

Abstract
Recent advances in computational protein design now enable the massively parallel de novo design and experimental characterization of small hyperstable binding proteins with potential therapeutic activity. By providing experimental feedback on tens of thousands of designed proteins, the design‐build‐test‐learn pipeline provides a unique opportunity to systematically improve our understanding of protein folding and binding. Here, we review the structures of mini‐protein binders in complex with Influenza hemagglutinin and Bot toxin, and illustrate in the case of disulfide bond placement how analysis of the large datasets of computational models and experimental data can be used to identify determinants of folding and binding.
Keywords: binding, computational, de novo, design, disulfide, folding, protein, stability
Introduction
Two iterations of massively parallel design‐build‐test‐learn cycles resulted in hundreds of high‐affinity designed mini‐protein binders 1 against influenza H1 hemagglutinin (HA) and the receptor‐binding domain of botulinum neurotoxin B (BoNT HCB). Mini‐proteins Bot.671.2 (PDB: http://www.rcsb.org/pdb/search/structidSearch.do?structureId=5VID) and HB1.6928.2.3 (PDB: http://www.rcsb.org/pdb/search/structidSearch.do?structureId=5VLI) obtained in these experiments were co‐crystallized with their respective targets. For Bot.671.2 (see Fig. 1A), the asymmetric unit of the crystal contains five copies of the target HCB, four in complex with the mini‐protein binder. All the copies of Bot.671.2 match the design closely (RMSD‐monomerCα < 1.0 Å). For HB1.6928.2.3 (see Fig. 1B), the asymmetric unit contains one copy of the molecule in complex with HA which closely recapitulates the computational design (RMSD‐monomerCα < 1.1 Å). Deep mutational scanning 2 revealed that the mini‐protein binders can tolerate a large number of mutations without negatively affecting binding; the exceptions are amino acids at the binding interface and polar amino acids in the vicinity of the binding surface (likely due to electrostatic effects 3). In agreement with control experiments in which lower design success was obtained with simple gly/ser loops than with Rosetta designed loops, amino acids in many of the loop positions cannot be indiscriminately replaced by glycines or serines.
Figure 1.
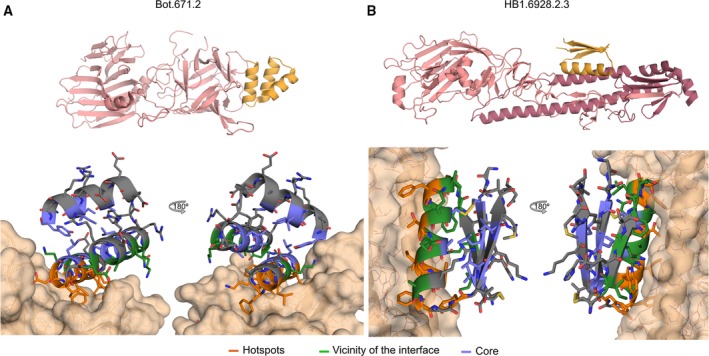
Structure of de novo designed binders. (A) The co‐crystal structure of the binder Bot.671.2 in complex with BoNT HCB. Top: The binders (orange color) in the context of the target (pink color). Bottom: Close‐up view of binding site. One (chains G and D) of the four copies of the complex present in the asymmetric unit of the crystal is shown. (B) Same as ‘A’, for the co‐crystal structure of mini‐protein binder HB1.6928.2.3 in complex with HA.
We studied the effect of systematically introducing all geometrically allowable disulfide bridges 4 into each of the BoNT designs (3193 combinations of disulfides for 141 initial nondisulfided designs). We found that 23% of all the pairs tested improved binding (or stability), 51% did not affect binding, and 26% were detrimental. We found a strong correlation between the number of interaction hotspot residues spanned by the disulfide and binding (see Fig. 2B). For example, ~ 64% of the designs that have average binding energy per‐residue < −3.25 kcal·mol−1 and whose disulfides enclose at least four hotspots improved protein binding or stability. Starting from Bot.728.3 (see Fig. 2A), we used this information to synthesize an improved disulfide protein variant named Bot.2110.4 (disulfide C5‐C28) and co‐crystallized it with HCB (PDB: http://www.rcsb.org/pdb/search/structidSearch.do?structureId=5VMR, see Fig. 2C). The experimental X‐ray structure closely resembles the design model (RMSD‐monomerCα < 1.4 Å), has a correctly formed disulfide bond, and thermal denaturation experiments show that it is hyperstable (Tm > 95 °C). Thus, even for small mini‐proteins 4, 5, there are many potential ways to introduce covalent staples to further enhance stability and function.
Figure 2.
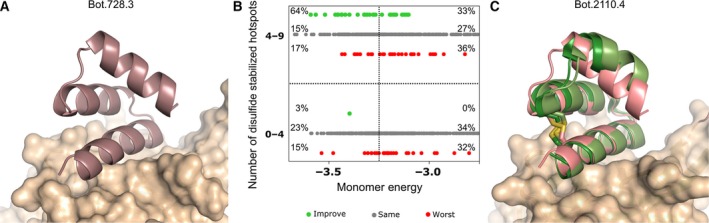
Disulfide stabilization of de novo designed mini‐protein binders. (A) The designed structure (cartoon representation) for the binder Bot.728.3 (the nondisulfide parent of Bot.2110.4, see ‘C’). (B) Effect on function of introduced disulfides on BoNT. X‐axis, the Rosetta monomer energy (kcal·mol−1 per‐residue). Y‐axis, the number of hotspot residues between the two cysteines forming the disulfide, green, disulfide designs with improved binding (or stability), red, disulfide designs with decreased binding, grey, designs with no change in binding. Disulfide designs with lower energy and more hotspots enclosed are more likely to be improved. (C) Co‐crystal structure of binder Bot.2110.4 (cartoon representation) bound to HCB (surface representation). Two copies of Bot.2110.4 in the asymmetric unit are shown in different shades of green, the computational design is in pink. The disulfide (sticks representation) is very similar in the design model and crystal structure.
Methods
Crystallographic structures are available in the RCSB protein databank (http://www.rcsb.org) 6. The dataset of computational (Rosetta) and experimental metrics is publicly available in the zenodo repository (http://www.zenodo.org, https://doi.org/10.5281/zenodo.838815). Data analysis was performed in Python (http://www.python.org) using the iPython shell 7 and the NumPy module 8. For the systematic scan of disulfides in BoNT binders (see Fig. 2B), a given design was considered to improve (or vice‐versa) if the change in binding was at least two categories (where the possible binding categories are as follows: not binding, 100, 10, 1, and 1 nm + protease resistant).
Acknowledgements
D‐AS thanks MJ Countryman for help editing this manuscript. D‐AS acknowledges support from the PEW Latin‐American Fellowship in the biomedical sciences and Mexico's CONACyT. The authors acknowledge funding support from NIH NIAID grants R01 AI125704, R01 AI091823, and R21 AI123920 to RJ, and the Washington Research Foundation, the Life Science Discovery Fund grant #9598385, and the Henrietta and Aubrey Davis Endowed Professorship in Biochemistry support to DB.
References
- 1. Chevalier A, Silva D‐A, Rocklin GJ, Hicks DR, Vergara R, Murapa P, Bernard SM, Zhang L, Lam K‐H, Yao G et al (2017) Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Whitehead TA, Chevalier A, Song Y, Dreyfus C, Fleishman SJ, De Mattos C, Myers CA, Kamisetty H, Blair P, Wilson IA et al (2012) Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat Biotechnol 30, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu Z, Ma B, Wolfson H & Nussinov R (2000) Conservation of polar residues as hot spots at protein interfaces. Proteins 39, 331–342. [PubMed] [Google Scholar]
- 4. Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A et al (2016) Accurate de novo design of hyperstable constrained peptides. Nature 538, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rocklin GJ, Chidyausiku TM, Goreshnik I, Ford A, Houliston S, Lemak A, Carter L, Ravichandran R, Mulligan VK, Chevalier A et al (2017) Global analysis of protein folding using massively parallel design, synthesis, and testing. Science 357, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN & Bourne PE (2006) The protein data bank, 1999– International Tables for Crystallography. John Wiley & Sons, Ltd, Hoboken, NJ, 675–684. [Google Scholar]
- 7. Perez F & Granger BE (2007) IPython: a system for interactive scientific computing. Comput Sci Eng 9, 21–29. [Google Scholar]
- 8. Millman KJ, Jarrod Millman K & Aivazis M (2011) Python for Scientists and Engineers. Comput Sci Eng 13, 9–12. [Google Scholar]