co‐workers. They efficiently decarboxylated lysine electrochemically and with bromide assistance. In this manner, the selectivity of the carboxylation could be tuned, depending on the cathode material used, to obtain nitrile, amine, or amide groups (Scheme 38).127
Scheme 38.
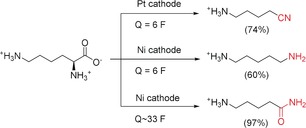
Electrochemical decarboxylation of lysine to the corresponding nitrile, amine, or amide.127
Onomura and co‐workers published an electrochemical method for the direct α‐cyanation of N‐protected cyclic amines on graphite electrodes. For example, the direct cyanation of a proline derivative at position 5 was reported (Scheme 39).128
Scheme 39.

Electrochemical α‐cyanation of an N‐protected proline derivative.128 Tr=trityl.
7. Electrochemical Reactions in Flow Cells
The development and evolution of electrochemical processes under continuous flow conditions has occurred in the last two decades. Usually, electrosynthesis in flow cells is associated with large‐scale operations, since scale‐up is viable by simply increasing the number of electrolyzing devices. On the one hand, the use of flow cells for electroconversion requires significantly more electrical equipment, such as pumps. On the other hand, continuous synthesis under well‐defined conditions can be achieved. In comparison with classical flask chemistry, flow chemistry offers the possibility to reduce the amounts of solvents and substrates as well as making process optimization much easier. The generation of a homogeneous electric field represents a big advantage. In this regard, a variety of electrooxidations and electroreductions, as well as a combination of them, have been developed over the years.
7.1. Electrooxidations in Flow Cells
One of the very first examples of electrooxidation was carried out by Yoshida and co‐workers. They reported a direct electrooxidative C−C bond formation using a low‐temperature electrochemical microflow system to combine carbamates with allylsilanes by exploiting the “cation‐pool” method described in Section 3. Different carbamates as well as allylsilanes proved to be suitable for the reaction (Scheme 40).47 A few examples involving vinyl ethers were also described.129
Scheme 40.
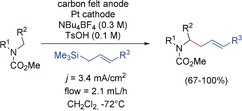
Continuous electrochemical α‐allylation of carbamates using allylsilanes as electrophiles.47
Common electroconversions were used to demonstrate the power of electrosynthetic methods and devices under continuous flow conditions. Most of these transformations exhibit a technical significance. One such anodic conversion is the dimethoxylation of toluene derivatives. These ketals are important intermediates for condensation reactions. The first experiments on this transformation were described by Löwe and Ehrfeld.130 Yoshida and co‐workers developed an electrochemical oxidation of p‐methoxytoluene to afford the corresponding ketal under flow conditions, without using a supporting electrolyte (Scheme 41, top). Several other examples of the electrochemical methoxylation of organic compounds were also reported.131 This reaction was also tested in the development of new microreactors and flow cells.132 A similar process was described by the Roth research group. In this case, they were able to expand the scope of substituents appended to the arene moiety, as well as synthesize not only ketals and subsequent aldehydes, but also methyl esters of certain substrates by using a polyvinylidene fluoride (PVDF) anode (Scheme 41, bottom).133
Scheme 41.

Electrochemical oxidation of different para‐substituted toluene derivatives in flow cells.131, 133
The installation of two methoxy groups at the α‐positions of a furan ring was demonstrated by Atobe and co‐workers.134 They used a thin‐layer flow cell and just a single pass of the furan solution (Scheme 42). A supporting electrolyte was not needed, since the methoxide anions generated provided sufficient conductivity.
Scheme 42.

Electrodimethoxylation of furan without a supporting electrolyte in a flow cell.134
Another example of an anodic methoxylation was developed by the Brown research group. In a Shono‐type reaction, N‐formylpyrrolidine was methoxylated at the α‐position (Scheme 43).135 The process was optimized later136 and expanded to other electroconversions, including the formation of quinone ketals, fluorinations, generation of CeIV, and synthesis of esters and amides from aldehydes.137
Scheme 43.

α‐Methoxylation of N‐formylpyrrolidine in a flow cell.135
The Atobe research group have made significant contributions to the field of electrooxidation processes. The electrogeneration of o‐benzoquinone from catechol, and a subsequent Michael‐type addition of benzenethiols led to the formation of diaryl thioethers (Scheme 44).138 It is noteworthy that the yields under flow cell conditions proved to be much higher than the yields achieved for the same process in a batch‐type cell. This finding could be explained by the fact that benzoquinone could be generated effectively without interference of thiol oxidation. In addition, the generated benzoquinone could be used directly without decomposition. The Atobe research group also performed aromatic C−C cross‐coupling between naphthalene and differently substituted methylbenzene derivatives (Scheme 45).139
Scheme 44.

Formation of aryl thioethers by electrooxidation of catechol.138
Scheme 45.
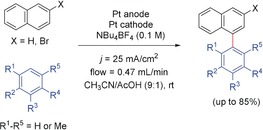
Continuous C−C cross‐coupling reaction between naphthalenes and methylbenzenes.139
However, not only simple and direct oxidation processes have been described using electrochemistry in flow cells. Very recently, some more elaborate examples of electrooxidations in flow cells have also been reported. Wirth and co‐workers described the electrosynthesis of a broad variety of diaryliodonium salts in flow by using a microreactor (Scheme 46),140 as well as the installation of CF3 and CF2H groups on electron‐deficient alkenes such as acrylates and acrylamides.141 In this second case, the radicals were produced by Kolbe electrolysis of di‐ and trifluoroacetic acid at the platinum anode and subsequently reacted with the alkene to afford the final products (Scheme 47).
Scheme 46.

Electrosynthesis of diaryliodonium salts in flow cells.140
Scheme 47.

Continuous electrochemical introduction of CF3 and CF2H groups in electron‐deficient alkenes.141 EWG=electron‐withdrawing group.
The Atobe group, in collaboration with the Waldvogel group, developed an electrochemical flow process for an aryl‐phenol cross‐coupling (Scheme 48).142 Depending on the additive and on the solvent, a mixture of the desired phenolic product and the undesired homocoupling product was obtained, but the selectivity for the production of the cross‐coupling product could be easily controlled and tuned.
Scheme 48.

Electrochemical anodic aryl‐phenol cross‐coupling in a flow cell.142
The Brown research group described a catalytic TEMPO‐mediated electrooxidation of primary and secondary alcohols in a microfluidic electrolytic cell.143 The TEMPO radical was oxidized at the PVDF anode, and the resulting oxoammonium cation subsequently oxidized the alcohol to yield the corresponding aldehyde or ketone (Scheme 49).
Scheme 49.

Oxidation of alcohols by electrogeneration of an oxammonium cation in a flow cell.143
Electrosynthesis in flow cells by oxidation reactions has also been used to synthesize natural products and different metabolites. Nishiyama and co‐workers were able to transform isoeugenol into licarin A in just one reaction step (Scheme 50).144 The very low yield of the reaction mainly results from the reaction between MeOH and the formed radical.
Scheme 50.

Electrosynthesis of licarin A from isoeugenol.144
A representative example was described by Stalder and Roth. They performed different electrochemical transformations of five commercial drugs in flow cells.145 The aim of this study was to investigate the versatility of continuous flow electrosynthesis for the generation, isolation, and full characterization of drug metabolites on a preparatory scale (Scheme 51).
Scheme 51.

Electrosynthetic generation of metabolites from different commercial drugs in flow cells.145
7.2. Electroreductions in Flow Cells
In addition to oxidations, several electroreduction processes in flow cells have been reported, most of them developed by Atobe and co‐workers. In a recent example, they performed the electrochemically assisted reduction of toluene to the corresponding methylcyclohexane by using hydrogen as a reducing agent.146 The formation of anions prior to electroreduction has also been utilized as a useful method in the development of some processes. In this regard, there are two similar examples. In the first one, a process was designed for the α‐alkylation of methyl phenylacetate,147 in which 2‐pyrrolidone was electroreduced in flow, with the formed anion acting as a base. The base could abstract the α‐proton of methyl phenylacetate, thereby forming a carbanion which reacted subsequently with iodomethane to afford the methylated product (Scheme 52, top). In the second example, the pyrrolidone anion was used to deprotonate chloroform to form a trichloromethyl anion, which could easily attack benzaldehyde to yield 2,2,2‐trichloro‐1‐phenylethanol (Scheme 52, bottom).148
Scheme 52.

