

Summary
Hypoxia (i.e. oxygen deprivation) activates the hypoxia‐signalling pathway, primarily via hypoxia‐inducible transcription factors (HIF) for numerous target genes, which mediate angiogenesis, metabolism and coagulation, among other processes to try to replenish tissues with blood and oxygen. Hypoxia signalling dysregulation also commonly occurs during chronic inflammation. We sampled gingival tissues from rhesus monkeys (Macaca mulatta; 3–25 years old) and total RNA was isolated for microarray analysis. HIF1A, HIF1B and HIF2A were significantly different in healthy aged tissues, and both HIF1A and HIF3A were positively correlated with aging. Beyond these transcription factor alterations, analysis of patterns of gene expression involved in hypoxic changes in tissues showed specific increases in metabolic pathway hypoxia‐inducible genes, whereas angiogenesis pathway gene changes were more variable in healthy aging tissues across the animals. With periodontitis, aging tissues showed decreases in metabolic gene expression related to carbohydrate/lipid utilization (GBE1, PGAP1, TPI1), energy metabolism and cell cycle regulation (IER3, CCNG2, PER1), with up‐regulation of transcription genes and cellular proliferation genes (FOS, EGR1, MET, JMJD6) that are hypoxia‐inducible. The potential clinical implications of these results are related to the epidemiological findings of increased susceptibility and expression of periodontitis with aging. More specifically the findings describe that hypoxic stress may exist in aging gingival tissues before documentation of clinical changes of periodontitis and, so, may provide an explanatory molecular risk factor for an elevated capacity of the tissues to express destructive processes in response to changes in the microbial biofilms characteristic of a more pathogenic microbial challenge.
Keywords: aging, hypoxia, mucosal tissues, non‐human primates, periodontitis
Introduction
We have recently implemented an investigation using a non‐human primate model of periodontitis that has been demonstrated to have extensive similarities in clinical, microbiological and immunological features of human periodontitis. The model enables us to document characteristics of the transcriptome in gingival tissues, as a representative mucosal tissue, obtained from animals representing young individuals (approximately 10‐year‐old humans) to aged individuals (approximately 70‐ to 80‐year‐old humans).1, 2, 3, 4 We have shown significant differences in apoptosis pathway gene expression profiles associated with aging, even in healthy gingival tissues.2, 4 Differences were also noted in inflammasome gene pathways, including both receptors critical for signalling, and downstream effector functions,5 in antigen processing and presentation pathways,6 and differential expression of immune system genes.7
The human subgingival ecology has been shown to accommodate over 700 species of bacteria8 that differ both qualitatively and quantitatively in health, gingivitis and periodontitis.9 These difference have been documented to relate to the capacity of pioneer microorganisms to bind to host surfaces through receptor–ligand interactions,10 asaccharolytic species of bacteria that develop in inflamed sites related to a different nutritional microenvironment,11 anaerobic species that emerge at diseased sites presumably reflecting environmental changes of low oxygen levels, probably resulting from inflammation‐induced processes generating reactive oxygen intermediates,12 and species of bacteria that produce a range of volatile sulphur compounds.13 However, much of the research in this area has focused on the physiology and metabolism of the bacterial species related to their ability to survive and expand, as well as new knowledge of their genomes delineating unique pathways enabling them a competitive edge and probably contributing to the virulence potential of the pathogenic biofilms.14
Generally overlooked in these investigations is that the cellular response to oxygen is a central process in mammalian cells. Oxygen is required for aerobic energy metabolism and is required by the cells to produce adequate amounts of ATP necessary for metabolic activities. Hypoxia (i.e. oxygen deprivation) is an event in a diverse range of human tissues and cells with disease states including neoplastic development, ischaemia and chronic inflammation.15, 16 Organisms have evolved a range of adaptive mechanisms for hypoxic conditions including activation or repression of certain homeostatic genes that enable survival of tissues and cells. Specifically, low oxygen conditions activate the hypoxia signalling pathway, primarily via the hypoxia‐inducible factor‐1 (HIF‐1). The gene for HIF‐1 is among the primary genes involved in the homeostatic processes in hypoxia by increasing vascularization.17, 18, 19 It is also a transcription factor for numerous target genes, which mediate multiple biological functions, such as angiogenesis, haematopoiesis and the maintenance of vascular tone to provide or replenish tissues with blood and oxygen. HIF‐1 further mediates cellular responses to hypoxia by regulating glucose uptake and anaerobic respiration in oxygen‐depleted environments.17, 20
Hypoxia signalling dysregulation commonly occurs during chronic inflammation, at least in part through the activation and/or potentiation of nuclear factor‐κB (NF‐κB). Nuclear factor‐κB can be engaged through a classical pathway that is activated through the inhibitor of κB kinase (IKKα/β/γ) complex, and an alternative pathway that involves NF‐κB‐inducing kinase‐mediated activation of IKKα.21 Both sustained and intermittent hypoxia enhance NF‐κB activity using the classical pathway.22
The analyses in this report were designed to test the hypothesis that gingival tissues in aging animals demonstrate enhanced expression of genes in physiological pathways consistent with hypoxic stress in the tissues before a disease process. These changes might be anticipated to provide a risk for the tissues to express destructive processes in response to the microbial challenge, as occurs during periodontitis.
Methods
Non‐human primate model and oral clinical evaluation
Rhesus monkeys (Macaca mulatta) (n = 34; 14 female and 20 male) housed at the Caribbean Primate Research Center at Sabana Seca, Puerto Rico, were used in these studies. Healthy animals (five to seven per group) were distributed by age into four groups: ≤ 3 years (young), 3–7 years (adolescent), 12–16 years (adult) and 18–23 years (aged). Only adult (n = 5) and aged (n = 6) additional animals with periodontitis were used, as periodontitis does not occur naturally in younger animals. The non‐human primates were typically fed a 20% protein, 5% fat, and 10% fibre commercial monkey diet (diet 8773, Teklad NIB primate diet modified: Harlan Teklad). The diet was supplemented with fruits and vegetables, and water was provided ad libitum in an enclosed corral setting.
A protocol approved by the Institutional Animal Care and Use Committee of the University of Puerto Rico enabled anaesthetized animals to be examined for clinical measures of periodontal health including probing pocket depth and bleeding on probing as we have described previously.23 Periodontitis was defined as mean mouth values of probing pocket depth ≥ 3 mm and bleeding on probing ≥ 1.
Tissue sampling and gene expression microarray analysis
A buccal gingival sample from either healthy or periodontitis‐affected tissue from the premolar/molar maxillary region of each animal was taken using a standard gingivectomy technique, and maintained frozen in RNAlater solution. Total RNA was isolated from each gingival tissue using a standard procedure as we have described and tissue RNA samples were submitted to the microarray core to assess RNA quality and analyse the transcriptome using the GeneChip® Rhesus Macaque Genome Array (Affymetrix, Santa Clara, CA).4, 24 Individual samples were used for gene expression analyses. Table 1 lists the hypoxia‐related gene set examined in this report.
Table 1.
Hypoxia pathway genes and associated functions
| Gene ID | Fxn | Gene title |
|---|---|---|
| ARNT (HIF1B) | T | Aryl hydrocarbon receptor nuclear translocator |
| COPS5 | T | COP9 constitutive photomorphogenic homologue subunit 5 |
| EPAS1 (HIF2A) | T | Endothelial PAS domain protein 1 |
| HIF1A | T | Hypoxia inducible factor 1, α subunit (basic helix‐loop‐helix transcription factor) |
| HIF1AN | T | Hypoxia inducible factor 1, α subunit inhibitor |
| HIF3A | T | Hypoxia inducible factor 3, α subunit |
| HNF4A | T | Hepatocyte nuclear factor 4 , α |
| NCOA1 | T | Nuclear receptor coactivator 1‐like |
| PER1 | T | Period homologue 1 |
| APEX1 | I | APEX nuclease (multifunctional DNA repair enzyme) 1 |
| EGLN1 | I | Egl nine homologue 1 |
| EGLN2 | I | Egl nine homologue 2 |
| NFKB1 | I | Nuclear factor of κ light polypeptide gene enhancer in B‐cells 1 |
| P4HA1 | I | Prolyl 4‐hydroxylase, α polypeptide I |
| P4HB | I | Protein disulphide‐isomerase‐like |
| TP53 | I | Tumour protein p53 |
| ADORA2B | A | Adenosine A2b receptor |
| ANGPTL4 | A | Angiopoietin‐related protein 4 |
| ANXA2 | A,C | Annexin A2 |
| BTG1 | A,AP,CP | B‐cell translocation gene 1, anti‐proliferative |
| EGR1 | A,CP | Early growth response 1 |
| EDN1 | A | Endothelin 1 |
| EPO | A | Erythropoietin |
| F3 | A,C | Coagulation factor 3, thromboplastin |
| GPI | A,M | Glucose‐6‐phosphate isomerase |
| HMOX1 | A | Haem oxygenase (decycling) 1 |
| HMOX2 | A | Haem oxygenase (decycling) 2 |
| JMJD6 | A | Jumonji domain containing 6 |
| LOX | A | Lysyl oxidase |
| MMP9 | A | Matrix metallopeptidase 9 (gelatinase B) |
| PGF | A,CP | Placental growth factor |
| PLAU (uPA) | A,C | Plasminogen activator, urokinase |
| SEMA7A | A | Semaphorin 7A, GPI membrane anchor |
| SERPINE1 (PAI1) | A,C | Plasminogen activator inhibitor 1 |
| VEGFA | A | Vascular endothelial growth factor A |
| ALDOA | C,M | Aldolase A |
| F10 | C | Coagulation factor X |
| LIS1 (PAFAH1) | C | Platelet‐activating factor acetylhydrolase 1b |
| SLC16A3 | C,TR | Solute carrier family 16, member 3 |
| ATR | D | Ataxia telangiectasia and Rad3 related |
| MIF | D,AP,CP | Macrophage migration inhibitory factor |
| NDRG1 | D | N‐myc downstream regulated 1 |
| RUVBL2 | D | RuvB‐like 2 (Escherichia coli) |
| DDIT4 | M,AP | DNA‐damage‐inducible transcript 4 |
| CP | M | Ceruloplasmin |
| ENO1 | M | Enolase 1, (α) |
| CTSA | M | Cathepsin A |
| EROL1 | M | ERO1‐like (Saccharomyces cerevisiae) |
| GBE1 | M | Glucan (1,4‐α‐), branching enzyme 1 |
| GYS1 | M | Glycogen synthase 1 (muscle) |
| HK2 | M | Hexokinase 2 |
| LDHA | M | Lactate dehydrogenase A |
| PDK1 | M | Pyruvate dehydrogenase kinase, isozyme 1 |
| PFKFB3 | M | 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase 3 |
| PRKFB4 | M | 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphosphatase 4 |
| PFKP | M | Phosphofructokinase, platelet |
| PGAM1 | M | Phosphate glycerate mutase 1 |
| PGK1 | M | Phosphoglycerate kinase 1 |
| PKM2 | M | Pyruvate kinase, muscle |
| SLC2A1 | M,TR | Solute carrier family 2 (facilitated glucose transporter), member 1 |
| SLC2A3 | M,TR | Solute carrier family 2 (facilitated glucose transporter), member 3 |
| TPI1 | M | Similar to Triosephosphate isomerase (TIM) |
| ADM | AP,CP | Adrenomedullin |
| BNIP3 | AP | BCL2/adenovirus E1B 19 000 interacting protein 3‐like |
| IER3 | AP | Immediate early response 3 |
| NOS3 | AP,CP | Nitric oxide synthase 3 (endothelial cell) |
| PIM1 | AP,CP | Pim‐1 oncogene |
| BLM | CP | Bloom syndrome, RecQ helicase‐like |
| CCNG2 | CP | Cyclin G2 |
| IGFBP3 | CP | Insulin‐like growth factor binding protein 3 |
| MET | CP | Met proto‐oncogene (hepatocyte growth factor receptor) |
| MXI1 | CP | MAX interactor 1 |
| NAMPT | CP | Nicotinamide phosphoribosyltransferase |
| ODC1 | CP | Ornithine decarboxylase 1 |
| TXNIP | CP | Thioredoxin interacting protein |
| BULHE40 | CD | Basic helix‐loop‐helix family, member e40 |
| FOS | CD | FBJ murine osteosarcoma viral oncogene homologue |
| RBPJ | CD | Recombination signal binding protein for immunoglobulin κ J region |
| USF2 | CD | Upstream transcription factor 2 |
| TRFC | TR | Transferrin receptor (p90, CD71) |
| VDAC1 | TR | Similar to voltage‐dependent anion‐selective channel protein 1 |
| ANKRD37 | OR | Ankyrin repeat domain 37 |
| CA9 | OR | Carbonic anhydrase IX |
| DNAJC5 | OR | DnaJ (Hsp40) homologue, subfamily C, member 5 |
| EIF4EBP1 | OR | Eukaryotic translation initiation factor 4E binding protein 1 |
| LGALS3 | OR | Lectin, galactoside‐binding, soluble, 3 |
| MAP3K1 | OR | Mitogen‐activated protein kinase kinase kinase 1 |
A, angiogenesis; AP, regulation of apoptosis; C, Coagulation; CD, cell differentiation; CP, regulation of cell proliferation; D, DNA damage and repair; I, HIF interactors; M, metabolism; OR, other response genes; T, transcription/co‐transcription factors; TR, transporters, channels, receptors.
Based upon the microarray outcomes we selected five genes and performed a quantitative PCR analysis using a standard technique in our laboratory employing a Roche 480 LightCycler.25 Quantitative PCR primers were designed using software primerquest at the Integrated DNA Technologies website (http://www.idtdna.com) and were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). Primers were prepared for LDHA (forward: AGATTCCAGTGTGCCTGTATG; reverse: ACCTCTTTCCACTGTTCCT TATC), PGAM1 (forward: CATCTGGAGGGTCTCTCTGAA; reverse: AACTGCA TGGGCTTGATAGG), PGK1 (forward: GATGATTATTGGTGGCGGAATG; reverse: GACAATCTTGGCTCCCTCTT), ENOL1 (forward: GAGGTTTACCACAACCTGAAGA; reverse: AGCTCCAGGCCTTCTTTATTC) and EPAS1 (forward: GAAGCGACAGC TGGAGTATG; reverse: TGAGGTTCTTCATCCGTTTCC) genes. The level of message was determined according to our previously published methods25 and those levels were compared across the RNA samples prepared from each of the healthy groups and the two periodontitis groups.
Data analysis
Normalization of values across the chips was accomplished through signal intensity standardization across each chip using the Affymetrix PLIER algorithm. The arrays contained matched and mismatched pairs allowing the MAS 5 algorithm to be used. For each gene we first determined differences in expression across the groups using analysis of variance (version 9.3, SAS Inc., Cary, NC). The healthy aged tissues were then compared with the other age groups using a t‐test and accepting a P‐value ≤ 0·05 for significance. Because of the cost of these types of non‐human primate experiments and the availability of primates of the various ages, we did not have sufficient samples to identify whether the relationship between age and gene expression could be treated using a linear model, so the subjects were classified and analysis of variance was used for analysis. The choice of least significant difference for multiple comparisons (analysis of variance followed by t‐tests) provided maximum power given our necessarily small sample sizes. We determined a correlation with aging in healthy tissues using a Spearman Rank correlation analysis that was fitted to the gene expression by age. A P‐value ≤ 0·05 was used to evaluate the significance of the correlation. Volcano plots were prepared to visualize outlier gene expression profiles in the aging animals compared with the other age group.26 JMP (version 10.0, SAS Inc.) was used to create metagenes independently of group classification using principal components based on the correlation matrix. The plots are of the first two principal components analysis scores across the healthy tissues. The variability is explained by each of the scores indicated on the plots. The data have been uploaded into the arrayexpress data base (http://www.ebi.ac.uk) under accession number: E‐MTAB‐1977.
Results
The results in Fig. 1 depict a principal components analysis of both healthy tissues from the four age groups, and a comparison of gene expression between healthy and periodontitis tissues in the adult and aged groups (young and adolescent animals do not exhibit naturally occurring periodontitis). The points denote a composite expression profile for all hypoxia genes for each individual animal. The results demonstrated a grouping of the aged healthy tissues with 26·1% of the variation in hypoxia gene expression related to the age of the animal (Fig. 1a). Similarly, the aged periodontitis group members (five of six) were clustered, with aging and disease accounting for 29·6% of the variation in hypoxia gene expression patterns (Fig. 1b). These results suggested that a profile of these genes related to the hypoxia pathway was indicative of healthy gingival tissues that were differentiated by aging, and that periodontitis significantly alters the environment contributing to the altered gene expression patterns.
Figure 1.

Principal components analysis of hypoxia gene expression in healthy gingival tissues from four age groups (a), or comparing expression in healthy to periodontitis tissues from adult and aged animals (b). Each point denotes the relative position using two principal component factors for each animal. The matching coloured triangles denote the ‘group means’ of the principal component factors.
Figure 2 provides a heat map of gene expression levels in the healthy and periodontitis tissues. The results depict a group of hypoxia pathway genes that were elevated in gingival tissues from healthy aged animals. These are identified by increases in Fold‐Δ in the figure and tended to cluster in transcription factors, angiogenesis/coagulation, metabolic (M) and other response genes. Expanding this analysis to the periodontitis tissues demonstrated an array of genes whose expression was even more elevated in periodontitis, compared with healthy tissues from similar age groups of animals. These genes clustered in the angiogenesis/coagulation, metabolic, cellular differentiation and other reponse gene groups.
Figure 2.
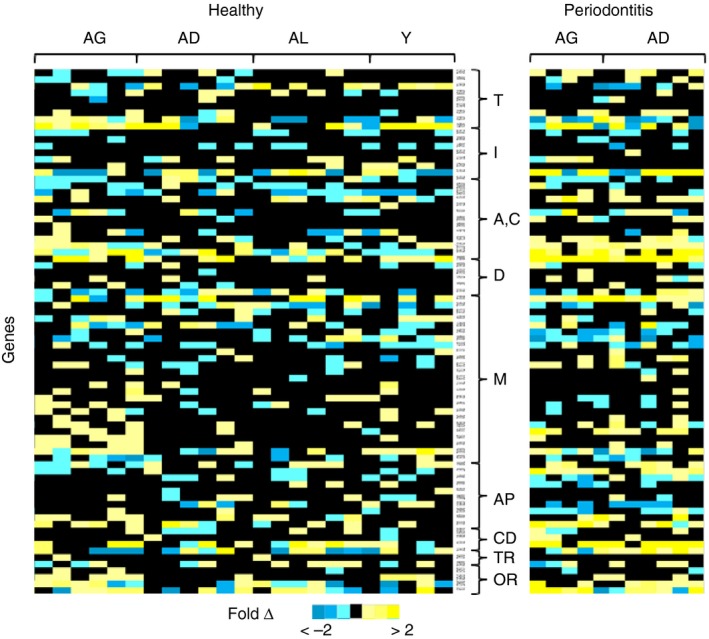
Heatmap of gene expression profiles in gingival tissues from different age groups and periodontitis tissues from adult and aged animals. The fold change in expression for each individual animal is based upon the median level in the adult healthy gingival tissues. Groups of genes are described as: T, transcription/co‐transcription factors; I, hypoxia‐inducible factor interactors; A,C, angiogenesis/coagulation; D, DNA damage and repair; M, metabolism; AP, regulation of apoptosis/regulation of cell proliferation; CD, cellular differentiation; TR, transporters, channels, receptors; and OR, other response genes.
Figure 3 provides Volcano plots of hypoxia gene expression patterns with a visualization of differential gene expression in the healthy aging tissues and how periodontitis alters the gingival tissue profiles. The points signify the level of expression of individual hypoxia pathway genes in aged healthy gingiva compared with levels expressed in the other age groups (Fig. 3a). Those genes significantly altered at least at P < 0·05 are provided with gene IDs. Points with a fold change < 0 are at lower levels in aged tissues and > 0 appear to be up‐regulated. As was predicted from the heat map of individual genes, in healthy aged tissues gene expression was altered. This was represented by some primary hypoxia regulatory factors, as well as those related to angiogenesis and metabolism. Interestingly, those genes most highly expressed in aging periodontitis tissues were generally unique compared with those altered in healthy aging, with substantially greater changes in expression levels versus those noted in healthy gingival tissues (Fig. 3b).
Figure 3.

Volcano plot of gene expression in healthy tissues of aging animals compared with other age groups (a) and comparison of aging periodontitis compared with all healthy age groups (b). The points denote each gene comparing the expression values as statistically different and as a fold difference (Fold Δ) in expression. The red dashed line denotes a P‐value of < 0·05.
Figure 4 focuses on primary biomolecules involved in sensing and response to low oxygen conditions, including the various hypoxia‐inducible transcription factors. The results demonstrate significant elevations in both HIF1A and HIF1B gene expression in the gingival tissues from aged animals, even though the tissues were deemed clinically healthy. Interestingly, HIF3A and HIF4A were also elevated, albeit, they did not reach statistical significance. In contrast, HIF2A (EPAS1) was significantly decreased in the aged gingival tissues. An additional analysis also evaluated up‐regulation of the 88 genes, focusing on gene expression that was at least twofold in aged animals compared with the median value of the adult group of animals. These altered genes included, c‐Fos, MMP9, HNF4A and CP (ceruloplasmin) (see Fig. 3).
Figure 4.

Expression of hypoxia transcription factor genes in aging healthy gingival tissues compared with levels in healthy gingival tissues from all other age groups. Bars denote the means for aged (AG, n = 6) and other age groups (ALL, n = 17). The vertical brackets enclose one SD. The asterisk (*) identifies a significant difference at least at P < 0·05.
Table 2 provides a summary of the genes that were altered and helped to characterize the age and disease status of the gingival tissues. Of the 88 genes examined, approximately 30% of the genes demonstrated some differences with aging. In health, aging gene expression differences or correlations were observed with various HIFs and HIF interactors (e.g. PH4B) (9 of 18), as well as numerous angiogenesis (13 of 20) and metabolic (13 of 28) genes.
Table 2.
Alterations in gene expression in healthy and periodontitis tissues
| Gene ID | Hypoxia pathway function | Aging healtha | Correl health | Correl PD | Health versus PD | Aged PD versus All |
|---|---|---|---|---|---|---|
| ARNT (HIF1B) | HIF/co‐transcription | + | + | |||
| HIF1A | HIF/co‐transcription | + | + | + | + | |
| HIF3A | HIF/co‐transcription | + | ||||
| EPAS1 (HIF2A) | HIF/co‐transcription | + | − | − | ||
| PER1 | HIF/co‐transcription | + | ||||
| HNF4A | HIF/co‐transcription | |||||
| EGLN2 | HIF interactors | − | + | |||
| APEX1 | HIF interactors | − | ||||
| P4HB | HIF interactors | + | + | |||
| ADORA2B | Angiogenesis | + | ||||
| EDN1 | Angiogenesis | + | ||||
| MMP9 | Angiogenesis | + | + | + | + | |
| HMOX1 | Angiogenesis | − | + | |||
| HMOX2 | Angiogenesis | + | + | |||
| ANXA2 | Angiogenesis, coagulation | + | + | |||
| PLAU | Angiogenesis, coagulation | + | + | + | ||
| EGR1 | Angiogenesis, cell proliferation | + | + | |||
| F10 | Coagulation | + | ||||
| LIS1 (PAFAH1) | Coagulation | + | − | + | ||
| SLC16A3 | Transporter | + | ||||
| ENO1 | Metabolism | + | + | |||
| ERO1L | Metabolism | + | ||||
| GYS1 | Metabolism | + | ||||
| LDHA | Metabolism | + | + | + | ||
| PGK1 | Metabolism | + | + | |||
| GPI | Metabolism | + | + | |||
| TPI1 | Metabolism | + | ||||
| GBE1 | Metabolism | + | + | |||
| DDIT4 | Metabolism, apoptosis | + | ||||
| TXNIP | Cell proliferation | + | − | − | ||
| MET | Cell proliferation | + | + | |||
| MXI1 | Cell proliferation | + | ||||
| ODC1 | Cell proliferation | + | ||||
| FOS | Apoptosis, transcription factors | + | + | |||
| MIF | DNA damage/repair, apoptosis | + | ||||
| RUVBL2 | DNA damage/repair | + | + | |||
| LGALS3 | Response genes | + | ||||
| CTSA | Response genes | + | ||||
| DNAJC5 | Response genes | + | ||||
| EIF4EBP1 | Response genes | + | + |
Aging Health denotes expression values in aged tissues compared with all other age groups; Correl Health denotes correlation in gene expression in healthy tissues across all age groups; Correl PD denotes correlation in gene expression in periodontitis tissues with age; Health versus PD denotes comparison of expression values in healthy adult and aged tissues with adult and aged periodontitis tissues; and Aged PD versus All denotes comparison of expression values in aged periodontitis tissues with healthy tissues from non‐aged groups. (+) denotes significant difference (t‐test at P < 0·05 or lower), or significant positive correlation (Spearman Rank Correlation, P < 0·05 or lower); (−) denotes significant negative correlation.
Table 3 provides a summary of the fold‐difference in selected gene expression levels (LDHA, PGAM1, PGK1, ENOL1, EPAS1) determined using the microarray analysis compared with results from a quantitative PCR analysis. The results of this comparison identified similar directions for the altered gene expression, although the magnitude varied between these two independent analyses.
Table 3.
Comparison of gene expression profiles using quantitative PCR and microarray analyses. Values represent fold‐difference compared with Adult Healthy tissue message levels assigned a value of 1·0. Perio denotes periodontitis tissues from aged animals
| Gene ID | Young | Adolescent | Aged | Perio |
|---|---|---|---|---|
| LDHA | ||||
| qPCR | 1·33 | 1·58 | 1·47 | 1·44 |
| GeneChip | 1·19 | 1·44 | 1·24 | 1·02 |
| PGAM1 | ||||
| qPCR | 1·24 | 1·65 | 1·96 | 1·54 |
| GeneChip | 1·05 | 1·38 | 1·78 | 1·06 |
| PGK1 | ||||
| qPCR | 1·93 | 2·23 | 2·03 | 1·94 |
| GeneChip | 1·11 | 1·01 | 1·25 | 1·08 |
| ENOL1 | ||||
| qPCR | 1·56 | 1·38 | 1·60 | 1·71 |
| GeneChip | 1·46 | 1·28 | 1·79 | 1·06 |
| EPAS1 | ||||
| qPCR | 3·38 | 1·54 | 1·69 | 0·81 |
| GeneChip | 1·96 | 1·57 | 0·81 | 1·17 |
Discussion
Periodontal disease manifests as a persistent inflammatory response of the local tissues that has been suggested to reflect changes in the characteristics of the subgingival microbial ecology at diseased sites.27, 28, 29 Additional findings in studies of periodontitis report the increased frequency and severity of disease with aging,30, 31, 32 leading to the consideration that periodontitis is a disease of aging related to altered immune functions that occur with increasing prevalence coincident with decades of life in the general population,33 or potentially a reflection of changing oral environments that select for a microbial ecology with greater pathogenic potential.
Using a non‐human primate oral model to explore the responses of mucosal tissues constantly exposed to a polybacterial challenge of mucosal tissues with responses in these tissues we have shown significant differences in apoptosis pathway gene expression profiles associated with aging, even in healthy gingival tissues.2, 4 This report evaluated gene expression patterns that would reflect changes in the gingival microenvironment in healthy gingival tissues of aging non‐human primates, to address aging effects on this milieu that could presage susceptibility to tissue destruction, a hallmark of periodontitis. We specifically targeted gene expression patterns related to hypoxia, as genes in this pathway could reflect alterations in the subgingival area that could enhance the emergence of pathogenic anaerobic bacteria in the ecology, as well as the recognition that chronically inflamed tissues are increasingly hypoxic.17, 34, 35, 36 The results demonstrated that sets of hypoxia pathway genes are altered in healthy tissues from aged animals. These included all of the major HIF transcription factors [HIF1A, HIF1B (ARNT), HIF2A (EPAS1), HIF3A] that are critical for altering gene expression profiles in cells under hypoxic stress.17, 37 Additionally, at least one member of the PHD (prolyl hydroxylase)–HIF signalling pathway, i.e. P4HB was increased in aged healthy tissues. This gene produces a protein that senses the external hypoxic environment and hydroxlates and activates the HIF molecules enabling nuclear translocation.38, 39, 40 These findings support that clinically healthy gingival tissues from aging individuals demonstrated a response to an altered gingival microenvironment with lower oxygen availability, without gross inflammation. One interpretation of these findings is that the aging gingival tissues, although clinically healthy, may be reflecting a profile of gene expression alterations that are indicative of enhanced tissue susceptibility to bacteria that emerge in more anaerobic pathogenic biofilms. Alternatively, these changes could reflect altered characteristics of the microbial biofilms in the aging animals that continually stress the tissues contributing to increased risk for disease initiation and progression. This is consistent with recent suggestions that proposed periodontopathogens, in particular Porphyromonas gingivalis, have metabolic characteristics that enable the pathogen to propel the development of more pathogenic biofilms in which the commensal bacteria may play a role in inflammation and tissue destruction.41 Additionally, these transcription factors were further increased in periodontitis versus healthy gingival tissues.
Beyond the more direct reflection of cellular responses to hypoxic stress, we identified a substantial array of altered gene expression associated with metabolic changes in the cells, as well as a pattern of genes consistent with increases in angiogenesis activities consistent with downstream signalling of the hypoxia pathway. These processes are coordinated by HIF‐1 in concert with regulation by the Von Hippel–Lindau tumour suppressor protein (pVHL).42, 43 Under hypoxic conditions the HIF‐1α subunit in the cytoplasm is not recognized by the pVHL protein, allowing it to accumulate and form a heterodimer with HIF‐1β enabling translocation to the nucleus. Here the active HIF‐1 interacts with a range of cofactors, including CBP (CREB Binding Protein) and the Pol II (DNA polymerase II) complex to bind to hypoxia‐responsive elements that activate transcription of various target genes.19, 38, 44, 45 In the presence of oxygen, HIF is destroyed in the cytoplasm by ubiquitination through a ubiquitin ligase.42, 46, 47, 48 HIF‐1 is a transcription factor that transactivates genes encoding proteins controlling metabolism, cell proliferation and vascularization [e.g. erythropoietin, lactate dehydrogenase, vascular endothelial growth factor (VEGF)].43, 49, 50, 51 Overexpression of HIF‐1 in vivo results in increased localized inflammation, whereas loss of the HIF‐1 gene decreases immune cell functions, in these tissues related to their physiological anaerobic respiration for energy production.34, 52, 53 Hence, an inability to produce HIF‐1 minimizes emigration to injured tissues and destruction of infectious microbes.54, 55, 56, 57 HIF‐1 also impacts the differentiation of haematopoietic cells into monocytes and macrophages.52
An important function of the HIF‐1 pathway is to promote angiogenesis and so increase oxygenation of the hypoxic tissues. The HIF‐1 transcription factor activates genes to direct migration of mature endothelial cells toward a hypoxic environment through VEGF transcription.58, 59, 60 These endothelial cells ultimately help to form new vessels, enhancing delivery of oxygenated blood to the hypoxic tissues.61 Additionally, HIF‐1 activates other genes related to angiogenesis including GLUT1 (for glucose transporter‐1), which activates glucose transport; and EPO (for erythropoietin), which induces erythropoiesis; activation of transcription of nitric oxide synthase also promotes angiogenesis and vasodilatation. ARNT (HIF1B) is another protein that forms heterodimers with HIF‐1α to activate other angiogenesis genes with some tissue specificity.46 Our findings demonstrated a range of angiogenesis genes that were significantly altered or correlated with aging in healthy gingival tissues. These include, as examples, HMOX2, (haem oxygenase‐2 isozyme) a constitutive enzyme essential in haem catabolism,62 MMP9 (matrix metalloprotease 9), which degrades type IV and V collagens and enhances release of VEGF,63 EDN1 (endothelin 1) as a potent vasoconstrictor produced by vascular endothelial cells,64 PLAU (urokinase‐type plasminogen activator) a serine protease involved in degradation of extracellular matrix and converts plasminogen to plasmin,65 and ANXA2 (annexin 2), which is involved in diverse cellular processes, such as cell motility and fibrinolysis.66 Fewer of the angiogenic genes were further up‐regulated with periodontitis in the aged animals.
HIF‐1 activation can also regulate cellular anaerobic metabolism. In the presence of oxygen, most cells produce ATP via oxidative phosphorylation. However, under hypoxic conditions anaerobic metabolism is engaged to provide cellular energy production. HIF‐1 is a crucial factor in this metabolic shift. Nuclear translocation induces a variety of glycolytic enzymes and glucose transporters [e.g. ENO1 (enolase 1), PGK1 (phosphoglycerate kinase 1), LDHA (lactate dehydrogenase), GYS1 (glycogen synthase)], which support efficient energy production in a hypoxic environment.58, 67 Beyond increasing the expression of these enzymes to generate energy, elevated HIF‐1 can decrease mitochondrial oxygen consumption [e.g. ERO1L (ERO1‐like protein α, oxidoreductase), PDK1 (pyruvate dehydrogenase kinase isozyme 1)] as was noted in samples from the aged animals. Within those genes related to altered metabolism under hypoxic conditions we also observed an increased level of CP (ceruloplasmin) in the aged healthy tissues. This acute‐phase reactant is the major serum ferrioxidase enzyme that converts toxic ferrous iron (Fe2+) to ferric iron (Fe3+) in the presence of oxygen.68 Hence, up‐regulation of this protein enhances protection of cells from toxic iron accumulation.
Altered expression of DDIT4 (DNA‐damage‐inducible transcript 4, REDDI) interfaces the metabolic changes with the apoptosis pathway.69 These changes observed in both healthy aged and periodontitis tissues were also supported by alterations in expression of TXNIP (thioredoxin‐interacting protein); c‐MET (hepatocyte growth factor receptor, proto‐oncogene); c‐FOS (proto‐oncogene); MIF (macrophage migration inhibition factor) that can protect from redox‐stress‐induced apoptosis; and, RUVBL2 (RuvB‐like 2) for DNA repair, as examples.
Within the intracellular hypoxia pathway HIF‐1α can be regulated by extracellular signal‐regulated kinase 2 (i.e. MAPK1) as a biochemical signal transducer in the cells, which phosphorylate HIF‐1α.70 HIF‐1α can associate with heat‐shock protein 90, which appears to enhance HIF‐1α transcriptional activity, increasing both hypoxia‐induced accumulation of VEGF and hypoxia‐dependent angiogenesis.42 More recently, a factor inhibiting HIF‐1α activation, FIH or HIF1AN (hypoxia‐inducible factor 1α inhibitor) has been described that provides another level of regulation of this important transcription factor, although it was not altered in the aging tissues.71
In periodontitis, the local availability of oxygen and consumption by gingival tissues has been suggested to be decreased, reflecting the chronic inflammatory process, and an environment that appears to select for more anaerobic bacteria. The microbial ecology at disease sites also appears to physiologically confer a low oxygen tension in juxtaposition to the periodontal tissue lesion. This oxygen shortage would be predicted to lead to stabilization of HIF‐1α transcription factor and to control specific downstream genes that modulate varied cellular functions that influence the process of periodontitis. In this regard, HIF‐1α has been shown to be up‐regulated in tissues from periodontal pockets.72 Ng et al.73 demonstrated HIF1A‐containing nuclei in both epithelial and endothelial cells in gingival tissues accompanied by clear increases in this gene expression in fibroblasts and leucocytes of periodontitis lesions. They also identified increased levels of HIF‐1α, VEGF and tumour necrosis factor‐α proteins in the diseased tissues. Extending these observations are an array of in vitro studies that demonstrate the ability of P. gingivalis lipopolysaccharide under hypoxic conditions to alter the responses of periodontal ligament cells related to levels of VEGF, interleukin‐1β (IL‐1β), and MMP‐1. These alterations occurred through effects on NF‐κB and HIF‐1α activation in the cells.74 Hypoxia also augmented P. gingivalis lipopolysaccharide induction of tumour necrosis factor‐α, IL‐1β and IL‐6 expression by periodontal ligament cells,75 all markers of tissue destructive inflammation and apparently functioning through TLR4 engagement.76 Interestingly, HIF‐1α is up‐regulated by IL‐1β.77 Hypoxia was shown to regulate periodontal ligament stem cell functions by altering their osteogenic potential and mineralization capacity through alteration of ERK1/2 and p38 kinase activities78 and during culture of peripheral blood mononuclear cells and osteoblasts it lead to the formation of functional osteoclasts following up‐regulation of HIF, VEGF and RANKL.79 Similarly, HIF‐2α, a key regulator of cartilage destruction, was up‐regulated in periodontal ligament cells from diseased tissues. In vitro, HIF‐2α appeared to contribute to nicotine and lipopolysaccharide induction of inflammatory mediators through multiple transcription factors.80 We also observed altered ceruloplasmin mRNA levels. As ceruloplasmin is induced by hypoxia and inflammation, Iwata et al.81 noted that polymorphonuclear cells from patients with aggressive periodontitis were primed for production of ceruloplasmin that would enhance hypoxia‐mediated O2 generation and increased oxidative stress.
Hence, an interpretation of the results of this study suggests that gingival tissue characteristics in aging are significantly different even in health. Hypoxia gene pathways appeared to be up‐regulated in healthy gingival tissues of aging non‐human primates. Major characteristics of the gene changes in health were related to angiogenesis and metabolism responses to hypoxia, in addition to the primary hypoxia transcription genes. Generally, hypoxia pathway gene expression profiles in both adult and aged periodontitis tissues were substantively different from those that were noted to change in healthy tissues as a reflection of an aging process. These molecular alterations may presage mucosal tissues in the aging individual to an enhanced risk of dysregulated responses, breakdown in homeostasis, and subsequent destruction from the chronic periodontal infections. The changes in gingival tissues with aging may signal a predisposition to a subgingival environment that can enrich for anaerobic species. It remains unclear whether a hypoxic/anaerobic tissue environment, particularly with inflammation, selects for characteristic pathogenic biofilms, or members of these biofilms ‘trigger’ these hypoxic changes.
Disclosures
The authors claim no conflict of interest regarding the conduct of the study or any results presented in the report.
Acknowledgements
This work was supported by National Institute of Health grants P20GM103538 and UL1TR000117. We express our gratitude to the Caribbean Primate Research Center supported by grant P40RR03640, and the Microarray Core of University Kentucky for their invaluable technical assistance. We thank M. Kirakodu for data management support.
References
- 1. Ebersole JL, Cappelli D, Mathys EC, Steffen MJ, Singer RE, Montgomery M et al Periodontitis in humans and non‐human primates: oral‐systemic linkage inducing acute phase proteins. Ann Periodontol 2002; 7:102–11. [DOI] [PubMed] [Google Scholar]
- 2. Gonzalez O, Novak MJ, Orraca L, Martinez‐Gonzalez J, Stromberg AJ, Ebersole JL. Apopotosis gene expression in healthy and oral mucosal tissues with aging. Apoptosis 2013; 18:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gonzalez O, Tobia C, Ebersole J, Novak MJ. Caloric restriction and chronic inflammatory diseases. Oral Dis 2012; 18:16–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gonzalez OA, Stromberg AJ, Huggins PM, Gonzalez‐Martinez J, Novak MJ, Ebersole JL. Apoptotic genes are differentially expressed in aged gingival tissue. J Dent Res 2011; 90:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kleinberg I, Wolff MS. Gingival crevicular fluid monitoring in the treatment of gingivitis‐periodontitis. Compendium 1987; 8:624–7, 30–1, 33–5. [PubMed] [Google Scholar]
- 6. Gonzalez OA, Novak MJ, Kirakodu S, Orraca L, Chen KC, Stromberg AJ et al Comparative analysis of gingival tissue antigen presentation pathways in aging and periodontitis. J Clin Periodontal 2014; 41:327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gonzalez OA, Nagarajan R, Novak MJ, Orraca L, Gonzalez‐Martinez JA, Kirakodu SS et al Immune system transcriptome in gingival tissues of young nonhuman primates. J Periodontal Res 2015; 51:152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z et al Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One 2011; 6:e22788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paster BJ, Dewhirst FE. Molecular microbial diagnosis. Periodontol 2000 2009; 51:38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kolenbrander PE, Palmer RJ Jr, Rickard AH, Jakubovics NS, Chalmers NI, Diaz PI. Bacterial interactions and successions during plaque development. Periodontol 2000 2006; 42:47–79. [DOI] [PubMed] [Google Scholar]
- 11. Periasamy S, Kolenbrander PE. Mutualistic biofilm communities develop with Porphyromonas gingivalis and initial, early, and late colonizers of enamel. J Bacteriol 2009; 191:6804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Battino M, Bullon P, Wilson M, Newman H. Oxidative injury and inflammatory periodontal diseases: the challenge of anti‐oxidants to free radicals and reactive oxygen species. Crit Rev Oral Biol Med 1999; 10:458–76. [DOI] [PubMed] [Google Scholar]
- 13. Torresyap G, Haffajee AD, Uzel NG, Socransky SS. Relationship between periodontal pocket sulfide levels and subgingival species. J Clin Periodontol 2003; 30:1003–10. [DOI] [PubMed] [Google Scholar]
- 14. Nogueira‐Filho GR, Peruzzo D, Sallum AW. Relationship between the formation of volatile sulfur compounds (VSC) and the severity of the periodontal disease: a pilot study. J Breath Res 2008; 2:017005. [DOI] [PubMed] [Google Scholar]
- 15. Prabhakar NR. Sensing hypoxia: physiology, genetics and epigenetics. J Physiol 2013; 591:2245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sica A, Melillo G, Varesio L. Hypoxia: a double‐edged sword of immunity. J Mol Med (Berl) 2011; 89:657–65. [DOI] [PubMed] [Google Scholar]
- 17. Shay JE, Celeste Simon M. Hypoxia‐inducible factors: crosstalk between inflammation and metabolism. Semin Cell Dev Biol 2012; 23:389–94. [DOI] [PubMed] [Google Scholar]
- 18. Gao L, Chen Q, Zhou X, Fan L. The role of hypoxia‐inducible factor 1 in atherosclerosis. J Clin Pathol 2012; 65:872–6. [DOI] [PubMed] [Google Scholar]
- 19. Semenza GL. Hypoxia‐inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 2011; 1813:1263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Semenza GL. Regulation of metabolism by hypoxia‐inducible factor 1. Cold Spring Harb Symp Quant Biol 2011; 76:347–53. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki J, Ogawa M, Muto S, Itai A, Isobe M, Hirata Y et al Novel IκB kinase inhibitors for treatment of nuclear factor‐κB‐related diseases. Expert Opin Investig Drugs 2011; 20:395–405. [DOI] [PubMed] [Google Scholar]
- 22. Nanduri J, Yuan G, Kumar GK, Semenza GL, Prabhakar NR. Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol 2008; 164:277–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ebersole JL, Steffen MJ, Gonzalez‐Martinez J, Novak MJ. Effects of age and oral disease on systemic inflammatory and immune parameters in nonhuman primates. Clin Vaccine Immunol 2008; 15:1067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meka A, Bakthavatchalu V, Sathishkumar S, Lopez MC, Verma RK, Wallet SM et al Porphyromonas gingivalis infection‐induced tissue and bone transcriptional profiles. Mol Oral Microbiol 2010; 25:61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirakodu SS, Govindaswami M, Novak MJ, Ebersole JL, Novak KF. Optimizing qPCR for the quantification of periodontal pathogens in a complex plaque biofilm. Open Dent J 2008; 2:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li W. Volcano plots in analyzing differential expressions with mRNA microarrays. J Bioinform Comput Biol 2012; 10:1231003. [DOI] [PubMed] [Google Scholar]
- 27. Ji S, Choi Y. Innate immune response to oral bacteria and the immune evasive characteristics of periodontal pathogens. J Periodontal Implant Sci 2013; 43:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discov 2012; 11:234–50. [DOI] [PubMed] [Google Scholar]
- 29. Kinane DF, Bartold PM. Clinical relevance of the host responses of periodontitis. Periodontol 2000 2007; 43:278–93. [DOI] [PubMed] [Google Scholar]
- 30. Hajishengallis G. Too old to fight? Aging and its toll on innate immunity. Mol Oral Microbiol 2010; 25:25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huttner EA, Machado DC, de Oliveira RB, Antunes AG, Hebling E. Effects of human aging on periodontal tissues. Spec Care Dentist 2009; 29:149–55. [DOI] [PubMed] [Google Scholar]
- 32. Gonsalves WC, Wrightson AS, Henry RG. Common oral conditions in older persons. Am Fam Physician 2008; 78:845–52. [PubMed] [Google Scholar]
- 33. Chung HY, Lee EK, Choi YJ, Kim JM, Kim DH, Zou Y et al Molecular inflammation as an underlying mechanism of the aging process and age‐related diseases. J Dent Res 2011; 90:830–40. [DOI] [PubMed] [Google Scholar]
- 34. Thompson AA, Binham J, Plant T, Whyte MK, Walmsley SR. Hypoxia, the HIF pathway and neutrophilic inflammatory responses. Biol Chem 2013; 394:471–7. [DOI] [PubMed] [Google Scholar]
- 35. Schaible B, McClean S, Selfridge A, Broquet A, Asehnoune K, Taylor CT et al Hypoxia modulates infection of epithelial cells by Pseudomonas aeruginosa . PLoS One 2013; 8:e56491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dietz I, Jerchel S, Szaszak M, Shima K, Rupp J. When oxygen runs short: the microenvironment drives host–pathogen interactions. Microbes Infect 2012; 14:311–6. [DOI] [PubMed] [Google Scholar]
- 37. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med 2011; 364:656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myllyharju J. Prolyl 4‐hydroxylases, master regulators of the hypoxia response. Acta Physiol (Oxf) 2013; 208:148–65. [DOI] [PubMed] [Google Scholar]
- 39. Sen Banerjee S, Thirunavukkarasu M, Tipu Rishi M, Sanchez JA, Maulik N, Maulik G. HIF‐prolyl hydroxylases and cardiovascular diseases. Toxicol Mech Methods 2012; 22:347–58. [DOI] [PubMed] [Google Scholar]
- 40. Myllyharju J, Koivunen P. Hypoxia‐inducible factor prolyl 4‐hydroxylases: common and specific roles. Biol Chem 2013; 394:435–48. [DOI] [PubMed] [Google Scholar]
- 41. Hajishengallis G, Darveau RP, Curtis MA. The keystone‐pathogen hypothesis. Nat Rev Microbiol 2012; 10:717–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Semenza GL. Hypoxia‐inducible factor 1 (HIF‐1) pathway. Sci STKE 2007; 2007:cm8. [DOI] [PubMed] [Google Scholar]
- 43. Ke Q, Costa M. Hypoxia‐inducible factor‐1 (HIF‐1). Mol Pharmacol 2006; 70:1469–80. [DOI] [PubMed] [Google Scholar]
- 44. Werth N, Beerlage C, Rosenberger C, Yazdi AS, Edelmann M, Amr A et al Activation of hypoxia inducible factor 1 is a general phenomenon in infections with human pathogens. PLoS One 2010; 5:e11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Loboda A, Jozkowicz A, Dulak J. HIF‐1 and HIF‐2 transcription factors–similar but not identical. Mol Cells 2010; 29:435–42. [DOI] [PubMed] [Google Scholar]
- 46. Semenza GL. Hypoxia. Cross talk between oxygen sensing and the cell cycle machinery. Am J Physiol Cell Physiol 2011; 301:C550–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thompson SJ, Loftus LT, Ashley MD, Meller R. Ubiquitin‐proteasome system as a modulator of cell fate. Curr Opin Pharmacol 2008; 8:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hellwig‐Burgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: hypoxia‐inducible factor‐1 (HIF‐1): a novel transcription factor in immune reactions. J Interferon Cytokine Res 2005; 25:297–310. [DOI] [PubMed] [Google Scholar]
- 49. Giatromanolaki A, Sivridis E, Simopoulos C, Polychronidis A, Gatter KC, Harris AL et al Hypoxia inducible factors 1α and 2α are associated with VEGF expression and angiogenesis in gallbladder carcinomas. J Surg Oncol 2006; 94:242–7. [DOI] [PubMed] [Google Scholar]
- 50. Giatromanolaki A, Sivridis E, Fiska A, Koukourakis MI. Hypoxia‐inducible factor‐2 α (HIF‐2α) induces angiogenesis in breast carcinomas. Appl Immunohistochem Mol Morphol 2006; 14:78–82. [DOI] [PubMed] [Google Scholar]
- 51. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL et al HIF‐1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest 2005; 115:1806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Riboldi E, Porta C, Morlacchi S, Viola A, Mantovani A, Sica A. Hypoxia‐mediated regulation of macrophage functions in pathophysiology. Int Immunol 2013; 25:67–75. [DOI] [PubMed] [Google Scholar]
- 53. Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE et al Fundamental role for HIF‐1α in constitutive expression of human β defensin‐1. Mucosal Immunol 2013; 6:1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V et al NF‐κB links innate immunity to the hypoxic response through transcriptional regulation of HIF‐1α . Nature 2008; 453:807–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peyssonnaux C, Boutin AT, Zinkernagel AS, Datta V, Nizet V, Johnson RS. Critical role of HIF‐1α in keratinocyte defense against bacterial infection. J Invest Dermatol 2008; 128:1964–8. [DOI] [PubMed] [Google Scholar]
- 56. Hartmann H, Eltzschig HK, Wurz H, Hantke K, Rakin A, Yazdi AS et al Hypoxia‐independent activation of HIF‐1 by Enterobacteriaceae and their siderophores. Gastroenterology 2008; 134:756–67. [DOI] [PubMed] [Google Scholar]
- 57. Kuhlicke J, Frick JS, Morote‐Garcia JC, Rosenberger P, Eltzschig HK. Hypoxia inducible factor (HIF)‐1 coordinates induction of toll‐like receptors TLR2 and TLR6 during hypoxia. PLoS One 2007; 2:e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M et al Role of HIF‐1α in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998; 394:485–90. [DOI] [PubMed] [Google Scholar]
- 59. Dery MA, Michaud MD, Richard DE. Hypoxia‐inducible factor 1: regulation by hypoxic and non‐hypoxic activators. Int J Biochem Cell Biol 2005; 37:535–40. [DOI] [PubMed] [Google Scholar]
- 60. Hewitson KS, Schofield CJ. The HIF pathway as a therapeutic target. Drug Discov Today 2004; 9:704–11. [DOI] [PubMed] [Google Scholar]
- 61. Richard DE, Berra E, Pouyssegur J. Nonhypoxic pathway mediates the induction of hypoxia‐inducible factor 1α in vascular smooth muscle cells. J Biol Chem 2000; 275:26765–71. [DOI] [PubMed] [Google Scholar]
- 62. Mazurek B, Amarjargal N, Haupt H, Fuchs J, Olze H, Machulik A et al Expression of genes implicated in oxidative stress in the cochlea of newborn rats. Hear Res 2011; 277:54–60. [DOI] [PubMed] [Google Scholar]
- 63. Tartour E, Pere H, Maillere B, Terme M, Merillon N, Taieb J et al Angiogenesis and immunity: a bidirectional link potentially relevant for the monitoring of antiangiogenic therapy and the development of novel therapeutic combination with immunotherapy. Cancer Metastasis Rev 2011; 30:83–95. [DOI] [PubMed] [Google Scholar]
- 64. Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin‐1 gene regulation. FASEB J 2011; 25:16–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jankun J, Al‐Senaidy A, Skrzypczak‐Jankun E. Can inactivators of plasminogen activator inhibitor alleviate the burden of obesity and diabetes? (Review). Int J Mol Med 2012; 29:3–11. [DOI] [PubMed] [Google Scholar]
- 66. Hedhli N, Falcone DJ, Huang B, Cesarman‐Maus G, Kraemer R, Zhai H et al The annexin A2/S100A10 system in health and disease: emerging paradigms. J Biomed Biotechnol 2012; 2012:406273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vaupel P. The role of hypoxia‐induced factors in tumor progression. Oncologist 2004; 9(Suppl 5):10–7. [DOI] [PubMed] [Google Scholar]
- 68. Chepelev NL, Willmore WG. Regulation of iron pathways in response to hypoxia. Free Radic Biol Med 2011; 50:645–66. [DOI] [PubMed] [Google Scholar]
- 69. Gleason JE, Corrigan DJ, Cox JE, Reddi AR, McGinnis LA, Culotta VC. Analysis of hypoxia and hypoxia‐like states through metabolite profiling. PLoS One 2011; 6:e24741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seta KA, Spicer Z, Yuan Y, Lu G, Millhorn DE. Responding to hypoxia: lessons from a model cell line. Sci STKE 2002; 2002:re11. [DOI] [PubMed] [Google Scholar]
- 71. Coleman ML, Ratcliffe PJ. Signalling cross talk of the HIF system: involvement of the FIH protein. Curr Pharm Des 2009; 15:3904–7. [DOI] [PubMed] [Google Scholar]
- 72. Kim YS, Shin SI, Kang KL, Chung JH, Herr Y, Bae WJ et al Nicotine and lipopolysaccharide stimulate the production of MMPs and prostaglandin E2 by hypoxia‐inducible factor‐1α up‐regulation in human periodontal ligament cells. J Periodontal Res 2012; 47:719–28. [DOI] [PubMed] [Google Scholar]
- 73. Ng KT, Li JP, Ng KM, Tipoe GL, Leung WK, Fung ML. Expression of hypoxia‐inducible factor‐1α in human periodontal tissue. J Periodontol 2011; 82:136–41. [DOI] [PubMed] [Google Scholar]
- 74. Golz L, Memmert S, Rath‐Deschner B, Jager A, Appel T, Baumgarten G et al Hypoxia and P. gingivalis synergistically induce HIF‐1 and NF‐κB activation in PDL cells and periodontal diseases. Mediators Inflamm 2015; 2015:438085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jian C, Li C, Ren Y, He Y, Li Y, Feng X et al Hypoxia augments lipopolysaccharide‐induced cytokine expression in periodontal ligament cells. Inflammation 2014; 37:1413–23. [DOI] [PubMed] [Google Scholar]
- 76. Li JP, Li FY, Xu A, Cheng B, Tsao SW, Fung ML et al Lipopolysaccharide and hypoxia‐induced HIF‐1 activation in human gingival fibroblasts. J Periodontol 2012; 83:816–24. [DOI] [PubMed] [Google Scholar]
- 77. Thornton RD, Lane P, Borghaei RC, Pease EA, Caro J, Mochan E. Interleukin 1 induces hypoxia‐inducible factor 1 in human gingival and synovial fibroblasts. Biochem J 2000; 350(Pt 1):307–12. [PMC free article] [PubMed] [Google Scholar]
- 78. Wu Y, Yang Y, Yang P, Gu Y, Zhao Z, Tan L et al The osteogenic differentiation of PDLSCs is mediated through MEK/ERK and p38 MAPK signalling under hypoxia. Arch Oral Biol 2013; 58:1357–68. [DOI] [PubMed] [Google Scholar]
- 79. Dandajena TC, Ihnat MA, Disch B, Thorpe J, Currier GF. Hypoxia triggers a HIF‐mediated differentiation of peripheral blood mononuclear cells into osteoclasts. Orthod Craniofac Res 2012; 15:1–9. [DOI] [PubMed] [Google Scholar]
- 80. Bae WJ, Shin MR, Kang SK, Jun Z, Kim JY, Lee SC et al HIF‐2 inhibition supresses inflammatory responses and osteoclastic differentiation in human periodontal ligament cells. J Cell Biochem 2015; 116:1241–55. [DOI] [PubMed] [Google Scholar]
- 81. Iwata T, Kantarci A, Yagi M, Jackson T, Hasturk H, Kurihara H et al Ceruloplasmin induces polymorphonuclear leukocyte priming in localized aggressive periodontitis. J Periodontol 2009; 80:1300–6. [DOI] [PMC free article] [PubMed] [Google Scholar]