

Abstract
Small‐molecule inhibitors of hypoxia‐inducible factor prolyl hydroxylases (HIF‐PHs) are currently under clinical development as novel treatment options for chronic kidney disease (CKD) associated anemia. Inhibition of HIF‐PH mimics hypoxia and leads to increased erythropoietin (EPO) expression and subsequently increased erythropoiesis. Herein we describe the discovery, synthesis, structure–activity relationship (SAR), and proposed binding mode of novel 2,4‐diheteroaryl‐1,2‐dihydro‐3H‐pyrazol‐3‐ones as orally bioavailable HIF‐PH inhibitors for the treatment of anemia. High‐throughput screening of our corporate compound library identified BAY‐908 as a promising hit. The lead optimization program then resulted in the identification of molidustat (BAY 85‐3934), a novel small‐molecule oral HIF‐PH inhibitor. Molidustat is currently being investigated in clinical phase III trials as molidustat sodium for the treatment of anemia in patients with CKD.
Keywords: BAY 85-3934, HIF-PH, inhibitors, metalloenzymes, molidustat
Introduction
The production of red blood cells in the bone marrow is a tightly controlled homeostatic process that depends on the availability of the hematopoietic growth hormone erythropoietin (EPO). EPO is primarily produced by specialized cells in the kidneys, from where it is released into the bloodstream. These cells respond to any decrease in oxygen supply with an increased production of EPO. Systematic analysis of EPO gene regulatory elements ultimately led to the discovery of the hypoxia‐inducible factors (HIFs) HIF‐1 and HIF‐2, which are constituents of the oxygen‐sensing pathway that enables higher organisms to adapt to changes in oxygen levels.1 While HIFs are the transcriptional activators of a plethora of hypoxia‐inducible genes, the mRNA of EPO shows by far the highest inducibility in response to hypoxia. HIFs are heterodimers consisting of an α‐ and a β‐subunit that bind to distinct hypoxia‐responsive elements in the regulatory sequences of hypoxia‐inducible genes.2 The HIFs are regulated by a family of three oxygen‐dependent and 2‐oxoglutarate (2‐OG)‐consuming dioxygenases, known as the HIF‐prolyl hydroxylases (HIF‐PHs) or prolyl hydroxylase domain‐containing proteins 1–3 (PHD 1–3).3 In the presence of oxygen, HIF‐PHs hydroxylate two distinct proline residues within the HIF‐α subunit. The hydroxylated HIF‐α subunit is recognized by an E3 ubiquitin ligase multiprotein complex (consisting of the Von Hippel–Lindau protein, elongins B and C, cullin 2 and RING‐box protein 1), and subsequently subjected to polyubiquitination and proteasomal degradation.4 Thus, HIF‐PHs function as oxygen sensors, shutting off hypoxia‐sensitive gene transcription by tagging HIFs for degradation under normoxia. In contrast, under hypoxia, HIF‐PH activity is decreased; HIF accumulates and migrates to the nucleus, activating EPO gene transcription (Figure 1).
Figure 1.
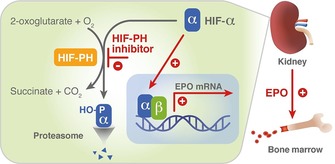
A promising novel therapeutic approach for the treatment of anemia: stabilization of HIF via inhibition of HIF‐PH translates into EPO induction in the kidney and formation of red blood cells in the bone marrow. EPO: erythropoietin; HIF: hypoxia‐inducible transcription factor; HIF‐PH: HIF‐prolyl hydroxylase; HO‐P: hydroxylated proline residues.
Anemia is common in patients with chronic kidney disease (CKD) because insufficient EPO is produced by failing kidneys.5 Consequently, increasing production of endogenous EPO in the kidneys by mimicking hypoxia via inhibition of HIF‐PHs is an attractive and novel approach for the treatment of anemia (Figure 1). The current standard of care consists of parenteral administration of recombinant human EPO (rhEPO). In controlled clinical studies, administration of rhEPO was associated with an increased incidence of cardiovascular side effects, which were attributed to highly supraphysiological plasma EPO levels.6 Small‐molecule HIF‐PH inhibitors may provide an alternative treatment. In addition to their oral route of administration, they have the potential to treat anemia in a more physiological manner by maintaining endogenous EPO levels close to the normal physiological range. Several programs are currently underway to identify small‐molecule HIF‐PH inhibitor drug candidates (Figure 2).7
Figure 2.
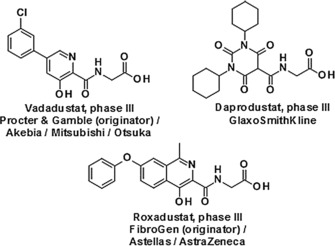
Other advanced small‐molecule oral HIF‐PH inhibitors currently in clinical development.
Herein we describe the discovery, SAR, and proposed binding mode of novel 2,4‐diheteroaryl‐1,2‐dihydro‐3H‐pyrazol‐3‐ones as HIF‐PH inhibitors, starting with the high‐throughput screening (HTS) hit BAY‐908 (1). Lead optimization efforts resulted in the discovery of molidustat (BAY 85‐3934, 45), a novel, small‐molecule, orally bioavailable HIF‐PH inhibitor currently being investigated in clinical phase III trials for the treatment of anemia in patients with CKD.
Results and Discussion
Screening cascade and hit evaluation
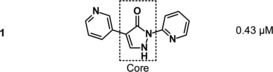
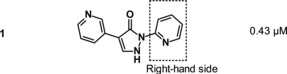
Almost one million compounds were tested for their potential activity as HIF‐PH inhibitors in a biochemical HTS, designed in‐house for 96‐ and 384‐well plate formats.8, 9 This assay was based on the interaction between a hydroxylated HIF‐derived peptide (comprising amino acids 556–574) and a europium‐labeled recombinant VBC complex. Potential HIF‐PH inhibitors that suppressed the hydroxylation of HIF‐proline 564 by human HIF‐PH, and thereby attenuating the binding of VBC, were detected by reduction of fluorescence intensity. Optimization of cofactor concentrations 2‐OG, Fe2+ and ascorbate led to a robust and highly reproducible assay.9 Almost 500 confirmed screening hits were identified in the HTS, among which BAY‐908 (1, Figure 3) was the most promising, showing sub‐micromolar potency.
Figure 3.
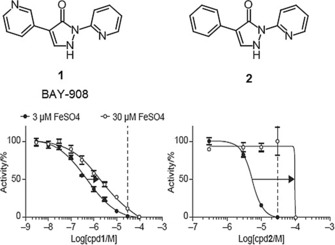
Concentration‐response curves of HIF‐PH activity at Fe2+ concentrations of 3 μm and 30 μm (30 μm indicated by dotted lines). While IC50 values of BAY‐908 (1) remained below the concentration of Fe2+, concentrations of compound 2 beyond the free iron concentration were required to achieve inhibition of the enzyme (IC50 shifts indicated by arrows).
A HIF‐reporter cell‐based assay measuring stabilization of endogenous HIF was used as a secondary assay. This assay used a recombinant A549 lung carcinoma cell, which was stably transfected with a construct expressing firefly luciferase under the control of a hypoxia‐responsive element tandem from the vascular endothelial growth factor (VEGF) promoter (HIFRE2).8 BAY‐908 (1) displayed an EC50 of 4.9 μm in this assay, being 5‐ and 10‐fold more potent than the nonspecific reference compounds deferoxamine (DFO, EC50≈25 μm) and cobalt chloride (CoCl2, EC50≈50 μm), respectively.
The induction of HIF‐1α was examined in human HeLa cell cultures. HIF‐1α was detected in cells exposed to concentrations of compound 1 >0.25 μm.
In human hepatoma HuH cells, compound 1 induced EPO mRNA with an EC50 of 1.9 μm and led to a threefold increase in EPO concentration in the supernatant compared with baseline.
In mice, BAY‐908 (1) increased EPO mRNA and plasma EPO levels after intraperitoneal (i.p.) and oral (p.o.) administration. Plasma EPO increased from ≈120 pg mL−1 to ≈900 pg mL−1 after p.o. treatment with 100 mg kg−1 of compound 1. Plasma EPO concentration also increased in rats after oral dosing with BAY‐908 50 mg kg−1.
It was assumed that BAY‐908 (1) preferentially binds to the Fe2+ in the active site of HIF‐PHs, thereby competing with 2‐OG. This hypothesis was corroborated by competition experiments with compound 1 and 2‐OG, in which the potency of compound 1 decreased with increasing 2‐OG concentration and vice versa. By contrast, large variations in Fe2+ concentration in the reaction buffer resulted in only minor changes in the potency of the inhibitor (Figure 3). IC50 values of compound 1 always remained below the concentration of Fe2+. Thus it could be concluded that the inhibitory activity of compound 1 was not occurring through the removal of the iron cation cofactor by stoichiometric chelation.10 Structurally, this iron independency of compound 1 could be attributed to the 3‐pyridyl moiety, because the corresponding phenyl derivative 2 behaved more like a typical iron chelator, with a shift in IC50 strongly dependent on the concentration of free iron in the media. For compound 2, concentrations greater than the free iron concentration were required to achieve inhibition of the enzyme (Figure 3).
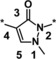
The compounds of our chemical series can exist principally as the pyrazolone or pyrazolol tautomer (Figure 4).11 In the solid state, a small‐molecule X‐ray crystal structure of BAY‐908 (1) revealed the pyrazolol tautomer, indicated by the presence of a hydroxy hydrogen atom and a stabilizing intramolecular hydrogen bond from O−H to pyridine‐N with O⋅⋅⋅N distance of 2.62 Å (see Supporting Information). In solution, a solvent‐dependent tautomeric equilibrium between the pyrazolone and pyrazolol tautomers can be expected, with the pyrazolol tautomer stabilized by a hydrogen bond similar to that observed in the solid state (Figure 4).
Figure 4.
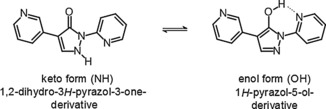
Pyrazolone–pyrazolol tautomerism of compound 1.11
To gain more insight into the different iron‐chelating properties of compounds 1 and 2, pK a values12 were determined and iron(II) coordination studies were conducted in aqueous solution.13 Compound 2 had a pK a of 6.0, whereas pK a values for compound 1 were 4.5 and 6.5 (Table 1).
Table 1.
Summary of key in vitro parameters for the screening hit BAY‐908 (1) (X=N) compared with the phenyl analogue 2 (X=CH).
| |||||
|---|---|---|---|---|---|
| Compd | IC50 [μm][a] | EC50 [μm][b] | logD 7.5 | LipE[c] | pK a |
| 1 | 0.43 | 4.9 | 0.6 | 5.8 | 4.5/6.5 |
| 2 | 5.4 | 5.9 | 2.7 | 2.6 | 6.0 |
The binding behavior of the deprotonated compound 2 toward Fe2+ was similar to that of typical iron(II) chelators such as 2,2′‐bipyridine and 1,10‐phenanthroline,16 whereas BAY‐908 (1) did not show the typical iron(II)‐chelating properties, but a more complex, pH‐dependent behavior (Supporting Information). It was concluded from these experiments that both compounds needed to be deprotonated for efficient iron chelation. However, these observations in aqueous solution were not sufficient to explain the different shapes of the concentration‐response curve in vitro (Figure 3). The main reason for the smaller iron‐dependent IC50 shift of BAY‐908 (1) compared with compound 2 (Figure 3) was assumed to be the more favorable interaction of BAY‐908 (1) with the HIF‐PH target enzyme, resulting in a minimized impact of free iron on potency.
BAY‐908 (1) and compound 2 also showed different behaviors in in vivo experiments in mice. BAY‐908 (1) induced an increase in hematocrit and reticulocytes in mice similar to that induced by rhEPO. In contrast, compound 2 was inactive with respect to an increase in hematocrit, although a small increase in plasma EPO was seen 4 h after administration. Oral administration of BAY‐908 (1) to rats did not result in any increased elimination of iron via urine, in contrast to the iron chelator DFO administered parenterally (data not shown).
Proposed binding mode of screening hit BAY‐908 (1)
Four X‐ray crystal structures of the catalytic domain of human PHD2 in complex with small‐molecule inhibitors were available in the Protein Data Bank at the time of searching.17 Like other 2‐OG oxygenases, PHD2 shows a double‐stranded α‐helix fold. The binding site contains a ferrous iron coordinated by two histidine and one aspartate residue (Figure 5). Because we were unable to produce an X‐ray structure of PHD2 with BAY‐908, docking studies were performed using a published structure (PDB ID: https://www.rcsb.org/structure/2G19).18
Figure 5.

Proposed binding mode: structure of BAY‐908 (1) docked into X‐ray structure of PHD2. Docking was performed with the electron‐neutral pyrazolol tautomer for technical reasons. Dotted lines indicate iron coordination (additional coordination to His313 and H2O not shown).
After protein preparation and ligand extraction, docking studies using Glide SP (Schrödinger LLC)19 were performed, as well as manual docking combined with energy minimizations. BAY‐908 (1) was docked as the electron‐neutral pyrazolol tautomer.
Docking results suggested bidentate iron binding, involving one core nitrogen and the nitrogen of the neighboring 2‐pyridyl ring, forming a five‐membered ring with the Fe2+ cation. Because technical limitations prevented the docking of deprotonated structures, there was no further indication as to whether binding to enzymatic iron was in fact taking place via the more plausible pyrazolate anion species, as suggested by the iron‐binding studies performed in aqueous solution.
An alternative binding mode, in which the core hydroxy group and the nitrogen of the neighboring 2‐pyridyl ring formed a six‐membered ring with the Fe2+ cation, seemed unlikely owing to steric hindrance of the resulting conformer within the binding pocket. The deep part of the PHD2 active site was assumed to be occupied by the 3‐pyridyl ring of BAY‐908 (1) (Figure 5).
It was initially assumed that the nitrogen in the 3‐pyridyl ring was mimicking the 5‐carboxylate of the 2‐OG co‐substrate by binding to a basic residue of the prolyl hydroxylase enzyme via a hydrogen bond.20 However, to accommodate the 3‐pyridyl moiety, the conformation of the Arg383 side chain had to be manually adapted. Consequently, an electrostatic interaction with Arg383 involving the π‐system of the 3‐pyridyl ring seemed more plausible. The 2‐pyridine moiety was assumed to be pointing toward the opening of the binding pocket, possibly stabilized by an edge‐to‐face interaction with Trp389 (Figure 5). No direct hydrogen bond between Tyr303 and the pyrazolol hydroxy group could be observed for compound 1, but the distance and angles suggested a possible indirect hydrogen bond mediated by water molecules (Figure 5).
Lead optimization
Screening hit BAY‐908 (1) was chosen as a starting point for further lead optimization. Apart from showing largely iron‐independent in vitro potency (IC50=0.43 μm, EC50=4.9 μm, Table 1) and promising in vivo efficacy (EPO induction in mice, see above), it displayed a clean cytochrome P450 (CYP) profile and a favorable selectivity profile in a lead candidate profiling panel covering a wide range of targets.
Pharmacokinetic (PK) studies of BAY‐908 (1) in rats revealed a moderate to high blood clearance of 2.9 L (h kg)−1 and an oral bioavailability of 35 %.21 Owing to the very low aqueous solubility of <1 mg L−1 from crystalline material, bioavailability dropped to only 9 % when the compound was administered as a methyl‐hydroxyethyl cellulose (Tylose) suspension.22 However, the low molecular weight (238 g mol−1) and low lipophilicity (logD 7.5=0.6, Table 1)23 resulted in a high ligand binding efficiency index (BEI=26.7)24 and lipophilic binding efficiency (LipE=5.8), making BAY‐908 (1) an attractive starting point for derivatizations. Derivatives were generally found to be pan‐HIF‐PH inhibitors, showing little selectivity for PHD subtypes. IC50 values presented here refer to PHD1 inhibition, for which the most comprehensive dataset was obtained.
SAR of the core moiety
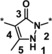
Structural changes at the core of the molecule turned out to have a strong influence on potency (showing a steep SAR). A set of variations is shown in Table 2.
Table 2.
SAR of the core moiety of the HIF‐PH inhibitors. The pyridyl moieties were kept constant, as shown for compound 1.
| ||
|---|---|---|
| Compd | Core structure | IC50 [μm][a] |
| 3 |
|
3.5 |
| 4 |
|
24 |
| 5 [b] |
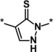
|
7.5 |
| 6 |
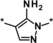
|
>50 |
| 7 |
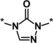
|
>50 |
| 8 |
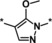
|
>50 |
| 9 |
|
>50 |
| 10 |
|
>50 |
[a] IC50 values refer to PHD1. [b] The compound was isolated and tested as the HCl salt.
The introduction of a methyl group in the 5‐pyrazolone position resulted in a significant loss in activity of one magnitude (compound 3, IC50=3.5 μm), and the introduction of an extra nitrogen in the ring further decreased inhibitory activity (1,2,3‐triazol‐4‐one 4, IC50=24 μm). Attempts to introduce bioisosteres as replacements for the pyrazolone keto group led to the weakly active thione 5 (IC50=7.5 μm) and amine 6 (IC50>50 μm). This might be due to the decreased ability to form a water‐mediated hydrogen bond with Tyr303. Furthermore, the modification of the electronic properties of the pyrazolone core associated with the keto group replacement might be detrimental to the coordination between the iron ion and the inhibitor. In general, the ability of the core moiety to form an anion and the distribution of a negative charge in the core seems to be important for activity. Likewise, the 1,2,4‐triazol‐3‐one 7 and methyl ether 8 both showed IC50 values >50 μm. Methylation of the nitrogen in the 1‐position (example 9) led to an IC50>50 μm, plausibly owing to loss of iron‐binding ability. Pyrimidine derivative 10 with an IC50 value >50 μm underlined the steepness of the SAR of the core moiety.
SAR of the 2‐pyridyl moiety of the molecule (“right‐hand side”)
A set of variations on the right‐hand side of the molecule is depicted in Table 3. A wider range of structural changes, including polar substituents, was tolerated. However, the position of the 2‐pyridyl nitrogen proved to be crucial for activity. A partial methyl scan around the 2‐pyridyl group indicated a preference for substituents at position 4 (11, IC50=0.34 μm), followed by position 5 (12, IC50=1.7 μm). Introduction of a methyl group in position 6 (compound 13), next to the pyridine nitrogen, resulted in a weakly active compound (IC50=8.9 μm), possibly owing to steric hindrance. Other rings containing a nitrogen in position 2, such as 1,3,4‐thiadiazole 14 (IC50=4.3 μm) and pyrazine 15 (IC50=0.93 μm), were tolerated. Various substituents with different steric and electronic properties were tolerated at position 4 of the pyridine ring (examples 16–19).
Table 3.
SAR of the 2‐pyridyl moiety of the HIF‐PH inhibitors (“right‐hand side”). The core and 3‐pyridyl moiety of the molecule were kept constant, as shown for compound 1.
| ||
|---|---|---|
| Compd | Right‐hand side | IC50 [μm][a] |
| 11 |

|
0.34 |
| 12 [b] |
|
1.7 |
| 13 |
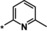
|
8.9 |
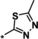
| 14 |
|
4.3 |
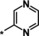
| 15 [b] |
|
0.93 |
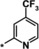
| 16 |
|
1.9 |
| 17 [b] |
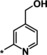
|
1.5 |
| 18 [b] |
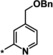
|
2.9 |
| 19 [b] |
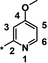
|
0.86 |
| 20 |
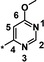
|
0.88 |
| 21 [b] |
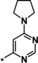
|
0.92 |
| 22 [b] |
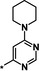
|
1.5 |
| 23 [b] |
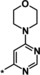
|
0.18 |
| 24 [b] |
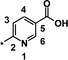
|
0.49 |
| 25 |
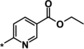
|
0.65 |
| 26 [b] |
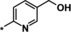
|
0.86 |
| 27 [b] |
|
1.1 |
[a] IC50 values refer to PHD1. [b] The compound was isolated and tested as the HCl salt.
The trifluoromethyl derivative 16 (IC50=1.9 μm) was less potent than the methyl analogue 11 (IC50=0.34 μm), whereas the hydroxymethyl derivative 17 (IC50=1.5 μm) displayed higher potency than the corresponding benzyloxymethyl analogue 18 (IC50=2.9 μm). In general, heteroatom substitution at position 4 of the pyridyl moiety often provided potent compounds, as illustrated by examples 19–23. Notably, analogously substituted pyridines and pyrimidines appeared to be similarly potent in the case of methoxy‐substituted pyridine 19 (IC50=0.86 μm) and pyrimidine 20 (IC50=0.88 μm). Amino substituents at this position were also well tolerated (21–23, IC50 values between 0.18 μm and 1.5 μm and EC50 values between 4 μm and 13 μm). Investigations into the in vivo activity of amines 21–23 revealed a marked increase in EPO plasma concentration in mice following i.p. application, but not after p.o. application, suggesting unfavorable oral PK properties.
Evaluation of position 5 of the pyridyl ring (examples 24–27) revealed that both lipophilic and hydrophilic groups were generally tolerated. Carboxylic acid 24 proved to be potent in the biochemical assay (IC50=0.49 μm), but showed significantly decreased potency in the cellular assay (EC50>20 μm), probably because of diminished cell permeability. Potency in the cellular assay was regained via formation of the corresponding ethyl ester 25 (IC50=0.65 μm, EC50=5.3 μm). The corresponding alcohol 26 (IC50=0.86 μm, EC50=13.4 μm) was of interest owing to a highly improved solubility (4.8 g L−1 as the hydrochloride) and was therefore selected for a more detailed characterization. The CYP profile of compound 26 proved to be favorable. In PK studies in rats, compound 26 displayed a moderate blood clearance of 1.5 L (h kg)−1 and a bioavailability of 55 %.25 The EPO concentration in mice (≈2000 pg mL−1) was higher than for compound 1 (≈900 pg mL−1).26 However, further investigations revealed that the corresponding carboxylic acid 24 was the main metabolite in human hepatocytes and in in vivo PK studies. In a monkey PK study, 70 % of compound 26 was converted into its metabolite 24 excreted with urine. Compound 26 was not pursued any further because of the complex PK‐pharmacodynamic relationship and a possible accumulation issue of active metabolite 24 in patients with CKD.
SAR of the 3‐pyridyl moiety of the molecule (“left‐hand side”)
A set of variations on the 3‐pyridyl moiety of the molecule is depicted in Table 4. Generally, only ring systems with a nitrogen atom with 1,3‐distance to the adjacent core moiety seemed to show the desired profile of iron‐independent potency. An initial methyl scan (examples 28–31) revealed a preference for substituents at positions 2 and 4 over positions 5 and 6. Larger groups than methyl were less well tolerated. This was in line with our assumption that the 3‐pyridyl moiety was pointing into the deep part of the HIF‐PH binding pocket, not permitting bulky substituents. The potencies of trifluoromethyl‐substituted compound 32 (IC50=2.1 μm) and methyl‐substituted compound 31 (IC50=3.2 μm) were in similar ranges. A marked increase in potency was observed after introducing methoxy and cyano groups at position 6 (IC50=0.19 μm for 33 and IC50=0.1 μm for 34). As an explanation for the improved potency of the electron‐deficient cyano compound 34, an enhanced electrostatic interaction between the electron‐withdrawing substituents at the left‐hand side aromatic ring and Arg383 was discussed. For electron‐donating methoxy derivative 33, an interaction of the π‐cation‐type might occur. Notably, a shift of the cyano group from position 6 (compound 34) to position 5 (compound 35) led to a tenfold drop in potency (IC50=1.0 μm for compound 35). This surprising SAR finding proved to be difficult to rationalize by means of molecular modeling in the absence of a co‐crystal structure.
Table 4.
SAR of the 3‐pyridyl moiety of the HIF‐PH inhibitors (“left‐hand side”). The core and 2‐pyridyl moiety of the molecule were kept constant, as shown for compound 1.
| ||
|---|---|---|
| Compd | Left‐hand side | IC50 [μm][a] |
| 28 |
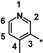
|
0.53 |
| 29 |
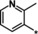
|
0.76 |
| 30 |
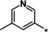
|
1.7 |
| 31 |
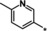
|
3.2 |
| 32 |
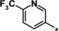
|
2.1 |
| 33 [b] |
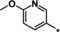
|
0.19 |
| 34 |
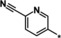
|
0.1 |
| 35 |
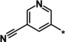
|
1.0 |
| 36 |
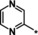
|
28 |
| 37 [b] |
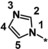
|
0.98 |
| 38 |
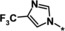
|
0.15 |
| 39 [b] |
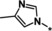
|
2.0 |
| 40 [b] |
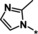
|
15 |
| 41 |
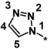
|
0.79 |
| 42 [b] |
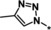
|
0.33 |
| 43 |
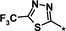
|
1.2 |
[a] IC50 values refer to PHD1. [b] The compound was isolated and tested as the HCl salt.
Other heteroaromatic rings were introduced as pyridine replacements on the left‐hand side of the molecule. Pyrazine derivative 36 was only weakly potent (IC50=28 μm), but five‐membered heteroaromatic rings (37–43) proved to be more potent in the biochemical assay. When imidazole 37 (IC50=0.98 μm) was substituted with trifluoromethyl and methyl groups at position 4 (compounds 38 and 39), a strong influence of electronegativity on potency was revealed. The introduction of the electron‐withdrawing trifluoromethyl group in the 4‐position resulted in the potent imidazole 38 (IC50=0.15 μm), whereas substitution with the electron‐donating methyl group led to the less potent example 39 (IC50=2.0 μm). Interestingly, moving the methyl group from position 4 to position 2 (example 40) further decreased potency (IC50=15 μm for compound 40). The potency of 1,2,3‐triazole 41 (IC50=0.79 μm) was in a similar range to that of the corresponding imidazole 37 (IC50=0.98 μm). However, both 37 and 41 showed far less potency in the cellular assay, with EC50 values slightly above 20 μm. EPO induction in mice after i.p. and p.o. application was investigated for imidazole 37, but proved to be less pronounced than for screening hit BAY‐908 (1). Surprisingly, the introduction of a methyl group at position 4 gave a more potent triazole derivative (example 42, IC50=0.33 μm) than compound 41. This contrasts with the effect of a methyl group in the analogous position on the corresponding imidazole 39 (IC50=2.0 μm). 1,2,4‐Thiadiazole 43 displayed an IC50 of 1.2 μm, completing the multifactorial SAR on the left‐hand side of the molecule.
SAR extension and discovery of molidustat (BAY 85‐3934, 45)
In addition to the single‐point modifications starting from screening hit BAY‐908 (1) (Tables 2–4), hundreds more diarylpyrazolone‐type analogues were synthesized by combining different left‐ and right‐hand side heteroaryl variations, while keeping the pyrazolone core moiety constant.27 After detailed investigations of the in vitro and in vivo pharmacological, physicochemical, PK, metabolic and early safety properties of these compounds (data not shown), certain combinations of imidazoles and 1,2,3‐triazoles (left‐hand side) and amino‐substituted pyrimidines (right‐hand side) were identified with improved overall profiles. Notably, EPO concentrations in mice after p.o. administration were generally higher for certain combinations (e.g., compounds 44 and 45, see below) than for the single‐point modifications described above (e.g., compounds 21–23 and 37). Piperidinyl derivative 44 (Figure 6) displayed an IC50 value of 0.5 μm (EC50=4.9 μm) and increased solubility in water (520 mg L−1), compared with compound 1 (<1 mg L−1). With a low molecular weight of 311 g mol−1 and a logD 7.5 of 1.0, the binding efficiencies of derivative 44 (LipE=5.3, BEI=20.2) were similar to those of BAY‐908 (1). PK studies of compound 44 in rats revealed a low to moderate blood clearance of 1.3 L (h kg)−1 and a bioavailability of 39 %.28 Notably, when administering compound 44 as a Tylose suspension, the bioavailability remained unchanged, reflecting the improved aqueous solubility of compound 44 compared with screening hit BAY‐908 (1).29 In rat plasma, an unbound fraction of 16 % was determined.
Figure 6.
Compounds 44 and 45 (molidustat, BAY 85‐3934), combining imidazole/1,2,3‐triazole (left‐hand side) with cyclic amino‐substituted pyrimidines (right‐hand side).
The EPO concentration in mice following oral administration of compound 44 was higher (≈3000 pg mL−1) than that observed after administration of compound 26 (≈2000 pg mL−1).26 Likewise, an improved EPO concentration in rats was observed (Figure 7). However, the early safety and CYP profiles of compound 44 were not optimal.
Figure 7.
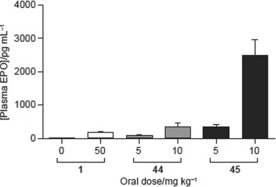
Plasma erythropoietin levels in rats at 4 h after oral dosing of compounds 1, 44 and 45 (molidustat).
A further optimized compound emerged with derivative 45 (BAY 85‐3934), as a combination of a 1,2,3‐triazole (left‐hand side) and a morpholinyl‐substituted pyrimidine (right‐hand side, Figure 6). Compound 45 displayed an IC50 value of 0.49 μm in the biochemical assay.30 The potency was independent of variations in the concentration of Fe2+ in the assay buffer.31 In the cellular assay, compound 45 showed an EC50 of 8.4 μm. A low molecular weight (314 g mol−1) and low lipophilicity (logD 7.5=0.27) resulted in high binding efficiencies (LipE=6.1, BEI=20.1). A pK a of 4.4 and an increased solubility of crystalline material in water (140 mg L−1) compared with BAY‐908 (1) (<1 mg L−1) were recorded.
Compound 45 showed a favorable CYP profile toward major CYP isoforms and a favorable selectivity profile in a lead profiling screen comprising a broad range of targets including 2‐OG‐dependent dioxygenases, metalloenzymes, nuclear hormone receptors and cell surface receptors.
PK studies of compound 45 in rats revealed a low to moderate blood clearance of 1.1 L (h kg)−1 and an oral bioavailability of 34 % (Table 5).32 The bioavailability remained unchanged when the compound was administered as a Tylose suspension.33 In rat plasma, an unbound fraction of 34 % was determined.
Table 5.
Pharmacokinetics of molidustat (BAY 85‐3934, 45).
| Species: | Rat | Dog | Monkey | |
|---|---|---|---|---|
| Sex: | Male | Female | Female | |
| Dose (i.v.) | [mg kg−1] | 5 | 3 | 1 |
| AUCnorm | [kg h L−1] | 1.3 | 1.3 | 1.4 |
| CL[a] | [L (h kg)−1] | 1.1 (b) | 1.1 (b) | 0.70 (p) |
| V ss | [L kg−1] | 0.62 | 0.78 | 0.52 |
| t 1/2 term[b] | [h] | 3.1 | 2.7 | N/A[d] |
| Dose (p.o.) | [mg kg−1] | 5 | 1.5 | 1 |
| AUCnorm | [kg h L−1] | 0.44 | 0.92 | 0.87 |
| F | [%] | 34 | 71 | 61 |
| t 1/2 term[c] | [h] | 3.0 | 2.2 | N/A[d] |
[a] (b)=blood clearance; (p)=plasma clearance; [b] Terminal half‐life referring to intervals 5–9 h (rat) and 5–24 h (dog); t 1/2 term for monkey was not calculated because concentrations at late timepoints were below the limit of quantification. [c] Terminal half‐life referring to intervals 4–24 h (rat) and 5–15 h (dog); t 1/2 term for monkey was not calculated because concentrations at late timepoints were below the limit of quantification. [d] Not applicable.
In mice, compound 45 showed an unexpectedly high plasma EPO induction (≈4000 pg mL−1), which was even higher than previously observed for compound 44 (≈3000 pg mL−1).26 Very high EPO induction was also observed in rats (Figure 7, comparison with compounds 1 and 44).
Based on its improved overall profile, compound 45 was selected as a clinical drug candidate and later received the international nonproprietary name (INN) molidustat. A comprehensive pharmacological characterization of molidustat (BAY 85‐3934, 45) has recently been published.9
Proposed binding mode of molidustat (BAY 85‐3934, 45)
The pyrazolol tautomer of molidustat (BAY 85‐3934, 45) was docked into the active site of PHD2 (PDB ID: https://www.rcsb.org/structure/2G19, Figure 8, green carbon atoms).19, 20 Similar to BAY‐908 (1), bidentate iron binding was assumed to occur via the ring nitrogen atoms of the pyrazolol core and the adjacent pyrimidine ring, forming a five‐membered ring with the Fe2+ cation. As described above, technical limitations prohibited the docking of deprotonated structures. As for BAY‐908 (1) (Figure 5), the conformation of the Arg383 side chain had to be manually adapted to accommodate the left‐hand side of the ligand (triazole), which was suggested to point into the deep part of the binding cavity. The resulting, almost parallel arrangement of the triazole moiety with the planar head group of Arg383 suggested again a possible electrostatic interaction between the substituted triazole and Arg383 involving the π‐system of the 3‐pyridyl ring. On the right‐hand side of the ligand, the morpholine moiety seemed to point toward the solvent, possibly stabilized by an edge‐to‐face interaction of the pyrimidine with Trp389. This was in line with the observation that a wider range of substituents was tolerated on the pyrimidine part, including polar, water‐soluble groups (Table 3).
Figure 8.
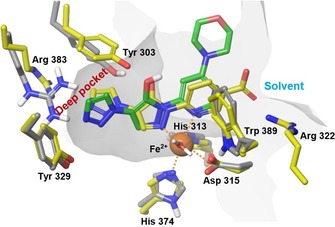
Docked structure of molidustat (BAY 85‐3934, 45, green carbon atoms) into the X‐ray crystal structure of PHD2 (PDB ID: https://www.rcsb.org/structure/2G19, grey carbon atoms) superimposed with a recently published X‐ray crystal structure of PHD2 with the pyrazolol derivative 6‐[5‐hydroxy‐4‐(1H‐1,2,3‐triazol‐1‐yl)‐1H‐pyrazol‐1‐yl]nicotinic acid (PDB ID: https://www.rcsb.org/structure/5A3U, yellow carbon atoms). Dotted lines indicate iron coordination (additional coordination to His313 and H2O not shown). Docking was performed with the electron‐neutral pyrazolol tautomer of molidustat.
As for BAY‐908 (1), no direct hydrogen bond between Tyr303 and the pyrazolol hydroxy group could be observed for molidustat (BAY 85‐3934, 45). However, a water‐mediated hydrogen bond would be possible.
Remarkably, key interactions of our proposed binding mode have been corroborated by a recently published X‐ray crystal structure of PHD2 (PDB ID: https://www.rcsb.org/structure/5A3U)34 with a HIF‐PH inhibitor of our compound class (Figure 8). The structure https://www.rcsb.org/structure/5A3U (Figure 8, yellow carbon atoms) confirms the main assumptions of our proposed binding mode: the general orientation of the ligand in the binding site, bidentate metal binding via the pyrazolol tautomer, flexibility of amino acids in the deep part of the binding cavity to accommodate the triazole moiety, and stabilization of the ligand by π‐interactions of the triazole (left‐hand side) and the pyrimidine/pyridine moieties (right‐hand side). Specifically, as assumed for BAY‐908 (1) and molidustat (BAY 85‐3934, 45), an edge‐to‐face interaction of the right‐hand side aromatic ring of the ligand with Trp389 can be found in https://www.rcsb.org/structure/5A3U. The π‐π interaction of the triazole is directed toward Tyr303 in the structure https://www.rcsb.org/structure/5A3U (and not toward Arg383, as assumed in our studies), revealing an even higher protein flexibility in the deep part of the binding cavity than expected. Furthermore, a direct hydrogen bond from Tyr303 to the pyrazolol core of the ligand can be found in the structure https://www.rcsb.org/structure/5A3U.
Syntheses
Synthesis of the examples of Table 2
Synthesis of BAY‐908 (1) was achieved starting from ethyl pyridin‐3‐ylacetate (46) via methods A and B (Scheme 1). Condensation with ethyl formate under basic conditions, followed by condensation of the resulting hydroxy‐pyridinyl‐acrylate 47 with 2‐hydrazinopyridine, gave the corresponding hydrazono derivative 48 as an isolated intermediate. Ring closure to compound 1 then proceeded under treatment with sodium ethanolate in ethanol at room temperature (RT), followed by acidification with 1 n hydrochloric acid (Scheme 1, method A). A shorter route reacted ethyl pyridin‐3‐ylacetate (46) with dimethyl formamide diethylacetal, and the resulting dimethylamino acrylate 49 was cyclized to compound 1 using camphorsulfonic acid (Scheme 1, method B).
Scheme 1.
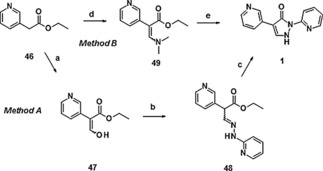
Synthesis of compound 1: a) NaH, 18‐K‐6, toluene, RT→85 °C, then HC(O)OEt, RT→90 °C, 67 %; b) 2‐hydrazinopyridine, EtOH, RT, 93 %; c) NaOEt, EtOH, RT; then HCl, 85 %; d) (Me)2NCH(OEt)2, 100 °C, 70 %; e) 2‐hydrazinopyridine, CSA, EtOH, reflux; then NaOEt, RT; then HCl, 83 %. CSA=camphorsulfonic acid.
Triazolone 4 was synthesized via cyclization of pyridinyl acetamide 51 with 4‐acetamidobenzenesulfonyl azide under non‐optimized conditions (Scheme 2).
Scheme 2.
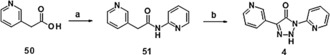
Synthesis of compound 4: a) pyridin‐2‐amine, CDI, acetone, RT, 56 %; b) NaOEt, 4‐acetamidobenzenesulfonyl azide, EtOH, 0 °C→RT, 3 %. CDI=N,N′‐dicyclohexylcarbodiimide.
Starting from dimethylamino acrylate 49, propenethioate 52 was obtained using Lawesson's reagent. Condensation with 2‐hydrazinopyridine yielded the thione compound 5. The synthesis of amino derivative 6 started from 2‐pyridin‐3‐ylacetonitrile 53, which was transformed into hydroxyacrylonitrile 54 before condensation with 2‐hydrazinopyridine to yield amino derivative 6 (Scheme 3).
Scheme 3.
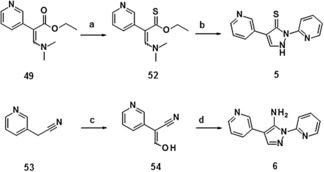
Synthesis of compounds 5 and 6: a) Lawesson's reagent, toluene, reflux, 17 %; b) 2‐hydrazinopyridine, EtOH, CSA, reflux, 4 %; c) NaH, 18‐K‐6, toluene, RT→90 °C, then HC(O)OEt, RT→90 °C, 46 %; d) 2‐hydrazinopyridine, EtOH, RT→80 °C, then NaOEt, RT, 13 %.
3H‐1,2,4‐Triazol‐3‐one 7 was obtained via initial formylation of 2‐hydrazinopyridine to give 55, followed by addition of 3‐isocyanatopyridine, yielding the open‐chained precursor 56. Subsequent cyclization under basic conditions gave 7 under non‐optimized conditions (Scheme 4).
Scheme 4.
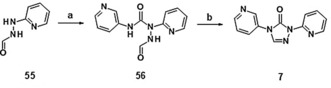
Synthesis of compound 7: a) 3‐isocyanatopyridine, CHCl3, 55 °C, 42 %; b) NaOH, MeOH, RT→45 °C, 19 %. Compound 55 was obtained from 2‐hydrazinopyridine under the following conditions: HCO2H, RT, 79 %.
Treatment of compound 1 with methanol under Mitsunobu conditions resulted in both O‐ and N‐alkylation. The methoxy derivative 8 and N‐methylated derivative 9 were obtained in moderate yields following chromatographic separation (Scheme 5).
Scheme 5.
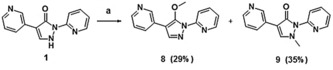
Synthesis of compounds 8 (29 %) and 9 (35 %): a) PPh3, MeOH, DIAD, RT, then separation of isomers via column chromatography on silica gel. DIAD=diisopropyl azodicarboxylate.
The synthesis of pyrimidine derivative 10 was achieved via consecutive palladium‐catalyzed arylation of the dichloro derivative 57, first with 2‐(tributylstannyl)pyridine and then with pyridin‐3‐ylboronic acid. Finally, the resulting methoxy pyrimidine 59 was hydrolyzed to the hydroxy pyrimidine 10 (Scheme 6).
Scheme 6.
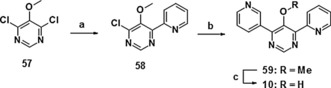
Synthesis of derivative 10: a) 2‐(tributylstannyl)pyridine, Pd(PPh3)4 toluene, reflux, 34 %; b) pyridin‐3‐ylboronic acid, Pd(PPh3)2Cl2, NaHCO3, dioxane, 80 °C, 43 %; c) NaOH, diethylene glycol, 200 °C, 95 %.
Synthesis of representative examples of Table 3
Most of the compounds shown in Table 3 were directly accessible according to the synthetic routes leading to compound 1 (via method A or method B, Scheme 1).35 Representative syntheses are detailed in Scheme 7. The 5‐hydroxymethyl derivative 26 was obtained by the direct cyclization of the dimethylamino acrylate 49 with (6‐hydrazinopyridin‐3‐yl) methanol, whereas the isomeric 4‐hydroxymethyl derivative 17 was synthesized via a route involving the cyano derivative 60, carboxylic acid 61 and methyl ester 62. The corresponding isomeric 5‐substituted carboxylic acid 24 was obtained by direct cyclization of the dimethylamino acrylate 49 yielding the ethyl ester 25, followed by saponification. Introduction of the methoxy group in the pyrimidine derivative 20 was achieved via nucleophilic substitution of the corresponding chloride 63 with methanol.
Scheme 7.
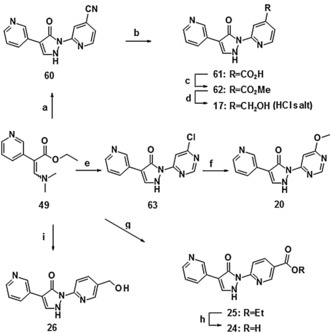
Synthesis of selected compounds of Table 3: a) 2‐hydrazino‐isonicotinonitrile, HOAc, RT, 83 %; b) NaOH, EtOH/H2O, reflux, 84 %; c) MeOH, H2SO4, reflux, 74 %; d) NaBH4, CaCl2, EtOH, 0 °C→RT, then 1 n HCl, 63 %; e) 4‐chloro‐6‐hydrazinopyrimidine, HOAc, RT, 77 %; f) MeOH, NaH, THF, TBAI, RT→reflux, 52 %; g) ethyl 6‐hydrazinonicotinate, CSA, EtOH, reflux, 54 %; h) LiOH, 1,4‐dioxane, reflux, 88 %; i) (6‐hydrazinopyridin‐3‐yl)methanol, CSA, EtOH, reflux, 66 %. CSA=camphorsulfonic acid, TBAI=tetrabutylammonium iodide.
Synthesis of representative examples of Table 4
Synthetic access to the derivatives shown in Table 4 was gained by adapting the synthetic routes leading to compound 1 via method A or method B. Synthesis of the cyano compounds 34 and 35 proceeded via the corresponding halide intermediates and subsequent palladium(0)‐catalyzed cyanation, as shown in Scheme 8.
Scheme 8.
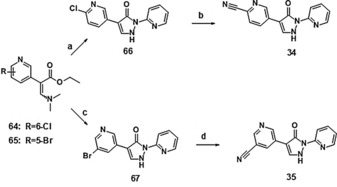
Synthesis of compounds 34 and 35: a) 2‐hydrazinopyridine, HOAc, RT, 59 %; b) Pd(PPh3)4, Zn(CN)2, DMF, microwave, 190 °C, 12 %; c) 2‐hydrazinopyridine, HOAc, RT, 31 %; d) Pd(PPh3)4 Zn(CN)2, DMF, microwave, 190 °C, 48 %. For syntheses of 64 and 65 see Supporting Information.
Synthesis of compounds 44 and 45 (molidustat, BAY 85‐3934)
The syntheses of compounds 44 and 45 (molidustat, BAY 85‐3934) started with ethyl bromoacetate 68 (Scheme 9). Nucleophilic substitution with imidazole and 1,2,3‐triazole, respectively, yielded the corresponding imidazolyl acetate 69 and a mixture of isomeric ethyl triazolyl acetates 70 and 71, respectively. The latter were separated via high‐vacuum distillation. Subsequent synthesis of the dimethylamino acrylates 72 and 73 was performed by addition of N,N‐dimethylformamide diethyl acetal at 100 °C. Cyclization to the final compounds 44 and 45 was achieved under acidic conditions via the condensation of dimethyl aminoacrylates 72 and 73 with the hydrazine derivatives 77 and 78.
Scheme 9.
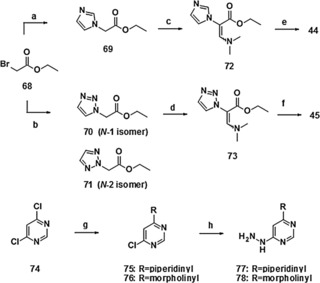
Synthesis of compounds 44 and 45 (molidustat, BAY 85‐3934): a) imidazole, NaOEt, EtOH, RT, 81 %; b) 1,2,3‐1H‐triazole, NaOEt, EtOH, RT, 50 % for 70 and 28 % for 71, followed by separation via high‐vacuum distillation; c) (Me)2NCH(OEt)2, 100 °C, 42 %; d) (Me)2NCH(OEt)2, 100 °C, 100 %; e) 77, PTS, EtOAc, reflux, 34 %; f) 78, TFA, EtOAc, reflux, 71 %; g) piperidine/morpholine, H2O, 115 °C/90 °C, 47 % for 75 and 85 % for 76; h) hydrazine hydrate, EtOH, RT→80 °C/reflux, 69 % for 77 and 68 % for 78. PTS=4‐toluenesulfonic acid.
Scale‐up syntheses of molidustat (45) and molidustat sodium
For the production of multi‐kilogram amounts, the synthetic route leading to compound 45 (molidustat, BAY 85‐3934) was modified and further improved (Scheme 10).36 Accordingly, the pyrimidine intermediate 78 was produced from the 4,6‐dichloropyrimidine 74, hydrazine hydrate and morpholine as a one‐pot procedure in water as the only solvent. Changing the sequence of processes compared with the initial synthesis (adding first hydrazine and then morpholine) allowed a drastic decrease in the excess of toxic hydrazine hydrate and resulted in a highly pure pyrimidine intermediate with yields of >70 %.
Scheme 10.
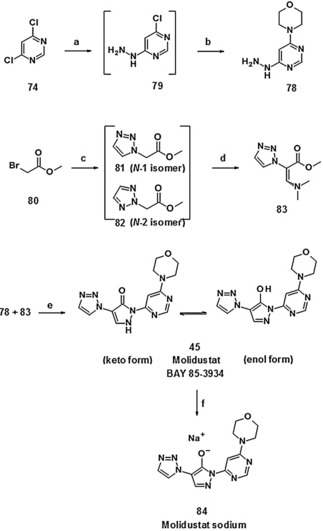
Large‐scale synthesis of molidustat (BAY 85‐3934, 45) and molidustat sodium (84): a) NEt3, hydrazine hydrate, H2O, no work‐up; b) morpholine, NaHCO3, H2O, heat, 72 % (one‐pot/two steps); c) 1,2,3‐triazole, N,N‐diisopropylethylamine (Hünig's base), EtOAc, filtration; d) N,N‐dimethylformamide dimethyl acetal, heat, acetone, crystallization from 2‐propanol, 76 % of 81; e) EtOAc, TFA, heat, H2O/HOAc, filtration, 85 %; f) NaOH, MeOH/H2O, NEt3, 98 %.
Following the optimization of reaction conditions for the formation of the acrylate intermediate, a ratio of 6:1 in favor of the N1 isomer (81) was achieved.37 The high‐vacuum distillation step could therefore be circumvented and the N‐1 acrylate 83 was crystallized from 2‐propanol in high purity, containing only <0.5 % of the N2 isomer. In summary, an improved overall yield of about 50 % for the synthesis of molidustat (BAY 85‐3934, 45) based on the starting material 1,2,3‐triazole was achieved (Scheme 10).
Molidustat (BAY 85‐3934, 45) and most of its salts were distinctly hygroscopic. Surprisingly, the crystalline sodium salt 84 (Scheme 10) proved to be an exception with regard to hygroscopicity.38 In addition, the sodium salt 84 offered highly increased solubility in water (28 g L−1 for the sodium salt 84 versus 143 mg L−1 for the parent compound 45).39 Consequently, molidustat sodium (84) was selected as a drug substance for clinical trials.
Metabolism and preclinical PK properties
The metabolism of compound 45 (molidustat) occurred via glucuronidation. The corresponding glucuronide metabolite 85 (Scheme 11) (metabolite M‐1, BAY‐348) was the only one present in human plasma and in the plasma of the other animal species investigated. Metabolite BAY‐348 is almost exclusively excreted via urine (85 % of dose).40
Scheme 11.
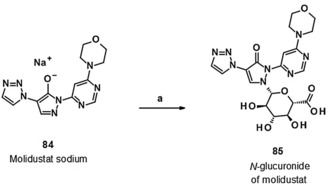
Biocatalytic transformation of molidustat sodium (84) into the N‐glucuronide 85 (BAY‐348, M‐1 metabolite of molidustat): a) S. griseochromogenes, sterile aqueous nutrient solution, DMF, 27 °C, 21 %.
A sample of this M‐1 metabolite was isolated and purified from supernatants of rat hepatocytes incubated with the parent compound 45. The inhibitory activity of M‐1 on HIF‐PH isoforms 1 and 2 was compared with that of the parent compound 45. No relevant HIF‐PH inhibitory activity and no relevant CYP inhibition of the M‐1 metabolite were detected.
Owing to pyrazolone–pyrazolol tautomerism (Figure 4), the glucuronidation of compound 45 could a priori yield either the O‐ or N‐glucuronide. NMR studies confirmed the structure of the M‐1 metabolite of compound 45 (molidustat) to be the N‐glucuronidated compound 85 (BAY‐348).
For further preclinical in vitro profiling and the development of a fully validated bioanalytical assay, a larger amount of the metabolite M‐1 was required. First, we tried to synthesize the glucuronide metabolite via chemical derivatization of compound 45. However, trichloroacetimidate‐promoted glucuronidation of compound 45 did not yield the desired metabolite, despite being a well‐established method. Possible reasons for the low chemical conversion might be the tautomeric equilibrium of compound 45 and the steric hindrance of corresponding O‐ and N‐nucleophilic positions.
Based on previous experience with biocatalysts,41 we investigated the use of UDP glucuronyltransferases (UGTs)42 recombinantly expressed in the fission yeast Schizosaccharomyces pombe 43 for the biosynthesis of metabolite 85. However, no turnover of compound 45 was observed upon incubation with S. pombe expressing relevant UGT, using either published or modified conditions.44
At that time a report using wild‐type Streptomyces strains for transforming drugs into phase I and phase II metabolites was published.45 Streptomyces are known to possess a wide array of sugar biosynthesis and transferase genes.46 We therefore initiated a systematic study using our corporate collection of microorganisms for the preparation of the desired phase II metabolite of compound 45.
After screening our internal collection of Streptomyces strains, we identified several strains that were able to convert compound 45 into a product with the molecular mass of the desired M‐1 metabolite. Notably, we were able to achieve a 20–50 % turnover on an analytical scale using a Streptomyces griseochromogenes strain and molidustat sodium (84). Successful scale‐up to multiple 20 L steel fermenters yielded a total of 1.2 g of the desired metabolite in analytically pure form. To the best of our knowledge, this successful preparation of the metabolite of compound 45 (molidustat) is the first example of a biocatalytic N‐glucuronidation on the gram scale.
The preclinical PK properties of molidustat (BAY 85‐3934, 45) were investigated in several animal species (Table 5). Taken together, the preclinical drug metabolism and PK properties of the drug candidate compound 45 were found to be well suited to further development.
Conclusions
In our pursuit of new therapies for renal anemia, a novel small‐molecule HIF‐PH inhibitor compound class (2,4‐diheteroaryl‐1,2‐dihydro‐3H‐pyrazol‐3‐ones) was discovered. Starting from high‐throughput screening hit BAY‐908 (1), medicinal chemistry optimization eventually led to the identification of BAY 85‐3934 (molidustat, 45), with an optimized compound profile including very high EPO induction in rodents. A binding hypothesis for key members of our compound class into the HIF‐PH pocket was developed by molecular modeling in the absence of a co‐crystal structure. Assumptions about general ligand orientation and bidentate iron binding were corroborated by a recently published X‐ray crystal structure of PHD2 with a HIF‐PH inhibitor of our compound class. The convergent synthesis leading to molidustat (45) and its non‐hygroscopic sodium salt 84 was largely optimized and resulted in ≈50 % overall yield on the multi‐kilogram scale. A whole‐cell microbial biotransformation on the gram scale was developed to synthesize the N‐glucuronide of molidustat (85, BAY‐348), the only metabolite in plasma through all animal species investigated. Clinical candidate molidustat (BAY 85‐3934, 45) is currently being investigated as molidustat sodium (84) in phase III clinical trials for the treatment of anemia in patients with CKD.
Experimental Section
The experimental data for the syntheses of BAY‐908 (1), compound 44, molidustat (BAY 85‐3934, 45), molidustat sodium (84), and the N‐glucuronide (BAY‐348, 85) can be found in the Experimental Section. All other experimental data, as well as general methods and materials, are provided in the Supporting Information.
Synthesis of BAY‐908 (1)
Ethyl 3‐hydroxy‐2‐(pyridin‐3‐yl)acrylate (47): Ethyl pyridin‐3‐ylacetate (46, 1.65 g, 10.0 mmol) was introduced into anhydrous toluene (20 mL) under argon. Sodium hydride suspension (410 mg, 10.3 mmol, 60 % in paraffin oil) and 18‐crown‐6 (130 mg, 0.50 mmol) were added in portions to the solution and the mixture was stirred at RT for 30 min and then at 85 °C for 30 min. After this time, the mixture was cooled and ethyl formate (1.48 g, 20.0 mol) was added dropwise at ≈20 °C. The mixture was stirred first at RT for 60 min and then at 90 °C for 60 min. After cooling, the reaction solution was introduced into ≈50 mL of saturated ammonium chloride solution and extracted with ethyl acetate (five times with 40 mL each time). The combined organic phases were washed with 50 mL of saturated sodium chloride solution, dried over sodium sulfate, filtered and concentrated. The solid obtained was washed with pentane and dried in vacuo. Yield: 1.3 g (67 %); 1H NMR (400 MHz, [D6]DMSO): δ=11.38 (br s, 1 H), 8.50 (d, 1 H), 8.39 (dd, 1 H), 7.97 (s, 1 H), 7.71 (d, 1 H), 7.35 (dd, 1 H), 4.12 (q, 2 H), 1.97 ppm (t, 3 H); MS (DCI) m/z: 194 [M+H]+.
Ethyl 2‐(pyridin‐3‐yl)‐3‐(pyridin‐2‐ylhydrazono)propanoate (48): Ethyl 3‐hydroxy‐2‐(pyridin‐3‐yl)acrylate (47, 2.90 g, 15.0 mmol) and 2‐hydrazinopyridine (1.72 g, 15.8 mmol) were dissolved in ethanol (75 mL) and the mixture was stirred at RT for 4 days. The reaction mixture was concentrated on a rotary evaporator and the residue was chromatographed over silica gel 60 (mobile phase: dichloromethane→dichloromethane/methanol 10:1→dichloromethane/methanol 2:1). The product fractions were combined and the solvent was removed on a rotary evaporator. After drying in vacuo, 3.95 g (93 %) of the title compound was obtained. MS (EI+) m/z: 285 [M+H]+.
Ethyl 3‐(dimethylamino)‐2‐(pyridin‐3‐yl)acrylate (49): Ethyl pyridin‐3‐ylacetate (46, 37.4 g, 226 mmol) was heated in dimethyl formamide diethylacetal (100 g, 679 mmol) at 100 °C overnight. After cooling, the mixture was concentrated and the residue was purified via flash chromatography (silica gel, gradient: cyclohexane/ethyl acetate 1:1→ethyl acetate/ethanol 9:1). The thus obtained product was further purified via vacuum distillation (1 mbar, 200 °C bath temperature). Yield: 35.0 g (70 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.37 (dd, 1 H), 8.31 (dd, 1 H), 7.59 (s, 1 H), 7.51 (dt, 1 H), 7.29 (ddd, 1 H), 4.00 (q, 2 H), 2.67 (s, 6 H), 1.11 ppm (t, 3 H); MS (EI+) m/z: 221 [M+H]+.
1‐(Pyridin‐2‐yl)‐4‐(pyridin‐3‐yl)‐1H‐pyrazol‐5‐ol (BAY‐908, 1): Method A: Ethyl 2‐(pyridin‐3‐yl)‐3‐(pyridin‐2‐ylhydrazono) propanoate (48, 3.95 g, 13.9 mmol) was introduced into ethanol (80 mL) under argon and sodium ethanolate (945 mg, 13.9 mmol) was added in portions at RT. After stirring for 30 min, 1 n hydrochloric acid (13.9 mL) was added dropwise. The precipitate obtained was filtered off with suction, washed with cold ethanol (20 mL) and with water (twice with 20 mL each time) and dried in vacuo; 2.80 g (85 %) of the title compound was obtained. Method B: To a mixture of 2‐hydrazinopyridine (83 mg, 0.76 mmol) and ethyl 3‐(dimethylamino)‐2‐(pyridin‐3‐yl)acrylate (49, 200 mg, 0.91 mmol) in ethanol (5 mL), CSA (18 mg, 0.08 mmol) was added and the reaction mixture was stirred overnight at reflux. The mixture was concentrated and the residue was again dissolved in ethanol (5 mL). After addition of sodium ethylate (0.34 mL, 1.14 mmol, 21 wt. % in ethanol) the mixture was stirred at RT for 1 h and then treated with 1 n hydrochloric acid until pH 5.5 was reached, then stirred overnight at RT. The precipitate was filtered off, washed with ether and dried in vacuo to yield 149 mg (83 %) of the title compound. 1H NMR (400 MHz, CDCl3): δ=13.5 (br s, 1 H), 8.97 (d, 1 H), 8.44 (dd, 1 H), 8.34 (d, 1 H), 8.05–7.91 (m, 3 H), 7.87 (s, 1 H), 7.31 (dd, 1 H), 7.25 ppm (m, 1 H); MS (DCI) m/z: 239 [M+H]+.
Synthesis of compound 44
Ethyl 1H‐imidazol‐1‐ylacetate (69): Sodium (118.2 g, 5.1 mol) was slowly added to ethanol (2.5 L). Imidazole (350.0 g, 5.1 mol) was then added and ethyl bromoacetate (570 mL, 5.1 mol) was added dropwise at an internal temperature of 20–25 °C. The mixture was stirred at RT for 24 h. The precipitate was filtered off and the filtrate was concentrated and filtered again. The residue was purified by column chromatography (silica gel, mobile phase: ethyl acetate). Yield: 639.0 g (81 %); MS (EI+) m/z: 155 [M+H]+.
Ethyl 3‐(dimethylamino)‐2‐(1H‐imidazol‐1‐yl)acrylate (72): Ethyl 1H‐imidazol‐1‐ylacetate (69, 400.0 g, 2.59 mol) was stirred in N,N‐dimethylformamide diethyl acetal (1.56 L, 9.08 mol) at a bath temperature of 100 °C for 16 h. After cooling, the mixture was concentrated in vacuo, and the residue was chromatographed over silica gel (mobile phase: dichloromethane/methanol 95:5). The product fractions were combined and the solvent was removed on a rotary evaporator. The residue was stirred with diisopropyl ether and the solid was filtered off and finally washed with diisopropyl ether to give the title compound (229.5 g, 42 %). 1H NMR (400 MHz, [D6]DMSO): δ=7.52 (s, 1 H), 7.49 (s, 1 H), 7.05 (s, 1 H), 6.91 (s, 1 H), 4.02 (q, 2 H), 2.63 (br s, 6 H), 1.12 ppm (t, 3 H); MS (EI+) m/z: 210 [M+H]+.
2‐Chloro‐4‐(piperidin‐1‐yl)pyrimidine (75): A mixture of 4,6‐dichloropyrimidine (10.0 g, 67.1 mmol) and piperidine (5.7 g, 67.1 mmol) in 100 mL of water was stirred at a bath temperature of 115 °C for 16 h. After cooling to RT, the precipitate was filtered off, washed with water and dried under reduced pressure. Yield: 6.4 g (47 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.29 (s, 1 H), 6.92 (s, 1 H), 3.65–3.58 (m, 4 H), 1.66–1.62 (m, 2 H), 1.60–1.48 ppm (m, 4 H); MS (EI+) m/z: 198 [M+H]+.
4‐Hydrazino‐6‐(piperidin‐1‐yl)pyrimidine (77): With stirring and at RT, hydrazine hydrate (17.7 mL, 364.2 mmol) was added dropwise to a solution of 4‐chloro‐6‐(piperidin‐1‐yl)pyrimidine (75, 6.0 g, 30.4 mmol) in ethanol (50 mL). The reaction solution was stirred at 80 °C for a further 16 h. For work‐up, the mixture was concentrated under reduced pressure, the residue was stirred in water, the precipitate was filtered off, the filter residue was washed twice with water (150 mL) and twice with diethyl ether (100 mL) and the product was dried under reduced pressure. Yield: 4.0 g (69 %); 1H NMR (400 MHz, [D6]DMSO): δ=7.91 (s, 1 H), 7.54 (br s, 1 H), 5.89 (s, 1 H), 4.11 (br s, 2 H), 3.50–3.47 (m, 4 H), 1.61–1.58 (m, 2 H), 1.51–1.46 ppm (m, 4 H); MS (EI+) m/z: 194 [M+H]+.
4‐(1H‐Imidazol‐1‐yl)‐1‐[6‐(piperidin‐1‐yl)pyrimidin‐4‐yl]‐1H‐pyrazol‐5‐ol (44): Ethyl 3‐(dimethylamino)‐2‐(1H‐imidazol‐1‐yl)acrylate (72, 55 g, 263 mmol) and 4‐hydrazino‐6‐(piperidin‐1‐yl)pyrimidine (77, 61 g, 315 mmol) were dissolved in ethyl acetate (550 mL) and TFA (12.0 g, 105 mmol) was added. The mixture was heated at reflux and was stirred at this temperature for 16 h. After cooling to RT, the precipitate was filtered off and washed with ethyl acetate. The solid was dissolved in water (500 mL) and the solution was adjusted to pH 7 with 1 n hydrochloric acid. The precipitate was filtered off, washed with water and diisopropyl ether and finally dried over phosphorus pentoxide. Yield: 78 g (95 %); 1H NMR (400 MHz, [D6]DMSO): δ=9.24 (s, 1 H), 8.53 (s, 1 H), 8.28 (s, 1 H), 8.04 (s, 1 H), 7.82 (s, 1 H), 7.45 (s, 1 H), 3.76–3.72 (m, 4 H), 1.74–1.52 ppm (m, 6 H); 13C NMR (125 MHz, [D6]DMSO): δ=23.9, 25.0, 44.8, 86.1, 102.4, 118.8, 126.3, 133.9, 134.6, 154.3, 155.0, 155.8, 161.6 ppm; MS (EI+) m/z: 312 [M+H]+.
Synthesis of molidustat (BAY 85‐3934, 45)
Ethyl 1H‐1,2,3‐triazol‐1‐ylacetate (70) and ethyl 2H‐1,2,3‐triazol‐2‐ylacetate (71): Sodium (129.2 g, 5.6 mol) was slowly added to ethanol (4.0 L). 1,2,3‐1H‐Triazole (400 g, 5.6 mol) was then added and ethyl bromoacetate (623 mL, 5.6 mol) was added dropwise at an internal temperature of 20–25 °C. The mixture was stirred at RT for 48 h. The precipitate was filtered off and the filtrate was concentrated and filtered again. The residue was taken up in ethyl acetate, the mixture was filtered and concentrated in vacuo and the residue was purified by distillation over a 30 cm Vigreux column. The product 71 was obtained at a bath temperature of 140 °C, an overhead temperature of 60–115 °C and a pressure of 1–7 mbar. Yield of compound 71: 28 % (purity 86 %); 1H NMR (400 MHz, [D6]DMSO): δ=7.85 (s, 2 H), 5.45 (s, 2 H), 4.16 (q, 2 H), 1.20 ppm (t, 3 H); MS (EI+) m/z: 156 [M+H]+. Compound 70 was obtained as distillation residue. Yield of compound 70: 440.0 g (50 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.14 (d, 1 H), 7.77 (d, 1 H), 5.42 (s, 2 H), 4.18 (q, 2 H), 1.22 ppm (t, 3 H); MS (EI+) m/z: 156 [M+H]+.
Ethyl 3‐(dimethylamino)‐2‐(1H‐1,2,3‐triazol‐1‐yl)acrylate (73): Ethyl 1H‐1,2,3‐triazol‐1‐ylacetate (70, 1.0 g, 6.45 mmol) was heated in N,N‐dimethylformamide diethyl acetal (3.31 mL, 19.34 mmol) overnight at 100 °C. After cooling, the mixture was concentrated, and the residue was purified by means of preparative HPLC (RP‐18 column, mobile phase: acetonitrile/water gradient). Yield: 1.4 g (100 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.10 (d, 1 H), 7.78 (d, 1 H), 7.65 (s, 1 H), 4.03 (q, 2 H), 3.06 (br s, 3 H), 2.10 (br s, 3 H), 1.12 ppm (t, 3 H); MS (EI+) m/z: 211 [M+H]+.
4‐(2‐Chloropyrimidin‐4‐yl)morpholine (76): 4,6‐dichloropyrimidine (45.0 g, 302.1 mmol) was initially charged in water (450 mL). Morpholine (26.3 g, 302.1 mmol) was added and the mixture was stirred at 90 °C for 16 h. The mixture was then cooled to 0 °C and the precipitate formed was filtered off. The precipitate was washed once with water (50 mL) and air‐dried. Yield: 51.0 g (85 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.35 (s, 1 H), 6.95 (s, 1 H), 3.62 ppm (s, 8 H); MS (EI+) m/z: 200 [M+H]+.
4‐(6‐Hydrazinopyrimidin‐4‐yl)morpholine (78): Method A (gram‐scale): 4‐(6‐chloropyrimidin‐4‐yl)morpholine (76, 53.0 g, 0.27 mol) was charged in ethanol (260 mL). Hydrazine hydrate (132.9 g, 2.70 mol) was added and the mixture was stirred under reflux for 16 h. The mixture was cooled to RT and half of the solvent was removed by distillation. The mixture was then cooled to 0 °C and the solid formed was filtered off. The solid was washed with cold ethanol, initially air‐dried and then dried under reduced pressure. Yield: 35.0 g (68 %); 1H NMR (400 MHz, [D6]DMSO): δ=7.94 (s, 1 H), 7.70 (s, 1 H), 5.91 (s, 1 H), 4.15 (s, 2 H), 3.66–3.60 (m, 4 H), 3.45–3.37 ppm (m, 4 H); MS (EI+) m/z: 196 [M+H]+. Method B (kilogram‐scale): 4,6‐dichloropyrimidine (74, 35.0 kg, 234.9 mol) was suspended in water (82 kg) at 20 °C in a stirred vessel, admixed with triethylamine (28.5 kg, 281.6 mol) and more water (5 kg), then cooled to 12 °C. Hydrazine hydrate (14.0 kg, 279.4 mol) was subsequently introduced at 12–14 °C over a period of about 1 h. More water (5 kg) was added, the mixture was stirred for another 3 h at 12 °C and was then warmed to 20 °C. After 16 h at 20 °C, sodium hydrogen carbonate (23.8 kg, 283.3 mol), then morpholine (23.6 kg, 270.9 mol) and water (5 kg) were added. The mixture was heated at a jacket temperature of 90 °C and stirred for 9 h. The solution obtained was cooled to 70 °C and seeded with 4‐(6‐hydrazinopyrimidin‐4‐yl)morpholine (78, 0.14 kg), then cooled to 3 °C over a period of 5 h. The suspension obtained was stirred at 3 °C for 3 h and filtered in a number of portions in a peeler centrifuge. The product cake was in each case washed with cold water and dried at 40 °C for about 8 h under reduced pressure in a mixer‐dryer. Yield: 32.9 kg (72 %).
Methyl 3‐(dimethylamino)‐2‐(1H‐1,2,3‐triazol‐1‐yl)acrylate (83): Kilogram‐scale: In a stirred vessel, methyl bromoacetate (80, 53.7 kg, 351 mol) was added dropwise to a solution of ethyldiisopropylamine (74.9 kg, 579.5 mol) and 1,2,3‐triazole (20 kg, 289.6 mol) in ethyl acetate (1843 kg) at 35 °C over a period of about 2 h in such a way that the internal temperature was kept at 35 to 37 °C. The reaction mixture was subsequently stirred at 40 °C for 8 h. After cooling to 10 °C, the suspension obtained was filtered, the filter cake was washed with ethyl acetate (53.3 kg) and the combined filtrates were concentrated at up to 60 °C under reduced pressure (255 kg of distillate). The evaporation residue (max. 289.6 mol) was admixed at 40 °C with N,N‐dimethyl formamide dimethyl acetal (53.1 kg, 445.6 mol) and stirred at 80 to 86 °C for 2 h. Low boilers formed were distilled off (16 kg of distillate). The mixture was subsequently cooled to 50 °C, acetone (122 kg) was added, the mixture was maintained at 50 °C and undissolved constituents were filtered off hot. The filter cake was washed once with acetone (19 kg, 50 °C). The combined filtrates were concentrated by distillation (97 kg of distillate), admixed with isopropanol (47 kg), once again concentrated by distillation at atmospheric pressure until a temperature of about 83 °C had been reached (60 kg of distillate), cooled to 70 °C and admixed with isopropanol (16 kg). The mixture was inoculated with methyl 3‐(dimethylamino)‐2‐(1H‐1,2,3‐triazol‐1‐yl)acrylate (83, 0.15 kg) and cooled to 0 °C over a period of 7 h. After a further hour at 0 °C, the mixture was filtered, the filter cake was washed twice with a mixture, which had been cooled to 0 °C, of tert‐butyl methyl ether (16 kg) and isopropanol (16 kg) and dried at 40 °C under reduced pressure. Yield: 43.0 kg (76 %, based on 1,2,3‐triazole); 1H NMR (500 MHz, [D6]DMSO): δ=8.10 (d, 1 H), 7.78 (d, 1 H), 7.67 (s, 1 H), 3.55 (s, 3 H), 3.4–2.4 (br s, 3 H), 2.4–1.7 ppm (br s, 3 H); MS (EI+) m/z: 197 [M+H]+.
1‐[6‐(Morpholin‐4‐yl)pyrimidin‐4‐yl]‐4‐(1H‐1,2,3‐triazol‐1‐yl)‐1H‐pyrazol‐5‐ol (molidustat, BAY 85‐3934, 45): Method A (gram‐scale): Ethyl 3‐(dimethylamino)‐2‐(1H‐1,2,3‐triazol‐1‐yl)acrylate (73, 1.98 g, 9.43 mmol) and 4‐(6‐hydrazinopyrimidin‐4‐yl)morpholine (78, 1.89 g, 9.70 mmol) were introduced into ethyl acetate (25 mL) and TFA (502 mg, 4.4 mmol) was added at RT. The mixture was stirred under reflux for 18 h, then cooled to 0–5 °C and subsequently stirred for a further 2 h. The solid formed was filtered off, washed with cold ethyl acetate and dried first in air and thereafter under a high vacuum. Yield: 2.13 g (71 %); 1H NMR (400 MHz, [D6]DMSO): δ=8.42 (s, 1 H), 8.38 (s, 1 H), 8.01 (s, 1 H), 7.73 (s, 1 H), 7.70 (s, 1 H), 3.71–3.65 (m, 4 H), 3.57–3.51 ppm (m, 4 H); 13C NMR (125 MHz, [D6]DMSO): δ=44.3, 65.6, 85.6, 102.8, 123.7, 132.9, 135.8, 152.4, 154.1, 154.7, 162.0 ppm; IR (KBr): =3441, 3135–3108, 2965–2884, 1636–1345, 1257 cm−1; UV/Vis (acetonitrile/water 1:1): λ max (ϵ)=249 nm (34 928 L (mol cm)−1); MS (EI+) m/z: 315 [M+H]+; Anal. calcd for C13H14N8O2: C 49.7, H 4.5, N 35.7, O 10.2, found: C 49.5, H 4.4, N 35.5, O 12.6. Method B (kilogram‐scale): In a stirred vessel, 4‐(6‐hydrazinopyrimidin‐4‐yl)morpholine (78, 42.0 kg, 215.1 mol) and methyl 3‐(dimethylamino)‐2‐(1H‐1,2,3‐triazol‐1‐yl)acrylate (83, 44.0 kg, 224.2 mol) were suspended in ethyl acetate (378 kg), admixed with TFA (12.1 kg, 106.1 mol) and heated under reflux (from 78 °C to 81 °C) at a jacket temperature of 90 °C for 26 h. The suspension obtained was cooled to 0 °C, stirred at 0 °C for 1 h and filtered. The filter cake was washed with ethyl acetate (53 kg) and dried under reduced pressure at up to 45 °C. The filter cake was admixed with a mixture of water (355 kg) and acetic acid (11.7 kg), then suspended and stirred at 50–54 °C for 1 h. After cooling to 24 °C, the suspension was filtered. The filter cake was washed first with water (90 kg), then twice with methanol (50 kg each time) and finally dried at 35–45 °C under reduced pressure. Yield: 57.4 kg (85 %).
Synthesis of molidustat sodium (84)
Sodium 1‐[6‐(morpholin‐4‐yl)pyrimidin‐4‐yl]‐4‐(1 H ‐1,2,3‐triazol‐1‐yl)‐1H‐pyrazol‐5‐olate (molidustat sodium, 84): Kilogram scale: In a stirred vessel, compound 45 (55 kg, 175.0 mol) was suspended in a mixture of methanol (200 kg) and water (30 kg), admixed with triethylamine (17.8 kg, 175.9 mmol), heated at 60 °C, stirred further for about 1 h and filtered hot to separate off undissolved constituents. The filter cake was washed with methanol (15 kg, 60 °C). Sodium hydroxide solution (18.7 kg, 210.4 mmol, 45 % strength) was slowly introduced at 60 °C and methanol (5 kg) was added. Sodium 1‐[6‐(morpholin‐4‐yl)pyrimidin‐4‐yl]‐4‐(1H‐1,2,3‐triazol‐1‐yl)‐1H‐pyrazol‐5‐olate (84, 0.12 kg) was added as seed crystals and the mixture was stirred at 60 °C for another 1 h and cooled to 24 °C over a period of about 2 h. The mixture was stirred for 8 h at this temperature, subsequently cooled to 0 °C over a period of about 1 h and filtered in portions by means of a centrifuge. The filter cake was washed with a mixture of water (24 kg) and methanol (168 kg) and also methanol (about 23 kg in each case) and dried all together at 40 °C under reduced pressure in a dryer for 8 h. Yield: 57.6 kg (98 %); 1H NMR (500 MHz, [D6]DMSO): δ=8.98 (d, J=1.4 Hz, 1 H), 8.72 (s, 1 H), 8.68 (s, 1 H), 8.64 (d, J=1.4 Hz, 1 H), 7.77 (s, 1 H), 4.25–4.00 ppm (m, 8 H); 13C NMR (125 MHz, [D6]DMSO): δ=48.2, 67.8, 91.5, 107.0, 129.6, 130.9, 138.0, 151.7, 152.0, 157.4, 159.9 ppm; IR (KBr): =3153–3006, 2976–2855, 1630–1439, 1241, 1112, 987 cm−1; UV/Vis (acetonitrile/water 1:1): λ max (ϵ)=284 nm (16 855 L [mol cm]−1); MS (EI+) m/z: 337 [M+Na]+, 315 [M+H]+; Anal. calcd for C13H13N8O2Na: C 46.4, H 3.9, N 33.3, found: C 46.1, H 4.0, N 33.1.
Synthesis of the N‐glucuronide 85 (BAY‐348)
1‐(Beta‐d‐glucopyranuronosyl)‐2‐[6‐(morpholin‐4‐yl)pyrimidin‐4‐yl]‐4‐(1H‐1,2,3‐triazol‐1‐yl)‐1,2‐dihydro‐3H‐pyrazol‐3‐one (85, BAY‐348): A 100 mL Erlenmeyer flask containing a sterile aqueous nutrient solution (20 mL) comprising malt extract (1.0 %, Oxoid L 39), yeast extract (0.4 %, Becton Dickinson 212720) and glucose monohydrate (0.4 %) at pH 7.2 was inoculated with a frozen DMSO‐sample of Streptomyces griseochromogenes (1 mL, ATCC 14511) and shaken (165 rpm) for 72 h at 27 °C. 10 mL of this culture was used to inoculate a 2 L Erlenmeyer flask containing 1000 mL of the same sterile medium as described above and shaken (165 rpm) for 48 h at 27 °C. This preculture was used to inoculate a 10 L steel fermenter which contained 8.3 L of a sterile aqueous nutrient solution comprising glucose monohydrate (1.0 %), yeast extract (0.1 %), meat extract (0.1 %, Becton Dickinson 212610) and tryptose (0.2 %) at pH 7.2. After 7 h at 27 °C, an aeration rate of 8 L min−1 and a stirring rate of 300 rpm, molidustat sodium (84, 250 mg) dissolved in DMF (20 mL) was added and cultivation was continued for another 120 h under unchanged conditions. LCMS analytics showed after this fermentation time a turnover of 25 % to the desired product with about 30 % unchanged starting material. The culture was harvested employing a continuous flow centrifuge (Contifuge) and the liquid fraction was concentrated in vacuo to provide 28 g of solid material. This material was levigated in a mixture of 620 mL methanol and 30 mL water and stirred under sonication for 5 days. Solids were removed, and the filtrate was concentrated in vacuo to yield 12.6 g of a foamy solid. HPLC purification (column: Reprospher C18 10 μm 125×30 mm; eluent A: H2O+0.1 % formic acid [99 %], eluent B: methanol; gradient: 0–0.5 min 25 mL min−1 to 70 mL min−1 15 % B; 0.5–5.5 min 5–50 % B at a flow rate of 70 mL min−1 at RT) provided the enriched target material (540 mg). A second round of HPLC purification (column: XBridge C18 5 μm 100×30 mm; eluent A: H2O+0.1 % formic acid [99 %], eluent B: methanol; gradient: 0‐0.5 min 25 mL min−1 to 70 mL min−1 5 % B; 0.5–5.5 min 5–35 % B, flow rate 70 mL min−1 at RT) provided 83 mg of analytically pure 85 (21 %). 1H NMR (400 MHz, [D6]DMSO): δ=9.10 (s, 1 H), 8.54 (d, 1 H), 8.52 (d, 1 H), 7.94 (d, 1 H), 7.08 (s, 1 H), 5.71 (d, 1 H), 5.42 (br s, 1 H), 5.39 (d, 1 H), 3.93–3.86 (m, 1 H), 3.72–3.60 (m, 9 H), 3.40–3.35 ppm (m, overlap with water signal); 1H NMR (500 MHz, CD3CN/D2O 70:30, reference CD3CN 1.9 ppm): δ=8.73 (s, 1 H), 8.43 (d, 1 H), 8.38 (d, 1 H), 7.83 (d, 1 H), 6.99 (d, 1 H), 5.29 (d, 1 H), 3.89–3.84 (1 H, obscured by HDO signal), 3.75–3.67 (5 H, partially obscured by TMB signal), 3.65–3.60 (m, 4 H), 3.49–3.46 ppm (m, 2 H); 13C NMR (125 MHz, CD3CN/D2O 70:30, reference CD3CN 1.3 ppm): δ=45.2, 66.9, 70.9, 71.8, 77.0, 77.5, 88.4, 97.5, 112.7, 125.5, 134.7, 137.3, 154.2, 158.9, 160.1, 164.0, 171.9 ppm; MS (EI+) m/z: 491 [M+H]+; TMB=1,3,5‐trimethoxybenzene (internal standard).
Animal studies
All anemia procedures conformed to national legislation (dt. Tierschutzgesetz v. 18.05.2006) and EU directives (86/609) for the use of animals for scientific purposes and were approved by the institutional animal care office of Bayer AG and by the competent regional authority (LANUV Recklinghausen).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Prof. Dr. K. Hegetschweiler for helpful discussions on iron coordination chemistry and interpretation of iron binding studies in aqueous solution, and M. Bureik for helpful discussions on the use of S. pombe‐derived biocatalysts. We are also grateful to Dr. G. Karig, Dr. A. Kuhl, Dr. L. Baerfacker, Dr. H. Paulsen, Dr. J. Gries, D. Schneider, Dr. M. Bauser, Dr. D. Bierer, Dr. R. Schohe‐Loop, Dr. J. Schuhmacher, Dr. P. Kolkhof, Dr. J. Hütter, and Prof. Dr. H. Wild for their contributions and discussions during the project phase; to Dr. P. Schmitt, H. Musche, R. Spang, Dr. H.‐C. Weiß, Dr. A. Harrenga, and Dr. M. Schäfer for analytical support including X‐ray analysis; and to U. Appel, M. Berger, P. Helfrich, A. Kischkel, I. Limberg, N. Lindner, S. Johnigk, T. Macek, K. Reimer, M. Ronge, R. Scherner, C. Siefert, N. Tesch, and S. Trenner for technical assistance. We thank Prof. Dr. A. Hillisch, Dr. K. Lustig, Dr. H. Schirok, Dr. Karl Collins, Prof. Dr. J. Mittendorf, Dr. G. Handke‐Ergueden, and Dr. F. von Nussbaum for critical reading and discussion of the manuscript. Medical writing assistance, funded by Bayer AG, was provided by Dr. Nicolas Bertheleme of Oxford PharmaGenesis, Oxford (UK). This study was funded by Bayer AG, Wuppertal, Germany.
H. Beck, M. Jeske, K. Thede, F. Stoll, I. Flamme, M. Akbaba, J.-K. Ergüden, G. Karig, J. Keldenich, F. Oehme, H.-C. Militzer, I. V. Hartung, U. Thuss, ChemMedChem 2018, 13, 988.
In memory of Jörg Keldenich
References
- 1.
- 1a. Semenza G. L., Cell 2012, 148, 399–408; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Wang G. L., Semenza G. L., Curr. Opin. Hematol. 1996, 3, 156–162. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Jelkmann W., J. Physiol. 2011, 589, 1251–1258; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Wenger R. H., Hoogewijs D., Am. J. Physiol. Renal Physiol. 2010, 298, F1287–F1296. [DOI] [PubMed] [Google Scholar]
- 3.PHD 1–3 are also known as C. elegans EGL9 homologues 1–3 (EGLN 1–3);
- 3a. Bruick R. K., McKnight S. L., Science 2001, 294, 1337–1340; [DOI] [PubMed] [Google Scholar]
- 3b. Epstein A. C. R., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J., Cell 2001, 107, 43–54. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Min J. H., Yang H., Ivan M., Gertler F., Kaelin W. G., Pavletich N. P., Science 2002, 296, 1886–1889; [DOI] [PubMed] [Google Scholar]
- 4b. Schofield C. J., Ratcliffe P. J., Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [DOI] [PubMed] [Google Scholar]
- 5. Nangaku M., Eckardt K. U., Semin. Nephrol. 2006, 26, 261–268. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Unger E. F., Thompson A. M., Blank M. J., Temple R., N. Engl. J. Med. 2010, 362, 189–192; [DOI] [PubMed] [Google Scholar]
- 6b. McCullough P. A., Barnhart H. X., Inrig J. K., Reddan D., Sapp S., Patel U. D., Singh A. K., Szczech L. A., Califf R. M., Am. J. Nephrol. 2013, 37, 549–558. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Muchnik E., Kaplan J., Expert Opin. Invest. Drugs 2011, 20, 645–656; [DOI] [PubMed] [Google Scholar]
- 7b. Rabinowitz M. H., J. Med. Chem. 2013, 56, 9369–9402; [DOI] [PubMed] [Google Scholar]
- 7c. Maxwell P., Eckardt K.-U., Nat. Rev. Nephrol. 2016, 12, 157–168; [DOI] [PubMed] [Google Scholar]
- 7d. Gupta N., Wish J. B., Am. J. Kidney Dis. 2017, 69, 815–826; [DOI] [PubMed] [Google Scholar]
- 7e. Locatelli F., Fishbane S., Block G. A., Macdougall I. C., Am. J. Nephrol. 2017, 45, 187–199. [DOI] [PubMed] [Google Scholar]
- 8. Oehme F., Jonghaus W., Narouz-Ott L., Huetter J., Flamme I., Anal. Biochem. 2004, 330, 74–80. [DOI] [PubMed] [Google Scholar]
- 9. Flamme I., Oehme F., Ellinghaus P., Jeske M., Keldenich J., Thuss U., PLoS One 2014, 9, e111838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iron(II) chelators such as 2,2′-bipyridine and deferoxamine (DFO) have been shown to inhibit enzyme activity, including HIF-PH activity, presumably by decreasing the amount of bound active site iron:
- 10a. Hewitson K. S., Schofield C. J., Drug Discovery Today 2004, 9, 704–711; [DOI] [PubMed] [Google Scholar]
- 10b. Ivan M., Haberberger T., Gervasi D. C., Michelson K. S., Gunzler V., Kondo K., Yang H. F., Sorokina I., Conaway R. C., Conaway J. W., Kaelin W. G., Proc. Natl. Acad. Sci. USA 2002, 99, 13459–13464; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Wang G. L., Semenza G. L., Blood 1993, 82, 3610–3615. [PubMed] [Google Scholar]
- 11.For detailed descriptions of pyrazolone–pyrazolol tautomerism see:
- 11a. Eicher T., Hauptmann S., Speicher A. in The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications, 3rd ed., Wiley-VCH, Weinheim, 2012, p. 247; [Google Scholar]
- 11b. Metwally M. A., Bondock S. A., El-Desouky S. I., Abdou M. M., Int. J. Mod. Org. Chem. 2012, 1, 19–54. [Google Scholar]
- 12.pK a values were determined via spectrophotometry using a Sirius Profiler SGA.
- 13.An acidified solution of Fe2+ and the ligand were treated with base and pH and absorbance spectra were recorded after each titrant addition. This was performed using a SIRIUS GLpKa instrument with dip probe absorption spectroscopy (DPAS) attachment. The experiments were conducted by Sirius Analytical Instruments Limited, UK.
- 14.Standard conditions for the biochemical HIF-PH assay included 20 μm 2-OG, 10 μm FeSO4, 2 mm ascorbate, 20 mm Tris (pH 7.5), 5 mm KCl, 1.5 mm MgCl2; Tris=tris(hydroxymethyl)aminomethane.
- 15.We calculated the lipophilic binding efficiency as LipE=pIC50−logD See also: Shultz M. D., Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991.24018190 [Google Scholar]
- 16. Marczenko Z., Balcerzak M. in Separation, Preconcentration and Spectrophotometry in Inorganic Analysis, 1st ed. (Ed.: E. Kloczko), Elsevier, Amsterdam, 2000, pp. 228–230. [Google Scholar]
- 17. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E., Nucl. Acids Res. 2000, 28, 235–242; [DOI] [PMC free article] [PubMed] [Google Scholar]; RCSB Protein Data Bank, http://www.rcsb.org. PDB IDs: 2HBT, 2HBU (not published), 2G19, 2G1M.
- 18. McDonough M. A., Li V., Flashman E., Chowdhury R., Mohr C., Liénard B. M., Zondlo J., Oldham N. J., Clifton I. J., Lewis J., McNeill L. A., Kurzeja R. J. M., Hewitson K. S., Yang E., Jordan S., Syed R. S., Schofield C. J., Proc. Natl. Acad. Sci. USA 2006, 103, 9814–9819; [DOI] [PMC free article] [PubMed] [Google Scholar]; PDB ID: 2G19 (RCSB Protein Data Bank, http://www.rcsb.org).
- 19. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749; [DOI] [PubMed] [Google Scholar]; Glide SP, Schrödinger LLC, http://www.schrodinger.com/Glide/.
- 20.In enzyme complexes with 2-OG, the 5-carboxylate of 2-OG is reported to bind to a basic residue (e.g., Arg383), often flanked by an additional hydrogen bond to an alcoholic or phenolic residue (e.g., Tyr329). For details see: Rose N. R., McDonough M. A., King O. N. F., Kawamura A., Schofield C. J., Chem. Soc. Rev. 2011, 40, 4364–4397. [DOI] [PubMed] [Google Scholar]
- 21.Determination of blood clearance of compound 1: i.v. dosing of 2 mg kg−1; vehicle: water/PEG400/EtOH 50:40:10. Determination of bioavailability of compound 1: p.o. dosing of 30 mg kg−1; vehicle: water/PEG400/EtOH/0.1 m NaOH 19:70:10:1 solution.
- 22.Determination of blood clearance of compound 1 from Tylose suspension: p.o. dosing of 30 mg kg−1 of compound 1; vehicle: 0.5 % Tylose in water. Relative bioavailability 26 %.
- 23.logD 7.5 values were determined by correlating HPLC retention times with compounds of known logD 7.5 values.
- 24.We calculated the binding efficiency index as BEI=pIC50 (MWcorr×1000)−1.
- 25.Determination of blood clearance of compound 26: i.v. dosing of 1.8 mg kg−1; vehicle: water/PEG400/EtOH 70:20:10. Determination of bioavailability of compound 26: p.o. dosing of 2 mg kg−1; vehicle: water/PEG400/EtOH 50:40:10.
- 26.Plasma EPO concentrations were determined at, or close to, expected peak values 4 h after p.o. dosing of 100 mg kg−1; baseline ≈120 pg mL−1.
- 27.For further details, including tables with IC50 values of representative examples, see for example: Jeske M., Flamme I., Stoll F., Beck H., Akbaba M. (Bayer Schering AG), Int. PCT Pub. No. WO2009129945 A1, 2009;; Thede K., Flamme I., Oehme F., Ergüden J.-K., Stoll F., Schuhmacher J., Wild H., Kolkhof P., Beck H., Keldenich J., Akbaba M., Jeske M. (Bayer Schering AG), Int. PCT Pub. No. WO2008067871 A1, 2008; and; Flamme I., Ergüden J.-K., Oehme F., Thede K., Karig G., Kuhl A., Wild H., Schuhmacher J., Kolkhof P., Bärfacker L., Hütter J. (Bayer Healthcare AG), Int. PCT Pub. No. WO2006114213 A1, 2006.
- 28.Determination of blood clearance of compound 44: i.v. dosing of 1 mg kg−1; vehicle: plasma/DMSO 99:1. Determination of bioavailability of compound 44: p.o. dosing of 5 mg kg−1; vehicle: saline/PEG400/EtOH/0.1 m NaOH 50:30:10:10).
- 29.Determination of blood clearance of compound 44 from Tylose suspension: p.o. dosing of 5 mg kg−1; vehicle: 0.5 % Tylose in water. Relative bioavailability 97 %.
- 30.The IC50 value of 0.49 μm refers to the HCl salt, as reported in: Thede K., Flamme I., Oehme F., Ergüden J.-K., Stoll F., Schuhmacher J., Wild H., Kolkhof P., Beck H., Keldenich J., Akbaba M., Jeske M. (Bayer Schering AG), Int. PCT Pub. No. WO2008067871 A1, 2008, example 72. For the salt-free compound, IC50 values of 0.48 μm, 0.28 μm and 0.45 μm (for PHD1, PHD2 and PHD3) were obtained; for a detailed description see Ref. [9].
- 31.Variation of the concentration of Fe2+ in the reaction buffer by a factor of 30 did not alter the potency of compound 45 by more than 2-fold. For a detailed description see Ref. [9].
- 32.Determination of blood clearance of compound 45: i.v. dosing of 5 mg kg−1; vehicle: water/PEG400/EtOH 30:60:10. Determination of bioavailability of compound 45: p.o. dosing of 5 mg kg−1; vehicle: PEG400/EtOH 80:20.
- 33.Determination of blood clearance of compound 45 from Tylose suspension: p.o. dosing of 1 mg kg−1; vehicle: 0.5 % Tylose in water. Relative bioavailability 102 %.
- 34. Chan M. C., Atasoylu O., Hodson E., Tumber A., Leung I. K. H., Chowdhury R., Gomez-Perez V., Demetriades M., Rydzik A. M., Holt-Martyn J., Tian Y.-M., Bishop T., Claridge T. D. W., Kawamura A., Pugh C. W., Ratcliffe P. J., Schofield C. J., PLoS One 2015, 10, e0132004; PDB ID: 5A3U (resolution 3.31 Å). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Method B proceeding via dimethylamino acrylate 49 was employed in most cases (compounds 12–15, 18, 19, 21–23, 25–27). Method A was used for compound 11 Both methods were applied for compound 16 (Supporting Information).
- 36. Militzer H.-C., Eggert J. (Bayer Pharma AG), US Pat. No. US20150087827 A1, 2015.
- 37.Alkylations of 1,2,3-triazole generally proceed with low selectivity for the N1-substituted product. Prior to process optimization ≈30–40 % of N2 isomer 71 were obtained as a by-product of the N1-isomer 70 (Scheme 9). Surprisingly, the ratio 81/82 could be improved robustly to give a ratio of >6:1 during process optimization. For detailed description see Ref. [36].
- 38.Under usual conditions (20–35 °C, normal pressure) hygroscopic compound 45 took up to 6 % w/w water already >20 % relative humidity. In contrast, the non-hygroscopic sodium salt 84 contained <0.5 % w/w water and changed water content only to a minimal extent, that is, <0.5 % w/w, even at a relative humidity of up to 90 %. For detailed description see Ref. [39].
- 39. Militzer H.-C., Gries J., Koep S. (Bayer Pharma AG), Int. PCT Pub. No. WO2012065967 A1, 2012.
- 40. Dib J., Mongongu C., Buisson C., Molina A., Schänzer W., Thuss U., Thevis M., Drug Test. Anal. 2017, 9, 61–67. [DOI] [PubMed] [Google Scholar]
- 41. Gottfried K., Klar U., Platzek J., Zorn L., ChemMedChem 2015, 10, 1240–1248. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Zöllner A., Buchheit D., Meyer M. R., Maurer H. H., Peters F. T., Bureik M., Bioanalysis 2010, 2, 1277–1290; [DOI] [PubMed] [Google Scholar]
- 42b. Cusack K. P., Koolman H. F., Lange U. E. W., Peltier H. M., Piel I., Vasudevan A., Bioorg. Med. Chem. Lett. 2013, 23, 5471–5483. [DOI] [PubMed] [Google Scholar]
- 43. Dragan C.-A., Buchheit D., Bischoff D., Ebner T., Bureik M., Drug Metab. Dispos. 2010, 38, 509–515. [DOI] [PubMed] [Google Scholar]
- 44.In our hands, CYP-expressing S. pombe biocatalysts have been used successfully to obtain phase I metabolites and an UGT-expressing S. pombe strain has been successfully used to convert a literature substrate into its gluruconide. Therefore, we exclude mere technical reasons for the lack of turnover observed in this case.
- 45.
- 45a. Marvalin C., Azerad R., Xenobiotica 2011, 41, 175–186; [DOI] [PubMed] [Google Scholar]
- 45b. Marvalin C., Denoux M., Perard S., S, Roy , Azerad R., Xenobiotica 2012, 42, 285–293. [DOI] [PubMed] [Google Scholar]
- 46. Walsh C. T., Fischbach M. A., J. Am. Chem. Soc. 2010, 132, 2469–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary