

Abstract
An efficient photoredox/nickel catalyzed sulfonylation reaction of aryl, heteroaryl, and vinyl halides has been achieved for the first time. This newly developed sulfonylation protocol provides a versatile method for the synthesis of diverse aromatic sulfones at room temperature and shows excellent functional group tolerance. The electrophilic coupling partners are not limited to aryl, heteroaryl, and vinyl bromides and iodides, but also includes less reactive aryl chlorides as suitable substrates for this transformation.
Keywords: aryl halides, cross-coupling, nickel, photochemistry, sulfone
Sulfones are highly important organic molecules because of their versatile synthetic utility in organic synthesis as well as the widespread presence of sulfonyl groups in pharmaceuticals, agrochemicals, biologically active compounds, and polymer materials.1, 2 Conventionally, these valuable compounds are synthesized by oxidation of sulfides,3 sulfonylation of arenes,4 or palladium or copper‐catalyzed arylation of sulfinate salts.5 These methods suffer from significant drawbacks, including the use of foul‐smelling thiols and strong oxidizing reagents, harsh acidic treatments, and high reaction temperatures, which can limit the functional group tolerance and substrate scope. Alternatively, SO2 surrogates such as DABCO⋅(SO2)2 and K2S2O5 have been applied to the synthesis of sulfones by fixation of sulfur dioxide to generate sulfinate anion intermediates which can then undergo (SO2)‐arylation/alkylation.6
In recent years, increasing attention has been devoted to the field of dual photoredox and transition‐metal catalysis, and useful transformations have been achieved by this strategy.7, 8, 9, 10 In particular, photoredox/nickel catalysis proved attractive because of the unique catalytic properties of nickel catalysts.8, 9, 10 Pioneering works in this field focused on C(sp3)−C(sp2) bond formations by coupling aryl halides with benzylic trifluoroborates and α‐carboxy sp3‐carbon atoms.8 Since then, progress has been made in the field of C−C bond formation.9 Moreover, this elegant strategy has also been applied to the synthesis of aryl ethers, aryl esters, aryl amines, indolines, triarylphosphine oxides, and thioethers by different C–heteroatom bond formations,10 thus confirming the enormous potential of dual photoredox/metal catalysis not only in the improvement of known reactions but also in the discovery of novel catalytic protocols.
As part of our continuing studies in the area of photoredox and transition‐metal catalysis, we herein report the first photoredox/metal catalyzed cross‐coupling of sulfinate salts with aryl, heteroaryl, and vinyl halides at room temperature (Scheme 1). This protocol provides a versatile approach to aromatic sulfones with a broad substrate scope and excellent functional group tolerance. Notably, less‐reactive aryl chlorides could also be converted into the corresponding sulfones.
Scheme 1.

Photoredox/nickel catalyzed synthesis of aromatic sulfones at room temperature.
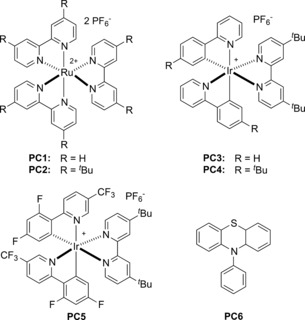
Our study commenced with 4‐bromobenzonitrile (1 a) and sodium 4‐methylbenzenesulfinate (2 a) as model substrates. Initially, the ratio of 1 a and 2 a was set as 1:1 with PC1 as the photocatalyst, NiCl2⋅glyme as the nickel source, dtbbpy as the ligand, and DMF as the solvent, which afforded the corresponding sulfone 3 a in 43 % yield only (Table 1, entry 1). The use of K2CO3 as a base decreased the yield dramatically (entry 2). The yield of 3 a increased to 70 % when two equivalents of 2 a were employed (entry 3). After screening several commonly used photocatalysts, including an organic dye (entries 4–8), the Ir complex PC4 was found to give the best result (entry 6). A series of nickel catalysts such as NiBr2⋅O(CH2CH2OCH3)2, Ni(OAc)2⋅4 H2O, Ni(acac)2, NiCl2, and Ni(cod)2 were subsequently examined, however no further improvement was observed (entries 9–13). The use of DMF as a solvent was found to be crucial for this transformation. When other solvents were utilized, the sulfonylation reaction either did not take place or occurred in low yield (entries 14–16). Performing the reaction under undegassed conditions provided the product in 18 % yield, thus indicating the importance of avoiding the presence of molecular oxygen in the reaction (entry 17). Control experiments confirmed the role of the photocatalyst, light, and nickel catalyst for the reaction (entries 18 and 19). In addition, a CF Lamp was tested and proved suitable for this transformation, thus providing the corresponding product in 57 % yield (entry 20).
Table 1.
Optimization of the reaction conditions.[a]

| Entry | PC | [Ni] | Solvent | Yield [%][b] |
|---|---|---|---|---|
| 1[c] | PC1 | NiCl2⋅glyme | DMF | 43 |
| 2[c,d] | PC1 | NiCl2⋅glyme | DMF | trace |
| 3 | PC1 | NiCl2⋅glyme | DMF | 70 |
| 4 | PC2 | NiCl2⋅glyme | DMF | 74 |
| 5 | PC3 | NiCl2⋅glyme | DMF | 75 |
| 6 | PC4 | NiCl2⋅glyme | DMF | 89(86[e]) |
| 7 | PC5 | NiCl2⋅glyme | DMF | 60 |
| 8 | PC6 | NiCl2⋅glyme | DMF | 63 |
| 9 | PC4 | NiBr2⋅O(CH2CH2OCH3)2 | DMF | 86 |
| 10 | PC4 | Ni(OAc)2⋅4 H2O | DMF | 44 |
| 11 | PC4 | Ni(acac)2 | DMF | 57 |
| 12 | PC4 | NiCl2 | DMF | 30 |
| 13 | PC4 | Ni(cod)2 | DMF | 69 |
| 14 | PC4 | NiCl2⋅glyme | CH3CN | 0 |
| 15 | PC4 | NiCl2⋅glyme | PhCF3 | 0 |
| 16 | PC4 | NiCl2⋅glyme | THF | trace |
| 17[f] | PC4 | NiCl2⋅glyme | DMF | 18 |
| 18[g] | – | NiCl2⋅glyme | DMF | 0 |
| 19 | PC4 | – | DMF | 0 |
| 20[h] | PC4 | NiCl2⋅glyme | DMF | 57 |
| ||||
[a] Reaction conditions: 1 a (0.1 mmol), 2 a (0.2 mmol), photocatalyst (0.001 mmol), [Ni] (0.01 mmol), ligand (0.01 mmol), and degassed solvent (1.0 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield determined by GC. [c] 2 a (1 equiv). [d] K2CO3 (2 equiv) was added. [e] Yield of isolated product. [f] Undegassed. [g] No light. [h] CF‐Lamp was used instead of blue LED strips. cod=1,5‐cyclooctadiene, dtbbpy=4,4′‐di‐tert‐butyl‐2,2′‐bipyridine, Ts=4‐toluenesulfonyl.
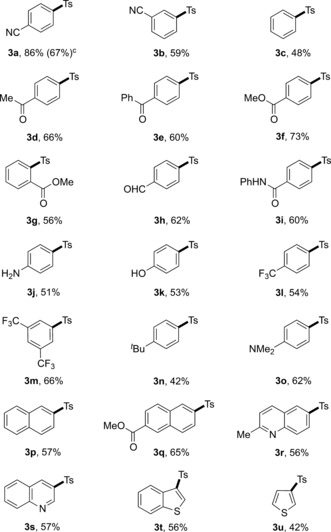
With the optimized reaction conditions in hand, the scope with respect to the aryl bromides was firstly evaluated (Table 2). A wide variety of aryl bromides bearing electron‐donating and electron‐withdrawing functional groups gave the corresponding products in moderate to excellent yields. The position of the substituents on the aryl ring had a minor effect on the efficiency of this transformation. For example, not only the aryl bromides 1 a and 1 f possessing para substituents but also the substrates 1 b and 1 g, bearing substituents in meta and ortho positions, afforded the corresponding products in good yields. In addition, our newly developed protocol tolerated a variety of functionalities, including ketone (3 d and 3 e), methylester (3 f, 3 g, and 3 q), aldehyde (3 h), amide (3 i), trifluoromethyl (3 l and 3 m), t‐butyl (3 n), and tertiary amine (3 o). Notably, a reactive primary amine and hydroxy group on the aromatic ring were also tolerated (3 j and 3 k). Moreover, naphthyl bromides (1 p and 1 q) also underwent this reaction efficiently, thus giving the corresponding products in good yields.
Table 2.
Scope of aryl bromides.[a,b]
|
[a] Reaction conditions: Aryl bromide 1 (0.1 mmol), 2 a (0.2 mmol), PC4 (0.001 mmol), NiCl2⋅glyme (0.01 mmol), dtbbpy (0.01 mmol), and degassed DMF (1 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield after purification. [c] Reaction performed on a 2.0 mmol scale.
Significantly, the scope of this protocol could be extended to pharmaceutically relevant heteroaromatic bromides such as quinoline (1 r and 1 s), benzothiophene (1 t), and thiophene (1 u) derivatives (Table 2). Noteworthy, 3 a was obtained in a good yield of 67 % when the reaction was performed on a 2.0 mmol scale in the presence of 0.5 mol % photocatalyst, thus indicating the scalability and practicability of this mild sulfonylation protocol.
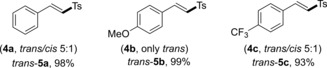
In addition, our newly developed protocol could also be readily extended to vinyl bromides (Table 3). Accordingly, substrates possessing electron‐donating and electron‐withdrawing functional groups gave the α,β‐unsaturated sulfones 5 a–c in excellent yields (93–99 %). It is important to note that only the trans products were generated, even when the vinyl bromides 4 a and 4 c, containing a trans/cis mixture, were employed.
Table 3.
Scope of vinyl bromides.[a,b]

|
[a] Reaction conditions: vinyl bromide 4 (0.1 mmol), 2 a (0.2 mmol), PC4 (0.001 mmol), NiCl2⋅glyme (0.01 mmol), dtbbpy (0.01 mmol), and degassed DMF (1 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield after purification.
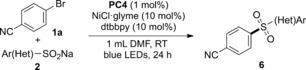
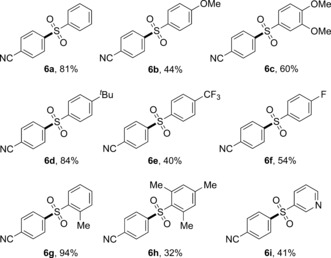
Next, the scope with respect to the sodium sulfinates 2 was explored (Table 4). A wide range of structurally diverse sodium sulfinates were suitable substrates for this transformation. For example, benzenesulfinate and sodium sulfinates bearing methoxy groups could generate the corresponding diarylsulfones 6 a–c in moderate to excellent yields. Also, functional groups such as t‐butyl (6 d), trifluoromethyl (6 e), and fluoro (6 f) were well tolerated under our reaction conditions. Strikingly, sodium sulfinate possessing a methyl group in the ortho position of the aromatic ring could undergo this reaction smoothly, thus affording the corresponding product 6 g in 94 % yield. However, continuously increasing steric hindrance led to diminished yield (6 h). Likewise, the reaction could also be applied to the heterocyclic sodium sulfinate 2 i, thus giving the heterocycle‐containing sulfone 6 i, which is important in medicinal chemistry.
Table 4.
Scope of sodium sulfinates.[a,b]
|
[a] Reaction conditions: 1 a (0.1 mmol), sodium sulfinate 2 (0.2 mmol), PC4 (0.001 mmol), NiCl2⋅glyme (0.01 mmol), dtbbpy (0.01 mmol), and degassed DMF (1 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield after purification.
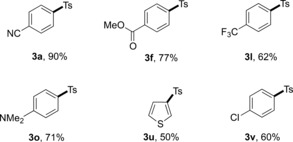
Moreover, aryl iodides were also suitable substrates for this transformation (Table 5). Likewise, substrates bearing electron‐donating and electron‐withdrawing functional groups both afforded the corresponding products in good to high yields. Interestingly, a chloro group was well tolerated under the present conditions.
Table 5.
Scope of aryl iodides.[a,b]

|
[a] Reaction conditions: aryl iodide 7 (0.1 mmol), 2 a (0.2 mmol), PC4 (0.001 mmol), NiCl2⋅glyme (0.01 mmol), dtbbpy (0.01 mmol), and degassed DMF (1 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield after purification.
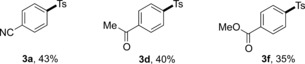
It is noteworthy that minor modification of the standard conditions, by just switching the dtbbpy ligand to the more electron rich 4,4′‐dimethoxy‐2,2′‐bipyridine (4,4′‐(MeO)2‐bpy) ligand, made this protocol applicable to more challenging aryl chloride substrates (8), thus indicating the potential of our newly developed sulfonylation protocol in applying less reactive electrophiles (Table 6).
Table 6.
Scope of aryl chlorides.[a,b]

|
[a] Reaction conditions: Aryl chloride 8 (0.1 mmol), 2 a (0.2 mmol), PC4 (0.001 mmol), NiCl2⋅glyme (0.01 mmol), 4,4′‐(MeO)2‐bpy (0.01 mmol), and degassed DMF (1 mL) at room temperature under irradiation with 2.6 W blue LED strips for 24 h. [b] Yield after purification.
To shed light on the mechanism of this protocol, a radical‐trapping experiment was conducted with two equivalents of TEMPO (2,2,6,6‐tetramethyl‐1‐piperidinyloxy; Scheme 2). The reaction was completely suppressed and no desired product was detected, thus suggesting the involvement of a sulfonyl radical in the transformation.
Scheme 2.
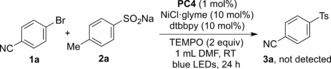
Radical‐trapping experiment.
Based on our results and previous studies9a, 11 a mechanism for this new photoredox/nickel catalyzed sulfonylation protocol is proposed in Scheme 3. Firstly, the IrIII complex absorbs visible light and gives a long‐lived triplet excited state (PC4, E 1/2 red [*IrIII/IrII]=+0.99 V versus SCE in CH3CN),12 which then reacts with the sodium sulfinate A (TsNa, E red=+0.45 V versus SCE in CH3CN)12 to afford the intermediate B along with the formation of the IrII reductant (PC4, E 1/2 red [IrIII/IrII]=−1.48 V versus SCE in CH3CN).12 B then resonates to the radical C, which subsequently adds to Ni0 to form the NiI intermediate D. The oxidative addition of aryl halide to D delivers the NiIII intermediate E, which is prone to undergoing reductive elimination to produce the coupling product F and a NiI complex. Finally, the NiI complex undergoes one‐electron reduction with the IrII reductant to regenerate Ni0 {E 1/2 red [NiI/Ni0] > E 1/2 red [NiII/Ni0]=−1.2 V versus SCE in DMF}8b along with the ground‐state IrIII complex.
Scheme 3.
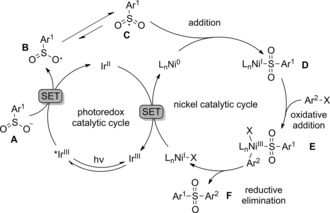
Proposed mechanism for the new photoredox/nickel catalyzed sulfonylation reaction.
In summary, we have developed a novel and efficient method for the synthesis of sulfones by photoredox/nickel catalysis for the first time. This protocol allows the cross‐coupling of a series of sodium sulfinates with a wide range of aryl, heteroaryl, and vinyl bromides and iodides, as well as more challenging aryl chlorides. Importantly neither sacrificial reagents nor organic electron mediators are necessary in this reaction. Moreover, the utility of sodium sulfinates as precursors of sulfonyl radicals and the generation of reactive NiIII intermediates promote this transformation at room temperature. Therefore, the reaction possesses a broad tolerance of functional groups, showing its advantages in comparison to the traditional methods.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
H.Y. acknowledges the China Scholarship Council. The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ ERC Grant Agreement no. 617044 (SunCatChem). We thank Prof. Aiwen Lei and Huamin Wang for fruitful discussions.
H. Yue, C. Zhu, M. Rueping, Angew. Chem. Int. Ed. 2018, 57, 1371.
References
- 1.
- 1a. Simpkins N. S., Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993; [Google Scholar]
- 1b. Stirling C. J. M., The Chemistry of Sulphones and Sulphoxides, Wiley, New York, 1988; [Google Scholar]
- 1c. Harrak Y., Casula G., Basset J., Rosell G., Plescia S., Raffa D., Cusimano M. G., Pouplana R., Pujol M. D., J. Med. Chem. 2010, 53, 6560–6571; [DOI] [PubMed] [Google Scholar]
- 1d. Smith D. A., Jones R. M., Curr. Opin. Drug Discovery Dev. 2008, 11, 72–79; [PubMed] [Google Scholar]
- 1e. Sun Z.-Y., Botros E., Su A.-D., Kim Y., Wang E., Baturay N. Z., Kwon C.-H., J. Med. Chem. 2000, 43, 4160–4168; [DOI] [PubMed] [Google Scholar]
- 1f. Silvestri R., De Martino G., La Regina G., Artico M., Massa S., Vargiu L., Mura M., Loi A. G., Marceddu T., La Colla P., J. Med. Chem. 2003, 46, 2482–2493; [DOI] [PubMed] [Google Scholar]
- 1g. Otzen T., Wempe E. G., Kunz B., Bartels R., Lehwark-Yvetot G., Hänsel W., Schaper K.-J., Seydel J. K., J. Med. Chem. 2004, 47, 240–253. [DOI] [PubMed] [Google Scholar]
- 2.For recent reviews on the synthesis of sulfones, see:
- 2a. Liu N.-W., Liang S., Manolikakes G., Synthesis 2016, 48, 1939–1973; [Google Scholar]
- 2b. Liu N.-W., Liang S., Manolikakes G., Adv. Synth. Catal. 2017, 359, 1308–1319. [Google Scholar]
- 3.For selected examples, see:
- 3a. Shaabani A., Mirzaei P., Naderi S., Lee D. G., Tetrahedron 2004, 60, 11415–11420; [Google Scholar]
- 3b. Griffin R. J., Henderson A., Curtin N. J., Echalier A., Endicott J. A., Hardcastle I. R., Newell D. R., Noble M. E., Wang L.-Z., Golding B. T., J. Am. Chem. Soc. 2006, 128, 6012–6013; [DOI] [PubMed] [Google Scholar]
- 3c. Sato K., Hyodo M., Aoki M., Zheng X.-Q., Noyori R., Tetrahedron 2001, 57, 2469–2476; [Google Scholar]
- 3d. Trost B. M., Curran D. P., Tetrahedron Lett. 1981, 22, 1287–1290; [Google Scholar]
- 3e. Kozak J. A., Dake G. R., Angew. Chem. Int. Ed. 2008, 47, 4221–4223; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4289–4291; [Google Scholar]
- 3f. Catarinella M., Grüner T., Strittmatter T., Marx A., Mayer T. U., Angew. Chem. Int. Ed. 2009, 48, 9072–9076; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9236–9240; [Google Scholar]
- 3g. Pritzius A. B., Breit B., Angew. Chem. Int. Ed. 2015, 54, 3121–3125; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3164–3168. [Google Scholar]
- 4.For early examples, see:
- 4a. Olah G. A., Kobayashi S., Nishimura J., J. Am. Chem. Soc. 1973, 95, 564–569; [Google Scholar]
- 4b. Effenberger F., Huthmacher K., Angew. Chem. Int. Ed. Engl. 1974, 13, 409–410; [Google Scholar]; Angew. Chem. 1974, 86, 409–410; [Google Scholar]
- 4c. Hyatt J. A., White A. W., Synthesis 1984, 1984, 214–217; [Google Scholar]
- 4d. Répichet S., Le Roux C., Hernandez P., Dubac J., Desmurs J.-R., J. Org. Chem. 1999, 64, 6479–6482. [Google Scholar]
- 5.
- 5a. Cacchi S., Fabrizi G., Goggiamani A., Parisi L. M., Org. Lett. 2002, 4, 4719–4721; [DOI] [PubMed] [Google Scholar]
- 5b. Baskin J. M., Wang Z., Org. Lett. 2002, 4, 4423–4425. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Emmett E. J., Hayter B. R., Willis M. C., Angew. Chem. Int. Ed. 2013, 52, 12679–12683; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12911–12915; [Google Scholar]
- 6b. Deeming A. S., Russell C. J., Willis M. C., Angew. Chem. Int. Ed. 2015, 54, 1168–1171; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1184–1187; [Google Scholar]
- 6c. Rocke B. N., Bahnck K. B., Herr M., Lavergne S., Mascitti V., Perreault C., Polivkova J., Shavnya A., Org. Lett. 2014, 16, 154–157; [DOI] [PubMed] [Google Scholar]
- 6d. Shavnya A., Hesp K. D., Mascitti V., Smith A. C., Angew. Chem. Int. Ed. 2015, 54, 13571–13575; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13775–13779; [Google Scholar]
- 6e. Shavnya A., Coffey S. B., Smith A. C., Mascitti V., Org. Lett. 2013, 15, 6226–6229; [DOI] [PubMed] [Google Scholar]
- 6f. Johnson M. W., Bagley S. W., Mankad N. P., Bergman R. G., Mascitti V., Toste F. D., Angew. Chem. Int. Ed. 2014, 53, 4404–4407; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4493–4496; [Google Scholar]
- 6g. von Wolff N., Char J., Frogneux X., Cantat T., Angew. Chem. Int. Ed. 2017, 56, 5616–5619; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5708–5711; For reviews, see: [Google Scholar]
- 6h. Deeming A. S., Emmett E. J., Richards-Taylor C. S., Willis M. C., Synthesis 2014, 46, 2701–2710; [Google Scholar]
- 6i. Liu G., Fan C., Wu J., Org. Biomol. Chem. 2015, 13, 1592–1599; [DOI] [PubMed] [Google Scholar]
- 6j. Emmett E. J., Willis M. C., Asian J. Org. Chem. 2015, 4, 602–611. [Google Scholar]
- 7.For reviews and examples of photoredox/transition metal catalysis, see:
- 7a. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Tellis J. C., Kelly C. B., Primer D. N., Jouffroy M., Patel N. R., Molander G. A., Acc. Chem. Res. 2016, 49, 1429–1439; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Gui Y.-Y., Sun L., Lu Z.-P., Yu D.-G., Org. Chem. Front. 2016, 3, 522–526; [Google Scholar]
- 7d. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969–1979; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Vila C., ChemCatChem 2015, 7, 1790–1793; [Google Scholar]
- 7f. Jahn E., Jahn U., Angew. Chem. Int. Ed. 2014, 53, 13326–13328; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13542–13544; [Google Scholar]
- 7g. Hopkinson M. N., Sahoo B., Li J. L., Glorius F., Chem. Eur. J. 2014, 20, 3874–3886; [DOI] [PubMed] [Google Scholar]
- 7h. Hopkinson M. N., Tlahuext-Aca A., Glorius F., Acc. Chem. Res. 2016, 49, 2261–2272; [DOI] [PubMed] [Google Scholar]
- 7i. Kalyani D., McMurtrey K. B., Neufeldt S. R., Sanford M. S., J. Am. Chem. Soc. 2011, 133, 18566–18569; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7j. Neufeldt S. R., Sanford M. S., Adv. Synth. Catal. 2012, 354, 3517–3522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7k. Ye Y., Sanford M. S., J. Am. Chem. Soc. 2012, 134, 9034–9037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7l. Ye Y., Künzi S. A., Sanford M. S., Org. Lett. 2012, 14, 4979–4981; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7m. Rueping M., Koenigs R. M., Poscharny K., Fabry D. C., Leonori D., Vila C., Chem. Eur. J. 2012, 18, 5170–5174; [DOI] [PubMed] [Google Scholar]
- 7n. Fabry D. C., Zoller J., Raja S., Rueping M., Angew. Chem. Int. Ed. 2014, 53, 10228–10231; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10392–10396; [Google Scholar]
- 7o. Zoller J., Fabry D. C., Ronge M. A., Rueping M., Angew. Chem. Int. Ed. 2014, 53, 13264–13268; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13480–13484; [Google Scholar]
- 7p. Fabry D. C., Ronge M. A., Zoller J., Rueping M., Angew. Chem. Int. Ed. 2015, 54, 2801–2805; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2843–2847; [Google Scholar]
- 7q. Sahoo B., Hopkinson M. N., Glorius F., J. Am. Chem. Soc. 2013, 135, 5505–5508; [DOI] [PubMed] [Google Scholar]
- 7r. Hopkinson M. N., Sahoo B., Glorius F., Adv. Synth. Catal. 2014, 356, 2794–2800; [Google Scholar]
- 7s. Shu X.-z., Zhang M., He Y., Frei H., Toste F. D., J. Am. Chem. Soc. 2014, 136, 5844–5847; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7t. Kim S., Rojas-Martin J., Toste F. D., Chem. Sci. 2016, 7, 85–88; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7u. Huo H., Harms K., Meggers E., J. Am. Chem. Soc. 2016, 138, 6936–6939; [DOI] [PubMed] [Google Scholar]
- 7v. Xie J., Zhang T., Chen F., Mehrkens N., Rominger F., Rudolph M., Hashmi A. S. K., Angew. Chem. Int. Ed. 2016, 55, 2934–2938; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2987–2991; [Google Scholar]
- 7w. Xie J., Rudolph M., Rominger F., Hashmi A. S. K., Angew. Chem. Int. Ed. 2017, 56, 7266–7270; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7372–7376. [Google Scholar]
- 8.
- 8a. Tellis J. C., Primer D. N., Molander G. A., Science 2014, 345, 433–436; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W., Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Examples:
- 9a. Gutierrez O., Tellis J. C., Primer D. N., Molander G. A., Kozlowski M. C., J. Am. Chem. Soc. 2015, 137, 4896–4899; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Primer D. N., Karakaya I., Tellis J. C., Molander G. A., J. Am. Chem. Soc. 2015, 137, 2195–2198; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Ryu D., Primer D. N., Tellis J. C., Molander G. A., Chem. Eur. J. 2016, 22, 120–123; [DOI] [PubMed] [Google Scholar]
- 9d. Karakaya I., Primer D. N., Molander G. A., Org. Lett. 2015, 17, 3294–3297; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. El Khatib M., Serafim R. A. M., Molander G. A., Angew. Chem. Int. Ed. 2016, 55, 254–258; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 262–266; [Google Scholar]
- 9f. Yamashita Y., Tellis J. C., Molander G. A., Proc. Natl. Acad. Sci. USA 2015, 112, 12026–12029; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Noble A., McCarver S. J., MacMillan D. W., J. Am. Chem. Soc. 2015, 137, 624–627; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9h. Chu L., Lipshultz J. M., MacMillan D. W., Angew. Chem. Int. Ed. 2015, 54, 7929–7933; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8040–8044; [Google Scholar]
- 9i. Le C. C., MacMillan D. W., J. Am. Chem. Soc. 2015, 137, 11938–11941; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9j. Corcé V., Chamoreau L. M., Derat E., Goddard J. P., Ollivier C., Fensterbank L., Angew. Chem. Int. Ed. 2015, 54, 11414–11418; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11576–11580; [Google Scholar]
- 9k. Jouffroy M., Primer D. N., Molander G. A., J. Am. Chem. Soc. 2016, 138, 475–478; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9l. Lévêque C., Chenneberg L., Corcé V., Ollivier C., Fensterbank L., Chem. Commun. 2016, 52, 9877–9880; [DOI] [PubMed] [Google Scholar]
- 9m. Fan L., Jia J., Hou H., Lefebvre Q., Rueping M., Chem. Eur. J. 2016, 22, 16437–16440; [DOI] [PubMed] [Google Scholar]
- 9n. Joe C. L., Doyle A. G., Angew. Chem. Int. Ed. 2016, 55, 4040–4043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4108–4111. [Google Scholar]
- 10.
- 10a. Terrett J. A., Cuthbertson J. D., Shurtleff V. W., MacMillan D. W., Nature 2015, 524, 330–334; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Welin E. R., Le C., Arias-Rotondo D. M., McCusker J. K., MacMillan D. W., Science 2017, 355, 380–385; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Oderinde M. S., Jones N. H., Juneau A., Frenette M., Aquila B., Tentarelli S., Robbins D. W., Johannes J. W., Angew. Chem. Int. Ed. 2016, 55, 13219–13223; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13413–13417; [Google Scholar]
- 10d. Corcoran E. B., Pirnot M. T., Lin S., Dreher S. D., DiRocco D. A., Davies I. W., Buchwald S. L., MacMillan D. W., Science 2016, 353, 279–283; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e. Oderinde M. S., Frenette M., Robbins D. W., Aquila B., Johannes J. W., J. Am. Chem. Soc. 2016, 138, 1760–1763. [DOI] [PubMed] [Google Scholar]
- 11. Wang H., Lu Q., Chiang C. W., Luo Y., Zhou J., Wang G., Lei A., Angew. Chem. Int. Ed. 2017, 56, 595–599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 610–614. [Google Scholar]
- 12.See the cyclic voltammetry measurements of the photocatalyst PC4 and TsNa in the Supporting Information in detail.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary