

Abstract
In this study, an epitope‐imprinting strategy was employed for the dynamic display of bioactive ligands on a material interface. An imprinted surface was initially designed to exhibit specific affinity towards a short peptide (i.e., the epitope). This surface was subsequently used to anchor an epitope‐tagged cell‐adhesive peptide ligand (RGD: Arg‐Gly‐Asp). Owing to reversible epitope‐binding affinity, ligand presentation and thereby cell adhesion could be controlled. As compared to current strategies for the fabrication of dynamic biointerfaces, for example, through reversible covalent or host–guest interactions, such a molecularly tunable dynamic system based on a surface‐imprinting process may unlock new applications in in situ cell biology, diagnostics, and regenerative medicine.
Keywords: cell adhesion, cell release, dynamic biointerfaces, epitopes, molecular imprinting
Cellular processes are crucially dependent on dynamic receptor–ligand interactions occurring at the interface between the cell membrane and the extracellular matrix (ECM).1 Changes in these interactions as a consequence of ECM remodeling give rise to specific cell‐signaling and intracellular cascades. These processes are central to physiology and pathological processes, such as tissue self‐repair and tumorigenesis.1b,1c As mimics of such dynamic interactions, artificial matrices with the reversible display of bioactive ligands have attracted much attention. Surfaces capable of modulating cell–biomaterial interactions are commonly exploited for in situ cell‐biology experimentation and in tissue engineering.1c, 2 Furthermore, a dynamic biointerface with reversibly immobilized ligands has also shown great promise in drug targeting and isolation methods for therapeutics and diagnostics.3
Current methods to control reversible ligand presentation on biomaterial interfaces mainly rely on surface functionalization with reversible linkers (e.g., noncovalent or reversible covalent interactions) to which the bioactive ligand is tethered. For example, by means of host–guest interactions,4 reversible covalent interactions,5 molecular assembly,6 or other multiple noncovalent interactions,7 the integrin‐targeted cell‐adhesive peptide RGD (Arg‐Gly‐Asp)8 could be dynamically and reversibly immobilized on biointerfaces to regulate cell‐adhesion behavior. These approaches towards the simulation of reversible ligand presentation in a biological system have greatly promoted the development of dynamic biointerfaces and a new generation of artificial ECM materials. To date, only a few reversible linkage chemistries have been exploited. In view of the vast number of natural receptor–ligand pairs in biology, further effort is warrented. Linkages mimicking the exquisite complementarity exhibited by natural ligand–receptor pairs are particularly attractive,9 but current strategies in this direction still leave significant room for improvement.
To attain molecularly tunable reversibility in analogy with natural receptor–ligand interactions we turned to molecular imprinting and molecularly imprinted polymers (MIPs).10 The recognition sites in MIPs, generally created by a template‐imprinting process, are spatially complementary to the shape and functionality of the template molecules.10a–10c Thus, molecular recognition in MIPs occurs by a “lock and key” mechanism similar to natural receptor–ligand interactions. In contrast to the scarce number of dynamic covalent or host–guest‐based linkages,11 imprinting can be used to tailor reversible affinity towards various ligands, including small molecules, peptides, and proteins.12 It has been shown that MIPs now can challenge and even exceed the performance of antibodies in affinity related applications both in vitro and in vivo.12a, 13 To date, however, few attempts have been made to use molecular imprinting for the reversible introducation of surface bioactivity.14
Recently, we designed an RGD‐peptide‐imprinted substrate to assist fast cell‐sheet harvesting.12c Given that we then used bioactive ligands as templates per se in that system, template binding would produce two counteracting effects: 1) it would concentrate the bioactive molecules near the interface, and 2) tightly bound bioactive molecules would be unaccessible for receptor binding.
To address the latter problem, we employed in this study an epitope‐imprinting strategy15 for introducing cell‐adhesive RGD in a fully exposed form. The principle is based on the use of a short peptide (the epitope) as template during the imprinting process and subsequently use of this epitope as an RGD tag for anchoring the RGD sequence to the substrate surface. In this design, the epitope peptide could act as a reversible anchor of RGD peptide, thus leaving the latter exposed for interaction with cell‐surface integrin receptors. Moreover, the addition of an appropriate amount of epitope peptide to the system would induce gradual release of the bound RGD‐based peptides through competitive molecular exchange; that is, the bioactivity at the material interface can be dynamically controlled.
To obtain high epitope affinity in the resultant MIPs for stable RGD anchoring, we focused on charged peptide sequences capable of engaging in multiple ionic interactions with a functional monomer. Exploitation of the amidine–carboxylate interaction seemed particularly worthwhile given its high stability in competitive solvents and previous success in molecular imprinting,16 for example, the use of benzamidine‐bearing monomers for stoichiometric imprinting.13a, 16b,16c
The epitope‐imprinted biointerface (EIB) was prepared by a microcontact imprinting method (Scheme 1).17 As a proof‐of‐concept, a structurally symmetrical and carboxyl‐rich peptide (DDDGGDDD) was used as the epitope peptide template for imprinting. Meanwhile, a bioactive long peptide (DDDGGDDDSSSSSRGDS) consisting of the epitope tag (DDDGGDDD) at the N‐terminus, a hydrophilic spacer (SSSSS) in the middle, and an integrin‐targeted peptide (RGDS) at the C‐terminal end, was designed for subsequent peptide rebinding and cell‐recognition experiments. Before microcontact imprinting, the epitope template was covalently immobilized on a cover glass at the C‐terminal end (see Figure S1 in the Supporting Information). The imprinting was subsequently performed between the cover glass with the immobilized epitope and a methacrylate‐modified quartz substrate (10 mm in diameter). The polymerization reaction was photochemically initiated by a UV lamp after dripping 3 μL of a phosphate buffer solution (PBS, 0.02 m, pH 7.4) containing 1 m 2‐hydroxyethyl acrylamide (hydrophilic backbone monomer), 0.01 m 4‐acrylamidophenyl(amino)methaniminium chloride (benzamidine‐bearing monomer; see Figure S2),13d and 0.05 m methylenebisacrylamide (cross‐linker) onto the cover glass. By polymerization and peeling of the cover glass, a peptide‐imprinted layer (ca. 4 μm; see Figure S3) on the quartz substrate with oriented recognition sites for DDDGGDDD was readily obtained (see the Supporting Information). This epitope‐imprinted biointerface (EIB) was subsequently employed to reversibly anchor the epitope‐tagged bioactive peptide DDDGGDDDSSSSSRGDS.
Scheme 1.
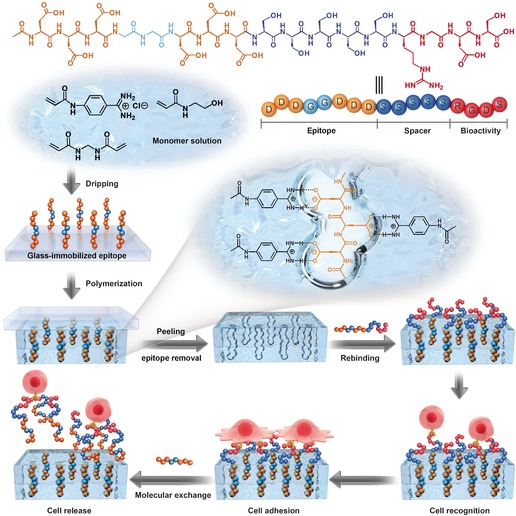
Generation of an epitope‐imprinted biointerface (EIB) and dynamic cell adhesion.
The epitope‐peptide affinity of EIB and its control NIB (non‐imprinted biointerface) was first studied by isothermal adsorption experiments. By using C‐terminally fluorescein isothiocyanate (FITC) labeled peptides (see Figures S4–S11) the amount of bound peptide could be indirectly determinated by recording the changes in fluorescence intensity in solution. Thus, for isothermal adsorption, EIB or NIB was incubated in PBS solutions of FITC‐labeled epitope (DDDGGDDDSSSSSK‐FITC) at different concentrations. The hydrophilic spacer SSSSS we anticipated would reduce the nonspecific binding of the labeled peptide to the EIB and NIB. After incubation for 12 h to ensure equilibrium adsorption, the amount of bound epitope peptide on EIB and NIB was quantified. EIB showed significantly higher peptide‐binding capacity as compared to NIB over a wide range of peptide concentrations (Figure 1 a). Scatchard analysis also revealed a higher association constant of EIB (K a=9.75×107 m −1) than that of NIB (K a=0.81×107 m −1), thus preliminarily indicating the successful imprinting of the epitope peptide on EIB (see Figure S12 and Table S1). Meanwhile, the apparent maximum number of recognition sites (i.e., imprinting sites) on EIB was estimated to be 7.35 pmol cm−2. Although the theoretical amount of imprinting sites on EIB was only one third of those of a previously reported epitope‐imprinted film, the K a value in this study was seven times higher.15 Such strong binding affinity could be ascribed to the multiple electrostatic interactions between carboxy and benzamidine groups in the imprinting sites during peptide rebinding. The selectivity of EIB and NIB towards the epitope peptide was also examined to further confirm the imprinting mechanism (Figure 1 b). The binding capacity of EIB and NIB towards the FITC‐labeled epitope (DDDGGDDDSSSSSK‐FITC), a FITC‐labeled scrambled peptide with a similar symmetrical structure (GGGDDGGGSSSSSK‐FITC), and a FITC‐labeled spacer peptide (SSSSSK‐FITC) was compared. In line with the results of isothermal adsorption, EIB showed significantly higher binding capacity towards the epitope peptide than NIB. However, the binding capacity of EIB towards the scrambled peptide and the spacer peptide was significantly lower as compared to the epitope peptide. As expected, there was no significant difference in the binding capacity of NIB towards the three different peptides. The high selectivity featured by EIB towards the epitope peptide confirms the presence of specific recognition sites for the epitope as imparted by the imprinting process.
Figure 1.
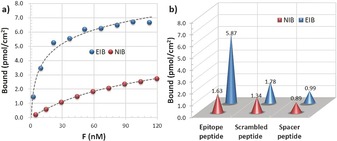
a) Binding isotherms for the epitope peptide to EIB and NIB in PBS. Peptide‐binding capacity was measured by incubating EIB or NIB in a solution of FITC‐labeled epitope (DDDGGDDDSSSSSK‐FITC, FITC‐epitope) over a concentration range of 0–124 nm at 25 °C for 12 h. F(nm) refers to the equilibrium concentration of epitope peptide after binding experiments. b) Selective binding of EIB and NIB towards different FITC‐labled peptides at a concentration of 51.8 nm in PBS. Epitope peptide, scrambled peptide, and spacer peptide refer to DDDGGDDDSSSSSK‐FITC, GGGDDGGGSSSSSK‐FITC, and SSSSSK‐FITC, respectively.
To investigate the dynamic binding properties, we immersed FITC‐peptide‐loaded EIB and NIB in PBS and subsequently followed peptide release by monitoring the substrate fluorescence intensity (Figure 2 a). After incubation for 12 h, the fluorescence intensity of EIB had decreased by only 27 %, whereas the fluorescence on NIB had decreased by nearly 50 %. More importantly, the fluorescence on EIB at 12 h was still more than three times stronger than that on NIB, thus demonstrating the stability of the bound epitope on EIB. In contrast, when it was incubated in PBS with free epitope peptide (0.1 mg mL−1), the fluorescence intensity on EIB decreased dramatically (only 25 % left after overnight incubation). Also, we found that EIB and NIB both exhibited weak fluorescence intensity after incubation with free epitope peptide for 12 h (Figure 2 b). This phenomenon is similar to observations in previously reported studies, in which reversible covalent or host–guest interactions could be switched by a competitive molecular exchange.4b, 5 According to the binding isotherms, the surface‐bound amount of epitope peptide on EIB (initial epitope concentration: 51.8 nm) was estimated to be 8.9 ng, which is far less than the amount of free epitope in solution (0.1 mg mL−1 in 1 mL of PBS). Thus, the surface‐bound peptide could be readily exchanged by the free epitope peptide, thus leading to a dramatic decrease in surface fluorescence intensity. These results demonstrated that EIB could not only bind the epitope peptide with high affinity but also release it through an epitope‐molecule‐exchange process.
Figure 2.
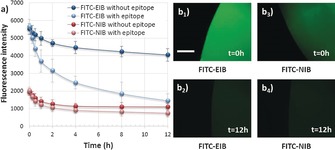
a) Fluorescence intensity changes of FITC‐epitope‐bound EIB and NIB (denoted as FITC‐EIB and FITC‐NIB, respectively) after incubation in PBS with or without the epitope peptide (0.1 mg mL−1). FITC‐EIB and FITC‐NIB were obtained by incubation in FITC‐epitope solution (51.8 nm in PBS) for 12 h. b) Representative fluorescence microscope photographs of FITC‐EIB and FTC‐NIB before and after incubation with the epitope. Scale bar: 500 μm.
EIB and NIB were then incubated in a solution with the epitope‐tagged RGD peptide DDDGGDDDSSSSSRGDS (51.8 nm in PBS) to introduce integrin‐targeted peptide RGD on the surface. The surface bioactivity of RGD was checked with integrin αvβ3, a cell‐membrane receptor.8a Atomic force microscopy (AFM) demonstrated that integrin αvβ3 could be efficiently recognized and bound to the RGD‐modified surface (see Figure S13), thus confirming the existence and accessibility of the surface‐bound RGD for cell recognition. We then checked the cell‐adhesion behavior of the RGD‐bound EIB and NIB by seeding mouse 3T3 fibroblasts on them (Figure 3). Without binding with RGD, the cell morphology only showed a typical non‐adhesive round shape (Figure 3 a,c) after culture for 3 h in α‐minimal essential medium (α‐MEM) supplemented with 10 % fetal bovine serum (FBS). After rinsing, almost no cells could adhere to either EIB or NIB. This cell‐repellant property is presumably due to the high hydrophilicity of EIB and NIB (see Figure S14) and thus reduced nonspecific protein adsorption. In contrast, the cell‐adhesion behavior on EIB was dramatically increased following RGD introduction. We observed a significant increase in adhered cells with a typical spreading shape on the RGD‐modified EIB (Figure 3 d; see also Figure S15). Also, EIB modified with different amounts of RGD all exhibited enhanced cell adhesion (see Figure S16). However, cells on the RGD‐modified NIB still featured the non‐adhered round shape. Such a significant difference between EIB and NIB can be ascribed to the markedly lower epitope‐binding constant of NIB, which resulted in weak affinity and low uptake of the peptide by this substrate. Poor cell adhesion was also observed on EIB modified with epitope‐tagged non‐adhesive RGE peptide DDDGGDDDSSSSSRGES, thus further confirming the RGD/integrin‐triggered cell‐adhesion mechanism (Figure 3 e).
Figure 3.
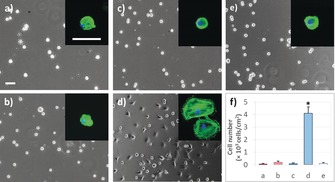
Representative micrographs of mouse 3T3 cells after culture for 3 h on a) NIB, b) NIB with RGD‐based peptide, c) EIB, d) EIB with RGD‐based peptide, and e) EIB with non‐adhesive RGE‐based peptide. RGE=Arg‐Gly‐Glu. The RGD‐ or RGE‐bound surfaces were obtained by incubation in peptide solutions (51.8 nm in PBS) for 12 h. Insets are the representative fluorescence micrographs of attached cells on differrent surfaces. Scale bar: 50 μm. f) Cell‐adhesion efficiency on different surfaces. Statistically significant differences are indicated by * p<0.001 as compared with others.
We further applied 4′‐6‐diamidino‐2‐phenylindole (DAPI) and fluorescein phalloidin to stain the nuclei and F‐actin of the adhered cells. The F‐actin networks of cells on RGD‐modified EIB exhibited typical focal adhesion patterns and spreading shape (inset in Figure 3 d; see also Figure S17). However, only sporadic round cells with no stress fibers were found for the other groups after cell staining. Taken together, these results demonstrated that the peptide‐recognition sites created by epitope imprinting could be employed to introduce an integrin‐targeted RGD peptide on a material surface and thereby induce specific cell adhesion.
The above peptide‐release experiments verified that the bound epitope on EIB could be released by an epitope‐triggered molecule‐exchange process. Thus, cell detachment from EIB was further examined to determine whether the cell‐adhesion behavior could be reversed by adding free epitope peptide to the medium. First, EIB was modified with DDDGGDDDSSSSSRGDS. After incubation for 3 h to allow cell spreading, the initial cell‐culture medium α‐MEM was changed to another α‐MEM medium containing 0.1 mg mL−1 free epitope DDDGGDDD. A gradual transition of the cell morphology from a spread‐out shape to a round shape was clearly observed in the first 4.5 h (Figure 4). After incubation for 12 h, more than 90 % of the adhered cells on EIB had been released (Figure 4). In contrast, no significant cell‐morphology change was observed on EIB without addition of the free epitope (see Figure S18). This result demonstrated the molecule‐exchange‐induced cell‐release property of our system. The cell‐release rate was much slower than for our previously reported biointerface based on monosaccharide‐responsive dynamic covalent bonds.5 We believe that the slower rate is due to the high peptide affinity of EIB and mass‐transfer limitations at the imprinted sites. The released cells could re‐adhere on a new petri dish, thus implying that the molecule‐exchange‐induced cell release from EIB occurs in a non‐invasive manner.
Figure 4.
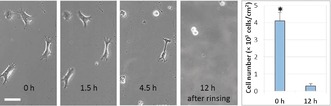
Time‐dependent detachment of the adhered cells from RGD‐bound EIB by incubation in α‐MEM with free epitope peptide (0.1 mg mL−1). Scale bar: 50 μm. The histogram on the right‐hand side indicates that the number of adhered cells had significantly decreased after incubation for 12 h. Statistically significant differences are indicated by * p<0.001 as compared with others.
In summary, we have developed an epitope‐imprinted biointerface for the reversible presentation of bioactivity and dynamic control of cell–material interactions. The surface molecular‐recognition sites, prepared by the epitope‐imprinting process, could be used to bind an epitope‐tagged bioactive peptide featuring the epitope tag at one terminus and a cell‐adhesive peptide RGD at the other terminus. Owing to the reversible‐binding property, the epitope‐imprinted biointerface exhibited dynamic RGD presentation and subsequently controllable cell‐adhesive behavior. As compared to the limited chemical means to achieve dynamic biointerfaces, such a biomimetic interaction may unlock new applications in cell biology, diagnostics, and regenerative medicine.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge financial support from Marie Skłodowska‐Curie Actions (H2020‐MSCA‐IF‐2014‐EF, 658953), the National Natural Science Foundation of China (21574091), and the Natural Science Foundation of Jiangsu Province (BK20160056).
G. Pan, S. Shinde, S. Y. Yeung, M. Jakštaitė, Q. Li, A. G. Wingren, B. Sellergren, Angew. Chem. Int. Ed. 2017, 56, 15959.
Contributor Information
Prof. Dr. Guoqing Pan, Email: tepid2010@gmail.com, Email: panguoqing@ujs.edu.cn.
Prof. Dr. Börje Sellergren, Email: borje.sellergren@mah.se.
References
- 1.
- 1a. Huebsch N., Mooney D. J., Nature 2009, 462, 426; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Daley W. P., Peters S. B., Larsen M., J. Cell Sci. 2008, 121, 255–264; [DOI] [PubMed] [Google Scholar]
- 1c. Robertus J., Browne W. R., Feringa B. L., Chem. Soc. Rev. 2010, 39, 354–378. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Uto K., Tsui J. H., DeForest C. A., Kim D.-H., Prog. Polym. Sci. 2017, 65, 53–82; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Rosales A. M., Anseth K. S., Nat. Rev. Mater. 2016, 1, 15012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Shen Q., Xu L., Zhao L., Wu D., Fan Y., Zhou Y., OuYang W. H., Xu X., Zhang Z., Song M., Adv. Mater. 2013, 25, 2368–2373; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Zhang Z., Chen N., Li S., Battig M. R., Wang Y., J. Am. Chem. Soc. 2012, 134, 15716–15719. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sheng W., Chen T., Tan W., Fan Z. H., ACS Nano 2013, 7, 7067–7076; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Boekhoven J., Rubert Pérez C. M., Sur S., Worthy A., Stupp S. I., Angew. Chem. Int. Ed. 2013, 52, 12077–12080; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12299–12302; [Google Scholar]
- 4c. An Q., Brinkmann J., Huskens J., Krabbenborg S., de Boer J., Jonkheijm P., Angew. Chem. Int. Ed. 2012, 51, 12233–12237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12399–12403; [Google Scholar]
- 4d. Seo J.-H., Kakinoki S., Inoue Y., Yamaoka T., Ishihara K., Yui N., J. Am. Chem. Soc. 2013, 135, 5513–5516; [DOI] [PubMed] [Google Scholar]
- 4e. Sánchez-Cortés J., Bähr K., Mrksich M., J. Am. Chem. Soc. 2010, 132, 9733–9737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pan G., Guo B., Ma Y., Cui W., He F., Li B., Yang H., Shea K. J., J. Am. Chem. Soc. 2014, 136, 6203–6206. [DOI] [PubMed] [Google Scholar]
- 6. Sur S., Matson J. B., Webber M. J., Newcomb C. J., Stupp S. I., ACS Nano 2012, 6, 10776–10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Li W., Wang J., Ren J., Qu X., Angew. Chem. Int. Ed. 2013, 52, 6726–6730; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6858–6862; [Google Scholar]
- 7b. Guillaume-Gentil O., Akiyama Y., Schuler M., Tang C., Textor M., Yamato M., Okano T., Vörös J., Adv. Mater. 2008, 20, 560–565. [Google Scholar]
- 8.
- 8a. Ruoslahti E., Pierschbacher M. D., Science 1987, 238, 491–497; [DOI] [PubMed] [Google Scholar]
- 8b. Pan G., Sun S., Zhang W., Zhao R., Cui W., He F., Huang L., Lee S.-H., Shea K. J., Shi Q., J. Am. Chem. Soc. 2016, 138, 15078–15086. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Moras D., Gronemeyer H., Curr. Opin. Cell Biol. 1998, 10, 384–391; [DOI] [PubMed] [Google Scholar]
- 9b. Koh J. T., Chem. Biol. 2002, 9, 17–23. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Vlatakis G., Andersson L. I., Müller R., Mosbach K., Nature 1993, 361, 645–647; [DOI] [PubMed] [Google Scholar]
- 10b. Shi H., Tsai W.-B., Garrison M. D., Ferrari S., Ratner B. D., Nature 1999, 398, 593; [DOI] [PubMed] [Google Scholar]
- 10c. Chen W., Ma Y., Pan J., Meng Z., Pan G., Sellergren B., Polymer 2015, 7, 1689–1715; [Google Scholar]
- 10d. Haupt K., Mosbach K., Chem. Rev. 2000, 100, 2495–2504. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Yang H., Yuan B., Zhang X., Scherman O. A., Acc. Chem. Res. 2014, 47, 2106–2115; [DOI] [PubMed] [Google Scholar]
- 11b. Rowan S. J., Cantrill S. J., Cousins G. R., Sanders J. K., Stoddart J. F., Angew. Chem. Int. Ed. 2002, 41, 898–952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 938–993; [Google Scholar]
- 11c.X. Zeng, G. Liu, W. Tao, Y. Ma, X. Zhang, F. He, J. Pan, L. Mei, G. Pan, Adv. Funct. Mater 2017, DOI: https://doi.org/10.1002/adfm.201605985.
- 12.
- 12a. Urraca J. L., Aureliano C. S., Schillinger E., Esselmann H., Wiltfang J., Sellergren B., J. Am. Chem. Soc. 2011, 133, 9220–9223; [DOI] [PubMed] [Google Scholar]
- 12b. Pan G., Zhang Y., Ma Y., Li C., Zhang H., Angew. Chem. Int. Ed. 2011, 50, 11731–11734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11935–11938; [Google Scholar]
- 12c. Pan G., Guo Q., Ma Y., Yang H., Li B., Angew. Chem. Int. Ed. 2013, 52, 6907–6911; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7045–7049; [Google Scholar]
- 12d. Whitcombe M. J., Chianella I., Larcombe L., Piletsky S. A., Noble J., Porter R., Horgan A., Chem. Soc. Rev. 2011, 40, 1547–1571; [DOI] [PubMed] [Google Scholar]
- 12e. Bie Z., Chen Y., Ye J., Wang S., Liu Z., Angew. Chem. Int. Ed. 2015, 54, 10211–10215; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10349–10353; [Google Scholar]
- 12f. Xing R., Wang S., Bie Z., He H., Liu Z., Nat. Protoc. 2017, 12, 964–987; [DOI] [PubMed] [Google Scholar]
- 12g. Bergmann N. M., Peppas N. A., Prog. Polym. Sci. 2008, 33, 271–288. [Google Scholar]
- 13.
- 13a. Cutivet A., Schembri C., Kovensky J., Haupt K., J. Am. Chem. Soc. 2009, 131, 14699–14702; [DOI] [PubMed] [Google Scholar]
- 13b. Hoshino Y., Kodama T., Okahata Y., Shea K. J., J. Am. Chem. Soc. 2008, 130, 15242–15243; [DOI] [PubMed] [Google Scholar]
- 13c. Kunath S., Panagiotopoulou M., Maximilien J., Marchyk N., Sänger J., Haupt K., Adv. Healthcare Mater. 2015, 4, 1322–1326; [DOI] [PubMed] [Google Scholar]
- 13d. Nestora S., Merlier F., Beyazit S., Prost E., Duma L., Baril B., Greaves A., Haupt K., Tse Sum Bui B., Angew. Chem. Int. Ed. 2016, 55, 6252–6256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6360–6364; [Google Scholar]
- 13e. Shinde S., El-Schich Z., Malakpour A., Wan W., Dizeyi N., Mohammadi R., Rurack K., Wingren A. G., Sellergren B., J. Am. Chem. Soc. 2015, 137, 13908–13912; [DOI] [PubMed] [Google Scholar]
- 13f. Panagiotopoulou M., Salinas Y., Beyazit S., Kunath S., Duma L., Prost E., Mayes A. G., Resmini M., Tse Sum Bui B., Haupt K., Angew. Chem. Int. Ed. 2016, 55, 8244–8248; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8384–8388; [Google Scholar]
- 13g. Zhang Y., Deng C., Liu S., Wu J., Chen Z., Li C., Lu W., Angew. Chem. Int. Ed. 2015, 54, 5157–5160; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5246–5249; [Google Scholar]
- 13h. Wang S., Wen Y., Wang Y., Ma Y., Liu Z., Anal. Chem. 2017, 89, 5646–5652; [DOI] [PubMed] [Google Scholar]
- 13i. Cecchini A., Raffa V., Canfarotta F., Signore G., Piletsky S., MacDonald M. P., Cuschieri A., Nano Lett. 2017, 17, 2307–2312; [DOI] [PubMed] [Google Scholar]
- 13j. Takeuchi T., Kitayama Y., Sasao R., Yamada T., Toh K., Matsumoto Y., Kataoka K., Angew. Chem. Int. Ed. 2017, 56, 7088–7092; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7194–7198. [Google Scholar]
- 14.
- 14a. Ciardelli G., Silvestri D., Cristallini C., Barbani N., Giusti P., J. Biomater. Sci. Polym. Ed. 2005, 16, 219–236; [DOI] [PubMed] [Google Scholar]
- 14b. Fukazawa K., Ishihara K., Biosens. Bioelectron. 2009, 25, 609–614. [DOI] [PubMed] [Google Scholar]
- 15. Nishino H., Huang C. S., Shea K. J., Angew. Chem. Int. Ed. 2006, 45, 2392–2396; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2452–2456. [Google Scholar]
- 16.
- 16a. Sellergren B., Anal. Chem. 1994, 66, 1578; [Google Scholar]
- 16b. Wulff G., Gross T., Schönfeld R., Angew. Chem. Int. Ed. Engl. 1997, 36, 1962–9164; [Google Scholar]; Angew. Chem. 1997, 109, 2050–2052; [Google Scholar]
- 16c. Vaidya A. A., Lele B. S., Kulkarni M. G., Mashelkar R. A., J. Appl. Polym. Sci. 2001, 81, 1075–1083. [Google Scholar]
- 17. Lin H.-Y., Rick J., Chou T.-C., Biosens. Bioelectron. 2007, 22, 3293–3301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary