

Abstract
Key points
The voltage‐gated sodium channel Nav1.7 is a key player in neuronal excitability and pain signalling. In addition to voltage sensing, the channel is also modulated by mechanical stress.
Using whole‐cell patch‐clamp experiments, we discovered that the sodium channel subunit β1 is able to prevent the impact of mechanical stress on Nav1.7.
An intramolecular disulfide bond of β1 was identified to be essential for stabilisation of inactivation, but not activation, against mechanical stress using molecular dynamics simulations, homology modelling and site‐directed mutagenesis.
Our results highlight the role of segment 6 of domain IV in fast inactivation.
We present a candidate mechanism for sodium channel stabilisation against mechanical stress, ensuring reliable channel functionality in living systems.
Abstract
Voltage‐gated sodium channels are key players in neuronal excitability and pain signalling. Precise gating of these channels is crucial as even small functional alterations can lead to pathological phenotypes such as pain or heart failure. Mechanical stress has been shown to affect sodium channel activation and inactivation. This suggests that stabilising components are necessary to ensure precise channel gating in living organisms. Here, we show that mechanical shear stress affects voltage dependence of activation and fast inactivation of the Nav1.7 channel. Co‐expression of the β1 subunit, however, protects both gating modes of Nav1.7 against mechanical shear stress. Using molecular dynamics simulation, homology modelling and site‐directed mutagenesis, we identify an intramolecular disulfide bond of β1 (Cys21‐Cys43) which is partially involved in this process: the β1‐C43A mutant prevents mechanical modulation of voltage dependence of activation, but not of fast inactivation. Our data emphasise the unique role of segment 6 of domain IV for sodium channel fast inactivation and confirm previous reports that the intracellular process of fast inactivation can be modified by interfering with the extracellular end of segment 6 of domain IV. Thus, our data suggest that physiological gating of Nav1.7 may be protected against mechanical stress in a living organism by assembly with the β1 subunit.
Keywords: 3D structural modelling, patch‐clamp, molecular dynamics simulation, heterologous expression, ion channel, voltage‐gated sodium channel
Key points
The voltage‐gated sodium channel Nav1.7 is a key player in neuronal excitability and pain signalling. In addition to voltage sensing, the channel is also modulated by mechanical stress.
Using whole‐cell patch‐clamp experiments, we discovered that the sodium channel subunit β1 is able to prevent the impact of mechanical stress on Nav1.7.
An intramolecular disulfide bond of β1 was identified to be essential for stabilisation of inactivation, but not activation, against mechanical stress using molecular dynamics simulations, homology modelling and site‐directed mutagenesis.
Our results highlight the role of segment 6 of domain IV in fast inactivation.
We present a candidate mechanism for sodium channel stabilisation against mechanical stress, ensuring reliable channel functionality in living systems.
Introduction
Voltage‐gated sodium channels (Navs) are key players in action potential propagation in neuronal and muscle tissues. The large α subunits (260 kDa) consist of 24 transmembrane α‐helices organised in four homologous domains (DI–DIV). Each domain is equipped with four voltage‐sensing segments (S1–S4) and two pore‐forming segments (S5–S6). Ten isoforms (SCN1A–SCN9A and SCNX) have been described, which vary in tissue expression and electrophysiological properties (Catterall, 2000; Catterall et al. 2005).
Although the α subunit alone is sufficient for ion selectivity and gating (Auld et al. 1988), it is associated with β subunits as a functional unit in vivo. Five β subunit isoforms have been described (Isom et al. 1992, 1995; Morgan et al. 2000; Qin et al. 2003; Yu et al. 2003). Except for the soluble alternative splicing variant of β1, all subunits consist of one transmembrane α‐helix and an extracellular immunoglobulin (IG) domain (Namadurai et al. 2015). Since their first description, the β subunits have been shown to be important parts of the functional Nav unit: β subunits shift the voltage dependence, alter the current density (Isom, 2001) or are involved in eliciting new gating states, such as resurgent currents (Grieco et al. 2005). β subunits have also been shown to alter Nav surface expression patterns (Chen et al. 2004) and to mediate functions independent of α subunits, such as formation of cell–cell contacts (Malhotra et al. 2000). β1 has an intramolecular disulfide bond (C21–C43), which is located close to the N‐terminal end of the protein in the extracellular IG domain. Using cryo‐electron microscopy (EM), this N‐terminal end was shown to interact with the extracellular loop region of the α subunit (Yan et al. 2017). The pore loop of DIV (S5–S6DIV) in particular is involved in this interaction, while the transmembrane part of β1 seems to connect to one of the voltage‐sensing domains (Yan et al. 2017). Disruption of this disulfide bond, via the β1 C43A mutation, was suggested to have an impact on the interaction with Nav1.4 (Islas et al. 2013).
Navs are not exclusively voltage gated, but can also be mechanically modified (Tabarean et al. 1999; Morris & Juranka, 2007; Beyder et al. 2010). The channels’ functions are reversibly affected by changes in the mechanical properties of the cellular environment, by altering the tension in the lipid bilayer or its shape, and by altering pipette pressure (Shcherbatko et al. 1999). Complex living organisms are regularly exposed to mechanical influences. Heart contraction, blood pulse waves and nerve bending due to body movements all have a mechanical influence. However, precise channel gating is essential for functional in vivo systems. Small changes in sodium current or shifts in the voltage dependence of Nav gating have been shown to cause severe pathological phenotypes from heart failure to pain syndromes (Abriel, 2010; Lampert et al. 2010). This raises the question of whether a stabilising co‐factor is required to ensure reliable Nav gating in vivo (Anishkin et al. 2014).
In this study, we propose the Nav β1 subunit as a candidate for such a stabilising role. Performing whole‐cell patch‐clamp experiments, we show that β1 protects Nav1.7 gating against mechanical shear stress. Using all‐atom molecular dynamics (MD) simulation of the β1 subunit IG domain, homology modelling of the human Nav1.7+β1 complex, and site‐directed mutagenesis, we identify the intramolecular disulfide bond C21–C43 to be especially important for protecting fast inactivation, but not activation against mechanical stress. β1 interacts with the extracellular end of S6DIV, thus stressing the role of this region in sodium channel fast inactivation.
Methods
Cell culture
Cells from the human embryonic kidney cell line HEK293, stably expressing Nav1.7, were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco–Life Technologies, Carlsbad, CA, USA) including 10% fetal bovine serum, 1.0 g l−1 glucose and 1% Geneticin (G‐418) (A&E Scientific, Marcq, Belgium). All cells were kept at 37°C and 5% CO2. Cells were transfected with either 0.3 μg human β1 subunit (pCLH‐SCN1b) in a pIRES vector containing green fluorescent protein (GFP), the β1 C43A mutation in the same vector or GFP alone in control groups using 1 μl JetPIe (Pyloplus Transfections, Illkirch, France) according to the manufacturer's instructions. Cells were recorded 1–2 days after transfection.
Electrophysiology
Whole‐cell voltage‐clamp experiments of transfected cells were performed using an EPC‐10USB amplifier (HEKA Elektronik, Lambrecht, Germany) at room temperature. Glass pipettes (tip resistance 1.0–2.4 MΩ) were manufactured with a DMZ puller (Zeitz Instruments GmbH, Martinsried, Germany) and filled with internal solution containing (in mm): 140 CsF, 10 NaCl, 10 Hepes, 1 EGTA, 18 sucrose (pH 7.33, adjusted with CsOH). The bath solution contained (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 Hepes, 20 glucose (pH 7.4, adjusted with NaOH).
Capacitive transients were cancelled and series resistance (always <5 MΩ) was compensated by at least 60%. Leak currents were subtracted online using the P/4 procedure following the test pulses. Signals were digitised at a sampling rate of 100 kHz. Low‐pass filter frequency was set to 2.873 kHz. Voltage protocols were carried out after current stabilisation and equilibration by a 3 min protocol in which the cells were held at −120 mV and stimulated at 0.1 Hz.
Current–voltage (I–V) relations of activation were obtained using 40 ms pulses to a range of test potentials in 10 mV steps at an interval of 5 s (see Fig. 1 for protocol). The voltage‐dependent Nav conductance G Na was calculated using the following equation: G Na = I Na/(V m − E rev), where I Na is the peak of the current at the voltage V m, and E rev is the reversal potential for sodium, which was determined for each cell individually.
Figure 1. β1 stabilises voltage dependence of Nav1.7 activation against mechanical stress.
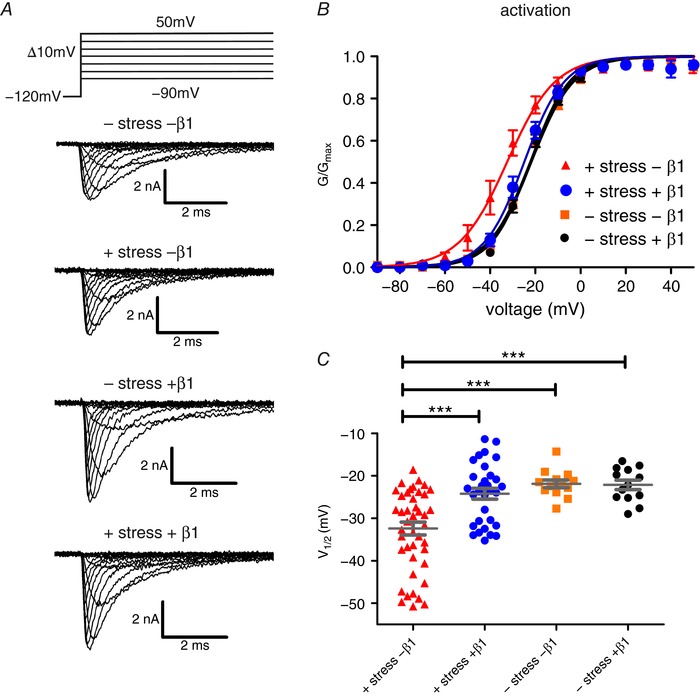
HEK cells, stably expressing Nav1.7, with and without β1 co‐expression, were exposed to mechanical shear stress via standardised perfusion flow. A, representative traces of sodium currents without and with mechanical stimulation (upper two panels), without mechanical stimulation but with β1 co‐expression (third from top) and with both mechanical stimulation and β1 co‐expression (bottom). The voltage protocol is displayed on top. B, voltage dependence of channel activation. Continuous lines are obtained by Boltzmann fits of normalised conductance–voltage curves. Application of mechanical stress leads to a negative shift of the activation curve and this is prevented by co‐expression of β1. C, values for half‐maximal voltage dependence of activation (V 1/2) obtained from Boltzmann fits for individual traces. Mean ± SEM values were −32.4 ± 1.5 mV for +shear stress –β1 (red triangles; P = 0.0004 vs. −shear stress −β1, P = 0.0006 vs. –shear stress +β1, P = 0.0002 vs. +shear stress +β1; n = 44), 24.2 ± 1.3 mV for +shear stress +β1 (blue circles, n = 31), −21.9 ± 0.9 mV for –shear stress –β1 (orange squares, n = 13) and −22.2 ± 1.1 mV for –shear stress +β1 (black circles, n = 13). *** P < 0.001 one‐way ANOVA with Bonferroni multiple comparisons test. [Color figure can be viewed at http://wileyonlinelibrary.com]
Activation curves were derived by plotting normalised G Na as a function of test potential and fitted with the Boltzmann equation: G Na = G Na,max/(1 + exp [(V m − V 1/2)/k]), where G Na,max is the maximum sodium conductance, V 1/2 is the membrane potential at half‐maximal activation, V m is the membrane voltage and k is the slope factor.
The voltage dependence of steady‐state fast inactivation was measured using a series of 500 ms pre‐pulses from −130 mV to −20 mV in 5 mV steps, each sweep followed by a 40 ms depolarisation to 0 mV that served as a test pulse to assess the available non‐inactivated channels. Normalised peak inward current amplitude (I Na/I Na,max) at each test pulse is displayed as a function of pre‐pulse potential and fitted using the following (Boltzmann) equation: I Na/I Na,max(V) = 1/(1 + exp[(V m − V 1/2)/k]), resulting in V 1/2 (the potential of half‐maximal inactivation) and the slope factor k.
Decay of fast inactivation was described using the single‐exponential fit function of the HEKA Fitmaster software for the inactivating part of a current trace (see Fig. 2).
Figure 2. β1 stabilises voltage dependence of Nav1.7 fast inactivation against mechanical stress.
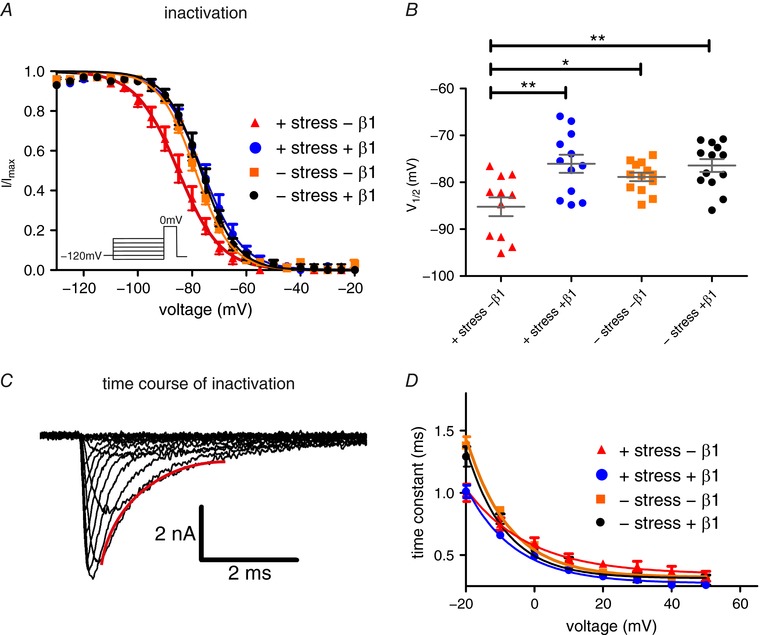
A, voltage dependence of steady‐state fast inactivation with and without β1 expression and exposure to mechanical shear stress. Continuous lines obtained by Boltzmann fits of normalised current–voltage traces. Co‐expression of β1 protects Nav1.7 from a negative shift of fast inactivation induced by mechanical stress. B, values for half‐maximal voltage‐dependent fast inactivation obtained from Boltzmann fits of individual traces. Mean ± SEM values were −85.2 ± 2.0 mV for +shear stress –β1 (red triangles, P = 0.0410 vs. –shear stress –β1, P = 0.0018 vs. –shear stress +β1, P = 0.0014 vs. +shear stress +β1; n = 11), −76.1 ± 1.9 mV for +shear stress +β1 (blue circles, n = 12), −78.9 ± 0.9 mV for –shear stress –β1 (orange squares, n = 13) and −76.4 ± 1.4 mV for –shear stress +β1 (black circles, n = 13). C, a single exponential fit of current decay was used to investigate the time constant of channel inactivation. A representative fit is displayed in red. D, time constant of current decay as obtained by single exponential fits of fast inactivation as shown in C. Mechanical shear stress leads to acceleration of fast inactivation at –20 mV. * P < 0.05; ** P < 0.01 one‐way ANOVA with Bonferroni multiple comparisons test. [Color figure can be viewed at http://wileyonlinelibrary.com]
Current density was calculated as the cell capacitance, divided by the maximal peak current amplitude, measured during the activation protocol.
Mechanical shear stress was applied via the flow of the bath solution through an AutoMate Scientific 8‐channel perfusion pencil (Automate Scientific, St. Berkeley, CA, USA) with a 250 μm wide outlet, placed at a constant distance (800 μm) to the measured cells. Flow was driven by hydrostatic pressure from the solution reservoir, placed 25 cm above the pencil, resulting in a flow of 270 μl min−1. This resulted in a tip flow of 28.8 cm s−1. Perfusion was started right before gigaseal formation.
Molecular dynamics simulation
Molecular dynamics (MD) simulations of the electric eel β1 IG domain in water were carried out using GROMACS 2016 (Abraham et al. 2015). Initial protein structures were taken from the electric eel Nav1.4–β1 complex structure (PDB ID 5XSY). The AMBER99SB‐ILDN force field (Lindorff‐Larsen et al. 2010) was used to model the protein using the residue range 24–152; all amino acids were modelled in their default ionisation state. Na+ ions were added to the simulation system to maintain neutrality using parameters from Joung and Cheatham (2008). Water was simulated using the SPC/E model (Berendsen et al. 1987). In a second simulation system, the Cys→Ala amino acid substitution was applied using MODELLER 9.19 (Webb & Sali, 2014).
Upon solvation of the protein, both simulation systems were equilibrated by 10 ns MD simulations in the canonical ensemble (NVT; constant number of atoms, volume, and temperature) and subsequent 150 ns in the isothermal–isobaric ensemble (NPT; constant number of atoms, pressure, and temperature) with position restraints on the protein heavy atoms. Finally, 10 ns NPT simulations with backbone‐only position restraints were performed, followed by 1 μs NPT production runs without any restraints.
A pressure of 1 bar was maintained by employing the semi‐isotropic Berendsen (Berendsen et al. 1984) and the Parrinello‐Rahman barostats (Parrinello & Rahman, 1981; Nosé & Klein, 1983) during the NPT equilibrations and the production simulations, respectively. Constant temperature was maintained by coupling protein and non‐protein separately to a heat bath at 310 K using the velocity rescaling algorithm (Bussi et al. 2007). Virtual sites for protein hydrogen atoms enabled simulation time steps of 4 fs.
Homology modelling
The cryo‐EM structure of the electric eel Nav1.4–β1 complex (PDB ID 5XSY) (Yan et al. 2017) served as the structural template to construct a human Nav1.7–human β1 complex homology model. Sequence alignment was performed using Jalview (Waterhouse et al. 2009) and 100 loop‐refined models were generated using MODELLER 9.19 (Webb & Sali, 2014). The best model was chosen based on the DOPE‐HR score implemented in MODELLER and visual inspection.
Data analysis
Data were analysed and graphed using Fitmaster software (HEKA Elektronik), Igor Pro 5.2 software (Wavemetrics), Prism versions 5 and 6 (Graphpad), Xmgrace and Corel Draw X3–X6 (Corel Corporation). Data are presented as means ± SEM.
For statistical testing, groups larger than two were compared by an ANOVA or a Kruskal–Wallis test in case of non‐parametric testing, followed by a Bonferroni post hoc analysis or Dunn's multiple comparison test in case of non‐parametric testing. Groups of two were compared by Student's t test or a Mann–Whitney test in case of non‐parametric testing (significance in every case at least P < 0.05, represented by *; P < 0.01 by ** and P < 0.001 by ***).
Results
The β1 subunit prevents mechanical impact on Nav1.7 activation
Nav gating is sensitive to mechanical influences. Here, we applied mechanical stress with a commonly used perfusion system during patch‐clamp recordings. Cells were perfused with extracellular solution, which exerts a force on the cell membrane. Mechanical stress was standardised by maintaining a constant distance (800 μm) between the cell and the perfusion pen and a stable solution flow (270 μl min−1; for further details see Methods). At first, we quantified the impact of the solution's flow on activation of Nav1.7, stably expressed in HEK cells (Fig. 1 A). Mechanical stimulation of the cells resulted in a significant left shift of the voltage dependence of activation (Fig. 1 B and C, Table 1). In the human body, mechanical stress occurs regularly and even though the impact of mechanical stress on cell membranes in vivo is difficult to quantify, it has been assumed that a stabilising momentum is likely to be necessary for proper function of ion channel complexes in vivo (Anishkin et al. 2014). Therefore, our aim was to look for a possible candidate for Nav channel stabilisation. Surprisingly, when we co‐expressed β1 together with Nav1.7, the impact of mechanical stress on Nav1.7 activation was completely abolished (Fig. 1 B and C, Table 1).
Table 1.
Activation and inactivation properties of Nav1.7 currents as a function of β1 co‐expression and mechanical stimulation
| −β1/−stim | −β1/+stim | +β1/−stim | +β1/+stim | +CA/−stim | +CA/+stim | |
|---|---|---|---|---|---|---|
| Activation | ||||||
| V 1/2 (mV) | −21.9 ± 0.9 | −32.4 ± 1.5 | 22.2 ± 1.1 | −24.2 ± 1.3 | −23.0 ± 2.7 | −24.8 ± 2.2 |
| Slope (1/mV) | 9.3 ± 0.3 | 8.5 ± 0.3 | 8.4 ± 0.5 | 7.6 ± 0.29 | 7.9 ± 0.5 | 8.6 ± 0.8 |
| Inactivation | ||||||
| V 1/2 (mV) | −78.9 ± 0.9 | −85.2 ± 2.0 | −76.4 ± 1.4 | 76.1 ± 1.9 | −74.1 ± 1.4 | −80.4 ± 1.8 |
| Slope (1/mV) | 6.4 ± 0.2 | 6.9 ± 0.5 | 6.0 ± 0.3 | 6.1 ± 0.3 | 5.2 ± 0.2 | 6.1 ± 0.3 |
Stim: mechanical stimulation; CA: β1‐C43A mutant. Data obtained by Boltzmann fits.
β1 protects Nav1.7 inactivation from mechanical influence
Upon activation, Navs quickly inactivate within milliseconds and we sought to investigate the impact of mechanical stress and β1 on steady‐state fast inactivation of Nav1.7.
In the absence of β1, mechanical stress led to a significant left shift of the voltage dependence of steady‐state fast inactivation (Fig. 2 A and B, and Table 1). The time constant of current inactivation was equally affected by mechanical stimulation, as identified by a single‐exponential fit of current decay (Fig. 2 C and D): the kinetics of inactivation were accelerated by mechanical stimulation at a potential of −20 mV. When we co‐expressed β1, the shift in the voltage dependence of steady‐state fast inactivation was completely prevented (Fig. 2 B and D).
These results suggest that β1 subunits not only play an important role in modulating the expression and voltage gating of Navs, but also function as a mechanical stabiliser in the cell membrane. This would ensure a constant channel response to depolarising stimuli, regardless of the mechanical stimuli present in the cell's environment.
The β1 mutation C43A alters β1 binding to S5–S6 loop of DIV
The extracellular IG domain of β1 is stabilised by disulfide bonds and in particular the link between the residues Cys21 and Cys43 is close to the interaction site with the α subunit (Yan et al. 2017). We wondered if removing this disulfide bond would affect the interaction between Nav1.7 and β1. In the electric eel β1, the disulfide bond is established between homologous residues 25 and 47. We investigated the impact of the disulfide bond on the conformational distribution of the β1 IG domain using unguided all‐atom MD simulations (Fig. 3 A and B). We used the available structure of the electric eel β1 wildtype IG domain and its C47A mutation in water based on the recent Nav1.4–β1 complex cryo‐EM structure (Yan et al. 2017). Over a simulation time of >1 μs, both the wildtype and the mutated IG domain retained an overall stable conformation. However, removing the disulfide bond of β1 resulted in a reduced conformational stability of the N‐terminus, as indicated by an increased number of sampled conformational clusters for the mutant (Fig. 3 A). The root‐mean‐square fluctuation (RMSF) for the N‐terminal residues of β1 was increased in the absence of the disulfide bond (Fig. 3 B). In summary, our simulations show that the N‐terminal disulfide bond of β1 confers conformational stability to the N‐terminus of β1. In the cryo‐EM structure, this N‐terminus forms several interactions with the pore loop of DIV (S5–S6DIV) of the α subunit, a part of the channel which has been implicated in fast inactivation processes because of its close relationship to S6DIV (Yan et al. 2017) (Fig. 3 C and D). Therefore, our simulations suggest that the specific interaction between the α and β subunit is likely to be disturbed by the C→A mutation.
Figure 3. β1‐C47A mutation perturbs binding to the S6 loop of DIV.
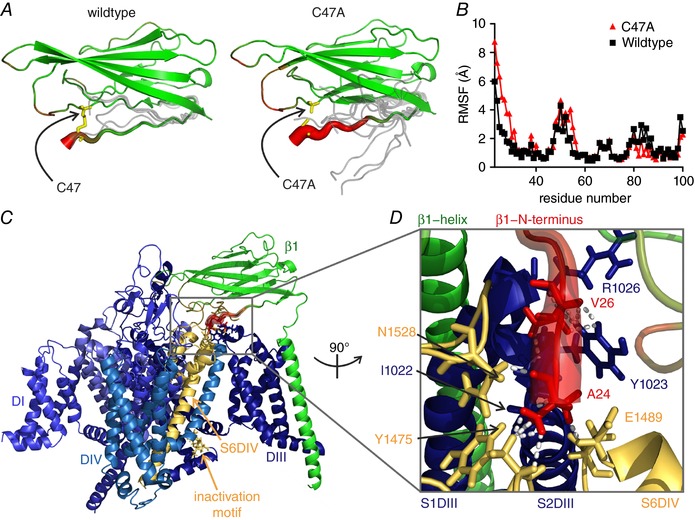
A and B, in the electric eel β1, the disulfide bond corresponding to C21–C43 in human β1 is formed by the corresponding residues 25 and 47. The C47A mutation increases the conformational flexibility of the electric eel β1 IG domain as observed in 1‐μs MD simulations of the wildtype and mutant IG domain. A, schematic illustration of the wildtype and mutant β1 IG domain backbone structure with representative conformations of the N‐terminus obtained by clustering of the MD trajectories (grey lines). Disulfide bond‐forming residues are shown as sticks. Root‐mean‐square fluctuation (RMSF) is encoded by tube thickness and colour (red: large RMSF; green: small RMSF; thus, thick and red areas indicate higher flexibility). B, per‐residue RMSF profile of the first 100 N‐terminal residues of β1‐wildtype (black squares) and β1‐C47A (red triangles) structural models. C, overview of the β1 subunit in complex with electric eel Nav1.4. The channel is shown in different blue shades for each subdomain, indicated by DI, DIV and DIII. S6DIV, the pore loop and the inactivation motif in the DIII–DIV linker are shown in yellow. Diameter and colouring of β1 IG domain represents conformational flexibility as in A. D, close‐up of the perturbed binding interface turned by 90° anti‐clockwise compared to C. Dashed lines represent interactions between the first 10 N‐terminal amino acids of β1 in the structure and the α subunit defined using a distance cut‐off of 2.3 Å. Ala24 of β1 interacts with Ile1022 of S2DIII, Glu1489 of S6DIV, and Tyr1475 and Asn1528 of S5–S6DIV of the α subunit; Val26 of β1 contacts Tyr1023 and Arg1026 in the S1–S2DIII. Interacting residues are displayed in stick representation. The decrease in conformational stability caused by the C47A mutation is shown in red and strongly affects the region of β1 interacting with the pore loop.
To transfer the results of the electric eel to the human α–β complex of Nav1.7 and β1, we constructed a Nav1.7 homology model in complex with a human β1 subunit, based on the electric eel Nav1.4 structure (Fig. 4). Both, the S5–6DIV and the N‐terminus of β1 are highly conserved across both organisms (Fig. 4 A). Several residues involved in interactions between the α subunit and the β1 IG domain are similar between human and eel and are modelled in close distance to each other (smaller than 2.3 Å, Fig. 4 B and C). In particular, three residues of the N‐terminus of β1 interact with hNav1.7: Gly20 is in contact with Tyr1657 and His1710 on the S5–S6DIV, and Lys1660 on S6DIV. Val22 of β1 is in contact with Tyr1204 and Arg1207 on S1–S2DIII, and Gly1712 on the S5–S6DIV. Val24 of β1 additionally contacts the backbone of Arg1207 on S1–S2DIII.
Figure 4. The DIV pore loop of the human Nav1.7 homology model interacts with β1.
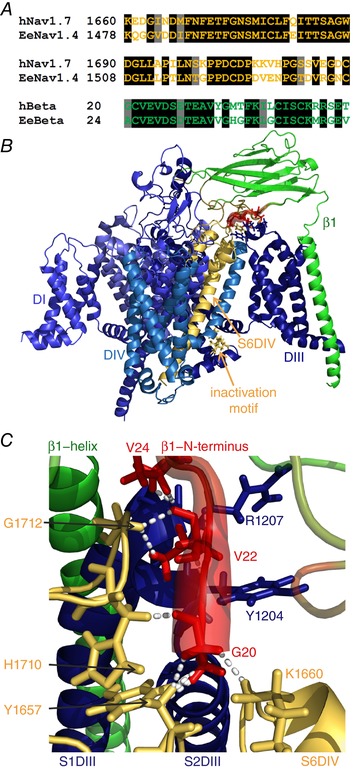
A, sequence alignment of the human Nav1.7 and the electric eel Nav1.4 in the pore loop (first two lines), and of the electric eel and human β1. The sequence identities for the shown parts are 72.8% for the pore loop and 66.6% for β1 (black background: identical residues; grey background: similar residues). B, overview of the homology model of the human Nav1.7 with the human β1 subunit. The colouring is identical to that in Fig. 3, with simulated electric eel β1 RMSF values being mapped onto the human β1 model. The overall structure resembles the electric eel structure. C, close‐up of β1 interface in contact with hNav1.7. Colouring and diameters are the same as in Fig. 3 to illustrate the flexible parts of β1 in red. Dashed lines represent interactions between the first 10 N‐terminal amino acids of β1 and the α subunit with a cut‐off defined at 2.3 Å. Gly20 of β1 interacts with Tyr1657 and His1710 on the S5–S6DIV, and Lys1660 on S6DIV; Val22 of β1 is in contact with Tyr1204 and Arg1207 on S1–S2DIII, and Gly1712 on the S5–S6DIV. Val24 of β1 additionally contacts the backbone of Arg1207 on S1–S2DIII. Interacting residues are displayed in stick representation. The human model of the Nav1.7–β1 interface strongly resembles the electric eel model. This is especially the case for the interaction between the N‐terminus and the pore loop (S5–S6DIV).
This indicates that the N‐termini of both the electric eel Nav1.4–β1 and the human Nav1.7–β1 complex have similar interactions with the pore loop of DIV (S5–S6DIV) and S6DIV. This suggests that the conclusions drawn from the MD simulations can be applied to the human channel complex, which we investigated functionally with patch‐clamp. In combination with the MD data, our results suggest that removing the disulfide bond by introducing the C43A mutation disturbs the binding of β1 to S5–S6DIV of human Nav1.7.
β1‐C43A prevents mechanical modulation of activation but not of fast inactivation
The MD simulations suggested that removing the disulfide bond in β1 by a C43A mutation disturbs the binding of β1 to the S5–S6DIV of Nav1.7. Thus, we investigated the functional impact of the β1‐C43A mutant on Nav1.7 and the mechano‐protective effect of β1. There was no influence of mechanical stimulation on voltage dependence of channel activation when β1‐C43A was co‐expressed: the mutant had the same capability of mechanical stabilisation as the β1 wildtype (Fig. 5 A and C, and Table 1). When investigating fast inactivation in the presence of β1‐C43A, we observed that mechanical stimulation led to a left shift of the voltage dependence just as in the experiments without co‐expression of β1 (Fig. 5 B and D, and Table 1). suggests that the mutant β1 subunit is not able to stabilise Nav1.7 fast inactivation. The inactivation kinetics of Nav1.7 with β1‐C43A seemed to be accelerated by mechanical stimulation at −20 mV, although this effect was not statistically significant (Fig. 5 E). Taken together these data suggest that Nav1.7 modulation of voltage dependence of activation and fast inactivation by β1 is dependent on distinct α–β interactions, and that β1 may interact with both via different structural components of the α subunit.
Figure 5. β1 mutation C43A can only partially stabilise Nav1.7.
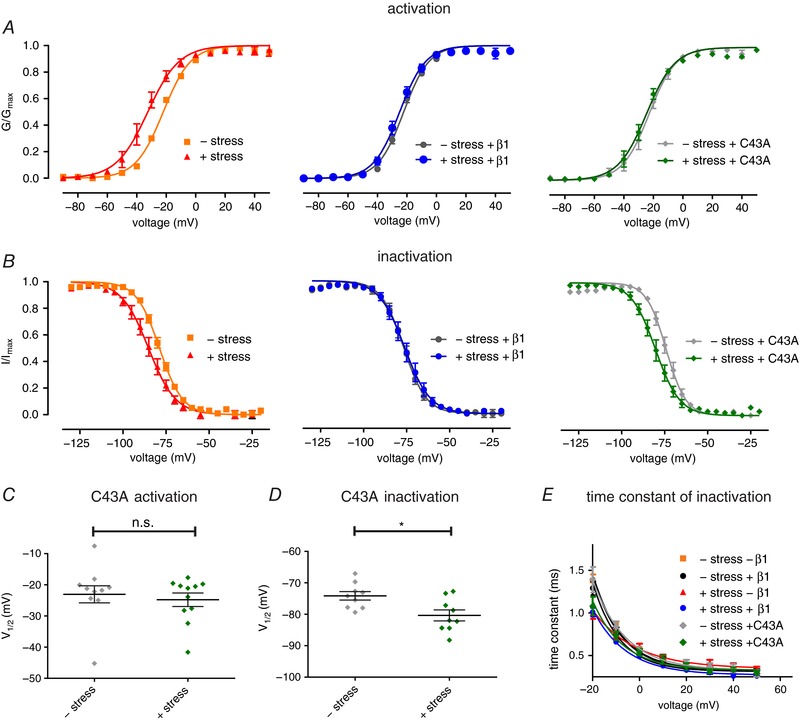
A and B, voltage dependence of channel activation and fast inactivation of either control cells (left) or cells expressing β1 wildtype (middle) or β1‐C43A (right). Cells were displayed according to expression pattern (wildtype, β1, C43A) for better clarity. Conductance–voltage curves with and without β1 (left and middle panels) are the same as in Figs 1 and 2 and shown for comparison. C, values for half‐maximal voltage‐dependent activation (V 1/2) obtained from Boltzmann fits of single traces. Mean ± SEM values were −23.0 ± 2.7 mV for –shear stress (grey, P = 0.8603 vs. +shear stress; n = 10) and −24.8 ± 2.2 mV for +shear stress (green, n = 11). D, values for half‐maximal voltage‐dependent fast inactivation (V 1/2) obtained from Boltzmann fits of single traces. Mean ± SEM values were −74.1 ± 1.4 mV for –shear stress (grey, P = 0.0124 vs. +shear stress +C43A; n = 9) and −80.4 ± 1.8 mV for +shear stress (green, n = 9). E, time constants of fast inactivation revealed by single exponential fit of inactivating current traces as shown in Fig. 2 C. * P < 0.05 with Mann–Whitney test for activation, two‐tailed unpaired t test for fast inactivation. [Color figure can be viewed at http://wileyonlinelibrary.com]
The β1 C21–C43 disulfide bond is necessary for enhancement of current density
It was reported before that β1 co‐expression enhances current density of Nav1.7 (Laedermann et al. 2013). Our experiments confirmed this effect when expressing the wildtype β1 subunit (Fig. 6 A). The β1‐C43A mutant, on the other hand, was not able to increase current density of Nav1.7 (Fig. 6 A). Mechanical stimulation had no impact on Nav1.7 current density (Fig. 6 B). These data suggest that the ability of β1 to increase Nav1.7 current density depends on the presence of the N‐terminal C21–C43 disulfide bond of β1.
Figure 6. C43A is unable to enhance Nav1.7 current density.
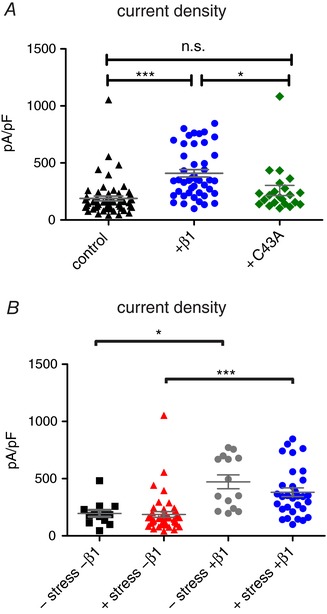
A, current density values for Nav1.7 control (black triangles, n = 59), co‐transfected with β1 (blue circles, P > 0.0001 vs. GFP, P = 0.0156 vs. C43A, n = 45) and C43A (green diamonds, n = 22). B, current density values for –shear stress –β1 (black squares, P = 0.0113 vs. –shear stress +β1; n = 13), +shear stress –β1 (red triangles, P < 0.0001 vs. +shear stress +β1; n = 44), –shear stress +β1 (grey circles, n = 14) and +shear stress +β1 (blue circles, n = 31). * P < 0.05; *** P < 0.001 with Kruskal–Wallis test with Dunn's post‐test for current density. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
Over recent years, Nav β subunits have gained increasing scientific interest because they appear to be the only known proteins that participate in cell adhesion, are part of a channel complex, and function as intra/extracellular signalling molecules (Hull & Isom, 2018). In this study, we reveal a novel feature of β subunit‐mediated ion channel modification: the β1 subunit is able to influence the mechanical susceptibility of Nav1.7.
β1 modulates mechanosensitivity of Nav1.7
It has been suggested that Navs interact with the membrane environment and are susceptible to changes in shape, constitution and lateral pressure (Shcherbatko et al. 1999; Tabarean et al. 1999; Lundbaek et al. 2004; Morris & Juranka, 2007; Beyder et al. 2010, p.5). Even just small changes in the voltage‐dependent gating of Nav1.7 lead to severe pain phenotypes (Lampert et al. 2014), suggesting that a regulator for mechanical stress susceptibility is needed in vivo (Anishkin et al. 2014). Here, we present β1 as a candidate for this role and assign a new function to these versatile proteins. We show that β1 compensates the effects of mechanical shear stress on channel activation and inactivation and thereby stabilises the channel's physiological response to membrane depolarisations.
With a combination of molecular simulations and electrophysiological methods, we show that the effect of mechanical protection cannot be attributed to one single α–β interaction. We generated a mutation of β1 which affects activation and inactivation differently, suggesting that the protective functions of β1 for these gating mechanisms are conferred by distinct molecular interactions. The first (and only) description of such a differential regulation of a voltage‐gated ion channel by an accessory subunit was provided for the structurally related L‐type calcium channel (Cav1.2, Olcese et al. 1994). The Cav β subunits are structurally very different to those of Navs, but nevertheless the results on this calcium channel support the concept of differential regulation of activation and inactivation by accessory subunits.
The Nav α–β subunit interaction is a complex phenomenon: different to β2 and β4, β1 and β3 are thought to non‐covalently bind to the α subunits (Namadurai et al. 2015). The recent cryo‐EM structure (Yan et al. 2017) shows the β1 subunit associated to the voltage‐sensing domain of DIII of the channel. In addition, recent studies using voltage‐clamp fluorometry show that, at least in Nav1.5, the voltage‐sensing domain of DIV is involved in β1 interaction as well (Zhu et al. 2017). Zimmer et al. have shown that different parts of β1 are necessary for interaction with Nav1.5 compared to Nav1.2 (Zimmer & Benndorf, 2002). Taken together, it seems that different effects on Nav subtypes have distinct underlying interaction sites. Our homology model of Nav1.7 is properly defined by the electric eel template (Yan et al. 2017) and seems to reliably describe the interaction with β1. The C43A mutation in the β1 subunit destabilises the interaction between the IG domain of β1 and the pore loop of DIV (S5–S6DIV) of the channel and has an impact on the protection of channel inactivation, but not on activation.
In a very recent proteomics study, β3 was identified as a strong interaction partner for Nav1.7 in mice (Kanellopoulos et al. 2018). Because of the high degree of sequence conservation and structural similarity of β1 and β3 subunits, one might expect similar mechanoprotective properties of β3. However, if mechanoprotection is a subtype‐specific feature of β1, or even conserved across all sodium channel β subtypes, awaits further research. It has recently been shown that the sodium channel β3 subunit oligomerises with itself (Namadurai et al. 2014). The authors solved the crystal structure of the β3 IG domain as a homotrimer, whose stability depends on a disulfide bond homologous to C21–C43 of β1. Using atomic force microscopy, the authors detected β3‐mediated clustering of Nav1.5. The interaction site of the three β3 subunits involves the N‐terminus of the IG domain (Namadurai et al. 2014). We wondered whether a similar clustering might be involved in the β1–Nav1.7 interaction. However, such a β1 trimer would be unable to bind to the α subunit because the N‐terminal end of β1 – which would be required for oligomerisation – would also be required for interaction with the α subunit (Yan et al. 2017). Consistently, however, in both cases the disruption of the N‐terminal disulfide bond prevents or at least perturbs oligomerisation of the β subunit with another β subunit or an α subunit, respectively, by preventing the formation of a stable binding interface.
Studying the mechanical impact on ion channel gating can be performed using different approaches. In previous studies, mechanical effects on Nav gating were investigated, e.g., by changing the membrane stretch via altering the recording pipette pressure (Shcherbatko et al. 1999). This technique has the disadvantage that altered interaction with the pipette glass and pressure‐dependent shape variations within the pipette may induce large variability (Bavi et al. 2014; Slavchov et al. 2014; Clausen et al. 2017). In our study, we therefore applied the mechanical stimulus by using the flow of a perfusion system. With this approach we aim to have constant pipette glass interactions. Concerning shape variations, both approaches (perfusion and pipette pressure variations) are dependent on the shape and size of the patched cell. The perfusion flow has a defined direction and will therefore lead to local maxima and minima of membrane stretch on the surface area of the cell. This may explain the larger standard deviation associated with the application of shear stress (e.g. the standard deviation increases from 3.2 mV to 10.1 mV due to mechanical stress for V 1/2 of activation). In order to overcome these limitations, we increased the number of experiments to obtain statistical consistency. Both approaches, pipette pressure alteration and perfusion, reveal similar effects on channel activation and inactivation, indicating the validity of both methods (Morris & Juranka, 2007).
Expression of β1 has no effect on Nav1.7 activation in the absence of mechanical stress, in agreement with Ho et al. (2012) and Laedermann et al. (2013). Whether β1 affects Nav1.7 fast inactivation seems to be less clear. While Ho et al. (2012) and Laedermann et al. (2013) reported a shift to more depolarised potentials, we did not observe such an effect. Furthermore, whereas we and Laedermann et al. (2013) describe an increased current density with β1 co‐expression, this was not reported by Ho et al. (2012). These differences in regulatory effects may be due to varied experimental settings: Laedermann et al. transiently transfected human Nav1.7 and Ho et al. used a cell line stably expressing rat Nav1.7. We, on the other hand, used a stable human Nav1.7 cell line. Also the HEK cells of each lab may present slight biological differences: β1 is known to be endogenously expressed in HEK cells (Moran et al. 2000), and it is possible that this expression level varies between laboratories. If our cells happen to have a higher β1 expression, which may be sufficient to induce gating changes, then these could have been masked in our experiments using mechanical stimulation. On the other hand, in order to shield Nav1.7 currents from mechanical stress it may be necessary that larger channel populations are associated with β1. Thus, the regulation of mechanical stress on Nav1.7 is likely to only be observable when β1 is co‐transfected.
Implications for sodium channel fast inactivation
After activation, Navs quickly inactivate within milliseconds. It is generally accepted that an amino acid sequence in the DIII–DIV linker – consisting of Ile, Phe and Met, the so‐called IFM motif – occludes the pore and thereby inactivates the mammalian Nav (Vassilev et al. 1988; West et al. 1992; Meents & Lampert, 2016). In the recently published structure of the electric eel Nav1.4–β1 complex (Yan et al. 2017), the authors discuss an alternative mechanism for fast inactivation. In contrast to previously published Nav structures (Shen et al. 2017), the inactivation particle of the electric eel Nav1.4 (here, Ile is replaced by Leu, therefore the inactivation sequence is established by an LFM motif) was found to be bound to the intracellular end on the non‐pore‐lining side of the S6DIV. The authors propose an allosteric fast inactivation mechanism in which the C‐terminal end of the S6 helix would occlude the pore, and not the IFM motif itself (Yan et al. 2017). Our experimental data suggest that β1 interacts with the extracellular end of S6DIV, thus potentially modulating this inactivation process. The β1‐C43A mutation selectively disrupts this interaction between the extracellular end of S6DIV and the β1 subunit (residues K1660, Y1657, the backbone of H1710 and G1712, Fig. 4 C), and it functionally only affects fast inactivation. Thereby, our data support a structural involvement of S6DIV in fast inactivation, and may explain how toxin binding to the extracellular part of the channel protein can potently affect fast inactivation. Site 3 neurotoxins have been shown to interact with the pore loop of DIV (Thomsen & Catterall, 1989) and to alter fast inactivation of Navs (Stevens et al. 2011). Thus, these toxins act at a similar site on Nav1.7 to our predicted β1 site. Nevertheless, in the classical “ball and chain” model the IFM motive interacts with S6DIV as well – but from its pore‐lining side, which might also be modulated by β1–S6 interactions. Thus, our findings do not permit a clear distinction between the classical direct plugging mechanisms or an allosteric fast inactivation mechanism comprising the inner S6 segment. In summary, our data stress the important role of S6DIV in sodium channel fast inactivation.
Conclusion
In this study, we have assigned a new function to Nav β1 subunits: the protection of Nav1.7 against mechanical stress. The voltage dependence of channel activation and inactivation are both shielded against shear stress. The protective mechanism of the β1–Nav1.7 interaction relies at least in part on a disulfide bond in β1 involving residues Cys21 and Cys43. Additionally, homology modelling and structural analyses are in accordance with a recently suggested model for fast inactivation of Nav channels. With the principle established here of mechanostabilisation of sodium channels by associated subunits, this study shows a new dimension of the complex regulation of these membrane proteins in a physiological environment in motion.
Additional information
Competing interests
None declared.
Author contributions
Experiments were performed in the laboratory of A.L., Institute of Physiology, RWTH Aachen University, Germany and in the group of J.‐P.M. at Forschungszentrum Jülich, Germany. J.K. participated in design of the study, designed and performed the patch‐clamp experiments, performed molecular dynamics simulations and homology modelling experiments, analysed the data and wrote the manuscript. J.M. designed and analysed patch‐clamp experiments, interpreted the results and critically revised the manuscript. J.‐P.M. contributed to study design, advised on the setup and analysis of molecular simulations, and critically revised the manuscript. A.L. conceived and designed the study, designed the patch‐clamp experiments, and critically revised the manuscript. All authors have read and approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) to J.‐P.M. (MA 7525/1‐1; as part of the Research Unit FOR 2518, DynIon) and to A.L. (LA 2740/3‐1). The authors gratefully acknowledge the computing time granted by the JARA‐HPC Vergabegremium and VSR Commission on the supercomputer JURECA (Krause & Thörnig, 2016) at Forschungszentrum Jülich.
Acknowledgements
We thank Petra Hautvast and Brigitte Hoch for excellent technical assistance.
Biography
In parallel with studying human medicine at RWTH Aachen, Jannis Körner pursued his interests in the field of ion channel and pain research which led to his very first paper being published in The Journal of Physiology. Since 2016 he has been part of the neurophysiology section of the department of physiology and will graduate as a physician by the end of 2018. His goal is to become a physician‐scientist, combining patient care with thoughtful basic research.

Edited by: Ian Forsythe & Reinhold Penner
References
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B & Lindahl E (2015). GROMACS: high performance molecular simulations through multi‐level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. [Google Scholar]
- Abriel H (2010). Cardiac sodium channel Nav1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol 48, 2–11. [DOI] [PubMed] [Google Scholar]
- Anishkin A, Loukin SH, Teng J & Kung C (2014). Feeling the hidden mechanical forces in lipid bilayer is an original sense. Proc Natl Acad Sci USA 111, 7898–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auld VJ, Goldin AL, Krafte DS, Marshall J, Dunn JM, Catterall WA, Lester HA, Davidson N & Dunn RJ (1988). A rat brain Na+ channel α subunit with novel gating properties. Neuron 1, 449–461. [DOI] [PubMed] [Google Scholar]
- Bavi N, Nakayama Y, Bavi O, Cox CD, Qin Q‐H & Martinac B (2014). Biophysical implications of lipid bilayer rheometry for mechanosensitive channels. Proc Natl Acad Sci USA 111, 13864–13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen HJC, Grigera JR & Straatsma TP (1987). The missing term in effective pair potentials. J Phys Chem 91, 6269–6271. [Google Scholar]
- Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A & Haak JR (1984). Molecular dynamics with coupling to an external bath. J Chem Phys 81, 3684–3690. [Google Scholar]
- Beyder A, Rae JL, Bernard C, Strege PR, Sachs F & Farrugia G (2010). Mechanosensitivity of Nav1.5, a voltage‐sensitive sodium channel. J Physiol 588, 4969–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D & Parrinello M (2007). Canonical sampling through velocity rescaling. J Chem Phys 126, 014101. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000). From ionic currents to molecular mechanisms: the structure and function of voltage‐gated sodium channels. Neuron 26, 13–25. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL & Waxman SG (2005). International Union of Pharmacology. XLVII. Nomenclature and structure‐function relationships of voltage‐gated sodium channels. Pharmacol Rev 57, 397–409. [DOI] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilaythong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA & Isom LL (2004). Mice lacking sodium channel β1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 24, 4030–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen MV, Jarerattanachat V, Carpenter EP, Sansom MSP & Tucker SJ (2017). Asymmetric mechanosensitivity in a eukaryotic ion channel. Proc Natl Acad Sci USA 114, E8343–E8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL & Raman IM (2005). Open‐channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron 45, 233–244. [DOI] [PubMed] [Google Scholar]
- Ho C, Zhao J, Malinowski S, Chahine M & O'Leary ME (2012). Differential expression of sodium channel β subunits in dorsal root ganglion sensory neurons. J Biol Chem 287, 15044–15053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull JM & Isom LL (2018). Voltage‐gated sodium channel β subunits: The power outside the pore in brain development and disease. Neuropharmacology 132, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas AA, Sánchez‐Solano A, Scior T, Millan‐PerezPeña L & Salinas‐Stefanon EM (2013). Identification of Navβ1 residues involved in the modulation of the sodium channel Nav1.4. PLoS One 8, e81995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL (2001). Sodium channel beta subunits: anything but auxiliary. Neuroscientist 7, 42–54. [DOI] [PubMed] [Google Scholar]
- Isom LL, DeJongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL & Catterall WA (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, DeJongh KS, Westenbroek RE, Reber BF, Scheuer T & Catterall WA (1995). Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83, 433–442. [DOI] [PubMed] [Google Scholar]
- Joung IS & Cheatham TE 3rd (2008). Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulos AH, Koenig J, Huang H, Pyrski M, Millet Q, Lolignier S, Morohashi T, Gossage SJ, Jay M, Linley JE, Baskozos G, Kessler BM, Cox JJ, Dolphin AC, Zufall F, Wood JN & Zhao J (2018). Mapping protein interactions of sodium channel NaV1.7 using epitope‐tagged gene‐targeted mice. EMBO J 37, 427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause D & Thörnig P (2016). JURECA: General‐purpose supercomputer at Jülich Supercomputing Centre. Journal of large‐scale research facilities 2, A62 https://doi.org/10.17815/jlsrf-2-121. [Google Scholar]
- Laedermann CJ, Syam N, Pertin M, Decosterd I & Abriel H (2013). β1‐ and β3‐ voltage‐gated sodium channel subunits modulate cell surface expression and glycosylation of Nav1.7 in HEK293 cells. Front Cell Neurosci 7, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert A, Eberhardt M & Waxman SG (2014). Altered sodium channel gating as molecular basis for pain: contribution of activation, inactivation, and resurgent currents. Handb Exp Pharmacol 221, 91–110. [DOI] [PubMed] [Google Scholar]
- Lampert A, O'Reilly AO, Reeh P & Leffler A (2010). Sodium channelopathies and pain. Pflugers Arch 460, 249–263. [DOI] [PubMed] [Google Scholar]
- Lindorff‐Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO & Shaw DE (2010). Improved side‐chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbaek JA, Birn P, Hansen AJ, Søgaard R, Nielsen C, Girshman J, Bruno MJ, Tape SE, Egebjerg J, Greathouse DV, Mattice GL, Koeppe RE & Andersen OS (2004). Regulation of sodium channel function by bilayer elasticity: the importance of hydrophobic coupling. Effects of micelle‐forming amphiphiles and cholesterol. J Gen Physiol 123, 599–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kazen‐Gillespie K, Hortsch M & Isom LL (2000). Sodium channel β subunits mediate homophilic cell adhesion and recruit ankyrin to points of cell‐cell contact. J Biol Chem 275, 11383–11388. [DOI] [PubMed] [Google Scholar]
- Meents JE & Lampert A (2016). Studying sodium channel gating in heterologous expression systems In Advanced Patch‐Clamp Analysis for Neuroscientists, (Neuromethods), ed. Korngreen A, pp. 37–65. Humana Press, New York, NY: Available at: https://link.springer.com/protocol/10.1007/978-1-4939-3411-9_3 [Accessed December 3, 2017]. [Google Scholar]
- Moran O, Nizzari M & Conti F (2000). Endogenous expression of the β1A sodium channel subunit in HEK‐293 cells. FEBS Lett 473, 132–134. [DOI] [PubMed] [Google Scholar]
- Morgan K, Stevens EB, Shah B, Cox PJ, Dixon AK, Lee K, Pinnock RD, Hughes J, Richardson PJ, Mizuguchi K & Jackson AP (2000). β3: an additional auxiliary subunit of the voltage‐sensitive sodium channel that modulates channel gating with distinct kinetics. Proc Natl Acad Sci USA 97, 2308–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CE & Juranka PF (2007). Nav channel mechanosensitivity: activation and inactivation accelerate reversibly with stretch. Biophys J 93, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namadurai S, Balasuriya D, Rajappa R, Wiemhöfer M, Stott K, Klingauf J, Edwardson JM, Chirgadze DY & Jackson AP (2014). Crystal structure and molecular imaging of the Nav channel β3 subunit indicates a trimeric assembly. J Biol Chem 289, 10797–10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namadurai S, Yereddi NR, Cusdin FS, Huang CL‐H, Chirgadze DY & Jackson AP (2015). A new look at sodium channel β subunits. Open Biol 5, 140192 https://doi.org/10.1098/rsob.140192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosé S & Klein ML (1983). Constant pressure molecular dynamics for molecular systems. Mol Phys 50, 1055–1076. [Google Scholar]
- Olcese R, Qin N, Schneider T, Neely A, Wei X, Stefani E & Birnbaumer L (1994). The amino terminus of a calcium channel beta subunit sets rates of channel inactivation independently of the subunit's effect on activation. Neuron 13, 1433–1438. [DOI] [PubMed] [Google Scholar]
- Parrinello M & Rahman A (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys 52, 7182–7190. [Google Scholar]
- Qin N, D'Andrea MR, Lubin M‐L, Shafaee N, Codd EE & Correa AM (2003). Molecular cloning and functional expression of the human sodium channel β1B subunit, a novel splicing variant of the β1 subunit. Eur J Biochem 270, 4762–4770. [DOI] [PubMed] [Google Scholar]
- Shcherbatko A, Ono F, Mandel G & Brehm P (1999). Voltage‐dependent sodium channel function is regulated through membrane mechanics. Biophys J 77, 1945–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zhou Q, Pan X, Li Z, Wu J & Yan N (2017). Structure of a eukaryotic voltage‐gated sodium channel at near‐atomic resolution. Science 355, eaal4326. [DOI] [PubMed] [Google Scholar]
- Slavchov RI, Nomura T, Martinac B, Sokabe M & Sachs F (2014). Gigaseal mechanics: creep of the gigaseal under the action of pressure, adhesion, and voltage. J Phys Chem B 118, 12660–12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens M, Peigneur S & Tytgat J (2011). Neurotoxins and their binding areas on voltage‐gated sodium channels. Front Pharmacol 2, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarean IV, Juranka P & Morris CE (1999). Membrane stretch affects gating modes of a skeletal muscle sodium channel. Biophys J 77, 758–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen WJ & Catterall WA (1989). Localization of the receptor site for alpha‐scorpion toxins by antibody mapping: implications for sodium channel topology. Proc Natl Acad Sci USA 86, 10161–10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T & Catterall WA (1988). Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M & Barton GJ (2009). Jalview Version 2 – a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B & Sali A (2014). Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics 47, 5.6.1–5.6.32. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL & Catterall WA (1992). A cluster of hydrophobic amino acid residues required for fast Na+‐channel inactivation. Proc Natl Acad Sci USA 89, 10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zhou Q, Wang L, Wu J, Zhao Y, Huang G, Peng W, Shen H, Lei J & Yan N (2017). Structure of the Nav1.4‐β1 complex from electric eel. Cell 170, 470–482.e11. [DOI] [PubMed] [Google Scholar]
- Yu FH, Westenbroek RE, Silos‐Santiago I, McCormick KA, Lawson D, Ge P, Ferriera H, Lilly J, DiStefano PS, Catterall WA, Scheuer T & Curtis R (2003). Sodium channel β4, a new disulfide‐linked auxiliary subunit with similarity to β2. J Neurosci 23, 7577–7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Voelker TL, Varga Z, Schubert AR, Nerbonne JM & Silva JR (2017). Mechanisms of noncovalent β subunit regulation of NaV channel gating. J Gen Physiol 149, 813–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer T & Benndorf K (2002). The human heart and rat brain IIA Na+ channels interact with different molecular regions of the β1 subunit. J Gen Physiol 120, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]