

Abstract
The purpose of the present two phase 1 studies was to assess the safety, tolerability and pharmacokinetics for topical application of a novel Janus kinase (JAK) inhibitor, JTE‐052, in Japanese healthy adult male volunteers and Japanese adult patients with atopic dermatitis (AD). Additionally, exploratory investigation was performed on the efficacy for disease severity and pruritus score in AD patients. In the QBX1‐1 study, the cutaneous safety of JTE‐052 ointment by a patch test and a photo patch test was assessed in an intra‐individual comparative study using placebo ointment, white petrolatum and non‐application as comparators. The study demonstrated that JTE‐052 ointment would be associated with a low potential for phototoxicity but had no potential for skin irritation or photoallergy. In the QBX1‐2 study, it was revealed that the systemic exposure to JTE‐052 in both healthy volunteers with normal skin and AD patients with inflamed skin was low in application of not only 1% but also 3% JTE‐052 ointment. JTE‐052 ointments of 1% and 3% were generally safe and well tolerated in both populations. In a repeated twice‐daily application for 7 days, the efficacy of JTE‐052 ointment to AD patients was observed with both 1% and 3% ointments in the exploratory investigations evaluated by Eczema Area and Severity Index, Investigator's Global Assessment and Numeric Rating Scale assessments. The mean scores for each assessment declined from the baseline throughout the study. These results suggest that the treatment of JTE‐052 ointment is generally safe and effective in AD patients, although further large confirmatory studies are needed.
Keywords: atopic dermatitis, JTE‐052, patch test, pharmacokinetics, topical
Introduction
Atopic dermatitis (AD) is a chronic relapsing pruritic inflammatory skin disease associated with skin barrier dysfunction, intense pruritus and eczematous skin lesions.1, 2 AD is estimated to affect 15–30% of children and 2–10% of adults in industrialized countries and causes significant impairments in health‐related quality of life from disease symptoms and the stigma associated with a highly visible skin condition.1, 3 Corticosteroids and calcineurin inhibitors are topically used for AD patients but these drugs have some limitations. Long‐term use of topical corticosteroids is associated with specific cutaneous adverse reactions such as atrophy and telangiectasia, frequently leading to steroid phobia due to the misunderstanding of the adverse reactions of systemic modalities.2 It causes poor treatment adherence and inadequate responses to treatments. Tacrolimus ointment is highly associated with skin irritation symptoms at the application site, such as a burning sensation and hot flushes, appearing often during the early phase of treatment.4 Also, tacrolimus ointment has a possibility of increased risk of local skin infections.4
A variety of cytokines exert their biological effects through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway, and several JAK inhibitors are approved or under development for the treatment of cytokine‐mediated diseases.5 JTE‐052 is a novel JAK inhibitor, which is being developed by Japan Tobacco Inc. (Tokyo, Japan; also in development for dermatological conditions by LEO Pharma, Ballerup, Denmark). JTE‐052 potently inhibited all of the JAK subtypes with half maximal inhibitory concentration values of 2.8 ± 0.6, 2.6 ± 0.2, 13 ± 0 and 58 ± 9 nmol/L for JAK1, JAK2, JAK3 and Tyk2, respectively.6 JTE‐052 has been investigated to test its ability to inhibit the activation of inflammatory cells such as T cells, B cells, monocytes and mast cells in vitro,6 and to suppress skin inflammation in animal dermatitis models.7 Not only immunological abnormalities but also skin barrier dysfunction and pruritus have recently been recognized as major factors involved in the pathogenesis and progression of AD.8 JTE‐052 improves the skin barrier function by promoting the production of keratinocyte proteins including filaggrin,9 and suppresses pruritus induced by interleukin (IL)‐31.10 In this context, JTE‐052 ointment is expected to be a novel drug for the treatment of AD, with an improving effect on skin barrier function and pruritus, as well as an anti‐inflammatory effect.
The primary objectives of these phase 1 studies were to evaluate the cutaneous safety of JTE‐052 ointment by a patch test and a photo patch test in Japanese healthy adult male volunteers (QBX1‐1) and the safety, tolerability and pharmacokinetics of JTE‐052 after single and during 7 days of repeated twice‐daily application of JTE‐052 ointment to healthy adult male volunteers and Japanese adult AD patients (QBX1‐2). In addition, an exploratory investigation was performed on the effect of JTE‐052 ointment on the dermatitis in AD patients in the QBX1‐2 study.
Methods
Both QBX1‐1 and QBX1‐2 studies were conducted at the Hosui General Medical Clinic (Hokkaido, Japan) according to Good Clinical Practices of the Japanese Ministerial Ordinance and the ethical principles of the Declaration of Helsinki. The study was reviewed and approved by the independent institutional review board. Written informed consent was obtained from participants prior to any study‐related assessment or procedures.
Study subjects
QBX1‐1 study
Eligible participants were healthy Japanese male volunteers aged 20–45 years with a body mass index (BMI) ranging 18.5–25.0 kg/m2 and in good health based on medical history, physical examination, vital signs, clinical laboratory measurements and 12‐lead electrocardiogram (ECG).
QBX1‐2 study
The study consisted of three parts. For part 1, Japanese male volunteers aged 20–45 years with a BMI ranging 18.5–25.0 kg/m2 and in good health based on medical history, physical examination, vital signs, clinical laboratory measurements and 12‐lead ECG were eligible. Subjects who had abnormal skin conditions that would have affected the study assessments were excluded. For parts 2 and 3, Japanese male and female patients aged from 18 to below 65 years with moderate, severe or very severe AD were studied. The diagnosis of AD was based on the criteria of the Japanese Dermatological Association; severity classification was the process of measuring the severity defined by the Research Group, granted by the Ministry of Health, Labor and Welfare.11 At baseline, patients had to have a Eczema Area and Severity Index (EASI)12 score of 10 or higher except the scores of head and neck. The following subjects were excluded: those with active infection or skin conditions that might have affected the study assessments, such as inflammation other than AD; those who had received systemic therapy such as corticosteroid or immunosuppressant therapy or phototherapy within 4 weeks before application of the investigational product; those who had changed the dosage regimen of topical steroids or tacrolimus hydrate ointment within 7 days before the day of hospitalization; those who had received an antihistamine drug, drug having antihistaminic action, antiallergic drug and Chinese herbal medicine for AD (excluding eye and nasal drops) within 7 days before the start of administration of the investigational product (except part 2); those who had received a live vaccine or hyposensitization therapy within 4 weeks before application of the investigational product; those who had received biological products within 24 weeks before application of the investigational product; and those who concurrently had an infection such as pneumonia, sepsis, urinary tract infection, herpes simplex and herpes zoster.
Study design
QBX1‐1 study
The study was a randomized, double‐blind, intra‐individual comparative study using placebo ointment, white petrolatum and non‐application as comparators. In the study, 22 subjects were enrolled. Subject disposition of the study is shown in Figure 1. The individual investigational products (0.03%, 0.1%, 0.3%, 1%, 3%, placebo ointment and white petrolatum) and the negative control (non‐application) were applied to the sites of A to H using a Finn Chamber® (SmartPractice, Phoenix, AZ, USA) for the patch test on the left paravertebral side and for the photo patch test on the right paravertebral side of the upper back in each subject (Fig. 2), according to the randomization table prepared using the circuit method.
Figure 1.
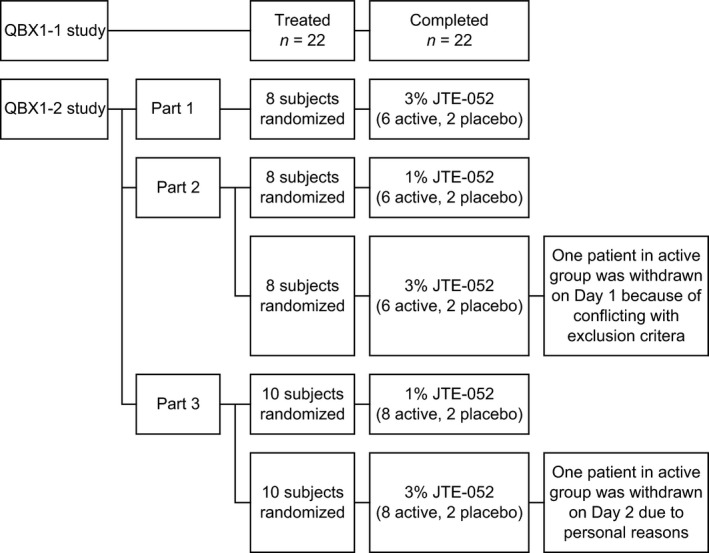
Subject disposition. Study design and subject allocation schema for QBX1‐1 study and QBX1‐2 study.
Figure 2.
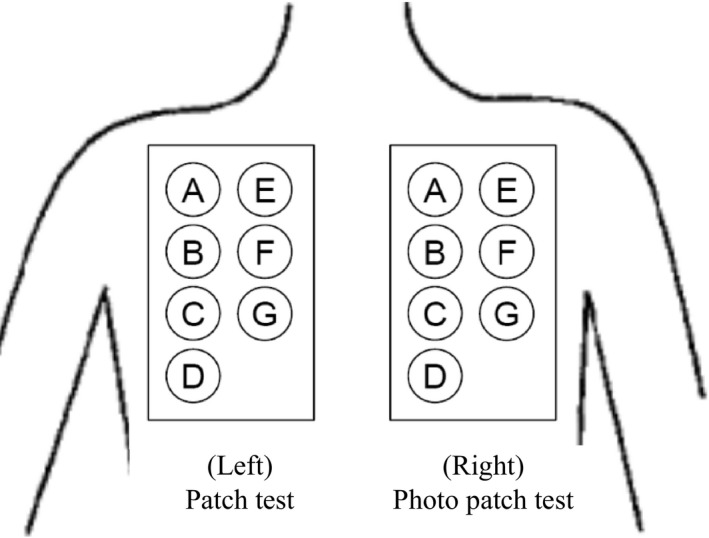
Application sites: 0.03%, 0.1%, 0.3%, 1%, 3% JTE‐052, placebo, white petrolatum and the negative control (non‐application) were applied using a Finn Chamber® for the patch test on the left paravertebral side and for the photo patch test on the right paravertebral side of the upper back. Individual investigational products (including the negative control) were randomly assigned to the application sites of each subject using the circuit method.
QBX1‐2 study
The study was a randomized, single‐blind study consisting of three parts. Subject disposition of the study is shown in Figure 1. In part 1, 3% JTE‐052 ointment was applied as a single application to Japanese healthy adult male volunteers; in part 2, 1% and 3% JTE‐052 ointments were applied as a single application to Japanese adult AD patients as a stepwise approach with two different dose groups; and in part 3, 1% and 3% JTE‐052 ointments were applied as a repeated application to Japanese adult AD patients twice daily for 7 days in the morning and the evening with a 12‐h interval in a stepwise approach with two different dose groups. After a screening period, eight subjects in parts 1 and 2 were randomized at a ratio of 3:1 to receive a single application of JTE‐052 ointment or placebo ointment, and 10 subjects in part 3 were randomized at a ratio of 4:1 to receive a repeated application of JTE‐052 ointment or placebo ointment. In part 1, 5 g of JTE‐052 ointment was applied to the back skin as a single application and then removed at 24 h post‐application. In part 2, 5 g of JTE‐052 ointment was applied to the areas affected by inflammatory eczema as a single application and then removed at 24 h post‐application. In part 3, 5 g of JTE‐052 ointment was applied twice daily for 7 days to the areas affected by inflammatory eczema, excluding the head, neck, palm, sole, groin and genitals. The application area had to be the same throughout the study and JTE‐052 ointment was removed in the morning on day 8. Rescue medication of hydrocortisone butyrate ointment 0.1% (Locoid®; Torii Pharmaceutical, Tokyo, Japan) could be used for the treatment of worsening of AD according to the investigator's judgment.
Assessments
QBX1‐1 study
The dermatologist assessed the results of: (i) the patch test; and (ii) the photo patch test as dermal safety in accordance with the Japanese Assessment Criteria.13
In (i), a Finn Chamber with disks containing the individual investigational products at a dose of approximately 20 mg was applied under occlusion to the assigned site on the left paravertebral side of the upper back for 48 h. After 48 h of application, the skin was observed and photographed at 1 and 24 h after removal of the investigational product. Erythema, edema, papule, pustule, blister, infiltration and other information on local reactions (e.g. reaction to the tape) were recorded. In (ii), a Finn Chamber with disks containing the individual investigational products was applied on the right paravertebral side of the upper back in the same way as the patch test. The Finn Chamber was removed at 24 h after application and the skin was then exposed to ultraviolet A radiation at a dose of 6.0 J/cm2 using a Dermaray 200 (Cannon Medical Supply, Tokyo, Japan). The skin was observed and photographed at 60 min after the start of irradiation, and a new Finn Chamber with empty disks was then applied for protection from light exposure. At 24 h after removal of the investigational product, the skin was then observed and photographed after a further 60 min and 24 h.
QBX1‐2 study
Pharmacokinetics, safety and exploratory efficacy evaluations of AD were assessed.
To assess the pharmacokinetics of JTE‐052, plasma and urine samples were collected. As for plasma, in parts 1 and 2, the plasma pharmacokinetic profiles of JTE‐052 after a single application of JTE‐052 ointment were assessed with the serial plasma samples collected at 0 (pre‐dose), 2, 4, 6, 8, 12, 24 and 48 h post‐dose. In part 3, the plasma pharmacokinetic profiles of JTE‐052 during 7 days of repeated twice‐daily application of JTE‐052 ointment were assessed with the serial plasma samples collected at 0 (pre‐dose), 2, 4, 6, 8, 12 and 24 h post‐dose after the morning application on days 1 and 7, and at 0 h (pre‐dose) on day 4 as a trough concentration. Also, for urine, in parts 1 and 2, urine samples were collected at the intervals of 0–4, 4–8, 8–12, 12–24 and 24–48 h to assess the urine pharmacokinetics of JTE‐052. In part 3, urine samples were collected at the intervals of 0–4, 4–8, 8–12 and 12–24 on day 7. Plasma and urine concentrations of JTE‐052 were measured using validated high‐performance liquid chromatography/tandem mass spectrometry methods with a lower limit of quantification of 1 ng/mL for plasma and 25 ng/mL for urine. Plasma pharmacokinetic parameters of JTE‐052 in the QBX1‐2 study were calculated from the plasma concentrations for each part and each dose group using a non‐compartmental method with WinNonlin Enterprise (version 5.3; Pharsight, Mountain View, CA, USA). Concentration values below the lower limit of quantification, less than 1 ng/mL, were dealt with zero (0). The pharmacokinetic parameters estimated for JTE‐052 were: Cmax, Tmax, area under the curve (AUC)0–48 and AUCtau. AUC0–48 was calculated by means of the linear‐up/log‐down trapezoidal method as the area under the concentration‐time curve from the time of application of JTE‐052 ointment to 48 h post‐dose in a single application. AUCtau was also calculated based on the dosing interval of 12 h in a repeated application. To assess the accumulation of plasma concentration of JTE‐052, the accumulation ratios of AUCtau (ARAUCtau), Cmax (ARCmax) and trough concentrations (ARCtrough) were calculated by taking a ratio of each parameter on day 7 to day 1 in the morning application. Urine concentrations and urine volumes from individual collection intervals were used to determine the urine pharmacokinetic parameters; the cumulative amount of JTE‐052 excreted in urine (Ae: 0–48 h post‐dose for parts 1 and 2, 0–12 and 12–24 h post‐dose on day 7 for part 3) was determined by the sum of the amount of JTE‐052 excreted in urine in each interval, which is obtained by multiplying the urine concentration by the urine volume per collection interval. The fraction of systemically available drug excreted into urine to the applied dose (fe: 0–48 h post‐dose for parts 1 and 2, 0–12 and 12–24 h post‐dose on day 7 for part 3) was calculated by dividing Ae by the amount of applied dose. Also, renal clearance (CLr) was determined by dividing Ae by AUC (AUC0–48 for parts 1 and 2, AUCtau for part 3).
Safety was assessed by subjective symptoms, objective findings, physiological tests (vital signs and 12‐lead ECG) and laboratory tests (hematology, blood chemistry profile and urinalysis). As for the QBX1‐1 study, adverse events were investigated from the start of study treatment on day 1 to day 4. While in the QBX1‐2 study, adverse events were investigated from the start of study treatment on day 1 to day 3 (parts 1 and 2) or day 9 (part 3). Laboratory tests were performed at screening and on days −1 and 3 (parts 1 and 2) or days −1, 4 and 9 (part 3).
Exploratory efficacy evaluations of AD by EASI, Investigator's Global Assessment (IGA) and pruritus Numeric Rating Scale (NRS) were assessed with the 1% and 3% JTE‐052 ointments in a twice‐daily application for 7 days (part 3). The assessments of EASI and IGA scores were performed at screening (only for EASI) and days −1, 1, 4 and 8. The assessment of NRS was performed twice daily from day −1 (hospitalization) to day 9 (1 day after the end of application); the period “from bedtime to morning awakening” was assessed at morning awakening and the period “from morning awakening to bedtime” was assessed at bedtime. In the efficacy assessments, subjects who discontinued the study and were treated with rescue medication were excluded.
Results
Demographics and baseline characteristics
Demographics and baseline characteristics of the 22 subjects (all men) for the QBX1‐1 study and the 44 randomized subjects (37 men and seven women) for the QBX1‐2 study are listed in Table 1. All subjects were Japanese and in good general health according to medical history, physical examination, vital signs and laboratory data, except AD in parts 2 and 3 of the QBX1‐2 study. The baseline conditions in AD patients in part 3 of the QBX1‐2 study, which was assessed efficacy exploratively, are also listed in Table 2.
Table 1.
Subject baseline demographics
| QBX1‐1 study | |||||
|---|---|---|---|---|---|
| n | Sex | Age,† years | Weight,† kg | BMI,† kg/m2 | |
| 22 | M | 21.9 ± 2.4 (20–30) | 60.6 ± 6.9 (50.5–81.8) | 20.7 ± 1.26 (18.6–24.3) | |
| QBX1‐2 study | ||||||
|---|---|---|---|---|---|---|
| Part | Group | n | Sex, n | Age, years | Weight, kg | BMI, kg/m2 |
| 1 | Placebo | 2 | M, 2; F, 0 | 24.5 | 64.2 | 23.1 |
| 3% | 6 | M, 6; F, 0 | 21.0 ± 1.3 (20–23) | 64.4 ± 7.4 (57.3–77.3) | 21.7 ± 1.5 (19.4–23.3) | |
| 2 | Placebo | 4 | M, 3; F, 1 | 30.8 ± 14.0 (21–51) | 67.1 ± 10.7 (55.5–78.1) | 24.6 ± 3.3 (21.1–28.8) |
| 1% | 6 | M, 4; F, 2 | 33.0 ± 13.1 (22–55) | 55.7 ± 6.6 (50.9–68.7) | 21.1 ± 2.0 (18.5–23.4) | |
| 3% | 6 | M, 4; F, 2 | 26.8 ± 9.6 (20–45) | 60.9 ± 7.1 (51.9–71.6) | 22.0 ± 1.8 (18.9–24.0) | |
| 3 | Placebo | 4 | M, 3; F, 1 | 24.8 ± 5.6 (21–33) | 68.7 ± 7.4 (61.6–79.0) | 24.2 ± 6.1 (19.9–33.1) |
| 1% | 8 | M, 8; F, 0 | 26.5 ± 5.4 (21–35) | 64.4 ± 8.6 (54.8–79.0) | 22.4 ± 2.0 (19.6–25.4) | |
| 3% | 8 | M, 7; F, 1 | 30.0 ± 8.2 (21–43) | 63.0 ± 9.7 (53.6–78.5) | 22.7 ± 3.2 (19.1–27.2) | |
†Mean ± standard deviation (range). BMI, body mass index; F, female; M, male; n, number of patients.
Table 2.
AD patient baseline demographics
| Part | Group | n | History of underlying disease,† year | Severity of AD, n | EASI score† | IGA† , ‡ |
|---|---|---|---|---|---|---|
| 3 | Placebo | 4 | 18.8 ± 10.8 (4–30) |
Moderate: 2 Severe: 2 Very severe: 0 |
21.2 ± 6.2 (11.9–24.9) | 3.0 ± 0.0 (3–3) |
| 1% | 8 | 20.3 ± 9.2 (2–32) |
Moderate: 8 Severe: 0 Very severe: 0 |
21.9 ± 9.7 (11.4–37.5) | 3.1 ± 0.6 (2–4) | |
| 3% | 8 | 26.4 ± 7.3 (17–38) |
Moderate: 4 Severe: 4 Very severe: 0 |
24.7 ± 6.7 (16.7–33.9) | 3.3 ± 0.5 (3–4) |
†Mean ± standard deviation (range). ‡The IGA is a 6‐point scale in severity; scales 0, 1, 2, 3, 4 and 5 are clear, almost clear, mild, moderate, severe and very severe, respectively. AD, atopic dermatitis; EASI, Eczema Area and Severity Index; IGA, Investigator's Global Assessment; n, number of patients.
Dermal safety (QBX1‐1 study)
Skin reactions at 60 min and 24 h after removal of the investigational product were assessed as “− (no reaction)” for all JTE‐052 ointments (0.03%, 0.1%, 0.3%, 1%, 3% and placebo ointment), white petrolatum and the negative control (non‐application) in all 22 subjects. No positive reactions of photoallergy were observed for any JTE‐052 ointment (0.03%, 0.1%, 0.3%, 1%, 3%, and placebo ointment), white petrolatum or the negative control (non‐application) in any of the 22 subjects assessed. Skin reactions at 60 min after removal of the Finn Chamber in the photo patch test were assessed as “+” for 0.03%, 0.1% and 0.3% JTE‐052 ointments each in one of the 22 subjects, and for 1% JTE‐052 ointment in two of the 22 subjects. For 3% JTE‐052 ointment, skin reactions were assessed as “+” and “±” each in one of the 22 subjects. For placebo ointment, white petrolatum and the negative control (non‐application), skin reactions were also assessed as “+” each in one of the 22 subjects. The phototoxicity index was calculated to be 4.5 for 0.03%, 0.1% and 0.3% JTE‐052 ointments, 9.1 for 1% JTE‐052 ointment, 6.8 for 3% JTE‐052 ointment, and 4.5 for placebo ointment, white petrolatum and the negative control (non‐application). Skin reactions at 24 h after removal of the Finn Chamber in the photo patch test were assessed as “− (no reaction or reaction equivalent to that at the application site)” for all JTE‐052 ointments (0.03%, 0.1%, 0.3%, 1%, 3% and placebo ointment), white petrolatum and the negative control (non‐application) in all 22 subjects.
Pharmacokinetics (QBX1‐2 study)
In part 1 with healthy volunteers, JTE‐052 plasma concentrations were all the lower limit of quantification (LLOQ, <1 ng/mL) after a single application of 3% JTE‐052 ointment. In part 2 with AD patients, JTE‐052 plasma concentrations after a single application of 1% JTE‐052 ointment were LLOQ in all subjects (n = 6) while 7.42 ng/mL of Cmax and 77 ng•h/mL of AUC0–48 was observed in one subject in the 3% JTE‐052 ointment group (n = 5). In part 3 with AD patients in a repeated application, the 1% JTE‐052 ointment group (n = 8) showed low levels of JTE‐052 concentrations, which were close to the LLOQ level and insufficient concentration data to calculate plasma pharmacokinetic parameters. While in the 3% JTE‐052 ointment group, JTE‐052 plasma concentrations were detected in all AD patients (n = 7) during the study, considering trough samples. Mean plasma concentration‐time profiles of JTE‐052 and its pharmacokinetic parameters are shown in Figure 3 and Table 3, respectively. JTE‐052 plasma concentration reached 3.75 ng/mL of Cmax at 4 h after the morning application on day 1 with 30.4 ng•h/mL of AUCtau. On day 7 after the morning application, 2.89 ng/mL of Cmax was obtained at 4 h with 25.2 ng•h/mL of AUCtau. The ARCmax, ARAUCtau and ARCtrough calculated by the ratio of day 7/day 1 for each exposure pharmacokinetic parameter were 0.797, 0.868 and 0.717, respectively.
Figure 3.
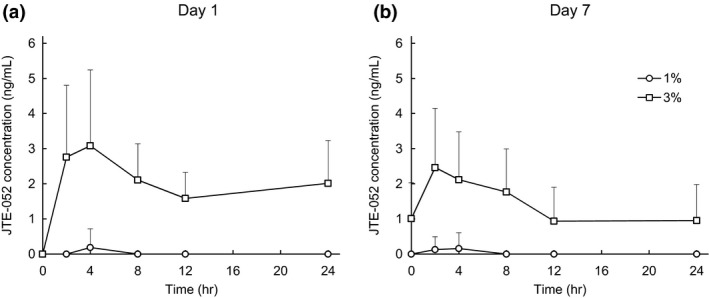
Mean (+ standard deviation) plasma concentration‐time profiles of JTE‐052 in atopic dermatitis (AD) patients on (a) day 1 and (b) day 7 after the application of 1% or 3% JTE‐052 ointment. Subjects were treated with 1% or 3% JTE‐052 ointment twice daily for 7 days in the morning and the evening with a 12‐h interval. Concentration values below the lower limits of quantification were dealt with zero (0) in the calculation of descriptive statistics for each time point.
Table 3.
Summary of plasma pharmacokinetic parameters of JTE‐052 in AD patients treated with 1% or 3% JTE‐052 ointment twice daily for 7 days in the morning and the evening with a 12‐h interval (part 3)
| Dose | Day 1 | Day 7 | ||
|---|---|---|---|---|
| 1% | 3% | 1% | 3% | |
| n | 8 | 7† | 8 | 7† |
| Tmax (h) | NC | 4.0 (2.0, 4.0)‡ | NC | 4.0 (2.0, 4.0)‡ |
| Cmax (ng/mL) | NC | 3.75 ± 1.78‡ | NC | 2.89 ± 1.39‡ |
| AUCtau (ng•h/mL) | NC | 30.4 ± 10.8‡ | NC | 25.2 ± 11.3‡ |
| ARCmax | NC | 0.797 ± 0.279§ | ||
| ARAUCtau | NC | 0.868 ± 0.347§ | ||
| ARCtrough | NC | 0.717 ± 0.269¶ | ||
†One subject withdrew due to personal reasons. ‡ n = 6: not detected JTE‐052 concentrations. § n = 5. ¶ n = 4. Parameters are expressed as mean ± standard deviation except for Tmax, median (minimum, maximum). ARAUCtau, accumulation ratio of AUCtau; ARCmax, accumulation ratio of Cmax; ARCtrough, accumulation ratio of trough concentrations; AUCtau, area under the concentration‐time curve during the dosing interval; Cmax, maximum plasma concentration; n, number of patients; Tmax, time to reach Cmax.
Urine pharmacokinetic parameters of JTE‐052 are shown in Table 4. In part 1, the fe in healthy volunteers after a single application was 0.016%. In part 2 with AD patients, the fe of JTE‐052 after a single application were 0.382% and 0.406% in 1% and 3% JTE‐052 ointment groups, respectively. Also, in part 3 with AD patients in a repeated application, the fe of JTE‐052 on day 7 after the morning application were 0.191% and 0.289% in 1% and 3% JTE‐052 ointment groups, respectively. The 19.8 L/h of CLr was obtained in the 3% JTE‐052 ointment group of part 3.
Table 4.
Summary of urine pharmacokinetic parameters of JTE‐052 in healthy Japanese male subjects and AD patients treated with 1% or 3% JTE‐052 ointment as a single application or twice daily for 7 days repeated application in the morning and the evening with a 12‐h interval
| Single application | Repeated application on day 7 | ||||
|---|---|---|---|---|---|
| Population | Healthy volunteers (part 1) | AD patients (part 2) | AD patients (part 3) | ||
| Dose | 3% | 1% | 3% | 1% | 3% |
| n | 6 | 6 | 5† | 8 | 7‡ |
| fe (%) | 0.016 ± 0.007 | 0.382 ± 0.341 | 0.406 ± 0.508 | 0.191 ± 0.174 | 0.289 ± 0.136 |
| CLr (L/h) | NC | NC | NC | NC | 19.8 ± 3.0§ |
†One subject withdrew due to meeting the exclusion criteria. ‡One subject withdrew due to personal reasons. § n = 6. Parameters are expressed as mean ± standard deviation. AD, atopic dermatitis; CLr, renal clearance; fe, fraction of systemically available drug excreted into urine; n, number of patients; NC, not calculated. Note that fe and CLr were calculated based on the 0–48‐h interval for single application or the 0–12‐h interval for repeated application.
These results demonstrated that there was low systemic exposure to JTE‐052 with no systemic accumulation when 1% or 3% JTE‐052 ointment was applied to both healthy volunteers with normal skin and AD patients with inflammatory eczema.
Safety
In the QBX1‐1 study, there were no reports of deaths, other serious adverse events (AE) or AE leading to discontinuation of observation after administration of the investigational product. There were also no reports of severe AE. One AE, protein urine present, was reported in one (4.5%) of the 22 subjects assessed. No adverse drug reactions were found in the study. As for the QBX1‐2 study, 1% and 3% JTE‐052 ointments were both generally safe and well tolerated in healthy volunteers and AD patients. Overall incidence of AE is listed in Table 5. In part 1, one (16.7%) of the six subjects in the 3% JTE‐052 ointment group reported one event (16.7%) of white blood cell count decreased. In part 2, one (16.7%) of the six subjects in the 3% JTE‐052 ointment group reported one event (16.7%) of white blood cell count decreased. One subject in the 3% JTE‐052 ointment group was withdrawn from the study 3 h after the first application of the investigational product on day 1 due to a revelation in conflict with exclusion criteria. In part 3, one (25.0%) of the four subjects in the placebo group reported one event of glucose urine present. Two (25.0%) of the eight subjects in the 1% JTE‐052 ointment group reported one event (12.5%) of erysipelas and one event (12.5%) of worsening of AD. One (12.5%) of the eight subjects in the 3% JTE‐052 ointment group reported one event (12.5%) of worsening of AD. All AE were mild in severity and erysipelas development in one subject in the 1% JTE‐052 ointment group was considered related to the study drug. Regarding the above two events of worsening of AD, those lesions were out of the application area of the investigational product and were treated with Locoid ointment as a rescue medication, and both the events subsequently resolved. There were no serious AE and no deaths occurred during the study. No consistent changes in laboratory values, vital signs or physical examinations were observed.
Table 5.
Overall incidence of adverse events
| Healthy volunteers | Patients with AD | |||||||
|---|---|---|---|---|---|---|---|---|
| Part 1 | Part 2 | Part 3 | ||||||
| Placebo | 3% | Placebo | 1% | 3% | Placebo | 1% | 3% | |
| MedDRA system organ class and preferred term† | (n = 2) | (n = 6) | (n = 4) | (n = 6) | (n = 6) | (n = 4) | (n = 8) | (n = 8) |
| Investigations | ||||||||
| White blood cell count decreased | 0 | 1 (16.7%) | 0 | 0 | 1 (16.7%) | 0 | 0 | 0 |
| Glucose urine present | 0 | 0 | 0 | 0 | 0 | 1 (25.0%) | 0 | 0 |
| Infections and infestations | ||||||||
| Erysipelas | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5%) | 0 |
| Skin and subcutaneous tissue disorders | ||||||||
| Dermatitis atopic | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5%) | 1 (12.5%) |
†Medical Dictionary for Regulatory Activities, version 16.1. AD, atopic dermatitis; n, number of patients.
Efficacy
Underlying disease condition of AD at baseline is presented in Table 2. AD patient baseline demographics were generally similar among groups except the severity of AD because there were no patients with severe AD in the group of 1% JTE‐052 ointment.
In both groups of 1% and 3% JTE‐052 ointments twice daily for 7 days, the efficacy to AD patients was observed. One patient who was withdrawn from the study and two patients who used rescue medication were excluded from the following efficacy analyses. As for EASI scores, the absolute changes from baseline (percentage of changes) on days 4 and 8 were −2.625 (−10.71%) and −7.000 (−32.71%) in the placebo group, −6.050 (−30.99%) and −10.857 (−53.12%) in the 1% JTE‐052 ointment group, and −3.700 (−17.27%) and −11.425 (−52.26%) in the 3% JTE‐052 ointment group, respectively (Fig. 4). In the distribution of subjects for absolute changes (percentage of subjects) in the IGA score on day 8, three (75%) and one (25%) out of the four subjects in the placebo group were zero (0) and −1; one (14.3%), five (71.4%) and one (14.3%) out of the seven subjects in the 1% JTE‐052 group were 0, −1 and −2; and one (16.7%), two (33.3%), two (33.3%) and one (16.7%) out of the six subjects in the 3% JTE‐052 group were 0, −1 −2 and −3, respectively (Fig. 5). The absolute changes in the NRS score at morning awakening (assessment of “during sleep”) on days 4 and 8 were −1.0 and −1.0 in the placebo group, −1.3 and −1.4 in the 1% JTE‐052 ointment group, and −2.2 and −2.7 in the 3% JTE‐052 ointment group, respectively. The absolute changes in the NRS score at bedtime (assessment of “daytime”) on days 4 and 8 were −1.0 and −1.5 in the placebo group, −0.4 and −1.0 in the 1% JTE‐052 ointment group, and −2.5 and −2.5 in the 3% JTE‐052 ointment group, respectively (Fig. 6).
Figure 4.
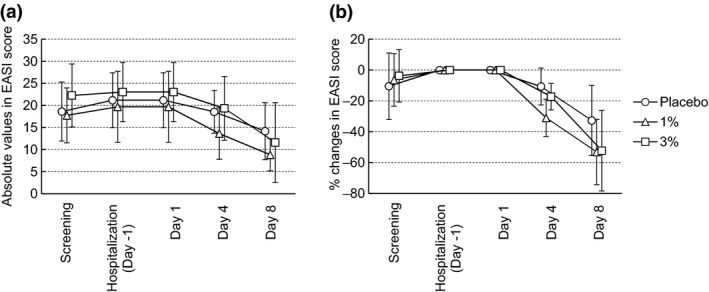
(a) Mean (± standard deviation) absolute changes in Eczema Area and Severity Index (EASI) score or (b) percentage changes in EASI score. Subjects were treated with placebo, 1% or 3% JTE‐052 ointment twice daily for 7 days in the morning and the evening with a 12‐h interval.
Figure 5.
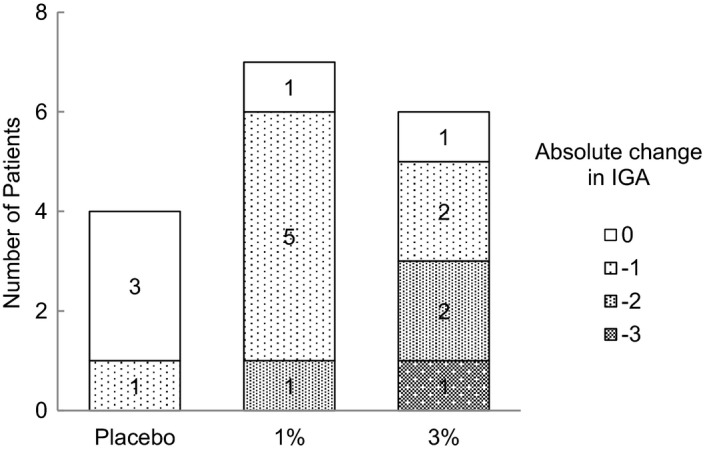
Patient distribution of absolute changes in the Investigator's Global Assessment (IGA) score on day 8. Subjects were treated with placebo, 1% or 3% JTE‐052 ointment twice daily for 7 days in the morning and the evening with a 12‐h interval.
Figure 6.
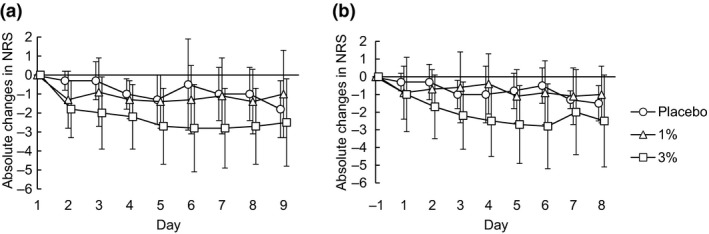
Mean (± standard deviation) absolute changes in Numeric Rating Scale (NRS). Subjects were treated with placebo, 1% or 3% JTE‐052 ointment twice daily for 7 days in the morning and the evening with a 12‐h interval. The assessment of NRS was performed twice daily from day −1 (hospitalization) to day 9 (1 day after the end of application); (a) the period “from bedtime to morning awakening” was assessed at morning awakening and (b) the period “from morning awakening to bedtime” was assessed at bedtime.
Discussion
This phase 1 study evaluated the safety, tolerability, pharmacokinetics and efficacy of JTE‐052. In the QBX1‐1 study, cutaneous safety was assessed in all 22 subjects. Based on the results of the phototoxicity index, the study demonstrated that JTE‐052 ointments up to 3% would be associated with a low potential for phototoxicity but had no potential for skin irritation or photoallergy.
In the QBX1‐2 study, the systemic exposure to JTE‐052 was investigated with 1% and 3% JTE‐052 ointments in both healthy volunteers with normal skin and AD patients with inflammatory eczema. Plasma concentrations of JTE‐052 in healthy volunteers were not detected at all even at 3% JTE‐052 ointment, while the systemic exposure to JTE‐052 in AD patients was obtained at 3% JTE‐052 ointment although its level was still low. The urine concentrations were detected in both populations. The fe after a single application of 3% JTE‐052 ointment in healthy volunteers and AD patients were 0.016 ± 0.007% (part 1) and 0.406 ± 0.508% (part 2), respectively. According to the pharmacokinetic characteristics of JTE‐052, JTE‐052 is stable to metabolism and mainly excreted into urine as JTE‐052 itself through drug transporters, explained by the result of 19.8 L/h of renal clearance, which is more than three‐times higher than the glomerular filtration rate in normal healthy subjects (Table 4). When considering the fe, the systemic exposure to JTE‐052 in AD patients was almost 20‐times higher than that in healthy volunteers, which indicates a significant higher absorption in AD patients. However, the systemic exposure to JTE‐052 in AD patients appeared to be still low.
In a repeated twice‐daily application for 7 days with 3% JTE‐052 ointment in AD patients, the pharmacokinetic parameters of JTE‐052 were obtained on both days 1 and 7 (Table 3). Cmax on days 1 and 7 were 3.75 ± 1.78 ng/mL and 2.89 ± 1.39 ng/mL, respectively. Also, AUCtau on days 1 and 7 were 30.4 ± 10.8 ng•h/mL and 25.2 ± 11.3 ng•h/mL, respectively. The accumulation ratios of day 7/day 1 for Cmax, AUCtau and Ctrough were 0.797 ± 0.279, 0.868 ± 0.347 and 0.717 ± 0.269, respectively. These findings indicate that there was no systemic accumulation despite repeated applications of JTE‐052 ointment over the 7‐day treatment period. In addition, any meaningful change of Ctrough of JTE‐052 during the study was not observed. Although the elimination half‐life was not obtained in the study, it was obviously longer than that in p.o. administration (~4 h half‐life, data not published). This longer elimination half‐life is more indicative of a slower rate of absorption rather than the actual rate of elimination. In spite of the similar patients with moderate, severe or very severe AD based on the background information, there was a difference on the systemic exposure levels between the 3% JTE‐052 ointment groups in part 2 (not detected in the most samples and subjects) and part 3 (obtained sufficient concentration data and enabled the calculation of plasma pharmacokinetic parameters in all subjects). The reason for that difference is not clear with the limited information obtained in this study, but the severity of AD and area of application site could be related. As in general knowledge, it was confirmed that the observed systemic exposure to JTE‐052 also increased accompanied by the increase of application area (data not shown).
The efficacy of JTE‐052, a JAK inhibitor, to AD patients was confirmed although the study included a small number of subjects and dosing duration was short because the study was designed primarily to assess safety and pharmacokinetics, which may limit interpretation of the results. However, 1% and 3% JTE‐052 ointments clearly and rapidly improved clinical signs and symptoms in patients with moderate, severe or very severe AD in this study. The improved change in EASI score in the placebo group was observed even though not as much as in the 1% and 3% JTE‐052 ointment groups. It was considered an effect of hospitalization in a well‐controlled condition because this study was an inpatient study. In a recent publication, it has been reported that type 2 cytokines directly activate sensory neurons in both mice and humans. Chronic itch is further dependent on neuronal IL‐4Rα and JAK1 signaling. Thus, signaling mechanisms previously ascribed to the immune system likely represent novel therapeutic targets for JAK inhibitors within the nervous system.14 The observed effect of JTE‐052 ointment, which is the reduction of NRS, is considered to be related to the inhibition of type 2 cytokine‐mediated JAK signaling activation of sensory neurons.
Observed AE were all mild in severity and high tolerability up to 3% of JTE‐052 ointment was confirmed. But one event of erysipelas in the 1% JTE‐052 ointment group in part 3 was judged as a study drug‐related AE. This AE could be caused by JAK inhibition through a mechanism of action of JTE‐052. Therefore, possible local infection due to an excessive immunosuppression will carefully be monitored in the future studies.
In conclusion, our findings indicated that JTE‐052 ointment shows considerable clinical improvements in AD patients with low systemic exposure to JTE‐052. Low systemic exposure to JTE‐052 is attractive for a JAK inhibitor due to the risk of adverse reactions. For these reasons, treatment with JTE‐052 ointment is a viable alternative to topical corticosteroids and tacrolimus in AD patients. Also, our findings support the concept that the inhibition of the JAK–STAT pathway has a potential for the treatment of AD. Further efficacy and safety results of JTE‐052 ointment will be investigated in further studies.
Conflict of Interest
H. N. is a consultant and/or has received research grants and/or honoraria from Japan Tobacco Inc., LEO Pharma and Maruho Co. Ltd. O. N. has received honoraria from Japan Tobacco Inc. H. Y., T. N. and N. N. are employees of Japan Tobacco Inc.
Acknowledgments
The authors are grateful to the subjects who participated in the study, the investigators, research staff and QBX1‐2 study team. The authors further thank Mr Koichi Maki for scientific advice about the bioanalysis part in the study. This study was sponsored by Japan Tobacco Inc.
References
- 1. Bieber T. Atopic dermatitis. N Engl J Med 2008; 358: 1483–1494. [DOI] [PubMed] [Google Scholar]
- 2. Saeki H, Nakahara T, Tanaka A et al Clinical practice guidelines for the management of atopic dermatitis 2016. J Dermatol 2016; 43: 1117–1145. [DOI] [PubMed] [Google Scholar]
- 3. Carroll CL, Balkrishnan R, Feldman SR, Fleischer AB, Manuel JC. The burden of atopic dermatitis: impact on the patient, family, and society. Pediatr Dermatol 2005; 22: 192–199. [DOI] [PubMed] [Google Scholar]
- 4. Japanese FK 506 Ointment Study Group . Clinical guidance for treatment of patients with atopic dermatitis by tacrolimus ointment 0.1% and 0.03%. Jpn J Clin Dermatol 2003; 57: 1217–1234 (In Japanese). [Google Scholar]
- 5. O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immunemediated disease. Immunity 2012; 36: 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanimoto A, Ogawa Y, Oki C et al Pharmacological properties of JTE‐052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo . Inflamm Res 2015; 64: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanimoto A, Shinozaki Y, Yamamoto Y et al A novel JAK inhibitor JTE‐052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: comparison with conventional therapeutic agent. Exp Dermatol 2017; 27: 22–29. [DOI] [PubMed] [Google Scholar]
- 8. Kabashima K. New concept of the pathogenesis of atopic dermatitis: interplay among the barrier, allergy, and pruritus as a trinity. J Dermatol Sci 2013; 70: 3–11. [DOI] [PubMed] [Google Scholar]
- 9. Amano W, Nakajima S, Kunugi H et al The Janus kinase inhibitor JTE‐052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol 2015; 136: 667–677. [DOI] [PubMed] [Google Scholar]
- 10. Yamamoto Y, Otsuka A, Nakashima C et al The effect of janus kinase inhibitor on pruritus in an atopic dermatitis murine model. J Invest Dermatol 2016; 136: S92. [Google Scholar]
- 11. Kohno Y, Yamamoto S. The study group for research on allergic disease and immunology by Ministry of Health, Labour and Welfare of Japan. Guidelines for the treatment of atopic dermatitis. 2008. (In Japanese).
- 12. Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol 2001; 10: 11–18. [DOI] [PubMed] [Google Scholar]
- 13. Kawamura T, Sasagawa S, Masuda T et al Basic studies on the standerdization of patch test. Jap J Dermatol 1970; 80: 301–314. (In Japanese). [Google Scholar]
- 14. Oetjen LK, Mack MR, Feng J et al Sensory neurons co‐opt classical immune signaling pathways to mediate chronic itch. Cell 2017; 171: 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]