

Abstract
Improvements in immunosuppression have modified short‐term survival of deceased‐donor allografts, but not their rate of long‐term failure. Mismatches between donor and recipient HLA play an important role in the acute and chronic allogeneic immune response against the graft. Perfect matching at clinically relevant HLA loci does not obviate the need for immunosuppression, suggesting that additional genetic variation plays a critical role in both short‐ and long‐term graft outcomes. By combining patient data and samples from supranational cohorts across the United Kingdom and European Union, we performed the first large‐scale genome‐wide association study analyzing both donor and recipient DNA in 2094 complete renal transplant‐pairs with replication in 5866 complete pairs. We studied deceased‐donor grafts allocated on the basis of preferential HLA matching, which provided some control for HLA genetic effects. No strong donor or recipient genetic effects contributing to long‐ or short‐term allograft survival were found outside the HLA region. We discuss the implications for future research and clinical application.
Keywords: basic (laboratory) research/science, genomics, graft survival, kidney transplantation/nephrology, rejection, translational research/science
Short abstract
The largest multicenter genome‐wide association study to date on short‐ and long‐term renal transplantation outcomes finds no loci outside the HLA region, highlighting challenges in phenotypic heterogeneity and sample size while also providing a reference dataset for future meta‐analyses.
Abbreviations
- NIHR
The National Institute for Health Research
- UKIRTC
The United Kingdom and Ireland Renal Transplant Consortium
- WTCCC3
Wellcome Trust Case Control Consortium 3
1. INTRODUCTION
Kidney transplantation is a highly successful treatment for end‐stage renal failure, with significant benefits for recipients both in survival and quality of life. Early outcomes have steadily improved over the last 10 years,1 with risk‐adjusted and death‐censored, 1‐year renal graft survival rates of 94% and 97% for deceased and living donor transplants, respectively.2 However, both late allograft loss and increased mortality among transplant recipients remain key challenges for the transplant community. There are a wide number of factors that are known to influence long‐term transplant outcome, including donor factors such as age and comorbidity, recipient factors such as comorbidity and response to immunosuppression, as well as allograft ischemic time, the degree of HLA mismatch, and the development of donor‐specific antibodies.3, 4, 5 However, a comprehensive understanding of the pathophysiology of graft failure has remained elusive, with the observed variation in patient outcomes still inadequately explained by our current understanding of risk factors. An improved understanding of the determinants of transplantation outcome would allow the development of truly personalized approaches to the management of transplant recipients.
The importance of genetic factors in transplantation has been clear since the inception of the technique, with the first successful kidney transplant having been performed between identical twins in 1954. Renal transplantation between identical twins continues to show excellent long‐term outcomes,6, 7 and HLA matching has a large impact on graft survival even in the modern era of immunosuppression.8
HLA genes are highly polymorphic, and demonstrate the importance of genetic variation in donor‐recipient pairing that impacts on long‐term outcomes. However, over recent decades, our ability to assay human genetic variation beyond the HLA region has increased considerably.
A number of studies have been published over recent years exploring the association between genotypes of interest and renal transplant outcomes.9, 10 A large proportion of these studies have concentrated on immune‐related genes, based on the hypothesis that the risk of acute rejection or late allograft loss may be modulated by genetic variation in the immune response. As summarized in Table S1, associations have been described between various transplant phenotypes and single nucleotide polymorphisms (SNPs) in a number of genes including those encoding tumor necrosis factor‐α, interleukins‐1, ‐6, and ‐10, and interferon‐γ. Of note, many of these studies have reported inconsistent findings. For example, analysis of DNA from donor‐recipient pairs in the Collaborative Transplant Study failed to replicate an earlier finding that particular combinations of C3 genotypes in the donor and recipient were associated with reduced graft survival.11, 12 While some of this discrepancy might be explained by methodological or populational differences between these studies, it is difficult to draw firm conclusions about the role of these genetic variations.13
More recently, attention has also focused on non‐immune‐related genetic risk variants. Donor genetic variation in CAV1 (caveolin‐1),14 APOL1 (apolipoprotein‐L1),15, 16 or ABCB1 (ATP‐binding cassette, subfamily‐B, member‐1, expressed in the kidney) genes 17, 18 has been reported to be associated with increased risk of allograft failure or poorer recipient survival. Recipient genetic variation effects on graft and patient survival have only been reported in 1 cohort for CAV1.14 In addition to effects of donor genetic variants, it might be expected that recipient genotypes in other pharmacometabolic pathways might also impact on transplant outcomes such as increased risk of acute rejection.19
In general, candidate gene studies in renal transplantation have so far failed to provide consistent and reproducible results. Some of the reasons for this may include small sample sizes, variations in genotyping methodology and strategy, and, perhaps most importantly, a lack of consistency in clinical phenotyping.20 Genome‐wide association studies (GWAS) have contributed greatly to an increased understanding of complex common conditions such as inflammatory bowel disease, hypertension, type 2 diabetes, and schizophrenia.21 A small number of GWAS have been reported in the field of renal transplantation, describing SNPs associated with cardiovascular adverse events in recipients taking calcineurin inhibitor immunosuppression,22 2 SNPs associated with serum creatinine levels at 5 years posttransplant,23 and a number of SNPs associated with the development of new‐onset diabetes after transplantation.24 Recently, a GWAS using pooled DNA of recipient‐only origin found variation in 2 new loci associated with acute rejection in both univariate and multivariate analysis.25 However, these studies were underpowered for discovery of genetic variants with small effect sizes.
The Wellcome Trust Case Control Consortium (http://www.wtccc.org.uk/ccc3) has led the deployment of GWAS in a wide range of conditions. As part of WTCCC‐3, all renal transplant centers in the United Kingdom and Ireland formed the United Kingdom and Ireland Renal Transplant Consortium (UKIRTC; http://www.ukirtc.org). Collaborative initiatives such as these are essential for the collection of adequate sample numbers, for the sharing of expertise, standardization of techniques, and building consensus on accurate phenotyping of clinical data. Through this consortium, 3936 samples comprising 2094 complete donor‐recipient pairs were tested in the GWAS discovery phase, and an additional 5866 complete donor‐recipient pairs in the replication phase, making this the largest GWAS conducted to date in renal transplantation outcomes.
2. METHODS
2.1. Discovery study participants
The large multicenter United Kingdom and Ireland Renal Transplant Consortium (http://www.UKIRTC.org), coordinated by King's College London in partnership with the WTCCC‐3 and the National Health Service Blood and Transplant database (NHS‐BT), sourced all available good‐quality stored DNA samples and pre‐existing GWAS data from both recipients and their corresponding donors from all renal transplantation centers in the United Kingdom and the Republic of Ireland (listed in Table S2). The study was approved by the Hammersmith and Queen Charlotte's & Chelsea Research Ethics Committee REC No 08/H0707/1, on October 14, 2009. Third‐party anonymized clinical data were provided by NHS‐BT UK. All samples and anonymized data for the replication cohort were provided by the University of Heidelberg.
Following genotyping and GWAS quality control (see below), there were 2689 unique recipients, 2204 unique donors, and 2094 complete donor‐recipient transplantation pairs available for analysis. Figure 1 describes the study design and analysis steps. Table 1 provides additional information for the complete donor‐recipient pairs (discrepancies between Figure 1 and Table 1 are because some recipients received more than 1 graft, some donors donated 2 kidneys to different recipients, and not all recipients had matching donor GWAS data and vice versa). The samples and data referred to transplants that took place between December 1981 and December 2007.
Figure 1.
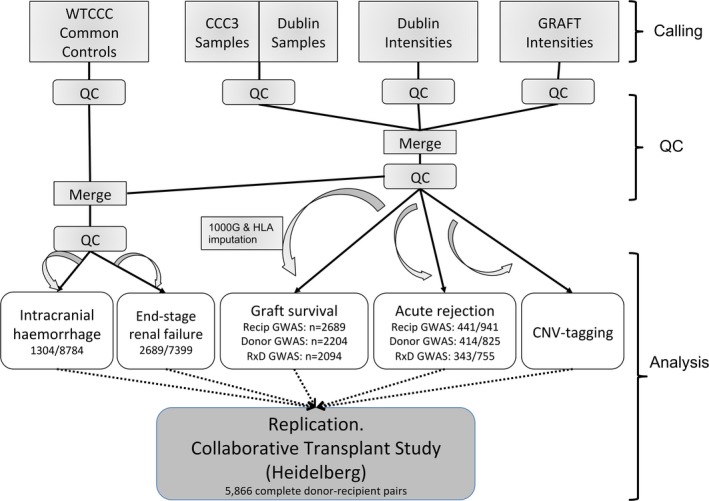
Analysis workflow and strategy. Main input cohorts, analysis methods, and sample sizes are indicated. For binary traits, numbers indicate samples with/without the trait. For further details, see Methods and Supplementary Methods. GWAS, genome‐wide association studies; WTCCC, Wellcome Trust Case Control Consortium
Table 1.
Indicative renal transplant demographics from WTCCC3 and the validation cohort. For consistency, numbers refer to transplants where both donors and recipients passed QC (“complete” donor‐recipient pairs)
| WTCCC3 (post QC) | % | Replication cohort | % | |
|---|---|---|---|---|
| Total transplants where both donors and recipients passed QC | 2094 | 100 | 5866 | 100 |
| Total unique donors with a paired recipient | 1850 | 100 | 5027 | 100 |
| Total unique recipients with a paired donor | 2086 | 100 | 5866 | 100 |
| Mean donor age ± SD | 43 ± 15.4 | 43 ± 16.7 | ||
| Mean recipient age ± SD | 45 ± 13.3 | 48 ± 13.6 | ||
| 0 previous grafts | 1864 | 89 | N/A | |
| 1 previous graft | 204 | 9.7 | N/A | |
| 2 or more previous grafts | 26 | 1.2 | N/A | |
| Graft survival time (days‐to‐uncensored‐event, 25% quartile) | 902 | 442 | ||
| Graft survival time (days‐to‐uncensored‐event, median) | 1866 | 1769 | ||
| Graft survival time (days‐to‐uncensored‐event, 75% quartile) | 3165 | 3162 | ||
| 0 HLA mismatches | 223 | 10.7 | N/A | |
| 1 or 2 Class I HLA mismatches | 839 | 40.1 | N/A | |
| 1 or 2 Class II HLA mismatches | 20 | 0.96 | N/A | |
| 1 or 2 mixed Class I/II HLA mismatches | 124 | 5.9 | N/A | |
| 3 to 5 HLA mismatches | 612 | 29.2 | N/A | |
| 6 HLA mismatches | 8 | 0.4 | N/A | |
| N/A HLA mismatches | 268 | 12.8 | N/A | |
| Graft survival: total uncensored | 495 | 23.6 | 2951 | 50.3 |
| Graft survival: total censored | 1599 | 76.4 | 2915 | 49.7 |
| Total double‐kidney transplants | 3 | 0.14 | N/A | |
| Total en bloc kidney transplants | 1 | 0.05 | N/A | |
| Total kidney+pancreas transplants | 16 | 0.76 | N/A | |
| Total kidney‐only transplants | 2074 | 99.0 | N/A | |
| Total rejections (first 3 mo) | 259 | 12.4 | N/A | |
| Total no rejections (first 3 mo) | 915 | 43.7 | N/A | |
| N/A rejections (first 3 mo) | 920 | 43.9 | N/A | |
| Total rejections (3‐12 mo) | 221 | 10.6 | 575 | 9.8 |
| Total no rejections (3‐12 mo) | 946 | 45.2 | 2573 | 43.9 |
| N/A rejections (3‐12 mo) | 927 | 44.3 | 2718 | 46.3 |
N/A, data not available; QC, quality control; WTCCC3, Wellcome Trust Case Control Consortium‐3.
Inclusion criteria for the study were as follows: (1) deceased donor kidney transplants only; (2) recipient is an adult (>16 years old); (3) reported European ancestry for recipients; and (4) graft survival time >3 months. Donor‐recipient allocation followed NHS‐BT standard UK‐protocols during the study period.
2.2. Replication phase participants
A cohort of 5866 complete donor‐recipient pairs, with similar ethnicity to that of the discovery study, were obtained from the Collaborative Transplant Study DNA Biobank held at the University of Heidelberg, Germany (Table 1).
Clinical variables, datasets, and analysis are described in Supplementary Methods.
2.3. Discovery phase genotyping and analysis
A whole genome amplification step was undertaken (Source BioScience, Nottingham) for samples containing 5‐40 μL of DNA (n = 990 samples). Samples were assayed via Illumina 670 Quad Custom GWAS chips, and subjected to standard postgenotyping quality control procedures before being imputed to the 1000‐genomes reference dataset. Imputation of HLA alleles from SNP genotype data was undertaken using HLA*IMP software26 and compared to serologically typed alleles. A series of GWAS analyses were performed to investigate different genetic models and the renal transplant outcomes of interest. Graft survival genome‐wide analyses were performed (using Cox proportional hazards modeling) for (1) donor SNP genotype main effects; (2) recipient SNP genotype main effects; (3) donor*recipient SNP genotype interaction effects (1df and 3df tests); and (4) CNV‐tag‐SNPs genotype mismatch effects (2 different models). Acute rejection genome‐wide analyses were performed (using logistic regression) for models (1)‐(3). An end‐stage renal failure genome‐wide analysis was performed (using logistic regression) for model (2), and to take advantage of the opportunity, an intracranial hemorrhage genome‐wide analysis was performed (using logistic regression) for model (1).
2.4. Replication‐phase genotyping and analysis
Replication DNA samples were received at King's College London and sent to the Wellcome Trust Sanger Institute for replication analysis. A replication panel of 139 SNPs (post–quality control) was tested based on a combination of low P value (<10−6) from the discovery phase, plus good support of association signals from SNPs in local linkage disequilibrium (LD), or on prior candidature from previous association studies. SNPs were tested according to the same model as motivated their inclusion in the replication panel (for example, if nominated based on a low P value for acute rejection in recipients, then that was also the test of interest in the replication analysis). Meta‐analysis of discovery and replication results was carried out using inverse variance meta‐analysis.27
For further details see Figure 1 and the Methods section in the Supplementary Material.
2.5. Role of the funding source
The funding sources did not participate in the study design, collection, analysis or interpretation of the data, nor did they have a role in writing the report or the decision to submit for publication.
3. RESULTS
3.1. SNP association analysis
Despite the large size of our study (Table 1 and Table S2), none of the phenotypes and genetic models tested in the discovery phase produced any LD‐supported single‐SNP results of genome‐wide significance (P ≤ 5 × 10−8). We also performed a partitioned heritability analysis via stratified LD score regression,28 which failed to reveal any significant enrichment of heritability in genomic regions marking tissue‐ or cell‐type‐specific activity (Figure 2).
Figure 2.
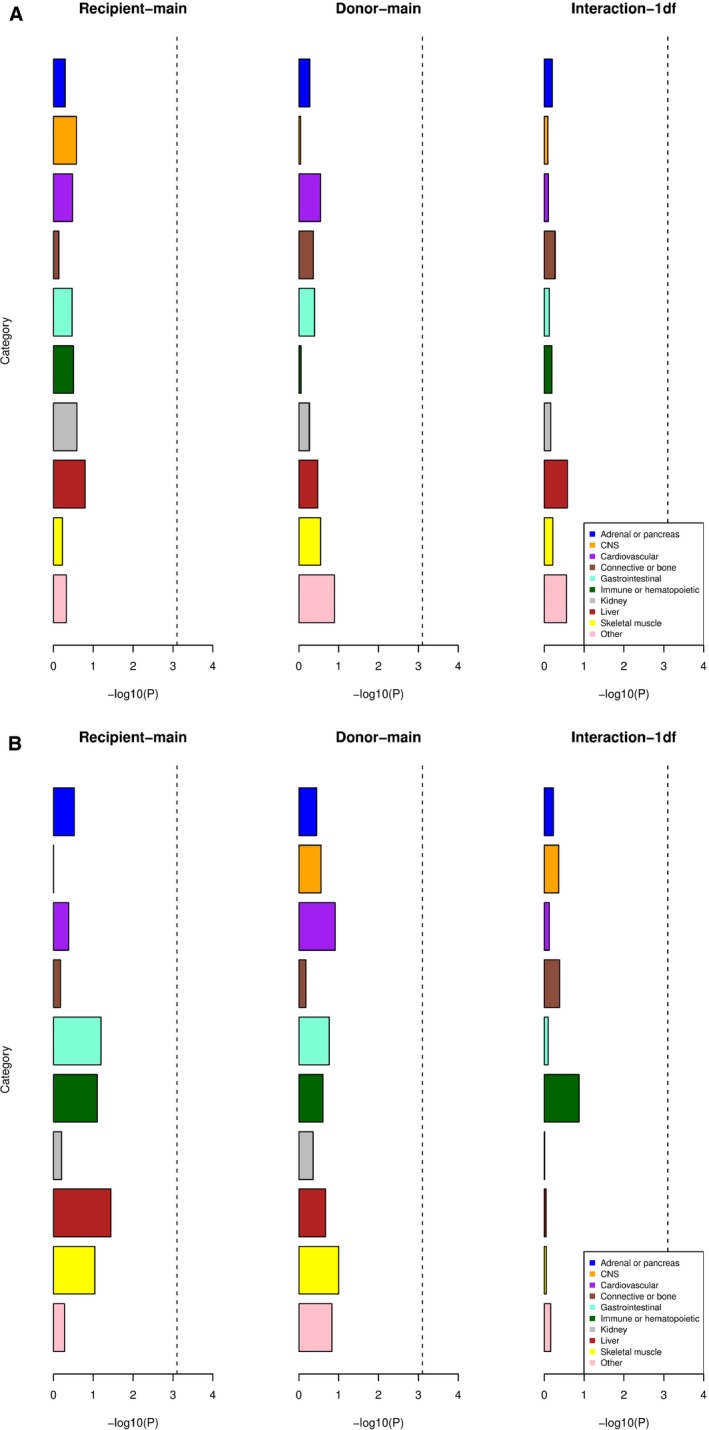
Partitioned heritability analysis of graft survival GWAS results. X‐axis indicates –log10 (P value) for a test for heritability enrichment within 10 cell/tissue‐type categories of genomic annotations, marking tissue‐ or cell‐type‐specific activity. Dotted lines indicate Bonferroni significance level. (A) Death treated as a censored event; (B) death treated as a failure event. CNS, central nervous system
We pursued a second “replication” phase in the hope that the most significant SNPs would be enriched for true association signals. One hundred thirty‐nine SNPs (post–quality control) were tested, based either on a low P value from the discovery phase plus LD support or on prior candidature from previous association studies. In general, the distribution of replication P values for all the tests did not depart appreciably from that expected under a global null hypothesis (Table S3). There was some enrichment for low P values in the recipient genotype main effect tests for acute rejection in the 12 months following transplantation. However, the P values from meta‐analysis (across both discovery and replication phases) did not reach genome‐wide significance (P ≤ 5 × 10−8). The single SNP with strongest evidence for association was rs2289887 (Figure S1 and Table S4), which had consistent effects in both cohorts but a meta‐analysis P value of only 0.00011, indicating that further studies are needed to establish the validity of this signal.
We collated all previously published association signals for early graft rejection and long‐term allograft survival and none of these replicated in our study (Table S1).
3.2. HLA mismatch analysis
We took advantage of recorded serological information to check for known associations with donor‐recipient mismatch levels and to compare recorded information with imputed mismatches based on SNP genotype information (Figure 3). As expected for deceased donor transplants, the number of non‐zero mismatch transplants in our data was low, reducing our power to detect associations. Nevertheless, we confirmed significant associations with HLA‐A (P = .022) and HLA‐DRB (P = .00049) mismatches using the recorded data.
Figure 3.
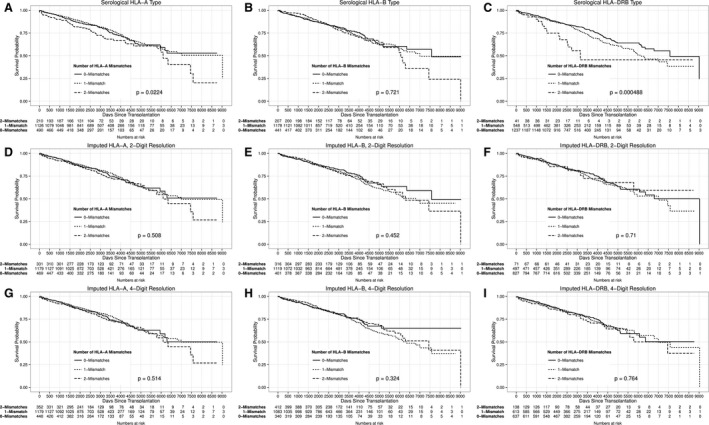
Kaplan‐Meier plots of graft survival by number of mismatches by serological typing (A, B, and C), imputed 2‐digit resolution (D, E, and F), and imputed 4‐digit resolution (G, H, and I). P values were obtained from likelihood ratio tests on Cox proportional hazards models
On the other hand, the imputed mismatch results did not reproduce the expected associations. We attribute this to 2 factors: (1) the true mismatch = 2 frequency was low, due to donor selection; (2) as with any statistical noise, the imputation inaccuracy tended to act with disproportionate effect in the extremes of the distribution (here, the mismatch = 2 group), resulting in higher relative errors in that group. Thus, although the overall imputation accuracy was high (Tables S5 and S6), the rate of false positives was disproportionately high in the mismatch = 2 categories, swamping our ability to detect the true association signals (Table S7).
4. DISCUSSION
In this article, we report the results of the first large‐scale GWAS in renal transplantation. Despite our considerable sample size, we did not replicate any proposed findings from previous candidate gene studies nor did we discover any convincing new variants in our own analyses. There are a number of plausible reasons that may explain this.
Firstly, although this was a study involving thousands of individuals, by GWAS standards it was at the lower end of the range of sample sizes that have been employed for other human traits.29, 30 A decade of GWAS across multiple complex traits has shown that single effect sizes for any 1 causal variant are typically low, and thus for some traits even bigger sample sizes than ours are needed to discover them. For example, the first robustly associated locus for schizophrenia was found in a discovery GWAS cohort of 3322 cases and 3587 controls.31 The number of reliably associated signals for schizophrenia has now grown to 108, providing new biological insights into the disease, thanks to a meta‐analysis that involved 40 000 cases and 113 000 controls.32
Secondly, part of our study sought to look for recipient‐donor genetic interactions. Interaction effects require even larger sample sizes to be reliably discovered. With a few notable exceptions,33 GWAS studies on other traits have been unsuccessful in discovering reliable interaction effects.
Thirdly, and perhaps most importantly, the transplantation outcomes we considered were relatively crude measures obtained retrospectively from national registry data, collected over many years for reasons other than for acting as endpoints in a genome‐wide association study. Our outcomes were therefore heterogeneous, subject to missingness, and subject to environmental factors that likely weakened the genetic signals. Graft survival time is expected to be subject to a range of factors including graft quality, drug regimen, patient compliance, changes in standard‐of‐care over time, between‐center differences, and underlying biological causes of renal dysfunction. Additionally, a substantial proportion of the survival time data were right‐censored (60.5%‐76.4%, depending on model being fitted), reducing the power for this endpoint.
Acute graft rejection is also a heterogeneous phenotype arising from various immune and nonimmune biological causes. Furthermore, this phenotype was derived from a field that in many records was left blank, resulting in a high degree of missingness (56.7%‐58.3%, depending on model being fitted). The high missingness rate reduced the power of our study, and nonrandom missingness would complicate the interpretation of any positive association signals we might have found (although we note the nonrandom missingness bias would need to be the same in both the discovery and replication cohorts for a signal to be replicated).
Batch effects are also a concern. Both the discovery and replication data were obtained from multiple different collection points in different countries over many years, and thus our phenotypes may be subject to batch effects, for example, arising from different treatment protocols over time and space. Genetic data can also be subject to batch effects, for example, arising from differences in sample collection protocols and unknown differences in population structure. We applied both covariate selection and genetically derived principal component axes to try to mitigate such effects. We also note that, as with nonrandom missingness, the batch effect structure would need to be the same in both the discovery and replication cohorts in order for false‐positive association signals to be replicated. Nevertheless, together these extraneous factors may well have increased the noise in our data, and so reduced (and made less detectible) the genetic effects in our data.
HLA imputation from chip‐based genetic data might in time be of value as an adjunct to serological typing. However, our investigations found that the additional noise introduced by imputation uncertainty prevented the imputed data from picking up the well‐known mismatch signals at HLA‐A and HLA‐DRB. This suggests that the accuracy of HLA imputation will need to be improved before it can be used reliably as an alternative to serotyping.
In contrast, our serological data successfully detected the HLA‐A and HLA‐DRB mismatch signals, despite the reduced power to do so resulting from HLA‐matching of deceased donor allografts. This indicates that, despite the various shortcomings of our study, strong HLA signals were detectable in our data. The implication therefore, at least as far as our primary graft survival endpoint is concerned, is that signals outside the HLA region are weaker than those already established in the HLA region. We therefore anticipate that genetic effects outside of the HLA region are more likely to be of value in elucidating biological pathways than in direct clinical prediction.
We explored this last point further via formal power calculations. These indicated that we were well powered to detect any main effect graft survival association signals involving causal SNPs with allelic hazard ratios in the range 1.4‐1.9 (log‐additive risk model, alpha = 5 × 10−8, power = 0.8, minor allele frequency>0.05), and to detect main‐effect acute rejection association signals with allelic odds ratios in the range 1.7‐2.9. We emphasize that these effect sizes are applicable to the traits investigated in this study, but that larger effects might be found in future studies under more precise phenotyping.
In summary, while our study was able to replicate known mismatch signals in the HLA region, we failed to find convincing association signals outside of the HLA region. Both phenotype heterogeneity and sample size may have contributed to this result. Looking ahead, we note that the general lessons from GWAS applied to multiple human traits over more than a decade have brought home 3 clear messages.29, 30 The first is that all complex traits contain a genetic component, and harbor a large number of causal variants throughout the genome. The second is that larger GWAS studies, often obtained via meta‐analysis with previous studies, inevitably succeed in discovering some portion of these causal variants. The third is that when a large enough portion of these variants is discovered, new insights into the biology of the trait are gained. With these points in mind, we look forward to an international consortium (iGeneTrain34), which has been formed to share and meta‐analyze genetic and phenotypic data from most major transplant cohorts worldwide. We also look forward to efforts to collect more detailed phenotypes of relevance to transplant failure, which should provide greater genetic resolution. The data provided by our study form a foundation for ongoing efforts seeking to uncover the biology and improve the prospects for renal transplantation outcomes.
AUTHOR CONTRIBUTIONS
M.P.H. conducted a literature search, did data and sample collection, manuscript preparation and review, and grant provision and supervision. C.F. and I.R.M. conducted a literature search, did data analysis and interpretation, manuscript preparation and review, and grant provision and supervision. J.M. conducted a literature search. F.D. did data and sample collection. E.P. conducted a literature search, did data and sample collection, did data analysis and interpretation, and manuscript preparation and review. C.S. did data analysis and interpretation. R.B., C.B., B.C., M.C., J.F., S.F., S.G., A.H., R.H., A.J., M.K., T.L., I.Mc., P.B.M., J.M., P.M., W.McK., A.McL., C.H., T.A., P.P., S.P., P.R., H.S., R.T., P.T., did data and sample collection and data analysis and interpretation. N.S. and E.S. did grant provision and supervision. R.T. did study design and grant provision and supervision. R.V. and S.S. did study design, data and sample collection, and data analysis and interpretation. G.C. and P.C. conducted a literature search, did the study design, data and sample collection, manuscript preparation and review, and grant provision and supervision. G.O. did data sample and collection. N.S. did manuscript preparation and review and grant provision and supervision. M.W. did study design, data analysis and interpretation, manuscript preparation and review, and grant provision and supervision. G.L. did study design, manuscript preparation and review, and grant provision and supervision.
DISCLOSURE
The authors of this article have conflicts of interest to disclose as described by the American Journal of Transplantation. M.E.W. is an employee of Genomics plc, a company providing genomic analysis services to the pharmaceutical and health care sectors. M.H.F. and I.R.B. are employees of UCB Celltech, a pharmaceutical company. Their involvement in the conduct of this research was solely in their capacity as academics at King's College London.
Supporting information
ACKNOWLEDGMENT
Supported by grants awarded from the Wellcome Trust (090355/A/09/Z, 090355/B/09/Z and 088849/Z/09/Z, “WTCCC3”), the Medical Research Council (grants G0600698 and MR/J006742/1 to S.H.S.; G0802068 to G.M.L. and MR/K002996/1 to G.M.L; grants G0801537/ID: 88245), Guy's and St Thomas’ Charity (grants R080530 and R090782) to M.H.F. and G.M.L., from the European Union FP7 (grant agreement no HEALTH‐F5–2010–260687 to M.H.F. and project number 305147: BIO‐DrIM to M.H.F. and I.R.M.); and by the National Institute for Health Research Biomedical Research Centre at Guy's and St Thomas’ and King's College London. Anonymized clinical data were also provided NIHR Health Informatics Collaborative. N.S.'s research is supported by the Wellcome Trust (Grant Codes WT098051 and WT091310), the EU FP7 (EPIGENESYS Grant Code 257082 and BLUEPRINT Grant Code HEALTH‐F5‐2011‐282510) and the National Institute for Health Research Blood and Transplant Research Unit (NIHR BTRU) in Donor Health and Genomics at the University of Cambridge in partnership with NHS Blood and Transplant (NHSBT). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health, or NHSBT. The NIHR Health Informatics Collaborative includes Bradley JR, University of Cambridge; Channon KM, University of Oxford; Lord GM, King's College London; Weber J, Imperial College London; Williams B, University College London; Crichton C, University of Oxford; Davies J, University of Oxford; Welch J, University of Oxford; and Woods K, Oxford University Hospitals NHS Foundation Trust.
Hernandez‐Fuentes MP, Franklin C, Rebollo‐Mesa I, et al. Long‐ and short‐term outcomes in renal allografts with deceased donors: A large recipient and donor genome‐wide association study. Am J Transplant. 2018;18:1370–1379. https://doi.org/10.1111/ajt.14594
Maria P. Hernandez‐Fuentes, Christopher Franklin, and Irene Rebollo‐Mesa are equal contributors.
Michael E. Weale and Graham M. Lord are equal senior contributors.
Funding information
Primary funding was provided by the Wellcome Trust. Additional funding information is detailed in the Acknowledgment section.
REFERENCES
- 1. Opelz G. President's address. The transplantation society–Berlin 2012. Transplantation. 2013;95(1):4‐7. [DOI] [PubMed] [Google Scholar]
- 2. Bradbury L. Annual Report on Kidney Transplantation. Report for 2014/2015. UK: National Health Service ‐ Blood and Transplant; September 2015.
- 3. Johnson RJ, Bradbury LL, Martin K, Neuberger J, UK Transplant Registry . Organ donation and transplantation in the UK‐the last decade: a report from the UK national transplant registry. Transplantation. 2014;97(Suppl 1):S1‐S27. [DOI] [PubMed] [Google Scholar]
- 4. Legendre C, Canaud G, Martinez F. Factors influencing long‐term outcome after kidney transplantation. Transpl Int. 2014;27(1):19‐27. [DOI] [PubMed] [Google Scholar]
- 5. Nabokow A, Dobronravov VA, Khrabrova M, et al. Long‐term kidney allograft survival in patients with transplant glomerulitis. Transplantation. 2015;99(2):331‐339. [DOI] [PubMed] [Google Scholar]
- 6. Merrill JP, Murray JE, Harrison JH, Guild WR. Successful homotransplantation of the human kidney between identical twins. J Am Med Assoc. 1956;160(4):277‐282. [DOI] [PubMed] [Google Scholar]
- 7. Kessaris N, Mukherjee D, Chandak P, Mamode N. Renal transplantation in identical twins in United States and United Kingdom. Transplantation. 2008;86(11):1572‐1577. [DOI] [PubMed] [Google Scholar]
- 8. Süsal C, Opelz G. Current role of human leukocyte antigen matching in kidney transplantation. Curr Opin Organ Transplant. 2013;18(4):438‐444. [DOI] [PubMed] [Google Scholar]
- 9. Simmonds MJ. Using genetic variation to predict and extend long‐term kidney transplant function. Transplantation. 2015;99(10):2038‐2048. [DOI] [PubMed] [Google Scholar]
- 10. Chand S, McKnight AJ, Borrows R. Genetic polymorphisms and kidney transplant outcomes. Curr Opin Nephrol Hypertens. 2014;23(6):605‐610. [DOI] [PubMed] [Google Scholar]
- 11. Brown KM, Kondeatis E, Vaughan RW, et al. Influence of donor C3 allotype on late renal‐transplantation outcome. N Engl J Med. 2006;354(19):2014‐2023. [DOI] [PubMed] [Google Scholar]
- 12. Varagunam M, Yaqoob MM, Döhler B, Opelz G. C3 polymorphisms and allograft outcome in renal transplantation. N Engl J Med. 2009;360(9):874‐880. [DOI] [PubMed] [Google Scholar]
- 13. Goldfarb‐Rumyantzev AS, Naiman N. Genetic predictors of acute renal transplant rejection. Nephrol Dial Transplant. 2010;25(4):1039‐1047. [DOI] [PubMed] [Google Scholar]
- 14. Moore J, McKnight AJ, Simmonds MJ, et al. Association of caveolin‐1 gene polymorphism with kidney transplant fibrosis and allograft failure. JAMA. 2010;303(13):1282‐1287. [DOI] [PubMed] [Google Scholar]
- 15. Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freedman BI, Kopp JB, Langefeld CD, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. 2010;21(9):1422‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woillard JB, Rerolle JP, Picard N, et al. Donor P‐gp polymorphisms strongly influence renal function and graft loss in a cohort of renal transplant recipients on cyclosporine therapy in a long‐term follow‐up. Clin Pharmacol Ther. 2010;88(1):95‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moore J, McKnight AJ, Döhler B, et al. Donor ABCB1 variant associates with increased risk for kidney allograft failure. J Am Soc Nephrol. 2012;23(11):1891‐1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Yang JW, Zeevi A, et al. IMPDH1 gene polymorphisms and association with acute rejection in renal transplant patients. Clin Pharmacol Ther. 2008;83(5):711‐717. [DOI] [PubMed] [Google Scholar]
- 20. Pallet N, Thervet E. The genetics of kidney transplantation. Hum Genet. 2012;131(3):317‐323. [DOI] [PubMed] [Google Scholar]
- 21. Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363(2):166‐176. [DOI] [PubMed] [Google Scholar]
- 22. Mushiroda T, Saito S, Tanaka Y, et al. A model of prediction system for adverse cardiovascular reactions by calcineurin inhibitors among patients with renal transplants using gene‐based single‐nucleotide polymorphisms. J Hum Genet. 2005;50(9):442‐447. [DOI] [PubMed] [Google Scholar]
- 23. O'Brien RP, Phelan PJ, Conroy J, et al. A genome‐wide association study of recipient genotype and medium‐term kidney allograft function. Clin Transplant. 2013;27(3):379‐387. [DOI] [PubMed] [Google Scholar]
- 24. McCaughan JA, McKnight AJ, Maxwell AP. Genetics of new‐onset diabetes after transplantation. J Am Soc Nephrol. 2014;25(5):1037‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghisdal L, Baron C, Lebranchu Y, et al. Genome‐wide association study of acute renal graft rejection. Am J Transplant. 2017;17:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dilthey AT, Moutsianas L, Leslie S, McVean G. HLA*IMP–an integrated framework for imputing classical HLA alleles from SNP genotypes. Bioinformatics. 2011;27(7):968‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evangelou E, Ioannidis JP. Meta‐analysis methods for genome‐wide association studies and beyond. Nat Rev Genet. 2013;14(6):379‐389. [DOI] [PubMed] [Google Scholar]
- 28. Finucane HK, Bulik‐Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome‐wide association summary statistics. Nat Genet. 2015;47(11):1228‐1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manolio TA. In retrospect: As decade of shared genomic associations. Nature. 2017;546(7658):360‐361. [DOI] [PubMed] [Google Scholar]
- 30. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schizophrenia Working Group of the Psychiatric Genomics Consortium . Biological insights from 108 schizophrenia‐associated genetic loci. Nature. 2014;511(7510):421‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strange A, Capon F, Spencer CC, et al. A genome‐wide association study identifies new psoriasis susceptibility loci and an interaction between HLA‐C and ERAP1. Nat Genet. 2010;42(11):985‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. (iGeneTRAiN) IGTRiTN . Design and implementation of the international genetics and translational research in transplantation network. Transplantation. 2015;99(11):2401‐2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials