

Abstract
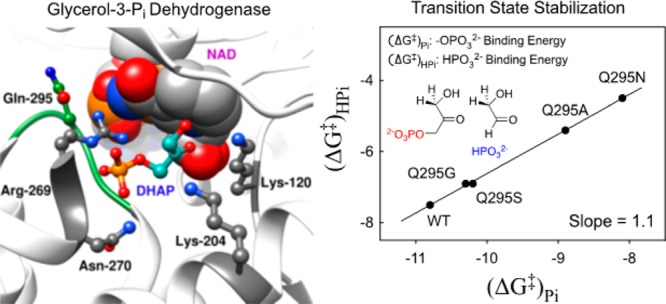
The side chain of Q295 of glycerol-3-phosphate dehydrogenase from human liver (hlGPDH) lies in a flexible loop, that folds over the phosphodianion of substrate dihydroxyacetone phosphate (DHAP). Q295 interacts with the side-chain cation from R269, which is ion-paired to the substrate phosphodianion. Kinetic parameters kcat/Km (M–1 s–1) and kcat/KGAKHPi (M–2 s–1) were determined, respectively, for catalysis of the reduction of DHAP and for dianion activation of catalysis of reduction of glycolaldehyde (GA) catalyzed by wild-type, Q295G, Q295S, Q295A, and Q295N mutants of hlGPDH. These mutations result in up to a 150-fold decrease in (kcat/Km)DHAP and up to a 2.7 kcal/mol decrease in the intrinsic phosphodianion binding energy. The data define a linear correlation with slope 1.1, between the intrinsic phosphodianion binding energy and the intrinsic phosphite dianion binding energy for activation of hlGPDH-catalyzed reduction of GA, that demonstrates a role for Q295 in optimizing this dianion binding energy. The R269A mutation of wild-type GPDH results in a 9.1 kcal/mol destabilization of the transition state for reduction of DHAP, but the same R269A mutation of N270A and Q295A mutants result in smaller 5.9 and 4.9 kcal/mol transition-state destabilization. Similarly, the N270A or Q295A mutations of R269A GPDH each result in large falloffs in the efficiency of rescue of the R269A mutant by guanidine cation. We conclude that N270, which interacts for the substrate phosphodianion and Q295, which interacts with the guanidine side chain of R269, function to optimize the apparent transition-state stabilization provided by the cationic side chain of R269.
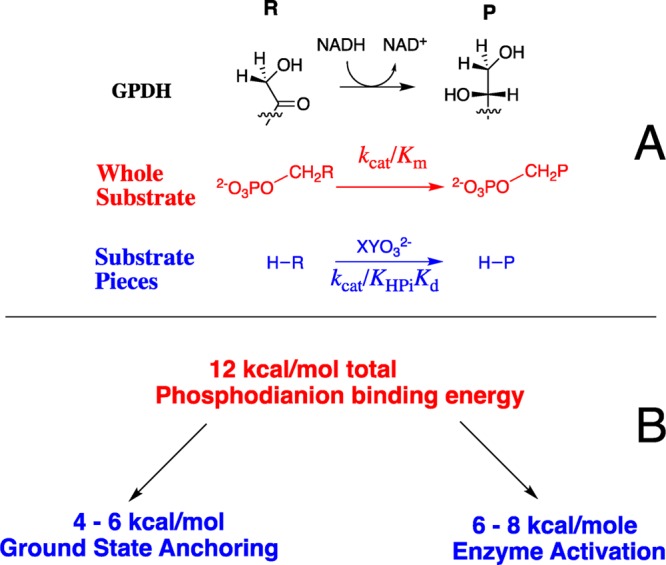
We are examining the proposition that a consideration of transition-state stabilization is sufficient to account for the rate acceleration of many or most enzymatic reactions.1−4 The transition states for the decarboxylation catalyzed by orotidine 5′-monophosphate decarboxylase (OMPDC),5 the proton transfer catalyzed by triosephosphate isomerase (TIM),6−8 and the hydride transfer catalyzed by glycerol-3-phosphate dehydrogenase (GPDH, Scheme 1A)9 are each stabilized by 11–12 kcal/mol by interactions between the protein and the substrate dianion (Scheme 1B). This is the intrinsic phosphodianion binding energy (IBE) that is utilized for catalysis of these reactions.3 From 4 to 6 kcal/mol of this IBE is expressed in the reaction ground state, where it serves to anchor the substrate to the protein catalyst.10 In the absence of the anchoring covalent connection between the dianion and substrate, from 6 to 8 kcal/mol of the dianion IBE is expressed as stabilization of the transition state for enzyme-catalyzed reactions of a truncated substrate by 1.0 M phosphite dianion (Scheme 1B).10 Similar results have been obtained in studies on the mechanism of action of β-phosphoglucomutase11 and 1-deoxy-d-xylulose-5-phosphate reductoisomerase.12
Scheme 1.
The observation of phosphite dianion activation of GPDH, OMPDC, and TIM for catalysis of the reactions of their respective truncated substrates shows that the active sites of these enzymes may be separated into a catalytic domain, which operates chemically on the bound truncated substrate, and a phosphodianion activation domain, where dianion binding interactions are utilized to promote chemistry at the catalytic domain.2,10 GPDH, OMPDC, and TIM are each activated by binding of a range of oxydianions [HPO32–, FPO32–, S2O32–, SO42–, and HOPO32–] to the respective dianion activation sites.13
We are working to characterize the dianion activation sites of TIM, OMPDC, and GPDH from human liver (hlGPDH) and to determine the common architectural features of these sites that enable dianion activation of the enzymes that catalyze a chemically diverse set or reactions. Dianion activation of TIM and OMPDC has been linked to enzyme conformational changes that are highlighted by closure of flexible loops over the phosphodianion of enzyme-bound substrate.14−19 The results of recent mutagenesis studies on OMPDC and TIM have provided considerable insight into the mechanism for activation of these enzymes by dianion driven loop closure.
Figure 1 shows the unliganded form of hlGPDH and the closed ternary hlGPDH·NAD·DHAP complex.10 The flexible loop [292-LNGQKL-297] that closes over the phosphodianion of DHAP at the ternary complex is shaded blue, and the side chain of R269 is shaded red. This cationic side chain acts to bridge the flexible enzyme loop to the substrate, through interactions with the loop amide side chain of Q295 and the phosphodianion of DHAP, which is shaded green. Closure of the flexible loop over the surface of hlGPDH locks DHAP in a protein cage,18,22 where the carbonyl group shows a high reactivity toward reduction by NADH.
Figure 1.

Representations, from X-ray crystal structures, of the surface of the open and closed forms of hlGPDH. (A) The open form of the unliganded enzyme (PDB entry 1X0V). (B) The closed form of the nonproductive E·NAD·DHAP ternary complex (PDB entry 1WPQ). In each the flexible loop [292-LNGQKL-297] is shaded blue, and the guanidine side chain of R269 is shaded red. The closed structure shows the phosphodianion at the surface of the protein shaded green and the cofactor shaded cyan.
We have proposed that the flexible “capping” loop (Figure 1) plays an important role in the activation of hlGPDH for catalysis, similar to that for the flexible loops at TIM and OMPDC.13,23 This proposal leads to the prediction that the amide side chain from Q295 plays a critical role in ensuring optimal enzyme activation by dianions. We report here the results of studies of the effect of site-directed mutations of Q295 on hlGPDH-catalyzed reactions of the whole substrate DHAP, of the substrate in pieces,13 and of the enzyme in pieces,23 which establish a role for the amide side chain of Q295 in optimizing the activating interactions between hlGPDH and the enzyme-bound dianions. We report additional results of the effect of R269A and N270A mutations on enzyme activity. These results are consistent with a high degree of cooperativity in the enzyme conformational change, which organizes the catalytic side chains at the enzyme active site.
Experimental Section
Materials
Water was purified using Milli-Q Academic purification system. Q-Sepharose and Sephacryl S-200 were purchased from GE Healthcare. Dowex 50WX4-200R (H+ form), nicotinamide adenine dinucleotide reduced (NADH, disodium salt), dihydroxyacetone phosphate hemimagnesium salt, glycolaldehyde dimer, 2-(N-morpholino)ethanesulfonic acid sodium salt (MES, ≥99.5%), triethanolamine hydrochloride (TEA, ≥99.5%), ampicillin, kanamycin sulfate, and d,l-dithiothreitol (DTT) were purchased from Sigma-Aldrich. Protease inhibitor tablets (Complete brand) and bovine serum albumin, fraction V (BSA), were purchased from Roche. Ammonium sulfate (enzyme grade), guanidinium hydrochloride (electrophoresis grade, min. 99%), sodium hydroxide (1.0 N), and hydrochloric acid (1.0 N) were purchased from Fisher. Sodium phosphite (dibasic, pentahydrate) was purchased from Fluka, and its water content was reduced to Na2HPO3·0.4H2O as previously described.7 Quikchange II Site-Directed Mutagenesis Kits were purchased from Agilent Technologies, and λDE3 Lysogenization Kits were purchased from Novagen. All other chemicals were reagent grade or better and were used without further purification.
Preparation of Solutions
Solution pH was determined at 25 °C using an Orion model 720A pH meter equipped with a Radiometer pHC4006-9 combination electrode that was standardized at pH 4.00, 7.00, and 10.00 at 25 °C. Stock solutions of NADH, prepared by dissolving the disodium form of the coenzyme in water, were stored at 4 °C. The concentration of NADH in aqueous solutions was determined from the absorbance at 340 nm using a value of ε = 6220 M–1 cm–1. Stock solutions of DHAP were prepared, starting with the hemimagnesium salt, and stirring over Dowex 50WX4-200R (H+ form) in water for 5–10 min at 25 °C to give the free acid. The Dowex was removed by filtration, the pH of the resulting solution was adjusted from ∼2.0 to 7.5 using 1 M NaOH, and the solution was then stored at −20 °C. The concentration of DHAP was determined spectrophotometrically at 340 nm, as the concentration of NADH oxidized upon quantitative conversion to glycerol-3-phosphate catalyzed by hlGPDH.
Stock solutions of glycolaldehyde dimer (200 mM monomer) were prepared by dissolving the dimer in water and waiting for 3 days at room temperature to allow for quantitative breakdown of the dimer to the monomer.7 Stock solutions of sodium phosphite were prepared by dissolving the salt in water and adjusting the pH to 7.5 with 1 M HCl. At pH 7.5, the dianion:monoanion ratio for sodium phosphite was 93:7. Stock solutions of guanidinium hydrochloride were prepared by dissolving the salt in water and adjusting the pH to 7.5 with 1 M HCl. MES and TEA buffers were prepared by addition of 1 M NaOH or 1 M HCl and solid NaCl to give the desired pH and final ionic strength. Stock solutions of Q295 mutant hlGPDH (10–20 mg/mL) were dialyzed exhaustively against 20 mM TEA buffer at 4 °C. Dilutions of the mutant enzymes were then prepared in 20 mM TEA buffer (pH 7.5) that contains 10 mM DTT and 0.1 mg/mL BSA. The enzyme concentration was determined from the absorbance at 280 nm using the extinction coefficient of 18 450 M–1cm–1 and a subunit molecular mass of 37 500 Da.24−26
Cloning and Site-Directed Mutagenesis of Human Liver Glycerol-3-phosphate Dehydrogenase
The plasmid pDNR-dual donor vector containing the gene for wild-type hlGPDH gene insert was purchased from the Harvard plasmid repository. The insert gene was subcloned into a bacterial expression vector pET-15b from Novagen and used for mutagenesis. Site-directed mutagenesis on pET-15b to introduce the mutations was carried out using the Quickchange II kit from Stratagene. The primers used to introduce base changes encoding Q295A, Q295G, Q295S, and Q295N mutations differ from the sequence for the wild-type gene as follows:
wild-type: 5′-GAAAGAGTTGCTGAATGGGCAGAAACTGCAGGGGCCCGAG-3′
Q295A: 5′-GAAAGAGTTGCTGAATGGGGCAAAACTGCAGGGGCCCGAG-3′
Q295G: 5′-GAAAGAGTTGCTGAATGGGGGAAAACTGCAGGGGCCCGAG-3′
Q295S: 5′-GAAAGAGTTGCTGAATGGGAGCAAACTGCAGGGGCCCGAG-3′
Q295N: 5′-GAAAGAGTTGCTGAATGGGAACAAACTGCAGGGGCCCGAG-3′
The Q295 mutants were constructed individually starting with 20 ng of plasmid pET-15b containing the gene for wild-type hlGPDH that had been purified from Escherichia coli BL21 (DE3) cells. This plasmid was added to a PCR mixture containing 5 μL of 10X Pfu Ultra Buffer, 125 ng each of the forward and reverse mutagenesis primers, 1 μL of 10 mM dNTP mixture, 2.5 units of Pfu Ultra HF DNA polymerase, and water to give a final volume of 50 μL. The parameters for PCR were 45 s at 95 °C followed by 17 cycles of 45 s at 95 °C, 90 s at 55 °C, and 10 min at 68 °C. The R269A and R269A/N270A mutations were performed by following published procedures.23 The R269A/Q295A mutant was constructed by following the same procedure, except that DNA from the R269A mutant plasmid was used as the template. In both cases, 20 units of the DpnI restriction enzyme was added to the product of the PCR reaction, and the solutions were incubated at 37 °C for 1 h to degrade the methylated template DNA. One microliter of each reaction mixture was transformed into E. coli DH5α cells, and in each case a single colony was used for mutant plasmid purification. In each case, the presence of the mutant sequence at the plasmid DNA was verified by sequencing at the Roswell Park Cancer Institute DNA Sequencing Facility.
Expression and Purification of Mutants of Glycerol-3-phosphate Dehydrogenase
The procedures for the preparation of the R269A23 and R269A/N270A27 mutant enzymes were described in earlier work. The GPDH-deficient glpD1 strain from the E. coli Keio collection was purchased from the Coli Genetic Stock Center at Yale University. Lysogenization of this strain was carried out using a λDE3 lysogenization kit from Novagen. The plasmids coding for single Q295 mutants and the R269A/Q295A double mutant of hlGPDH were transformed separately into freshly lysogenized competent E. coli glpD1 (DE3) cells. The cells containing the mutant plasmid were grown overnight in 200–300 mL of LB medium that contained 100 μg/mL ampicillin and 50 μg/mL kanamycin at 37 °C. This culture was diluted into 5 L of LB medium (100 μg/mL ampicillin and 50 μg/mL kanamycin), and grown at 37 °C to OD600 = 0.6, at which point 0.6 mM isopropyl-1-thio-d-galactoside was added to the culture and the temperature adjusted to 19 °C to induce protein expression. After 12 h of overexpression, the cells were harvested and stored in 20 mL of 25 mM MES buffer that contains 150 mM NaCl at pH 6.8.
The cell pellets were suspended in 25 mM MES at pH 6.8 in the presence of protease inhibitors (Complete brand) and lysed using a French press. The lysates were diluted to 40 mL with the same buffer and centrifuged at 18000g for 60 min. The resulting lysates were subject to fractional precipitation by ammonium sulfate. The mutant enzymes precipitated in the following fraction, where 100% is a saturated solution of ammonium sulfate: 0–40%, Q295S; 30–40%, Q295G and Q295N; and 40–50%, Q295A and R269A/Q295A. After 20 min the mixtures were centrifuged at 23000g for 20 min, and the resulting pellets were redissolved in 25 mL of 25 mM MES buffer at pH 6.8. The protein solutions were dialyzed overnight against 25 mM MES buffer pH 6.8 at 4 °C. The resulting dialysate was loaded onto a Q-Sepharose ion-exchange column previously equilibrated against 25 mM MES pH 6.8 that contains 30 mM NaCl. The column was eluted with 1.0 L of a linear 30–90 mM gradient of NaCl in the same buffer. The protein concentration of each column fraction was determined from the UV absorbance at 280 nm, using the extinction coefficient of 18 450 M–1cm–1. The fractions that contained the mutant protein were pooled, concentrated, and further purified over a Sephacryl S-200 column, equilibrated with 25 mM MES pH 6.8 that contains 120 mM NaCl, eluting with the same saline buffer solution. Fractions with A280 > 1 were pooled, concentrated, and stored at −80 °C in 20% glycerol, 25 mM MES buffer at pH 6.8, and 100 mM NaCl. The Q295 mutants of hlGPDH obtained by this procedure were judged to be homogeneous by SDS-PAGE electrophoresis. The following are the final yields of the Q295 mutants of hlGPDH purified from a 5 L bacterial culture: 120 mg, Q295A; 90 mg, Q295G; 140 mg, Q295S; 130 mg, Q295N; and 40 mg, R269A/Q295A.
Mutant hlGPDH-Catalyzed Reduction of DHAP by NADH
The reduction of DHAP catalyzed by Q295 mutants of hlGPDH was assayed in solutions that contain 20 mM TEA buffer (pH 7.5), 0.1 mg/mL BSA, 100 or 200 μM NADH, 0.04–15 mM DHAP at I = 0.12 (NaCl), and the following enzyme concentrations: 4.4 nM, Q295A; 2.6 nM, Q295G; 3.9 nM, Q295S; 4.6 nM, Q295N; and 20 μM, R269A/Q295A. Initial velocities for each assay were determined over a 5–10 min reaction time.
Assay mixtures for the R269A/N270A and R269A/Q295A mutant hlGPDH-catalyzed reduction of DHAP by NADH in the presence of guanidinium cation contained 20 mM TEA buffer (pH 7.5), 0.1 mg/mL BSA, 200 μM NADH, 0.5–5 mM DHAP, 10–80 mM guanidinium hydrochloride at I = 0.12 (NaCl) with the following mutant enzyme concentrations: 20 μM R269A/N270A; and 0.8 μM, R269A/Q295A. The change in absorbance at 340 nm was monitored, and the initial velocities for reactions catalyzed by R269A/Q295A and R269A/N270A mutants were determined over 5–10 and 5–50 min reaction times, respectively. The kinetic parameter Km for the hlGPDH-catalyzed reactions of DHAP was determined for the reactive form of DHAP, which is present as 55% of total DHAP.28
Mutant hlGPDH-Catalyzed Reduction of Glycolaldehyde by NADH
The reduction of GA catalyzed by Q295 mutants of hlGPDH was assayed in solutions that contain 10 mM TEA buffer (pH 7.5), 5–60 mM GA, 200 μM NADH, 0–30 mM phosphite dianion at I = 0.12 (NaCl) with the following enzyme concentrations: 14 μM, Q295A; 3 μM, Q295G and Q295S; 30 μM, Q295N; and 60 μM, R269A/Q295A. Initial velocities for the mutant hlGPDH-catalyzed reduction of GA by NADH were determined over 5–60 min reaction times.
The reduction of GA catalyzed by R269A, R269A/N270A, and R269A/Q295A mutants of hlGPDH was assayed in solutions that contain 10 mM TEA buffer (pH 7.5), 60 mM GA, 200 μM NADH at I = 0.12 (NaCl) with the following mutant enzyme concentrations: 20 μM, R269A hlGPDH; 40 μM, R269A/N270A; and 60 μM R269A/Q295A. The change in absorbance at 340 nm was monitored over a period of 60 min (ΔA340 ≈ 0.010) and compared with the change in absorbance for control reactions that contain no enzyme (ΔA340 ≤ 0.010). Upper limits for kcat/Km for the mutant hlGPDH-catalyzed reduction of GA by NADH were calculated for hlGPDH-catalyzed reduction of the carbonyl form of substrate (fcar = 0.06),7 with the assumption that ΔA340 ≤ 0.005 from this enzyme-catalyzed reaction.
Results
GPDH follows an ordered reaction mechanism with NADH (Kd = 7 μM)29 binding first, followed by DHAP.30 Mutations of R269, N270, and Q295 near the dianion binding site of DHAP are not expected to affect the binding of NADH at a distant site.10 This is consistent with the observation that the Michaelis–Menten plots of initial velocity data for wild-type, R269A, and N270A hlGPDH-catalyzed reduction of DHAP in the presence of 0.10 and 0.20 mM reducing agent NADH show a good fit to a single set of kinetic parameters kcat and Km.13,27
The Q295A and R269A/Q269A mutants of hlGPDH were prepared by standard methods,23,27 and their activity at 25 °C, pH 7.5 (20 mM TEA buffer), and I = 0.12 (NaCl) was determined by monitoring the reduction of DHAP by NADH. Figure 2A shows Michaelis–Menten plots of v/[E] against [DHAP] for reduction of DHAP by NADH (200 μM) catalyzed by Q295G, Q295S, Q295A, and Q295N mutants of hlGPDH (Scheme 1A). Figure 2B shows the Michaelis–Menten plot for reduction of DHAP by NADH (200 μM) catalyzed by R269A/Q295A hlGPDH. The rate data for Q295 mutants determined for reactions at 0.10 and 0.20 mM NADH show a good fit to the single set of kinetic parameters kcat and Km. The kinetic parameters obtained from nonlinear least-squares fits of data for reactions at 0.10 and 0.20 mM NADH to the Michaelis–Menten equation are reported in Table 1. Table 1 also reports kinetic parameters for N270A and R269A/N270A mutants of hlGPDH determined in earlier work.27
Figure 2.

Michaelis–Menten plots of v/[E] for reduction of DHAP by NADH (0.2 mM) catalyzed by Q295 mutants of hlGPDH at 25 °C, pH 7.5 (20 mM TEA buffer), and I = 0.12 (NaCl). (A) ▼, Q295G mutant; ◆, Q295S; ●, Q295A; and ▲, Q295N. (B) R269A/Q295A mutant.
Table 1. Kinetic Parameters for Wild-Type and Mutant hlGPDH-Catalyzed Reduction of Whole Substrate DHAP and Truncated Substrate Glycolaldehyde by NADH at pH 7.5 and I = 0.12.
| kcata | Kma | kcat/Kma | (kcat/Km)Eb | intrinsic dianion binding energy c | |
|---|---|---|---|---|---|
| enzyme | (s–1) | (M) | (M–1 s–1) | (M–1 s–1) | (kcal mol–1) |
| WTd | 240 ± 10 | (5.2 ± 0.3) × 10–5 | (4.6 ± 0.3) × 106 | (5.0 ± 0.6) × 10–2 | –10.8 |
| R269Ae | (5.9 ± 0.4) × 10–3 | (5.7 ± 0.5) × 10–3 | 1.0 ± 0.15 | ≤0.003 | |
| Q295G | 115 ± 5 | (1.5 ± 0.2) × 10–4 | (7.5 ± 1.0) × 105 | (1.9 ± 0.2) × 10–2 | –10.4 |
| Q295S | 92 ± 6 | (1.7 ± 0.2) × 10–4 | (5.4 ± 0.7) × 105 | (1.7 ± 0.1) × 10–2 | –10.2 |
| Q295A | 100 ± 10 | (1.6 ± 0.2) × 10–3 | (6.3 ± 1.0) × 104 | (1.4 ± 0.1) × 10–2 | –9.1 |
| Q295N | 60 ± 3 | (2.0 ± 0.3) × 10–3 | (3.0 ± 0.5) × 104 | (3.3 ± 0.3) × 10–2 | –8.1 |
| R269A/Q295A | (3.7 ± 0.2) × 10–3 | (4.9 ± 0.7) × 10–4 | 7.5 ± 0.2 | ≤0.003 | |
| N270Af | 9.0 ± 0.5 | (2.5 ± 0.2) × 10–2 | 360 ± 35 | 2.0 ± 0.2 | |
| R269A/N270Af | (2.8 ± 0.1) × 10–4 | (1.5 ± 0.1) × 10–2 | (1.7 ± 0.1) × 10–2 | ≤0.003 |
Kinetic parameters determined from the fit of the data to the Michaelis–Menten equation. The quoted errors are the average of the values determined from at least two sets of data for hlGPDH-catalyzed reduction of DHAP by 0.10 and 0.20 mM NADH (Figure 1).
Kinetic parameters for GPDH-catalyzed reduction of GA by 0.2 mM NADH determined from the fit of data to eq 1.
The energetic contribution of the phosphodianion to stabilization of the transition state for GPDH-catalyzed reduction of DHAP by NADH calculated from the data in this table using eq 2.3,10
Ref (13).
Ref (23).
Ref (27)
The Q295 mutants of hlGPDH catalyze the slow reduction of the truncated substrate GA by NADH (200 μM) at 25 °C, pH 7.5 (10 mM TEA buffer), and I = 0.12 (NaCl). The second-order rate constants (kcat/Km)E determined from the fit of plots of v/[E] against [GA] to a variant of the Michaelis–Menten equation that treats (kcat/Km)E and Km as variable parameters. (Figure S1, Supporting Information) for the reactions catalyzed by Q295G, Q295S, Q295A, and Q295N mutants are reported in Table 1. Figure 3A shows the dependence of vi/[E] (s–1) on [HPO32–] for the reduction of GA by NADH (200 μM) catalyzed by the Q295G mutant of hlGPDH. Figure 3B–D shows related data for reactions catalyzed by Q295S, Q295A, and Q295N mutants. The solid lines through the experimental data show the nonlinear least-squares fits of these data to eq 1, derived for Scheme 2, using the kinetic parameters reported in Table 2.13
| 1 |
Figure 3.

Dependence of v/[E] (s–1) on HPO32– for the reduction of GA by NADH (0.2 mM) catalyzed by Q295 mutants of hlGPDH at pH 7.5 (10 mM TEA buffer), 25 °C, and I = 0.12 (NaCl) on the concentration of HPO32– for reactions at different fixed concentrations of GA. (A) Q295G mutant: ●, 3.6 mM GA; ▼, 3.0 mM; ◆, 2.4 mM; ○, 1.8 mM; ■, 1.2 mM; ▲, 0.6 mM. (B) Q295S: ●, 3.6 mM GA; ▼, 3.0 mM; ◆, 2.4 mM; ○, 1.8 mM; ■, 1.2 mM; ▲, 0.6 mM. (C) Q295A: ●, 3.6 mM GA; ◆, 2.4 mM; ■, 1.2 mM; ▲, 0.6 mM. (D) Q295N: ●, 3.6 mM GA; ◆, 2.4 mM; ■, 1.2 mM; ▲, 0.6 mM.
Scheme 2.

Table 2. Kinetic Parameters for Activation of Wild-Type and hlGPDH by Phosphite Dianion and Derived Parameters for the Binding of Dianions to [E·S]⧧ (Scheme 3)a.
| (kcat)E·Xb | KGAc | KHPid | kcat/KGAKHPi | (K⧧)HPie | RT ln(K⧧)Xf | |
|---|---|---|---|---|---|---|
| enzyme | (s–1) | (mM) | (mM) | (M–2 s–1) | (M) | (kcal mol–1) |
| WT | 5.5 ± 0.3 | 4.9 ± 0.2 | 70 ± 4 | 16000 ± 1300 | 3.3 × 10–6 | –7.5 |
| Q295G | 1.0 ± 0.12 | 4.7 ± 0.32 | 110 ± 15 | 2100 ± 400 | 9.0 × 10–6 | –6.9 |
| Q295S | 1.0 ± 0.12 | 4.5 ± 0.30 | 110 ± 15 | 2100 ± 400 | 8.0 × 10–6 | –6.9 |
| Q295A | 0.07 ± 0.013 | 4.9 ± 0.04 | 130 ± 27 | 120 ± 30 | 1.1 × 10–4 | –5.4 |
| Q295N | 0.03 ± 0.006 | 4.9 ± 0.55 | 100 ± 24 | 63 ± 15 | 5.0 × 10–4 | –4.5 |
Reactions catalyzed by hlGPDH at pH 7.5 (10 mM TEA buffer), 25 °C, 0.2 mM NADH, and I = 0.12 (NaCl). The quoted uncertainty in these kinetic parameters is the standard error determined for the nonlinear least-squares fits of these data.
Dissociation constants for release of the oxydianion from the binary or ternary enzyme complex (Scheme 2).
Figure 4A shows the effect of increasing concentrations of guanidine cation [Gua+] on the second-order rate constant (kcat/Km)obs for R269A/Q295A hlGPDH-catalyzed reduction of DHAP. The values of (kcat/Km)obs for reactions in the presence of different fixed [Gua+] were determined as the slopes of these linear correlations. Figure 4B shows the effect of increasing [Gua+] on (kcat/Km)obs. The slope of the linear correlation is equal to the third-order rate constant (kcat/Km)Gua/Kd = 1400 M–2 s–1. Figure 4C shows the effect of increasing concentrations of guanidine cation [Gua+] on the second-order rate constant (kcat/Km)obs for R269A/N270A hlGPDH-catalyzed reduction of DHAP. The values of (kcat/Km)obs for reactions in the presence of different fixed [Gua+] were determined as the slopes of these linear correlations. Figure 4D shows the effect of increasing [Gua+] on (kcat/Km)obs. The slope of the linear correlation at [Gua+] is equal to the third-order rate constant (kcat/Km)Gua/Kd = 3.5 M–2 s–1.
Figure 4.

Effect of increasing [Gua+] on R269A/Q295A and R269A/N270A mutant hlGPDH-catalyzed reduction of DHAP by NADH for reactions at pH 7.5 (20 mM TEA buffer), 25 °C, saturating [NADH] = 0.2 mM, and I = 0.12 (NaCl). Top, for R269A/Q295A hlGPDH: (A) The increase in v/[E] (s–1), with increasing [DHAP], for reactions at different fixed [Gua+]: ●, 80 mM Gua+; ▼, 60 mM Gua+; ◆, 40 mM Gua+; ▲, 20 mM Gua+. (B) The effect of increasing [Gua+] on the values of (kcat/Km)obs from panel A. (C) The increase in v/[E] (s–1), with increasing [DHAP], for reactions at different fixed [Gua+]: ●, 80 mM Gua+; ▼, 60 mM Gua+; ■, 40 mM Gua+; ▲, 20 mM Gua+; ◆, 10 mM Gua. (D) The effect of increasing [Gua+] on the values of (kcat/Km)obs from panel C.
Discussion
The Q295 mutations result in up to a 150-fold decrease in (kcat/Km)DHAP for hlGPDH-catalyzed reduction of DHAP (Q295N), but no more than a 3-fold decrease in (kcat/Km)GA for hlGPDH-catalyzed reduction of GA (Table 1). The preferential effect of these mutations on (kcat/Km)DHAP shows that they cause a decrease in the intrinsic phosphodianion binding energy (IBE, (ΔG⧧)Pi) utilized in the stabilization of the transition state for hydride transfer (eq 2).3 The Q295 mutations likewise result in only small (<2-fold) changes in the KHPi and KGA for dianion activated, hlGPDH-catalyzed reduction of the substrate piece glycolaldehyde, but in large decreases in the third-order rate constant (kcat)E·X/KGAKHPi (M–2 s–1) dianion activation (Table 2). This reflects the large effect of Q295 mutations on the intrinsic phosphite dianion binding energy ((ΔG⧧)HPi, Scheme 3), which may be calculated from the kinetic parameters in Table 2 using eq 3. We conclude that Q295 promotes catalysis by optimizing transition-state stabilization from interactions with the enzyme-bound phosphodianion of substrate or with phosphite dianion.
| 2 |
| 3 |
Scheme 3.

The logarithmic plot (Figure S2) of second-order rate constants (kcat/Km)DHAP for wild-type and mutant hlGPDH-catalyzed reduction of the whole substrate DHAP, against the corresponding third-order rate constants (kcat)E·X/KGAKHPi for dianion activated hlGPDH-catalyzed reduction of GA, is linear with a slope of 1.1. This linear correlation provides strong evidence that the whole and truncated substrates for hlGPDH, which differ only by the covalent connection at the former, proceed through similar transition states that show similar changes in stability with changing side chain at residue 295. Similar linear free-energy correlations, with slopes of 1.0, of data for reactions catalyzed by wild-type and mutant enzymes have been observed for triosephosphate isomerase- and orotidine 5′-monophosphate decarboxylase-catalyzed reactions of whole and phosphodianion-truncated substrates.31−33
Figure 5 shows a related linear correlation, also with slope of 1.1, between the intrinsic phosphodianion binding energy (ΔG⧧)Pi (eq 2) that is utilized for stabilization of the transition state for hlGPDH-catalyzed reduction of the whole substrate DHAP, and the intrinsic phosphite dianion binding energy (ΔG⧧)HPi (eq 3) that is utilized in stabilization of the transition state for phosphite dianion-activated hlGPDH-catalyzed reduction of GA. This plot is similar to the direct logarithmic plot of second- and third-order rate constants, since mutations of Q295 cause only small changes in the second-order rate constant (kcat/km)GA for catalysis of the reaction of phosphodianion truncated substrate, which is used in the calculation of both (ΔG⧧)Pi (eq 2) and (ΔG⧧)HPi (eq 3).
Figure 5.

Linear correlation, with slope of 1.1 ± 0.02, between the intrinsic phosphite dianion binding energy (ΔG⧧)HPi expressed at the transition state for the reduction of the truncated substrate glycolaldehyde by NADH, and the intrinsic phosphodianion binding energy (ΔG⧧)Pi expressed at the transition state for the reduction of DHAP by NADH catalyzed by wild-type and Q295 mutants of hlGPDH.
The near-unit slope from Figure 5 shows that mutations of Q295 result in nearly identical falloffs in the intrinsic dianion binding energy utilized in the stabilization of the transition states for the hlGPDH-catalyzed reactions of the whole and the truncated substrates, so that this side chain promotes optimal stabilization of these different transition states through interactions with bound dianions. Q295 does not interact directly with the phosphodianion, but sits in a flexible loop that folds over the dianion.10 The loop, in turn, is anchored to the phosphodianion by interactions with the cationic side chain of R269, which is ion-paired with the phosphodianion (Figure 6). The network of interactions that runs from Q295 to the substrate phosphodianion functions to hold the flexible loop close to the substrate phosphodianion. We propose that Q295 mutations of hlGPDH give rise to a reorientation of this loop from its optimal “gripper” conformation, that results in a reduction in the intrinsic dianion binding energy.
Figure 6.
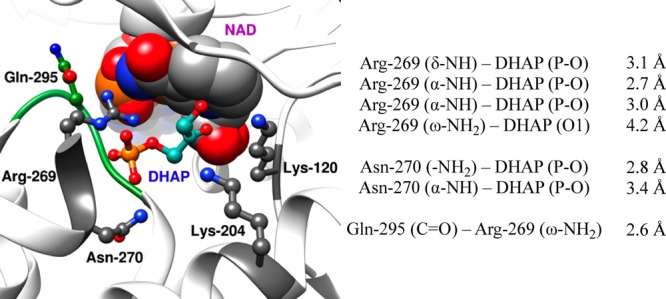
Representation of the X-ray crystal structure (PDB entry 1WPQ) of the nonproductive ternary Michaelis complex between wild-type hlGPDH, DHAP, and NAD. The distances separating the amino acid side chains (Arg-269, Asn-270, and Gln-295) from their neighboring side chains and the substrate phosphodianion DHAP are also listed above.
The Q295 mutations lead to ≤35-fold increases in the Michaelis constant Km, but a much smaller decrease in kcat for hlGPDH-catalyzed reduction of DHAP (Table 1). This shows that the protein-dianion interactions, which activate wild-type hlGPDH for catalysis of reduction of DHAP, are expressed at the Michaelis complex for the bound DHAP. These results may be rationalized by Scheme 4, where the binding energy of the substrate phosphodianion is utilized to drive a conformational change (Figure 1) that converts hlGPDH from the inactive open form (Eo = Eo·NADH) to the active closed form (Ec·DHAP).2,10 Our results are consistent with a value of Kc′ ≫ 1.0 for the reaction catalyzed by the wild-type enzyme, so that the dianion interactions are fully formed at the Michaelis complex. In this case, a weakening of dianion interactions that result in a decrease in Kc′ will result in an increase in (Km)obs but no change in (kcat)obs (eqs 4 and 5). A scheme similar to that shown by Scheme 4 was proposed to rationalize kinetic data reported for the decarboxylation of orotidine 5′-monophosphate by wild-type OMPDC and a series of mutants, with every possible single, double, and triple substitution of side chains Q215 (Q215A), Y217 (Y217F), and R235 (R235A) that interact the phosphodianion side chain of OMP.33
| 4 |
| 5 |
Scheme 4.

Role of N270 and Q295 in Activation of hlGPDH for Catalysis of Hydride Transfer
The amide side chains of N270 and Q295 interact, respectively, with the substrate phosphodianion and the cationic side chain of R269 (Figure 6). The former interaction has the effect of immobilizing the substrate phosphodianion, while the later has the effect of immobilizing the cationic side chain of R269. These interactions act to restrict the motion of the interacting cation and phosphodianion at the enzyme active site. The following observations provide strong evidence that such “preorganization” 34,35 of this ion pair has the effect of optimizing the strength of intermolecular ionic interactions.
(1) R269A mutation of wild-type hlGPDH results in a large (4.6 × 106)-fold decrease in kcat/Km for reduction of DHAP by the binary E·NADH complex, which corresponds to a 9.1 kcal/mol destabilization of the transition state for hlGPDH-catalyzed hydride transfer. By comparison, the R269A mutation of the Q295A mutant enzyme results in a smaller 4.9 kcal/mol transition-state destabilization (Figure 7A). These results are consistent with the conclusion that the loss of the interaction between R269 and Q295 at the Q295A mutant results in a (9.1 – 4.9) = 4.2 kcal/mol decrease in the stabilizing interaction between the cationic R269 side chain and the transition state for hlGPDH-catalyzed hydride transfer.
Figure 7.

Comparison of the effect of the R269A mutation on the stability of the transition state for wild-type hlGPDH-catalyzed reduction of DHAP (9.1 kcal/mol) with the effect of the same mutation on hlGPDH previously mutated at Q295 (A, 4.9 kcal/mol) and at N270 (B, 5.9 kcal/mol). These differences in the effect of an R269A mutation reflect the effect of the Q295A or N270A mutations on the interaction between the cationic side chain of R269 and the anionic transition state (see text).
(2) The R269A mutation at N270A mutant hlGPDH likewise results in a 5.9 kcal/mol transition-state destabilization that is smaller than the 9.1 kcal/mol effect of the R269A mutation of wild-type hlGPDH (Figure 7B). This is consistent with the conclusion that the loss of the interaction between R269 and N270 at the N270A mutant results in a (9.1 – 5.9) = 3.2 kcal/mol decrease in the stabilizing interaction between the cationic side chain for R269 and the transition state for hlGPDH-catalyzed hydride transfer. This and the previous result complement one another and provide strong support for the conclusion that the side chains of N270 and Q295 act to optimize the stabilizing interactions between the guanidine side chain of R269 and the transition state for hlGPDH-catalyzed reduction of DHAP by NADH.
(3) The addition of Gua+ results in rescue of the activity of R269A mutants for the reduction of DHAP.23,36,37 This rescue is characterized experimentally as the slope of a linear plot of observed second-order rate constants (kcat/Km)obs for mutant hlGPDH-catalyzed reduction of DHAP against the concentration of the Gua+ activator. The slope of the plot for rescue of the R269A mutant is the third-order rate constant [(kcat/Km)Gua/Kd] = 80 000 M–2 s–1.23 The smaller [(kcat/Km)Gua/Kd] = 1400 M–2 s–1 (Figure 4B) and 3.5 M–2 s–1 (Figure 4D) determined for the rescue of R269A/Q295A and R269A/N270A mutants by Gua+, respectively, show that N270A and Q295A mutations result in a falloff in the efficiency of Gua+ rescue of R269A mutants.
(4) The stabilizing interaction between Gua+ and the transition state for hydride transfer from NADH to DHAP, (ΔG⧧act)Gua, has been quantified using eq 6 derived for Scheme 5, where kcat/Km is the second-order rate constant for the unactivated mutant enzyme-catalyzed reaction (Table 1). These results are illustrated graphically by Figure 8, where −(ΔG⧧T)R269 is the effect of the R269A mutation on the reactivity of the whole substrate, −ΔG⧧Gua is the transition-state stabilization recovered upon addition of 1.0 M Gua+, and ΔG⧧S is the advantage from the covalent connection of guanidine cation to the whole enzyme.38 Substitution of the kinetic parameters for reactions catalyzed by R269A, R269A/Q295A, and R269A/N270A mutants of hlGPDH into eq 6 gives values of 6.7, 3.2, and 3.1 kcal/mol for stabilization of the respective transition states by interaction with Gua+ (Figure 8). We conclude that the Q295A and N270A mutations result in a 6.7 – 3.2 = 3.5 kcal/mol and 6.7 – 3.1 = 3.6 kcal/mol weakening in the interaction between exogenous Gua+ and the transition state for reduction of DHAP catalyzed by R269A mutants of hlGPDH. By comparison, the same mutations result in 3.2 kcal/mol (Q295A, Figure 7A) and 4.2 kcal/mol (N270A, Figure 7B) reductions in the stabilizing interaction between the cationic side chain for R269 and the transition state for hlGPDH-catalyzed hydride transfer to DHAP.
| 6 |
Scheme 5.

Figure 8.

Diagrams that show the effect of R269A mutations on the stability of the transition states for wild-type and mutant hlGPDH-catalyzed reduction of DHAP (−(ΔG⧧T)R269), the partitioning of this whole transition-state stabilization into the stabilization recovered upon addition of 1.0 M Gua+ (−ΔG⧧Gua), and the advantage obtained by connection of the guanidine cation to the whole enzyme (ΔG⧧S).38
(5) The R269A mutant of hlGPDH, guanidine cation, and phosphite dianion assemble spontaneously to form a functioning catalysis of the reduction of GA by NADH.39 The surprising observation of this fourth-order enzyme-catalyzed reaction reflects the efficient activation of hydride transfer from binding of the guanidine cation–phosphite dianion pair to the R269A mutant enzyme. The binding of this ion pair is assisted by the interactions of the guanidine cation with the amide side chain of Q295, and of phosphite dianion with the amide side of N270 (Figure 6). We propose that the function of this network of interactions is to stabilize an active closed form of hlGPDH.22
Conclusions and Speculations
The sum of the effect of R269A (9.1 kcal/mol), N270A (5.6 kcal/mol), and Q295A (3.0 kcal/mol) on the stability of the transition state for wild-type hlGPDH-catalyzed reduction of DHAP (18.0 kcal/mol) is much larger than expected for the total interactions between the transition state and the excised side chains. In particular, the side chain of Q295 does not interact directly with this transition state. We conclude that single mutations result in both the loss of the interaction from the excised side chain and a weakening in the stabilizing interactions with other participating side chains. The side chains of Q295A and N270A interact, respectively, with the side chain of R269 and the substrate phosphodianion:23Figure 7 shows that these interactions are required for the observation of the large 9.1 kcal/mol effect of the R269A mutation on transition-state stability.
It was previously assumed, for the sake of Occam’s razor, that the 9.1 kcal/mol effect of the R269A mutation is due entirely to the loss of electrostatic interactions between the side-chain cation or R269 and the highly anionic transition state for hydride transfer, in which case these electrostatic interactions provide a 2.8 kcal/mol stabilization of the Michaelis complex (effect of mutation on Km) and strengthen by 6.3 kcal/mol on proceeding to the transition state for hydride transfer (kcat effect). One explanation for the tightening of these electrostatic interactions on proceeding from the Michaelis complex to the transition state is that this reflects the buildup of additional negative charge at the carbonyl oxygen that occurs with transfer of a hydride anion to the carbonyl carbon of DHAP. We are skeptical of this explanation, because the large 5.7 Å distance (Figure 6) between the side-chain cation and the carbonyl oxygen does not favor a strong interaction between these sites. We suggest two possible complicating events that would favor the observation of a large effect of the R269A mutation on the stability of the transition state for hydride transfer:
(1) Figure 6 shows the nonproductive hlGPDH·NAD·DHAP complex. The cationic side chain may lie closer to the C-2 carbonyl at the productive hlGPDH·NADH·DHAP compared with the 5.7 Å separation observed at the nonproductive hlGPDH·NAD·DHAP complex.
(2) The C-2 carbonyl oxygen of DHAP at the nonproductive hlGPDH·NAD·DHAP complex lies nearer the cationic side chains of K120 (3.5 Å) and K204 (3.8 Å) than to the cationic side chain of R269 (ca. 6 Å). The optimal electrostatic stabilization of the transition state for hydride transfer by interactions from the K120 and K204 side chain will weaken if the R269A mutation results in a loosening in the “tight” structure of the ternary hlGPDH·NADH·DHAP complex and an increase in the separation between these side chains and the C-2 carbonyl oxygen. This proposal is consistent with the strong imperative for electrophilic assistance to hydride transfer to the carbonyl carbon,40,41 and with the notion that the conformational change of hlGPDH acts to organize catalytic active site side chains so that they provide optimal transition-state stabilization.34,42 This is similar to a previous proposal to rationalize the large 8.0 kcal/mol effect of the K12G mutation of the stability of the transition state for the isomerization reaction catalyzed by triosephosphate isomerase.8,43
We suggest that the ligand-driven conformational change of GPDH involves a network of side chains that includes those from R269, N270, and Q295, and possibly from K120 and K204, which lie close to the carbonyl group of DHAP. This conformational change is driven largely by the interactions between the substrate phosphodianion and the cationic side chain of R269. However, the effect of the conformational change on the stability of the transition state for hlGPDH-catalyzed hydride transfer will include interactions from side chains that are moved into a position to stabilize this transition state. If correct, then the complications that arise from this model will need to be dealt with when interpreting the effects of mutations of K120 and K204, and possibly other amino acids, on the stability of the transition state for hlGPDH-catalyzed hydride transfer.
Glossary
Abbreviations
- OMPDC
orotidine 5′-monophosphate decarboxylase
- TIM
triosephosphate isomerase
- GPDH
glycerol-3-phosphate dehydrogenase
- hlGPDH
glycerol-3-phosphate dehydrogenase from human liver
- DHAP
dihydroxyacetone phosphate
- GA
glycolaldehyde
- NADH
nicotinamide adenine dinucleotide, reduced form
- NAD
nicotinamide adenine dinucleotide
- MES
2-(N-morpholino)ethanesulfonic acid
- TEA
triethanolamine
- Gua+
guanidine cation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b01282.
Figure S1, plots showing the dependence of v/[E] (s–1) for the reduction of GA by NADH (0.2 mM) catalyzed by Q295A, Q295G, Q295S, and Q295N mutants of hlGPDH at pH 7.5; Figure S2, logarithmic plot of second-order rate constants (kcat/Km)DHAP for wild-type and mutant hlGPDH-catalyzed reduction of the whole substrate DHAP against the corresponding third-order rate constants (kcat)E·X/KGAKHPi for dianion-activated hlGPDH-catalyzed reduction of glycolaldehyde (PDF)
This work was generously supported by the following grants from the U.S. National Institutes of Health: GM116921 and GM039754.
The authors declare no competing financial interest.
Supplementary Material
References
- Pauling L. (1948) The nature of forces between large molecules of biological interest. Nature 161, 707–709. 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2013) Specificity in transition state binding: The Pauling model revisited. Biochemistry 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. (2006) Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Herschlag D.; Natarajan A. (2013) Fundamental Challenges in Mechanistic Enzymology: Progress toward Understanding the Rate Enhancements of Enzymes. Biochemistry 52, 2050–2067. 10.1021/bi4000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. (2005) Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; O’Donoghue A. C.; Richard J. P. (2001) Contribution of phosphate intrinsic binding energy to the enzymatic rate acceleration for triosephosphate isomerase. J. Am. Chem. Soc. 123, 11325–11326. 10.1021/ja016754a. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. (2007) Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. (2012) A Paradigm for Enzyme-Catalyzed Proton Transfer at Carbon: Triosephosphate Isomerase. Biochemistry 51, 2652–2661. 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. (2008) A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. (2017) Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J. Jr.; Long J. W.; Owens J. D. (1976) An analysis of the substrate-induced rate effect in the phosphoglucomutase system. Biochemistry 15, 4006–4017. 10.1021/bi00663a015. [DOI] [PubMed] [Google Scholar]
- Kholodar S. A.; Murkin A. S. (2013) DXP Reductoisomerase: Reaction of the Substrate in Pieces Reveals a Catalytic Role for the Nonreacting Phosphodianion Group. Biochemistry 52, 2302–2308. 10.1021/bi400092n. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. (2015) The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolis E.; Petsko G. A. (1990) Crystallographic analysis of the complex between triosephosphate isomerase and 2-phosphoglycolate at 2.5-Å resolution: implications for catalysis. Biochemistry 29, 6619–6625. 10.1021/bi00480a010. [DOI] [PubMed] [Google Scholar]
- Davenport R. C.; Bash P. A.; Seaton B. A.; Karplus M.; Petsko G. A.; Ringe D. (1991) Structure of the triosephosphate isomerase-phosphoglycolohydroxamate complex: an analog of the intermediate on the reaction pathway. Biochemistry 30, 5821–5826. 10.1021/bi00238a002. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Sugio S.; Komives E. A.; Liu K. D.; Knowles J. R.; Petsko G. A.; Ringe D. (1994) Crystal structure of recombinant chicken triosephosphate isomerase-phosphoglycolohydroxamate complex at 1.8-A resolution. Biochemistry 33, 2830–2837. 10.1021/bi00176a012. [DOI] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. (2000) Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. (2010) A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Gillon W.; Pai E. F. (2002) Mapping the Active Site-Ligand Interactions of Orotidine 5′-Monophosphate Decarboxylase by Crystallography. Biochemistry 41, 4002–4011. 10.1021/bi015758p. [DOI] [PubMed] [Google Scholar]
- Ou X.; Ji C.; Han X.; Zhao X.; Li X.; Mao Y.; Wong L.-L.; Bartlam M.; Rao Z. (2006) Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J. Mol. Biol. 357, 858–869. 10.1016/j.jmb.2005.12.074. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. (2014) Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Koudelka A. P.; Amyes T. L.; Richard J. P. (2015) Enzyme Architecture: Optimization of Transition State Stabilization from a Cation–Phosphodianion Pair. J. Am. Chem. Soc. 137, 5312–5315. 10.1021/jacs.5b02202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Gattiker A.; Hoogland C.; Ivanyi I.; Appel R. D.; Bairoch A. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud A.; Wilkins M. R.; Appel R. D.; Bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server. Proteomics Protocols Handbook 571–607. 10.1385/1-59259-890-0:571. [DOI] [Google Scholar]
- Hopkinson D. A.; Peters J.; Harris H. (1974) Rare electrophoretic variants of glycerol-3-phosphate dehydrogenase: evidence for two structural gene loci (GPD1 and GPD2). Ann. Hum. Genet. 37, 477–484. 10.1111/j.1469-1809.1974.tb01852.x. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. (2016) Enzyme Architecture: A Startling Role for Asn270 in Glycerol 3-Phosphate Dehydrogenase-Catalyzed Hydride Transfer. Biochemistry 55, 1429–1432. 10.1021/acs.biochem.6b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds S. J.; Yates D. W.; Pogson C. I. (1971) Dihydroxyacetone phosphate. Its structure and reactivity with α-glycerolphosphate dehdyrogenase, aldolase and triose phosphate isomerase and some possible metabolic implications. Biochem. J. 122, 285–297. 10.1042/bj1220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. (2016) Structure-Reactivity Effects on Intrinsic Primary Kinetic Isotope Effects for Hydride Transfer Catalyzed by Glycerol-3-Phosphate Dehydrogenase. J. Am. Chem. Soc. 138, 14526–14529. 10.1021/jacs.6b07028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black W. J. (1966) Kinetic studies on the mechanism of cytoplasmic L-α-glycerophosphate dehydrogenase of rabbit skeletal muscle. Can. J. Biochem. 44, 1301–1317. 10.1139/o66-150. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. (2015) Role of Loop-Clamping Side Chains in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. (2014) Enzyme Architecture: Remarkably Similar Transition States for Triosephosphate Isomerase-Catalyzed Reactions of the Whole Substrate and the Substrate in Pieces. J. Am. Chem. Soc. 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L. M.; Amyes T. L.; Goryanova B.; Gerlt J. A.; Richard J. P. (2014) Enzyme Architecture: Deconstruction of the Enzyme-Activating Phosphodianion Interactions of Orotidine 5′-Monophosphate Decarboxylase. J. Am. Chem. Soc. 136, 10156–10165. 10.1021/ja505037v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. (1998) Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Cannon W. R.; Benkovic S. J. (1998) Solvation, Reorganization Energy, and Biological Catalysis. J. Biol. Chem. 273, 26257–26260. 10.1074/jbc.273.41.26257. [DOI] [PubMed] [Google Scholar]
- Toney M. D.; Kirsch J. F. (1992) Bronsted analysis of aspartate aminotransferase via exogenous catalysis of reactions of an inactive mutant. Protein Sci. 1, 107–119. 10.1002/pro.5560010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney M. D.; Kirsch J. F. (1989) Directed Bronsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science 243, 1485–1488. 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- Jencks W. P. (1981) On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U. S. A. 78, 4046–4050. 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. (2016) Enzyme Architecture: Self-Assembly of Enzyme and Substrate Pieces of Glycerol-3-Phosphate Dehydrogenase into a Robust Catalyst of Hydride Transfer. J. Am. Chem. Soc. 138, 15251–15259. 10.1021/jacs.6b09936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V. P.; Thomas L. M.; Bobyk K. D.; Andi B.; Cook P. F.; West A. H. (2012) Evidence in Support of Lysine 77 and Histidine 96 as Acid–Base Catalytic Residues in Saccharopine Dehydrogenase from Saccharomyces cerevisiae. Biochemistry 51, 857–866. 10.1021/bi201808u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plapp B. V.; Ramaswamy S. (2012) Atomic-Resolution Structures of Horse Liver Alcohol Dehydrogenase with NAD + and Fluoroalcohols Define Strained Michaelis Complexes. Biochemistry 51, 4035–4048. 10.1021/bi300378n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamczyk A. J.; Cao J.; Kamerlin S. C. L.; Warshel A. (2011) Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc. Natl. Acad. Sci. U. S. A. 108, 14115–14120. 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Koudelka A.; Amyes T. L.; Richard J. P. (2010) Role of Lys-12 in Catalysis by Triosephosphate Isomerase: A Two-Part Substrate Approach. Biochemistry 49, 5377–5389. 10.1021/bi100538b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.