Continuous formation of a 2‐pyrrolidone anion by electroreduction and subsequent reactions.147, 148
The same idea of forming anions from allyl149 or benzyl halides150 and using them as nucleophiles in two different reactions has been described. In the first case, an aryl aldehyde was utilized to trap the anion and form the corresponding alcohol (Scheme 53, top). However, the use of hexamethylphosphoric triamide (HMPA) as solvent is less beneficial. In the second case, CO2 was used, which allowed the electrosynthesis of different benzyl carboxylic acids in a flow cell (Scheme 53, bottom).
Scheme 53.

Anions as nucleophiles in the synthesis of alcohols or carboxylic acids in flow cells.149, 150 HMPA=hexamethylphosphoric triamide.
Haswell and co‐workers reported a cathodic dimerization of 4‐nitrobenzyl bromide in a flow cell by using benzyl bromides.151 This process opened the possibility to functionalize this compound and some derivatives with other molecules. They were able to couple different benzyl bromides with acetic anhydride,152 as well as with different activated olefins.153 These reactions are depicted in Scheme 54.
Scheme 54.
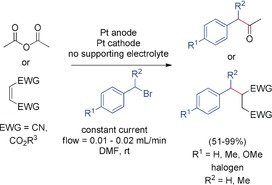
Cathodic coupling of benzyl bromides with acetic anhydride and activated olefins in a flow cell.151, 152, 153
A different electroreductive process in a flow cell was developed by Waldvogel and co‐workers. They were able to carry out a double dehalogenation in flow for the synthesis of a key intermediate for NS5A inhibitors (Scheme 55).154
Scheme 55.
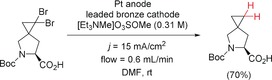
Electrochemical double dehalogenation of a cyclopropane derivative in a flow cell.154 Boc=tert‐butoxycarbonyl.
7.3. Electrooxidation and Electroreduction Sequences in Flow Cells
Finally, there are two flow processes in the literature in which both an electrooxidation and an electroreduction were involved at the same time. The first one was developed by Willians and co‐workers and consisted of the formation of copper‐NHC complexes.155 Different imidazolium cations were reduced to form carbenes, while Cu0 from a sacrificial anode was oxidized to CuI (Scheme 56). The effectiveness of the formed complexes was demonstrated, since they were used directly from the electrochemical flow cell in a hydrosilylation reaction.
Scheme 56.
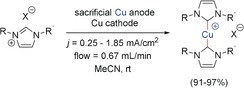
Electrosynthesis of copper‐NHC complexes in a flow cell.155
The other example, reported by Waldvogel and co‐workers, consisted of the oxidation of an ortho,ortho‐disubtituted aryl oxime to form the corresponding nitrile oxide, which was later reduced to afford a nitrile.156 In this example, the absence of a supporting electrolyte had a strong influence on the selectivity (Scheme 57).
Scheme 57.
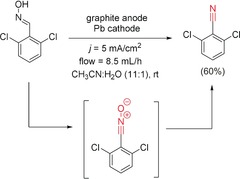
Domino oxidation‐reduction sequence of an oxime to the corresponding nitrile in a flow cell.156
8. Technical Significance
For a long time, the use of electricity in synthesis was focused on inorganic commodities such as the chloralkali process or aluminum production. The electrochemical synthesis of organic compounds represents rather niche applications because of their scale. However, some processes can generate up to several thousand tons per year.157 On this scale, only a few industrial organizations are interested (such as: BASF, Otsuka, Lanxess, and Clariant).158 The electrochemical production process has to be competitive with conventional transformations. On the one hand, the attractiveness of this technology is based on the avoidance of reagent waste and the use of electricity from regenerative resources. On the other hand, the electrochemical production technology is usually not carried out in a multipurpose plant and, therefore, rather specific know‐how is required. For the applicability of such processes, the current density should be in the range of 10–30 mA cm−2.159 This guarantees that the electrolysis cell will not be too large and the space‐time yield will be acceptable. However, for higher value‐added compounds, these numbers are less strict. The successful electrochemical conversion is the key step when establishing a technical process. Other crucial points are valid work‐up strategies and the recovery or recycling of electrolyte components. Since only partial conversion is often achieved by flow electrolyzers, a good separation of the desired compound from the electrolyte, intermediates, and starting materials is necessary. In addition, certain constraints exist for supporting electrolytes:160 They have be compatible with wastewater treatment facilities, and potentially explosive precipitates need to be avoided. Therefore, common supporting electrolytes, for example, perchlorates are not acceptable in industry. For a long time, the largest process was Monsanto's Baizer process. The cathodic reduction of acrylonitrile to adipontrile provides access to a central commodity for making polyamides such as nylon‐6.6. The beauty of the process consists of the use of water as the hydrogen source and molecular oxygen as the by‐product. At the end of the 1970s, this process reached 100 000 tons per year and the worldwide total capacity tripled some years later.161 However, the seminal scientific discovery happened almost 20 years earlier and was described as an inferior electroconversion in a divided cell.162 This impressively demonstrates the power of chemical engineering and process development. The Baizer process was initially conducted at cadmium cathodes, which created a significant environmental issue. After the technology came to BASF, the process was ameliorated and the cadmium cathodes were substituted by less‐hazardous copper‐lead alloys. By the end of the century, fossil energy was much less expensive than electricity and the uncertain cost of propylene led to a shutdown of most facilities. However, if large amounts of electricity have to be employed for the synthesis of valuable compounds, this process will be among the first candidates to be resumed (Scheme 58).
Scheme 58.
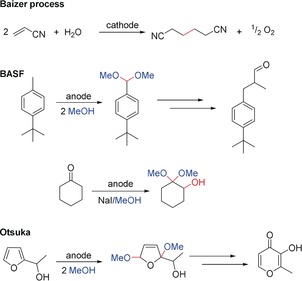
Examples of currently used technical electrochemical processes.
A major industrial user of electroorganic synthesis is BASF. Currently, the largest organic electroconversion is the dimethoxylation of 4‐tert‐butyltoluene to afford the protected benzaldehyde. This intermediate is used for a condensation reaction with propanal and subsequent hydrogenation to form lysmeral, which is a fragrance with the smell of lily of the valleys. By condensation, the methanol is recovered and the hydrogen formed during electrolysis could be used in the hydrogenation process. This electrochemical formation of benzaldehyde is conducted on several 10 000 tons per year. Another product of this process is anisaldehyde, which is used as a chemical intermediate and to cover strong odors. Another anodic conversion provides a methylketal of 2‐hydroxycyclohexanone, which is a common chemical intermediate. Starting from cyclohexanone in an iodide‐mediated process, this compound is selectively formed in very good yield on a ton scale. Another important fragrance is made by the Japanese company Otsuka: Starting from 2‐hydroxyethylfuran, which is readily accessible from furfural, an anodic dimethoxylation is carried out. Rearrangement of this intermediate provides maltol in about 150 tons per year.
These are only a few examples, but since several companies are now becoming interested in electrosynthesis there will undoubtedly be many more reported electroconversions on a ton scale in the near future.
9. Future Perspectives
The adoption of novel concepts and strategies from other fields has opened up new possibilities for electrosynthesis. The use of electricity for chemical conversions instead of using stoichiometric amounts of reagents provides an enormous potential for process development. In the future, tremendous advances will occur and subsequently push the field into broad applications:
Innovative electrolyte and electrode systems have to be elaborated to enable novel transformations. Thus, actual limits resulting from current electrode materials will be circumvented. Future electrode materials should avoid heavy metals such as lead or mercury. Synthetic carbon allotropes with tailor‐made surfaces seem to be a very promising option, as they provide larger over‐potentials for undesired side reactions, for example, hydrogen evolution. Moreover, such electrode systems should be highly resistant to fouling or corrosion processes and, therefore, be almost maintenance‐free. Initial steps in this direction have been made with boron‐doped diamond electrodes.163 In the electrolyte research, the solvent will not only be investigated as a reaction media, but also with regard to specific solvent effects that may tune the selectivity and enable novel electrosynthetic pathways. The supporting electrolytes are often considered as a significant drawback in electrochemical synthesis, since they cause significant costs and are mostly not compatible with modern wastewater treatments. The use of narrow gap cells in combination with the residual conductivity of the solvent may completely abolish the use of such supporting electrolytes.156, 164 In particular, protic solvents are splendid candidates and make such electrosynthetic approaches even more attractive.
In addition, very robust electrosyntheses are highly desired. Electrosynthetic conversions are commonly successful in a narrow current density range. If the required electrochemical conditions are not fulfilled because either the data reported are not precise enough or the geometry of the electrolysis cell is altered, problems with reproducibility will occur. On the one hand, the procedure should definitely be described in detail. This applies, in particular, to the electrode mounting and arrangement. On the other hand, the electrosyntheses should be developed as robust transformations, wherein the product is not prone to severe over‐conversion or the desired product is protected by solvent effects, for example.165 This will, in particular, be beneficial for newcomers in electrosynthesis, since small variations to the original procedures will not lead to dramatically inferior results or complete failure. In addition, this will open the opportunity to all‐rounder electrolysis cells, which can be employed as a standard setup in the laboratory.
Furthermore, the design and innovative concepts of electrolysis cells will allow the applicability of this technique to be expanded. When, for example, over‐reaction at the electrode turns out to be challenge, mass transport from the electrode regime into the bulk usually becomes an issue. Some approaches from the electrochemical generation of inorganic commodities might be adopted, for example zero‐gap flow cells166 or spinning‐disk electrodes.167 Great hope also lies in the combination of physical effects, such as ultrasonication168 or magneto‐electrochemistry,169 with the electrolysis. Indeed, the study of the influence of magnetic fields on chiral electrolytic events has just begun.170 However, the current research is still at a level far from being applicable in practical synthesis.
In conclusion, electroorganic synthesis will transform from a niche technology to a common synthetic method. Therefore, electrosynthesis also has to find its place in teaching and student training. The sustainability of this particular approach will make the use of this method inevitable at an academic and a technical level.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Sabine Möhle obtained her BSc in chemistry from the University of Regensburg in 2013. After an internship at the University of California, Santa Barbara (Prof. R. D. Little) in 2014, she finished her MSc in chemistry in the group of Prof. Dr. O. Reiser at the University of Regensburg in 2014. She is currently a PhD student under the supervision of Prof. S. R. Waldvogel, working on the electrochemical amination of arenes. Her PhD studies are supported by a Kekulé Fellowship of the Funds of the Chemical Industry.

Biographical Information
Michael Zirbes obtained his BSc in chemistry in 2014 from the Johannes Gutenberg University Mainz. After working there as an undergraduate research assistant in 2015, he obtained his MSc in organic chemistry in 2017 from the same university. Currently, he is a PhD student under the supervision of Prof. S. R. Waldvogel, working on the electrochemical conversion of renewable raw materials.

Biographical Information
Eduardo Rodrigo obtained his BSc in chemistry in 2009 and his MSc in organic chemistry in 2011 from the University Autónoma of Madrid. In 2014, he was a visitor at the University of Jyväskylä, Finland (Prof. P. M. Pihko). He finished his PhD in 2016 at the University Autónoma of Madrid in the field of organocatalysis under the supervision of Dr. M. B. Cid. Currently, he is as a postdoctoral researcher at the Johannes Gutenberg University Mainz in the group of Prof. S. R. Waldvogel, working on organic electrosynthesis.

Biographical Information
Tile Gieshoff obtained his Diploma in chemistry from the Johannes Gutenberg University Mainz in 2014, after conducting the Diploma work at Sanofi‐Aventis Deutschland GmbH. He is currently a PhD student under the supervision of Prof. S. R. Waldvogel and member of the MAINZ graduate school, working on the electroorganic synthesis of heterocycles. In 2016, he was a visiting researcher at the Washington University in St. Louis (Prof. K. D. Moeller).

Biographical Information
Anton Wiebe obtained his Diploma in chemistry from the Johannes Gutenberg University Mainz in 2014 in the group of Prof. S. R. Waldvogel, with a research internship at the University of Otago, New Zealand (Prof. Sally Brooker). He is currently a PhD student under the supervision of Prof. S. R. Waldvogel and member of the Max Planck Graduate Center, working on electrochemical oxidative coupling reactions of aromatic molecules.

Biographical Information
Siegfried R. Waldvogel studied chemistry in Konstanz and received his PhD in 1996 from the University of Bochum/Max‐Planck‐Institute for Coal Research with Prof. M. T. Reetz. After postdoctoral research at the Scripps Research Institute in La Jolla, California (Prof. J. Rebek, Jr.), he completed his habilitation in 1998 at the University of Münster. In 2004, he became professor for organic chemistry at the university of Bonn, and in 2010, full professor at the Johannes Gutenberg University Mainz. His main research interests are organic electrochemistry, oxidative coupling with Mo V reagents, and supramolecular sensing.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the DFG (Wa1276/14‐1 and Wa 1276/17‐1) for financial support. Support by the Advanced Lab of Electrochemistry and Electrosynthesis—ELYSION (Carl Zeiss Stiftung) is greatfully acknowledged. We highly appreciate the financial support from the Center for INnovative and Emerging MAterials (CINEMA). A.W. acknowledges the Max Planck Graduate Center for financial support. S.M. thanks the Fonds der Chemischen Industrie (FCI) for a Kekulé fellowship. T.G. is a recipient of a DFG fellowship by the Excellence Initiative by the Graduate School Materials Science in Mainz (GSC 266).
S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe, S. R. Waldvogel, Angew. Chem. Int. Ed. 2018, 57, 6018.
Contributor Information
Sabine Möhle, http://www.chemie.uni-mainz.de/OC/AK-Waldvogel/.
Prof. Dr. Siegfried R. Waldvogel, Email: waldvogel@uni-mainz.de.
References
- 1.
- 1a. Waldvogel S. R., Möhle S., Angew. Chem. Int. Ed. 2015, 54, 6398–6399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6496–6497; [Google Scholar]
- 1b. Waldvogel S. R., Selt M., Angew. Chem. Int. Ed. 2016, 55, 12578–12580; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12766–12768; [Google Scholar]
- 1c. Waldvogel S. R., Janza B., Angew. Chem. Int. Ed. 2014, 53, 7122–7123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7248–7249; [Google Scholar]
- 1d. Schäfer H. J., Angew. Chem. Int. Ed. 2017, 56, 15502–15503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15706–15708. [Google Scholar]
- 2.J. G. J. Olivier, G. Janssens-Maenhout, M. Muntean, J. A. H. W. Peters. Trends in Global CO2 Emissions: 2016 Report; The Hague, 2016.
- 3.Renewables 2017: Global Status Report; REN21, Paris, 2017.
- 4. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099–2119. [Google Scholar]
- 5. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoo S. J., Li L.-J., Zeng C.-C., Little R. D., Angew. Chem. Int. Ed. 2015, 54, 3744–3747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3815–3818. [Google Scholar]
- 7. Francke R., Little D., Chem. Soc. Rev. 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Herold S., Möhle S., Zirbes M., Richter F., Nefzger H., Waldvogel S. R., Eur. J. Org. Chem. 2016, 1274–1278; [Google Scholar]
- 8b. Waldvogel S. R., Elsler B., Boron-Doped Diamond for Green Electro-Organic Synthesis in Encyclopedia of Applied Electrochemistry (Eds.: G. Kreysa, K. Ota, R. F. Savinell), Springer, New York, 2014. [Google Scholar]
- 9.
- 9a. de Sarkar S., Biswas A., Samanta R. C., Studer A., Chem. Eur. J. 2013, 19, 4664–4678; [DOI] [PubMed] [Google Scholar]
- 9b. MacMillan D. W. C., Nature 2008, 455, 304–308. [DOI] [PubMed] [Google Scholar]
- 10. Ogawa K. A., Boydston A. J., Chem. Lett. 2015, 44, 10–16. [Google Scholar]
- 11. Mukherjee S., Yang J. W., Hoffmann S., List B., Chem. Rev. 2007, 107, 5471–5569. [DOI] [PubMed] [Google Scholar]
- 12. Bui N.-N., Ho X.-H., Mho S.-i., Jang H.-Y., Eur. J. Org. Chem. 2009, 5309–5312. [Google Scholar]
- 13.
- 13a. Kim H., MacMillan D. W. C., J. Am. Chem. Soc. 2008, 130, 398–399; [DOI] [PubMed] [Google Scholar]
- 13b. Sibi M. P., Hasegawa M., J. Am. Chem. Soc. 2007, 129, 4124–4125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Jang H.-Y., Hong J.-B., MacMillan D. W. C., J. Am. Chem. Soc. 2007, 129, 7004–7005. [DOI] [PubMed] [Google Scholar]
- 14. Ho X.-H., Mho S.-i., Kang H., Jang H.-Y., Eur. J. Org. Chem. 2010, 4436–4441. [Google Scholar]
- 15.
- 15a. Nagib D. A., Scott M. E., MacMillan D. W. C., J. Am. Chem. Soc. 2009, 131, 10875–10877; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Nicewicz D. A., MacMillan D. W. C., Science 2008, 322, 77–80; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Benfatti F., Capdevila M. G., Zoli L., Benedetto E., Cozzi P. G., Chem. Commun. 2009, 5919–5921. [DOI] [PubMed] [Google Scholar]
- 16. Jensen K. L., Franke P. T., Nielsen L. T., Daasbjerg K., Jørgensen K. A., Angew. Chem. Int. Ed. 2010, 49, 129–133; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 133–137. [Google Scholar]
- 17. Chiba T., Okimoto M., Nagai H., Takata Y., Bull. Chem. Soc. Jpn. 1982, 55, 335–336. [Google Scholar]
- 18. Corey E. J., Gilman N. W., Ganem B. E., J. Am. Chem. Soc. 1968, 90, 5616–5617. [Google Scholar]
- 19. Finney E. E., Ogawa K. A., Boydston A. J., J. Am. Chem. Soc. 2012, 134, 12374–12377. [DOI] [PubMed] [Google Scholar]
- 20. Ogawa K. A., Boydston A. J., Org. Lett. 2014, 16, 1928–1931. [DOI] [PubMed] [Google Scholar]
- 21. Green R. A., Pletcher D., Leach S. G., Brown R. C. D., Org. Lett. 2015, 17, 3290–3293. [DOI] [PubMed] [Google Scholar]
- 22. Green R. A., Pletcher D., Leach S. G., Brown R. C. D., Org. Lett. 2016, 18, 1198–1201. [DOI] [PubMed] [Google Scholar]
- 23. Breslow R., J. Am. Chem. Soc. 1958, 80, 3719–3726. [Google Scholar]
- 24. Barletta G., Chung A. C., Rios C. B., Jordan F., Schlegel J. M., J. Am. Chem. Soc. 1990, 112, 8144–8149. [Google Scholar]
- 25. Nakanishi I., Itoh S., Fukuzumi S., Chem. Eur. J. 1999, 5, 2810–2818. [Google Scholar]
- 26. Tam S. W., Jimenez L., Diederich F., J. Am. Chem. Soc. 1992, 114, 1503–1505. [Google Scholar]
- 27. Orsini M., Chiarotto I., Elinson M. N., Sotgiu G., Inesi A., Electrochem. Commun. 2009, 11, 1013–1017. [Google Scholar]
- 28. Feroci M., Chiarotto I., Orsini M., Pelagalli R., Inesi A., Chem. Commun. 2012, 48, 5361–5363. [DOI] [PubMed] [Google Scholar]
- 29. Yoshida J.-i., Suga S., Suzuki S., Kinomura N., Yamamoto A., Fujiwara K., J. Am. Chem. Soc. 1999, 121, 9546–9549. [Google Scholar]
- 30. Yoshida J.-i., Suga S., Chem. Eur. J. 2002, 8, 2650–2658. [Google Scholar]
- 31.
- 31a. Suga S., Yamada D., Yoshida J.-i., Chem. Lett. 2010, 39, 404–406; [Google Scholar]
- 31b. Suga S., Tsutsui Y., Nagaki A., Yoshida J.-i., Bull. Chem. Soc. Jpn. 2005, 78, 1206–1217; [Google Scholar]
- 31c. Suga S., Nishida T., Yamada D., Nagaki A., Yoshida J.-i., J. Am. Chem. Soc. 2004, 126, 14338–14339; [DOI] [PubMed] [Google Scholar]
- 31d. Suga S., Nagaki A., Tsutsui Y., Yoshida J.-i., Org. Lett. 2003, 5, 945–947; [DOI] [PubMed] [Google Scholar]
- 31e. Suga S., Okajima M., Yoshida J.-i., Tetrahedron Lett. 2001, 42, 2173–2176. [Google Scholar]
- 32. Suga S., Suzuki S., Yamamoto A., Yoshida J.-i., J. Am. Chem. Soc. 2000, 122, 10244–10245. [Google Scholar]
- 33.
- 33a. Nokami T., Watanabe T., Musya N., Morofuji T., Tahara K., Tobe Y., Yoshida J.-i., Chem. Commun. 2011, 47, 5575–5577; [DOI] [PubMed] [Google Scholar]
- 33b. Terao K., Watanabe T., Suehiro T., Nokami T., Yoshida J.-i., Tetrahedron Lett. 2010, 51, 4107–4109; [Google Scholar]
- 33c. Okajima M., Soga K., Watanabe T., Terao K., Nokami T., Suga S., Yoshida J.-i., Bull. Chem. Soc. Jpn. 2009, 82, 594–599; [Google Scholar]
- 33d. Okajima M., Soga K., Nokami T., Suga S., Yoshida J.-i., Org. Lett. 2006, 8, 5005–5007. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Saito K., Saigusa Y., Nokami T., Yoshida J.-i., Chem. Lett. 2011, 40, 678–679; [Google Scholar]
- 34b. Nokami T., Shibuya A., Tsuyama H., Suga S., Bowers A. A., Crich D., Yoshida J.-i., J. Am. Chem. Soc. 2007, 129, 10922–10928; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34c. Suzuki S., Matsumoto K., Kawamura K., Suga S., Yoshida J.-i., Org. Lett. 2004, 6, 3755–3758. [DOI] [PubMed] [Google Scholar]
- 35. Nokami T., Soma R., Yamamoto Y., Kamei T., Itami K., Yoshida J.-i., Beilstein J. Org. Chem. 2007, 3, No. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Kataoka K., Hagiwara Y., Midorikawa K., Suga S., Yoshida J.-i., Org. Process Res. Dev. 2008, 12, 1130–1136; [Google Scholar]
- 36b. Midorikawa K., Suga S., Yoshida J.-i., Chem. Commun. 2006, 3794–3796. [DOI] [PubMed] [Google Scholar]
- 37. Ashikari Y., Shimizu A., Nokami T., Yoshida J.-i., J. Am. Chem. Soc. 2013, 135, 16070–16073. [DOI] [PubMed] [Google Scholar]
- 38. Ashikari Y., Nokami T., Yoshida J.-i., J. Am. Chem. Soc. 2011, 133, 11840–11843. [DOI] [PubMed] [Google Scholar]
- 39. Hayashi R., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2016, 138, 8400–8403. [DOI] [PubMed] [Google Scholar]
- 40. Morofuji T., Shimizu A., Yoshida J.-i., Angew. Chem. Int. Ed. 2012, 51, 7259–7262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7371–7374. [Google Scholar]
- 41.
- 41a. Matsumoto K., Kozuki Y., Ashikari Y., Suga S., Kashimura S., Yoshida J.-i., Tetrahedron Lett. 2012, 53, 1916–1919; [Google Scholar]
- 41b. Fujie S., Matsumoto K., Suga S., Nokami T., Yoshida J.-i., Tetrahedron 2010, 66, 2823–2829. [Google Scholar]
- 42. Shimizu A., Takeda K., Mishima S., Saito K., Kim S., Nokami T., Yoshida J.-i., Bull. Chem. Soc. Jpn. 2016, 89, 61–66. [Google Scholar]
- 43.
- 43a. Yoshida J.-i., Ashikari Y., Matsumoto K., Nokami T., J. Synth. Org. Chem. Jpn. 2013, 71, 1136–1144; [Google Scholar]
- 43b. Yoshida J.-i., Kataoka K., Horcajada R., Nagaki A., Chem. Rev. 2008, 108, 2265–2299. [DOI] [PubMed] [Google Scholar]
- 44. Maruyama T., Mizuno Y., Shimizu I., Suga S., Yoshida J.-i., J. Am. Chem. Soc. 2007, 129, 1902–1903. [DOI] [PubMed] [Google Scholar]
- 45. Suga S., Suzuki S., Yoshida J.-i., J. Am. Chem. Soc. 2002, 124, 30–31. [DOI] [PubMed] [Google Scholar]
- 46. Suga S., Matsumoto K., Ueoka K., Yoshida J.-i., J. Am. Chem. Soc. 2006, 128, 7710–7711. [DOI] [PubMed] [Google Scholar]
- 47. Suga S., Okajima M., Fujiwara K., Yoshida J.-i., J. Am. Chem. Soc. 2001, 123, 7941–7942. [DOI] [PubMed] [Google Scholar]
- 48. Hayashi R., Shimizu A., Song Y., Ashikari Y., Nokami T., Yoshida J.-i., Chem. Eur. J. 2017, 23, 61–64. [DOI] [PubMed] [Google Scholar]
- 49. Schröder U., ChemSusChem 2008, 1, 281–282. [DOI] [PubMed] [Google Scholar]
- 50. Logan B. E., Hamelers B., Rozendal R., Schröder U., Keller J., Freguia S., Aelterman P., Verstraete W., Rabaey K., Environ. Sci. Technol. 2006, 40, 5181–5192. [DOI] [PubMed] [Google Scholar]
- 51. Harnisch F., Schröder U., Chem. Soc. Rev. 2010, 39, 4433–4448. [DOI] [PubMed] [Google Scholar]
- 52. Schröder U., Harnisch F., Angenent L. T., Energy Environ. Sci. 2015, 8, 513–519. [Google Scholar]
- 53. Kreysa G., Schenk K., Sell D., Vuorilehto K., Int. J. Hydrogen Energy 1994, 19, 673–676. [Google Scholar]
- 54.
- 54a. Cheng S., Logan B. E., Proc. Natl. Acad. Sci. USA 2007, 104, 18871–18873; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54b. Liu H., Grot S., Logan B. E., Environ. Sci. Technol. 2005, 39, 4317–4320; [DOI] [PubMed] [Google Scholar]
- 54c. Rozendal R., Buisman C. J. N., WO2005005981, 2005.
- 55. Rabaey K., Rozendal R. A., Nat. Rev. Microbiol. 2010, 8, 706–716. [DOI] [PubMed] [Google Scholar]
- 56. Sadhukhan J., Lloyd J. R., Scott K., Premier G. C., Eileen H., Curtis T., Head I. M., Renewable Sustainable Energy Rev. 2016, 56, 116–132. [Google Scholar]
- 57. Santoro C., Arbizzani C., Erable B., Ieropoulos I., J. Power Sources 2017, 356, 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shin H., Jain M., Chartrain M., Zeikus J., Appl. Microbiol. Biotechnol. 2001, 57, 506–510. [DOI] [PubMed] [Google Scholar]
- 59. Hollmann F., Schmid A., Biocatal. Biotransform. 2004, 22, 63–88. [Google Scholar]
- 60. Steckhan E., Top. Curr. Chem. 1994, 170, 83–111. [Google Scholar]
- 61.“Electroenzymatic Synthesis”: Kohlmann C., Lütz S. in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC, Boca Raton, 2016, pp. 1511–1541. [Google Scholar]
- 62. Hildebrand F., Kohlmann C., Franz A., Lütz S., Adv. Synth. Catal. 2008, 350, 909–918. [Google Scholar]
- 63. Hildebrand F., Lütz S., Tetrahedron: Asymmetry 2007, 18, 1187–1193. [Google Scholar]
- 64. Zhang N., Samanta S. R., Rosen B. M., Percec V., Chem. Rev. 2014, 114, 5848–5958. [DOI] [PubMed] [Google Scholar]
- 65. Ashby E. C., Acc. Chem. Res. 1988, 21, 414–421. [Google Scholar]
- 66. Bauld N. L., Pabon R., J. Am. Chem. Soc. 1983, 105, 633–634. [Google Scholar]
- 67. Chiba K., Miura T., Kim S., Kitano Y., Tada M., J. Am. Chem. Soc. 2001, 123, 11314–11315. [DOI] [PubMed] [Google Scholar]
- 68. Imada Y., Yamaguchi Y., Shida N., Okada Y., Chiba K., Chem. Commun. 2017, 53, 3960–3963. [DOI] [PubMed] [Google Scholar]
- 69.
- 69a. Forman M. A., Dailey W. P., J. Am. Chem. Soc. 1991, 113, 2761–2762; [Google Scholar]
- 69b. Grieco P. A., Nunes J. J., Gaul M. D., J. Am. Chem. Soc. 1990, 112, 4595–4596. [Google Scholar]
- 70. Okada Y., Nishimoto A., Akaba R., Chiba K., J. Org. Chem. 2011, 76, 3470–3476. [DOI] [PubMed] [Google Scholar]
- 71. Arata M., Miura T., Chiba K., Org. Lett. 2007, 9, 4347–4350. [DOI] [PubMed] [Google Scholar]
- 72. Miura T., Kim S., Kitano Y., Tada M., Chiba K., Angew. Chem. Int. Ed. 2006, 45, 1461–1463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1489–1491. [Google Scholar]
- 73. Ogawa K. A., Goetz A. E., Boydston A. J., J. Am. Chem. Soc. 2015, 137, 1400–1403. [DOI] [PubMed] [Google Scholar]
- 74. Okada Y., Yamaguchi Y., Ozaki A., Chiba K., Chem. Sci. 2016, 7, 6387–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dale B. E., J. Chem. Technol. Biotechnol. 2003, 78, 1093–1103. [Google Scholar]
- 76. Weissermel K., Arpe H.-J., Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2003. [Google Scholar]
- 77. Brandt C., Chem. Unserer Zeit 2002, 36, 214–224. [Google Scholar]
- 78.
- 78a. Schäfer H. J., C. R. Chim. 2011, 14, 745–765; [Google Scholar]
- 78b. Anastas P., Eghbali N., Chem. Soc. Rev. 2010, 39, 301–312; [DOI] [PubMed] [Google Scholar]
- 78c. Steckhan E., Arns T., Heineman W. R., Hilt G., Hoormann D., Jörissen J., Kröner L., Lewall B., Pütter H., Chemosphere 2001, 43, 63–73. [DOI] [PubMed] [Google Scholar]
- 79.
- 79a. Olah G. A., Goeppert A., Prakash G. K. S., J. Org. Chem. 2009, 74, 487–498; [DOI] [PubMed] [Google Scholar]
- 79b. Olah G. A., Prakash G. K. S., Goeppert A., J. Am. Chem. Soc. 2011, 133, 12881–12898. [DOI] [PubMed] [Google Scholar]
- 80. Jones J.-P., Prakash G. K. S., Olah G. A., Isr. J. Chem. 2014, 54, 1451–1466. [Google Scholar]
- 81. Yang N., Waldvogel S. R., Jiang X., ACS Appl. Mater. Interfaces 2016, 8, 28357–28371. [DOI] [PubMed] [Google Scholar]
- 82.
- 82a. Kortlever R., Shen J., Schouten K. J. P., Calle-Vallejo F., Koper M. T. M., J. Phys. Chem. Lett. 2015, 6, 4073–4082; [DOI] [PubMed] [Google Scholar]
- 82b. Lu X., Leung D. Y. C., Wang H., Leung M. K. H., Xuan J., ChemElectroChem 2014, 1, 836–849; [Google Scholar]
- 82c. Jhong H.-R., Ma S., Kenis P. J. A., Curr. Opin. Chem. Eng. 2013, 2, 191–199; [Google Scholar]
- 82d. Qiao J., Liu Y., Hong F., Zhang J., Chem. Soc. Rev. 2014, 43, 631–675; [DOI] [PubMed] [Google Scholar]
- 82e. Zhang R., Lv W., Li G., Mezaal M. A., Lei L., RSC Adv. 2015, 5, 68662–68667; [Google Scholar]
- 82f. Costentin C., Robert M., Savéant J.-M., Chem. Soc. Rev. 2013, 42, 2423–2436; [DOI] [PubMed] [Google Scholar]
- 82g. Chaplin R., Wragg A. A., J. Appl. Electrochem. 2003, 33, 1107–1123; [Google Scholar]
- 82h.“Electrochemical CO2 Reduction in Metal Electrodes”: Hori Y. in Modern Aspects of Electrochemistry, Vol. 42 (Eds.: C. G. Vayenas, M. E. Gamboa-Aldeco, R. E. White), Springer, New York, 2008, pp. 89–189; [Google Scholar]
- 82i. Hori Y., Wakebe H., Tsukamoto T., Koga O., Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar]
- 83.
- 83a. Shen J., Kortlever R., Kas R., Birdja Y. Y., Diaz-Morales O., Kwon Y., Ledezma-Yanez I., Schouten K. J. P., Mul G., Koper M. T. M., Nat. Commun. 2015, 6, 8177; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83b. Weng Z., Jiang J., Wu Y., Wu Z., Guo X., Materna K. L., Liu W., Batista V. S., Brudvig G. W., Wang H., J. Am. Chem. Soc. 2016, 138, 8076–8079; [DOI] [PubMed] [Google Scholar]
- 83c. Kortlever R., Peters I., Koper S., Koper M. T. M., ACS Catal. 2015, 5, 3916–3923; [Google Scholar]
- 83d. Kas R., Hummadi K. K., Kortlever R., de Wit P., Milbrat A., Luiten-Olieman M. W. J., Benes N. E., Koper M. T. M., Mul G., Nat. Commun. 2016, 7, 10748; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83e. Birdja Y. Y., Shen J., Koper M. T., Catal. Today 2017, 288, 37–47; [Google Scholar]
- 83f. Ooka H., Figueiredo M. C., Koper M. T. M., Langmuir 2017, 33, 9307–9313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.
- 84a. Zhang S., Kang P., Ubnoske S., Brennaman M. K., Song N., House R. L., Glass J. T., Meyer T. J., J. Am. Chem. Soc. 2014, 136, 7845–7848; [DOI] [PubMed] [Google Scholar]
- 84b. Wu J., Yadav R. M., Liu M., Sharma P. P., Tiwary C. S., Ma L., Zou X., Zhou X.-D., Yakobson B. I., Lou J., Ajayan P. M., ACS Nano 2015, 9, 5364–5371; [DOI] [PubMed] [Google Scholar]
- 84c. Liu Y., Chen S., Quan X., Yu H., J. Am. Chem. Soc. 2015, 137, 11631–11636; [DOI] [PubMed] [Google Scholar]
- 84d. Hara K., Kudo A., Sakata T., J. Electroanal. Chem. 1997, 421, 1–4; [Google Scholar]
- 84e. Eggins B. R., Brown E. M., McNeill E. A., Grimshaw J., Tetrahedron Lett. 1988, 29, 945–948; [Google Scholar]
- 84f. Christensen P. A., Hamnett A., Muir A., Freeman N. A., J. Electroanal. Chem. Interfacial 1990, 288, 197–215; [Google Scholar]
- 84g. Birdja Y. Y., Koper M. T. M., J. Am. Chem. Soc. 2017, 139, 2030–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Nakata K., Ozaki T., Terashima C., Fujishima A., Einaga Y., Angew. Chem. Int. Ed. 2014, 53, 871–874; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 890–893. [Google Scholar]
- 86.“Use of Diamond Films in Organic Electrosynthesis in Synthetic Diamond Films”: Waldvogel S. R., Kirste A., Mentizi S., Preparation, Electrochemistry, Characterization, and Applications (Eds.: C. A. Martínez-Huitle, E. Brillas), Wiley, Hoboken, 2011, pp. 483–510. [Google Scholar]
- 87.
- 87a. Rinaldi R., Jastrzebski R., Clough M. T., Ralph J., Kennema M., Bruijnincx P. C. A., Weckhuysen B. M., Angew. Chem. Int. Ed. 2016, 55, 8164–8215; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8296–8354; [Google Scholar]
- 87b. Pandey M. P., Kim C. S., Chem. Eng. Technol. 2011, 34, 29–41; [Google Scholar]
- 87c.“Renewable Raw Materials”: Ulber R., Sell D., Hirth T., New Feedstocks for the Chemical Industry, VCH, Weinheim, 2011; [Google Scholar]
- 87d. Fache M., Boutevin B., Caillol S., ACS Sustainable Chem. Eng. 2016, 4, 35–46. [Google Scholar]
- 88. Zakzeski J., Bruijnincx P. C. A., Jongerius A. L., Weckhuysen B. M., Chem. Rev. 2010, 110, 3552–3599. [DOI] [PubMed] [Google Scholar]
- 89. Smith C. Z., Utley J. H. P., Hammond J. K., J. Appl. Electrochem. 2011, 41, 363–375. [Google Scholar]
- 90. Pardini V. L., Smith C. Z., Utley J. H. P., Vargas R. R., Viertler H., J. Org. Chem. 1991, 56, 7305–7313. [Google Scholar]
- 91. Parpot P., Bettencourt A. P., Carvalho A. M., Belgsir E. M., J. Appl. Electrochem. 2000, 30, 727–731. [Google Scholar]
- 92.
- 92a. Schmitt D., Beiser N., Regenbrecht C., Zirbes M., Waldvogel S. R., Sep. Purif. Technol. 2017, 181, 8–17; [Google Scholar]
- 92b. Schmitt D., Regenbrecht C., Hartmer M., Stecker F., Waldvogel S. R., Beilstein J. Org. Chem. 2015, 11, 473–480; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92c. Schmitt D., Regenbrecht C., Schubert M., Schollmeyer D., Waldvogel S. R., Holzforschung 2017, 71, 35–41. [Google Scholar]
- 93. Stiefel S., Lölsberg J., Kipshagen L., Müller-Gulland R., Wessling M., Electrochem. Commun. 2015, 61, 49–52. [Google Scholar]
- 94. Tian M., Wen J., MacDonald D., Asmussen R. M., Chen A., Electrochem. Commun. 2010, 12, 527–530. [Google Scholar]
- 95. Smith C., Utley J. H. P., Petrescu M., Viertler H., J. Appl. Electrochem. 1989, 19, 535–539. [Google Scholar]
- 96.
- 96a. Zhu H., Chen Y., Qin T., Wang L., Tang Y., Sun Y., Wan P., RSC Adv. 2014, 4, 6232–6238; [Google Scholar]
- 96b. Zhu H., Wang L., Chen Y., Li G., Li H., Tang Y., Wan P., RSC Adv. 2014, 4, 29917–29924. [Google Scholar]
- 97. Wang Y.-s., Yang F., Liu Z.-h., Yuan L., Li G., Catal. Commun. 2015, 67, 49–53. [Google Scholar]
- 98. Liu M., Wen Y., Qi J., Zhang S., Li G., ChemistrySelect 2017, 2, 4956–4962. [Google Scholar]
- 99. Bosque I., Magallanes G., Rigoulet M., Kärkäs M. D., Stephenson C. R. J., ACS Cent. Sci. 2017, 3, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nguyen B. H., Perkins R. J., Smith J. A., Moeller K. D., J. Org. Chem. 2015, 80, 11953–11962. [DOI] [PubMed] [Google Scholar]
- 101. Schäfer H. J., Harenbrock M., Klocke E., Plate M., Weiper-Idelmann A., Pure Appl. Chem. 2007, 79, 2047–2057. [Google Scholar]
- 102.
- 102a. Weiper A., Schäfer H. J., Angew. Chem. Int. Ed. Engl. 1990, 29, 195–197; [Google Scholar]; Angew. Chem. 1990, 102, 228–230; [Google Scholar]
- 102b. Schnatbaum K., Schäfer H. J., Synthesis 1999, 864–872; [Google Scholar]
- 102c. Schämann M., Schäfer H. J., Electrochim. Acta 2005, 50, 4956–4972; [Google Scholar]
- 102d. Schämann M., Schäfer H. J., Eur. J. Org. Chem. 2003, 351–358. [Google Scholar]
- 103. Torto N., Bioelectrochemistry 2009, 76, 195–200. [DOI] [PubMed] [Google Scholar]
- 104.
- 104a. Matsumoto F., Harada M., Koura N., Uesugi S., Electrochem. Commun. 2003, 5, 42–46; [Google Scholar]
- 104b. Quan H., Park S.-U., Park J., Electrochim. Acta 2010, 55, 2232–2237. [Google Scholar]
- 105. James O. O., Sauter W., Schröder U., ChemSusChem 2017, 10, 2015–2022. [DOI] [PubMed] [Google Scholar]
- 106.
- 106a. Bin Kassim A., Rice C. L., Kuhn A. T., J. Appl. Electrochem. 1981, 11, 261–267; [Google Scholar]
- 106b. Park K., Pintauro P. N., Baizer M. M., Nobe K., J. Electrochem. Soc. 1985, 132, 1850–1855. [Google Scholar]
- 107. Piszczek L., Gieldon K., Wojtkowiak S., J. Appl. Electrochem. 1992, 22, 1055–1059. [Google Scholar]
- 108. Suástegui M., Matthiesen J. E., Carraher J. M., Hernandez N., Quiroz N. R., Okerlund A., Cochran E. W., Shao Z., Tessonnier J.-P., Angew. Chem. Int. Ed. 2016, 55, 2368–2373; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2414–2419. [Google Scholar]
- 109.
- 109a. Matthiesen J. E., Carraher J. M., Vasiliu M., Dixon D. A., Tessonnier J.-P., ACS Sustainable Chem. Eng. 2016, 4, 3575–3585; [Google Scholar]
- 109b. Matthiesen J. E., Suástegui M., Wu Y., Viswanathan M., Qu Y., Cao M., Rodriguez-Quiroz N., Okerlund A., Kraus G., Raman D. R., Shao Z., Tessonnier J.-P., ACS Sustainable Chem. Eng. 2016, 4, 7098–7109. [Google Scholar]
- 110. Nokami T., Tsuyama H., Shibuya A., Nakatsutsumi T., Yoshida J.-i., Chem. Lett. 2008, 37, 942–943. [Google Scholar]
- 111. Nokami T., Itoh T., Mong K.-K. T., Isr. J. Chem. 2015, 55, 297–305. [Google Scholar]
- 112.
- 112a. Kokoh K. B., Belgsir E. M., Tetrahedron Lett. 2002, 43, 229–231; [Google Scholar]
- 112b. Parpot P., Bettencourt A., Chamoulaud G., Kokoh K., Belgsir E., Electrochim. Acta 2004, 49, 397–403; [Google Scholar]
- 112c. Kwon Y., Schouten K. J. P., van der Waal J. C., de Jong E., Koper M. T. M., ACS Catal. 2016, 6, 6704–6717. [Google Scholar]
- 113.“Organic electrosyntheses in industry”: Degner D. in Topics in Current Chemistry. Electrochemistry III (Ed.: E. Steckhan), Springer, Berlin, 1988, pp. 1–95. [Google Scholar]
- 114.
- 114a. Nad S., Breinbauer R., Angew. Chem. Int. Ed. 2004, 43, 2297–2299; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2347–2349; [Google Scholar]
- 114b. Nad S., Breinbauer R., Synthesis 2005, 20, 3654–3665; [Google Scholar]
- 114c. Nad S., Roller S., Haag R., Breinbauer R., Org. Lett. 2006, 8, 403–406. [DOI] [PubMed] [Google Scholar]
- 115. Shono T., Matsumura Y., Tsubata K., Takata J., Chem. Lett. 1981, 10, 1121–1124. [Google Scholar]
- 116. Lestari S., Mäki-Arvela P., Beltramini J., Lu G. Q. M., Murzin D. Y., ChemSusChem 2009, 2, 1109–1119. [DOI] [PubMed] [Google Scholar]
- 117. Schäfer H. J., Eur. J. Lipid Sci. Technol. 2012, 114, 2–9. [Google Scholar]
- 118.
- 118a.“Recent contributions of Kolbe electrolysis to organic synthesis”: Schäfer H.-J. in Topics in Current Chemistry. Electrochemistry IV (Ed.: E. Steckhan), Springer, Berlin, 1990, pp. 91–151; [Google Scholar]
- 118b. Tanaka H., Kuroboshi M., Torii S., Oxidation of Carboxylic Acids and Derivatives in Organic Electrochemistry, 5th ed. (Eds.: O. Hammerich, B. Speiser), CRC, New York, 2016, pp. 1267–1307. [Google Scholar]
- 119. Weiper-Idelmann A., aus dem Kahmen M., Schäfer H. J., Gockeln M., Nielsen P. H., Püschl A., Mikkelsen K. V., Senning A., Acta Chem. Scand. 1998, 52, 672–682. [Google Scholar]
- 120. Adams C., Frankel E. N., Utley J. H. P., J. Chem. Soc. Perkin Trans. 1 1979, 353–356. [Google Scholar]
- 121. Breton T., Liaigre D., Belgsir E. M., Tetrahedron Lett. 2005, 46, 2487–2490. [Google Scholar]
- 122.
- 122a. dos Santos T. R., Harnisch F., Nilges P., Schröder U., ChemSusChem 2015, 8, 886–893; [DOI] [PubMed] [Google Scholar]
- 122b. dos Santos T. R., Nilges P., Sauter W., Harnisch F., Schröder U., RSC Adv. 2015, 5, 26634–26643; [Google Scholar]
- 122c. Nilges P., dos Santos T. R., Harnisch F., Schröder U., Energy Environ. Sci. 2012, 5, 5231–5235. [Google Scholar]
- 123. Maletz R., Hinkamp L., Zobel M., Kahmen M. A. D., Schäfer H. J., Perspektiven nachwachsender Rohstoffe in der Chemie (Ed.: H. Eierdanz), Wiley, Hoboken, 1996, pp. 259–262. [Google Scholar]
- 124.
- 124a. Baizer M. M., J. Electrochem. Soc. 1964, 111, 215–222; [Google Scholar]
- 124b. Baizer M. M., Tetrahedron Lett. 1963, 4, 973–977; [Google Scholar]
- 124c. Baizer M. M., Ann. N. Y. Acad. Sci. 1969, 147, 614–617. [Google Scholar]
- 125. Baldwin J. E., North M., Flinn A., Moloney M. G., Tetrahedron 1989, 45, 1453–1464. [Google Scholar]
- 126. Dai J.-J., Huang Y.-B., Fang C., Guo Q.-X., Fu Y., ChemSusChem 2012, 5, 617–620. [DOI] [PubMed] [Google Scholar]
- 127. Matthessen R., Claes L., Fransaer J., Binnemans K., de Vos D. E., Eur. J. Org. Chem. 2014, 6649–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Libendi S. S., Demizu Y., Onomura O., Org. Biomol. Chem. 2009, 7, 351–356. [DOI] [PubMed] [Google Scholar]
- 129.
- 129a. Suga S., Okajima M., Fujiwara K., Yoshida J.-i., QSAR Comb. Sci. 2005, 24, 728–741; [Google Scholar]
- 129b. Horii D., Ameniya F., Fuchigami T., Atobe M., Chem. Eur. J. 2008, 14, 10382–10387; [DOI] [PubMed] [Google Scholar]
- 129c. Horii D., Fuchigami T., Atobe M., J. Am. Chem. Soc. 2007, 129, 11692–11693. [DOI] [PubMed] [Google Scholar]
- 130. Löwe H., Ehrfeld W., Electrochim. Acta 1999, 44, 3679–3689. [Google Scholar]
- 131. Horcajada R., Okajima M., Suga S., Yoshida J.-i., Chem. Commun. 2005, 1303–1305. [DOI] [PubMed] [Google Scholar]
- 132.
- 132a. Bouzek K., Jiricný V., Kodým R., Kristal J., Bystron T., Electrochim. Acta 2010, 55, 8172–8181; [Google Scholar]
- 132b. Attour A., Dirrenberger P., Rode S., Ziogas A., Matlosz M., Lapique F., Chem. Eng. Sci. 2011, 66, 480–489. [Google Scholar]
- 133. Roth G. P., Stalder R., Long T. R., Sauer D. R., Djuric S. W., J. Flow Chem. 2013, 3, 34–40. [Google Scholar]
- 134. Horii D., Atobe M., Fuchigame T., Marken F., Electrochem. Commun. 2005, 7, 35–39. [Google Scholar]
- 135. Kuleshova J., Hill-Cousins J. T., Birkin P. R., Brown R. C. D., Pletcher D., Underwood T. J., Electrochim. Acta 2012, 69, 197–202. [Google Scholar]
- 136. Green R. A., Brown R. C. D., Pletcher D., J. Flow Chem. 2015, 5, 31–36. [Google Scholar]
- 137. Green R. A., Brown R. C. D., Pletcher D., J. Flow Chem. 2016, 6, 191–197. [Google Scholar]
- 138. Kashiwagi T., Ameniya F., Fuchigami T., Atobe M., Chem. Commun. 2012, 48, 2806–2808. [DOI] [PubMed] [Google Scholar]
- 139. Arai T., tateno H., Nakabayashi K., Kashiwagi T., Atobe M., Chem. Commun. 2015, 51, 4891–4894. [DOI] [PubMed] [Google Scholar]
- 140. Watts K., Gattrell W., Wirth T., Beilstein J. Org. Chem. 2011, 7, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Arai K., Watts K., Wirth T., ChemistryOpen 2014, 3, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kashiwagi T., Elsler B., Waldvogel S. R., Fuchigami T., Atobe M., J. Electrochem. Soc. 2013, 160, G3058–G3061. [Google Scholar]
- 143. Hill-Cousins J. T., Kuleshova J., Green R. A., Birkin P. R., Pletcher D., Underwood T. J., Leach S. G., Brown R. C. D., ChemSusChem 2012, 5, 326–331. [DOI] [PubMed] [Google Scholar]
- 144. Sumi T., Saitoh T., Natsui K., Yamamoto T., Atobe M., Einaga Y., Nishiyama S., Angew. Chem. Int. Ed. 2012, 51, 5443–5446; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5539–5542. [Google Scholar]
- 145. Stalder R., Roth G. P., ACS Med. Chem. Lett. 2013, 4, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Takano K., Tateno H., Matsumura Y., Fukazawa A., Kashiwagi T., Nakabayashi K., Nagasawa K., Mitsushima S., Atobe M., Bull. Chem. Soc. Jpn. 2016, 89, 1178–1183. [Google Scholar]
- 147. Matsumura Y., Kakizaki Y., Tateno H., Kashiwagi T., Yamaji Y., Atobe M., RSC Adv. 2015, 5, 96851–96854. [Google Scholar]
- 148. Matsumura Y., Yamaji Y., Tateno H., Kashiwagi T., Atobe M., Chem. Lett. 2016, 45, 816–818. [Google Scholar]
- 149. Amemiya F., Fuse K., Fuchigami T., Atobe M., Chem. Commun. 2010, 46, 2730–2732. [DOI] [PubMed] [Google Scholar]
- 150. Tateno H., Matsumura Y., Nakabayashi K., Senbokub H., Atobe M., RSC Adv. 2015, 5, 98721–98723. [Google Scholar]
- 151. He P., Watts P., Marken F., Haswell S. J., Electrochem. Commun. 2005, 7, 918–924. [Google Scholar]
- 152. He P., Watts P., Marken F., Haswell S. J., Green Chem. 2007, 9, 20–22. [Google Scholar]
- 153. He P., Watts P., Marken F., Haswell S. J., Angew. Chem. Int. Ed. 2006, 45, 4146–4149; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 4252–4255. [Google Scholar]
- 154. Gütz C., Bänziger M., Bucher C., Galvão T. R., Waldvogel S. R., Org. Process Res. Dev. 2015, 19, 1428–1433. [Google Scholar]
- 155. Chapman M. R., Shafi Y. M., Kapur N., Nguyen B. N., Willans C. E., Chem. Commun. 2015, 51, 1282–1284. [DOI] [PubMed] [Google Scholar]
- 156.
- 156a. Gütz C., Stenglein A., Waldvogel S. R., Org. Process Res. Dev. 2017, 21, 771–778; [Google Scholar]
- 156b. Gütz C., Grimaudo V., Holtkamp M., Werra J., Frensemeier L., Kehl A., Karst U., Broekmann P., Waldvogel S. R., ChemElectroChem 2018, 5, 247–252. [Google Scholar]
- 157. Cardoso D. S. P., Sljukic B., Santos D. M. F., Sequeira C. A. C., Org. Process Res. Dev. 2017, 21, 1213–1226. [Google Scholar]
- 158. Botte G. G., Electrochem. Soc. Inteface 2014, 49–55. [Google Scholar]
- 159. Pletcher D., Walsh F. C., Industrial Electrochemistry, 2nd ed., Springer, 1990. [Google Scholar]
- 160. Schmidt V. M., Elektrochemische Verfahrenstechnik: Grundlagen, Reaktionstechnik, Prozessoptimierung, Wiley-VCH, Weinheim, 2003. [Google Scholar]
- 161.
- 161a.“Electrochemistry, 3. Organic Electrochemisty”: Steckhan E. in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, pp. 316–348; [Google Scholar]
- 161b.“Industrial Electroorganic Chemistry”: Pütter H. in Organic Electrochemistry, 4th. ed. (Eds. H. Lund, O. Hammerich), Marcel Dekker, New York, 2001, pp. 1259–1307. [Google Scholar]
- 162. Knunyants I. L., Vyazankin N. S., Bull. Acad. Sci. USSR Div. Chem. Sci. 1957, 253–256. [Google Scholar]
- 163.“The Use of Conducting Diamond in Electrochemistry”: Macpherson J. V. in Electrochemistry of Carbon Electrodes (Eds.: R. C. Alkire, P. N. Bartlett, J. Lipkowski), Wiley-VCH, Weinheim, 2015. [Google Scholar]
- 164. Folgueiras-Amador A. A., Philipps K., Guilbaud S., Poelakker J., Wirth T., Angew. Chem. Int. Ed. 2017, 56, 15446–15450; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15648–15653. [Google Scholar]
- 165. Wiebe A., Riehl B., Lips S., Franke R., Waldvogel S. R., Sci. Adv. 2017, 3, eaao3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Pletcher D., Li X., Wang S., Int. J. Hydrogen Energy 2012, 37, 7429–7435. [Google Scholar]
- 167.
- 167a. Brost R., Schank W., Waldecker J., US 20070117002, 2007;
- 167b. Huang Z., Zhu Z., Chen X., Wang H., Xia Y., Wen Z., Mei C., Jiang Z., Wang X., CN 105970310, 2016;
- 167c.P. Granados Mendoza, 2016, Intensification of the chlor-alkali process using a rotor-stator spinning disc membrane electrochemical reactor Eindhoven: Technische Universiteit Eindhoven.
- 168.
- 168a. Cognet P., Wilheim A. M., Delmas H., Lyazidi H. A., Fabre P. L., Ultrason. Sonochem. 2000, 7, 163–167; [DOI] [PubMed] [Google Scholar]
- 168b.“Electrosynthesis under Ultrasound and Centrifugal Fields”: Atobe M. in Encyclopedia of Applied Electrochemistry (Eds.: G. Kreysa, K.-i. Ota, R. F. Savinell), Springer, New York, 2014, pp. 821–826, and references cited therein. [Google Scholar]
- 169.
- 169a. Clausmeyer J., Wilde P., Löffler T., Ventosa E., Tschulik K., Schuhmann W., Electrochem. Commun. 2016, 73, 67–70; [Google Scholar]
- 169b. Vock S., Tschulik K., Uhlemann M., Hengst C., Fähler S., Schultz L., Neu V., J. Appl. Phys. 2015, 118, 233901; [Google Scholar]
- 169c. Yang X., Tschulik K., Uhlemann M., Odenbach S., Eckert K., IEEE Trans. Magn. 2014, 50, 1–4. [Google Scholar]
- 170.
- 170a. Göhler B., Hamelbeck V., Markus T. Z., Kettner M., Hanne G. F., Vager Z., Naaman R., Zacharias H., Science 2011, 331, 894–897; [DOI] [PubMed] [Google Scholar]
- 170b. Xie Z., Markus T. Z., Cohen S. R., Vager Z., Gutierrez R., Naaman R., Nano Lett. 2011, 11, 4652–4655; [DOI] [PubMed] [Google Scholar]
- 170c. Naaman R., Waldeck D. H., J. Phys. Chem. Lett. 2012, 3, 2178–2187; [DOI] [PubMed] [Google Scholar]
- 170d. Ravi S., Sowmiya P., Karthikeyan A., Spin 2013, 3, 1350003; [Google Scholar]
- 170e. Mishra D., Markus T. Z., Naaman R., Kettner M., Göhler B., Zacharias H., Friedman N., Sheves M., Fontanesi C., Proc. Natl. Acad. Sci. USA 2013, 110, 14872–14876; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170f. Carmeli I., Kumar K. S., Hieflero O., Carmeli C., Naaman R., Angew. Chem. Int. Ed. 2014, 53, 8953–8958; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9099–9104; [Google Scholar]
- 170g. Kettner M., Göhler B., Zacharias H., Mishra D., Kiran V., Naaman R., Fontanesi C., Waldeck D. H., Sęk S., Pawłowski J., Juhaniewicz J., J. Phys. Chem. C 2015, 119, 14542–14547. [Google Scholar]
- 171.A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes, S. R. Waldvogel, Angew. Chem. Int. Ed 2018, 57, DOI: https://doi.org/10.1002/anie.201711060; Angew. Chem 2018, 130, DOI: https://doi.org/10.1002/ange.201711060.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary