

Summary
Recent studies report that loss and dysfunction of mitochondria and peroxisomes contribute to the myelin and axonal damage in multiple sclerosis (MS). In this study, we investigated the efficacy of a combination of lovastatin and AMP‐activated protein kinase (AMPK) activator (AICAR) on the loss and dysfunction of mitochondria and peroxisomes and myelin and axonal damage in spinal cords, relative to the clinical disease symptoms, using a mouse model of experimental autoimmune encephalomyelitis (EAE, a model for MS). We observed that lovastatin and AICAR treatments individually provided partial protection of mitochondria/peroxisomes and myelin/axons, and therefore partial attenuation of clinical disease in EAE mice. However, treatment of EAE mice with the lovastatin and AICAR combination provided greater protection of mitochondria/peroxisomes and myelin/axons, and greater improvement in clinical disease compared with individual drug treatments. In spinal cords of EAE mice, lovastatin‐mediated inhibition of RhoA and AICAR‐mediated activation of AMPK cooperatively enhanced the expression of the transcription factors and regulators (e.g. PPAR α/β, SIRT‐1, NRF‐1, and TFAM) required for biogenesis and the functions of mitochondria (e.g. OXPHOS, MnSOD) and peroxisomes (e.g. PMP70 and catalase). In summary, these studies document that oral medication with a combination of lovastatin and AICAR, which are individually known to have immunomodulatory effects, provides potent protection and repair of inflammation‐induced loss and dysfunction of mitochondria and peroxisomes as well as myelin and axonal abnormalities in EAE. As statins are known to provide protection in progressive MS (Phase II study), these studies support that supplementation statin treatment with an AMPK activator may provide greater efficacy against MS.
Keywords: autoimmunity, experimental autoimmune encephalomyelitis/multiple sclerosis, neurodegeneration, neuroinflammation
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
AMP‐activated protein kinase
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- HMG‐CoA
3‐hydroxy‐3‐methylglutaryl‐CoA
- IFN
interferon
- IL‐4
interleukin‐4
- i.p.
intraperitoneal
- LOVA
lovastatin
- MBP
myelin basic protein
- MnSOD
manganese superoxide dismutase
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NRF‐1
nuclear respiratory factor 1
- OXPHOS
oxidative phosphorylation
- PGC‐1α
peroxisome proliferator‐activated receptor‐γ coactivator‐1α
- PLP
proteolipid protein
- PMP70
70 000 MW peroxisomal membrane protein
- PPAR
peroxisome proliferator‐activated receptor
- PTX
pertussis toxin
- ROCK
Rho‐associated kinase
- ROS
reactive oxygen species
- SIRT‐1
Sirtuin 1
- TFAM
mitochondrial transcription factor A
- Th1
T helper type 1
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system (CNS). Although the aetiology of MS and its pathogenesis are not fully understood, the disease process starts with differentiation and expansion of myelin‐specific autoreactive lymphocytes, their infiltration into the CNS, and chronic encephalitogenic inflammation leading to damage to myelin, oligodendrocytes and axons.1 Currently, treatment of MS is based on immunomodulatory drugs, such as interferon‐β (IFN‐β), glatiramer acetate, mitoxantrone, natalizumab and fingolimod.2 However, these drugs provide limited efficacies against the CNS disease of MS.2 This underscores the need for an in‐depth understanding of the CNS disease mechanisms as well as identification of new drugs targeting these CNS disease processes in MS.
There is a growing body of evidence that mitochondrial dysfunction in oligodendrocytes and neurons contributes to the CNS disease mechanisms in MS as well as experimental autoimmune encephalomyelitis (EAE), an animal model for MS.3, 4, 5, 6 Mitochondria play a critical role in cellular homeostasis in energy and redox states. Therefore, dysfunction or loss of mitochondria could intrinsically be linked to the degeneration of oligodendrocytes/myelin and neurons/axons. Reported abnormalities in mitochondria in autopsy brains from patients with MS treated with immunomodulatory drugs indicate that these drugs are not effective against CNS pathology in MS.7 In addition to mitochondria, we previously also reported functional and structural loss of peroxisomes in the CNS of EAE animals.8 Peroxisomes are unique subcellular organelles that play a critical role in several key metabolic pathways, such as lipid (e.g. fatty acids and plasmalogens) and reactive oxygen species (ROS) metabolism.9 The importance of peroxisomes in the structural and functional integrity of the CNS was highlighted by the observation of extensive demyelination in disease conditions of a single gene mutation in peroxisomal protein (e.g. Pex5 or ALDP).10 Moreover, the observed loss and dysfunction of mitochondria in the CNS of peroxisomal diseases and in cells silenced for single peroxisomal protein also point to the importance of the functional relationship between mitochondria and peroxisomes in demyelinating disease.11, 12, 13, 14, 15, 16, 17 Indeed, peroxisomes and mitochondria share many aspects of the molecular machinery for their biogenesis [peroxisome proliferator‐activated receptor α/γ (PPARα/γ) and peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α)],18, 19, 20, 21 dynamics (fusion and fission)22, 23, 24 and autophagy.25, 26 In addition, peroxisomes and mitochondria have a significant degree of metabolic and redox (ROS) cross‐talk between them and this relationship was referred to as the ‘big brother and little sister’ relationship.27 However, the pathological interaction between mitochondrial and peroxisomal dysfunctions and their contribution to the CNS disease of MS and EAE are largely unknown.
Our laboratory was the first to report the anti‐inflammatory and immunomodulatory activities of statins, 3‐hydroxy‐3‐methylglutaryl‐CoA (HMG‐CoA) reductase inhibitors known as cholesterol‐lowering drugs, as well as their potential efficacy in patients with relapsing–remitting MS.28, 29, 30, 31, 32 These pleiotropic effects of statins are mediated by the inhibition of small GTPase signalling pathway (e.g. hRas and RhoA) via inhibiting its isoprenylation‐mediated membrane targeting, inhibition of nuclear factor‐κB‐mediated inflammatory gene expressions, induction of PPARα/γ‐mediated peroxisomal function and oligodendrocyte (OL) differentiation, and induction of neurotrophic factor release in astrocytes under inflammatory and autoimmune disease conditions.8, 28, 33, 34, 35, 36 Recently, a placebo‐controlled double‐blind study of 140 patients with progressive MS37 showing 50% reduction in brain atrophy over 2 years also supports the potential of statin as a possible candidate drug for MS. In these studies, however, statin therapy showed limited efficacy on the CNS disease,38 indicating a need for add‐on therapy to improve the efficacy of statin against the CNS disease of EAE/MS.
We have studied the regulation and role of AMP‐activated protein kinase (AMPK), an energy sensing metabolic switch, in EAE disease pathologies and reported for the first time that loss of AMPK in immune cells in EAE disease exacerbates disease severity.39 We also reported that AMPK activators, such as metformin and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) alleviate the disease severity of EAE by modulating pro‐inflammatory and immune responses and/or by elevating glial expression of neurotrophic factors.40, 41, 42, 43, 44 The combination of AMPK activators and statin treatment was even more effective, not only on immune and pro‐inflammatory modulations, but also on neurorepair and neuroregeneration.43, 45 However, the mechanistic basis for cooperative efficacy of statin and AMPK activators in EAE disease, particularly on peroxisomal and mitochondrial homeostasis and hence neurorepair and neuroregeneration, is not well understood. AMPK is known to play an essential role in PGC‐1α‐mediated peroxisomal and mitochondrial biogenesis.46 On the other hand, statin improves peroxisomal functions through PPARα/γ pathways by inhibiting RhoA/Rho‐associated kinase (ROCK).33, 34, 35 Statin is also known to increase sirtuin 1 (SIRT1) expression and hence activation of PGC‐1α.47, 48 Therefore, statin‐mediated mechanisms (RhoA/PPARα/γ and SIRT1) and AMPK‐mediated mechanisms (PGC‐1α) are expected to protect peroxisomal/mitochondrial function and hence efficient neuroprotection and neurorepair under EAE/MS conditions, in addition to their anti‐inflammatory and immunomodulatory activities. Based on the above rationale, the present study was undertaken to evaluate the efficacy of compounds targeting PPAR (statin) and AMPK activator (AICAR) on mitochondrial and peroxisomal integrities and functions in CNS of the EAE model.
Consistent with our previous study,40, 49 the treatment of EAE animals with statin (lovastatin; LOVA) or AICAR individually attenuated against clinical disease, and hence disability and demyelination. Accordingly, LOVA‐mediated PPARα/γ activation via inhibition of the Rho/ROCK signalling pathway or AICAR‐mediated AMPK activation partly protected against the EAE disease‐induced loss of peroxisomal and mitochondrial proteins and hence the CNS disease of EAE. However, greater protection of mitochondria and peroxisomes and hence clinical efficacy was observed in EAE animals treated with a combination of LOVA and AICAR. These studies provide evidence, for the first time, that a combination of LOVA and AICAR provides neuroprotection against the inflammatory mediator‐induced dysfunction in mitochondria and peroxisomes, and hence neuroprotection, and potentially promote neurorepair in EAE. As oral simvastatin is known to provide protection against MS,32, 37 these preclinical studies provide evidence that supplementation of statin therapy with AMPK activator is likely to provide greater efficacy for MS than statin alone.
Materials and methods
EAE induction
Female C57BL/6J mice, 8–12 weeks of age and weighing 18–22 g were purchased from Jackson Laboratory (Bar Harbor, ME) (RRID: IMSR_JAX:000664). They were provided with food and water ad libitum and kept in individually ventilated cages in the specific pathogen‐free animal care facility of the Medical University of South Carolina throughout the study. Temperature (21.1–22.2°), humidity (45–55%), and a 12/12‐hr light/dark cycle were controlled. All procedures were conducted in accordance with accepted standards of animal care as approved by the Institutional Animal Care and Use Committee in the Medical University of South Carolina (Approved number: AR#1644). EAE was induced as described previously.41 Briefly, mice were immunized subcutaneously in the flank regions with myelin oligodendrocyte glycoprotein 35–55 (MOG35‐55 peptide; 200 μg; Peptide International, Louisville, KY) emulsified (1 : 1) in 100 μl complete Freund's adjuvant on day 0 and day 7. Additionally, 200 ng of pertussis toxin (PTX; Sigma‐Aldrich, St Louis, MO) was given on day 0 and day 2 by intraperitoneal (i.p.) injection. Pertussis toxin was used as per the standardized protocol reported by us and other investigators for the induction of EAE.41 Similarly, the healthy control group received subcutaneous injection of complete Freund's adjuvant emulsion and PTX. Clinical signs of EAE were scored in the animal facility between 2 and 4 pm daily by examiners blinded to experimental treatments using the following scale: 0 = no clinical signs of disease; 1 = limp tail or waddling gait with tail tonicity; 2 = waddling gait with limp tail (ataxia); 2·5 = ataxia with partial limb paralysis; 3 = full paralysis of one limb; 3·5 = full paralysis of one limb with partial paralysis of second limb; 4 = full paralysis of two limbs; 4·5 = moribund stage; 5 = death. Starting the day of disease onset (with clinical score between 1 and 2) or peak of disease (with clinical score over 3), the animals were randomly grouped into 8–12 mice in each group in a blinded manner and given daily treatment with 100 μl of vehicle (PBS with 5% ethanol), AICAR (100 mg/kg body weight/100 μl vehicle/ per day, i.p.), LOVA (1 mg/kg body weight/100 μl vehicle/day, i.p.), or a combination of the same doses of AICAR and LOVA (100 mg/kg and 10 mg/kg body weight each/100 μl vehicle/per day, i.p. , respectively). Figure 1 depicts a time‐line diagram for EAE induction and drug treatments. The group size was determined by power analysis based on our previous data.41 Absolute ethanol was used for dissolving LOVA and AICAR and further diluted with PBS to produce a concentration of 5%. The drug treatments were continued till the termination of the study (day 30 post immunization). EAE animals without drug treatment received PBS. Likewise, healthy controls received vehicle. At the conclusion of the study, animals were anaesthetized with ketamine/xylazine (90/10 mg/kg, i.p.) and perfused with saline. The animals were further fixed with 4% paraformaldehyde for the morphological studies (n = 4) or dissected to harvest the fresh tissues for biochemical studies (n = 4).
Figure 1.
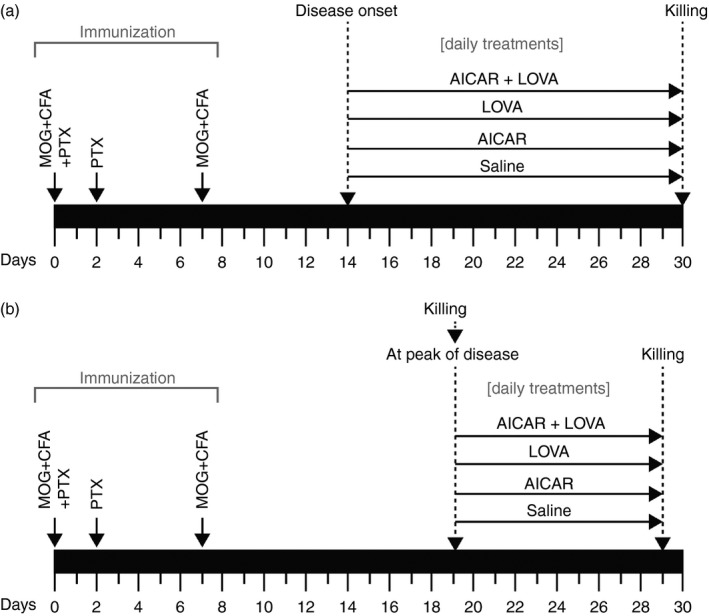
Time‐line diagram for experimental autoimmune encephalomyelitis (EAE) induction and drug treatments: Two sets of animal studies were performed: (a) To investigate the effects of 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR), lovastatin (LOVA), and their combination treatments, started after disease onset, on clinical and central nervous system (CNS) diseases of EAE. (b) To investigate the effects of AICAR, LOVA, and their combination treatments, started after peak of the disease, on clinical and CNS diseases of EAE.
Assay for mitochondrial complex and ATP levels
Mitochondria were isolated from the whole spinal cord using a mitochondria isolation kit as per the manufacturer's instructions (Abcam, Cambridge, MA). Mitochondrial complex I was assayed by using the Complex I microplate assay kit (Abcam). Briefly, the capture antibodies specific for complex I ATP were pre‐coated in the microplate wells. Mitochondria, isolated from spinal cord samples, were added to the pre‐coated microplate wells. Complex I activity was determined by following the oxidation of NADH to NAD+ and the simultaneous reduction of the provided dyes increased absorbance at optical density at 450 nm. Citrate synthase activity was measured by a citrate synthase assay kit (Sigma‐Aldrich) using spinal cord mitochondrial proteins. ATP in the spinal cord homogenate was measured by ATP assay kit (BioVision, Milpitas, CA).
Assay for ROCK activity
Rho‐associated kinase activity was measured in whole spinal cord lysates by using a ROCK assay kit according to the product manual (Millipore, Billerica, MA). Briefly, spinal cord tissue lysates were incubated in microplates pre‐coated with myosin phosphatase target subunit 1 (MYPT1) for 30 min at 30° and then the plates were washed and incubated with anti‐phosphoMYPT1 (Thr196) antibody for 1 hr at room temperature. After incubation, the plates were washed and incubated with horseradish peroxidase‐conjugated goat anti‐rabbit IgG secondary antibody for 1 hr. The amount of phosphorylated substrate is measured by addition of the chromogenic substrate tetramethylbenzidine. The absorbance signal at 450 nm reflects the relative amount of ROCK activity.
Western blot analysis
For Western blot analysis, whole spinal cords were lysed in RIPA buffer containing protease and phosphatase inhibitors and the lysates were spun at 15 000 g for 30 min at 4°. The resulting supernatant was analysed for protein content by Lowry assay using a DC protein assay kit (Bio‐Rad, Hercules, CA). For Western blot analysis, proteins were resolved by SDS–polyacrylamide gel (4–20%) electrophoresis and transferred to a nitrocellulose membrane (Amersham LifeSciences, Arlington Heights, IL). The membrane was blocked with 5% non‐fat milk in Tris‐buffered saline containing 0·05% Tween 20 (TBST) for 1 hr at room temperature. For analysis of phosphorylated protein, 5% bovine serum albumin was used instead of non‐fat milk. The membranes were incubated overnight at 4° with primary antibodies at 1 : 1000 dilutions in blocking buffer (TBST with 2% non‐fat milk). The following primary antibodies were used in the present study: proteolipid protein (PLP) (Santa Cruz Biotech # sc23570, Santa Cruz, CA; RRID: AB_2165797), myelin basic protein (MBP) (Santa Cruz Biotech # sc13914; RRID: AB_648798), mitochondrial transcription factor A (TFAM) (Santa Cruz Biotech # sc23588; RRID: AB_2303230), Phospho (Thr172) AMPKα (Cell Signaling # 2531L, Danvers, MA; RRID: AB_330330), AMPKα (Cell Signaling # 2532S; RRID: AB_330331), β‐actin (Cell Signaling # 4970; RRID: AB_2223172), nuclear respiratory factor 1 (NRF‐1) (Cell Signaling # 69432), cleaved caspase‐3 (Cell Signaling # 9664S; RRID: AB_2070042), manganese superoxide dismutase (MnSOD) (Millipore # 06‐984; RRID: AB_310325), PGC‐1 (Abcam # AB54481; RRID: AB_881987), SIRT1 (Abcam # AB110304; RRID: AB_10864359), PEX14 (Abcam # AB112942; RRID: AB_10860671) and oxidative phosphorylation (OXPHOS) (Abcam # AB110413; RRID: AB_2629281). Validation data for these antibodies are available at the companies’ websites. Following washing, the membranes were incubated with 1 : 10 000 diluted horseradish peroxidase‐conjugated secondary antibody (Jackson Immunoresearch Lab, West Grove, PA) for 1 hr at room temperature, washed and then incubated with ECL reagent (Amersham Life Science, Pittsbrugh, PA), and exposed to ECL film. Autoradiographs were scanned and the band intensity was quantified by NIH J image.
Histological and immunohistological analysis
Control, EAE and drug‐treated animals were anaesthetized and perfused first with saline and then with 4% paraformaldehyde and spinal cords were fixed in 10% formalin as described previously.49 Tissue samples (lumbar spinal cords) were paraffin‐embedded and sectioned transversely or longitudinally (4‐μm‐thick). Haemotoxylin & eosin staining was performed to assess infiltration of mononuclear cells and Luxol Fast Blue staining was performed to evaluate the status of myelin (demyelination) in spinal cord sections. To assess the status of myelin, axons, mitochondria and peroxisomes, the sections were stained with antibody specific to MBP, neurofilament H (Millipore #AB1991; RRID: AB_91203), OXPHOS, or 70 000 MW peroxisomal membrane protein (PMP70) (Abcam #AB85550; RRID: AB_10672335) and detected with secondary antibody conjugated with immunofluorescent analysis. DAPI (4′,6‐diamidino‐2‐phenylindole, dihydrochloride) was used for staining of nuclei. All digital images were taken using a BX‐60 microscope equipped with a DP70 camera unit (Olympus, Tokyo, Japan).
Electron microscopy study
All animal groups including controls were treated with pertussis toxin. Animals (control and EAE with or without drug treatment) were anaesthetized and perfused with 10 ml of normal saline containing 0·1% sodium nitrite followed by 15 ml of a mixture of 4% paraformaldehyde and 2% glutaraldehyde in 0·1 m phosphate buffer, pH 7·4. Then the spinal cords were fixed in the same fixative (above) and post‐fixed with 1% osmium tetroxide–1·5% ferrocyanide for 2 hr in the dark, then dehydrated and embedded in Epon LX 112 resin. Semi‐thin sections, approximately 1 μm thick, were cut and stained with toluidine blue. Ultrathin sections (70 nm thick) were stained with uranyl acetate and lead citrate and examined by electron microscopy.
ELISA for subset‐specific CD4+ T‐cell cytokines in the spinal cords
Enzyme‐linked immunosorbent assay was performed for analysis of CD4+ T cell [IFN‐γ for T helper type 1 (Th1), interleukin‐4 (IL‐4) for Th2, IL‐17 for Th17] and regulatory T (IL‐10) secreted cytokines. Briefly, control mice, EAE mice, EAE mice treated with LOVA, AICAR, or a LOVA + AICAR combination were killed for collection of spinal cords and brains. Following preparation of single‐cell suspensions, red blood cells were lysed with Pharma lyse buffer (BD Pharmingen, San Diego, CA) and the remaining cells were subjected to Percoll gradient. The isolated mononuclear cells were then re‐suspended in complete RPMI medium containing RPMI‐1640 (Life Technologies, Gaithersburg, MD), 10% fetal bovine serum (GE Healthcare Bio‐Sciences, Pittsburgh, PA), 100 μg/ml streptomycin and penicillin (Atlanta Biologicals, Norcross, GA), 1 mm glutamine, 1 mm non‐essential amino acids and 50 μm 2‐mercaptoethanol (Sigma‐Aldrich) in 12‐well plates (5 × 106 cells/2 ml per well) and stimulated with MOG peptide (25 μg/ml) for 48 hr. Following the centrifugation, the resulting supernatants were collected for ELISA for IFN‐γ (R&D Systems, Minneapolis, MN), IL‐4 (Biolegend, San Diego, CA), IL‐17 (R&D Systems), and IL‐10 (R&D Systems).
Cell culture study
MO3.13 human oligodendrocyte‐like cells50 were obtained from Cedarlane (#CLU301, Burlington, ON, Canada; RRID: CVCL_D357). This cell line is not listed in the International Cell Line Authentication Committee (ICLAC) database of cross‐contaminated or misidentified cell lines. The cells were maintained in Dulbecco's modified Eagle's medium (4·5 g/l glucose) containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and 1× penicillin/streptomycin (Invitrogen) at 37° in 5% CO2/95% air. At 60–70% confluency, the cells were incubated in serum‐free Dulbecco's modified Eagle's medium containing 0·1 μm of phorbol 12‐myristate 13‐acetate (Sigma‐Aldrich) for 3 days for differentiation as described in our previous publication.51 Following the differentiation, the cells were pretreated with LOVA (10 μm) and/or AICAR (200 μM) for 2 hr and treated with pro‐inflammatory cytokine mix (IL‐17; 25 ng/ml, tumour necrosis factor‐α; 10 ng/ml, and IL‐1β; 10 ng/ml) for 24 hr. The effects of LOVA and/or AICAR on pro‐inflammatory cytokine‐induced cytotoxicity (MTT assay and caspase‐3 activation), ROCK and AMPK activities, mitochondrial (mt‐ND1)/chromosomal (GAPDH) DNA ratio, mitochondrial membrane potential (JC‐1 staining), cellular ATP levels and expressions of PGC‐1α, NRF‐1, PPAR‐α/γ, OXPHOS, PMP70, catalase and β‐actin were analysed as described above and reported previously by our laboratory.52, 53
Statistical analysis
The data are given as mean ± standard error mean (SEM) and statistical analysis was performed using graphpad prism 5.0 software (GraphPad, San Diego, CA). Significance between the groups was determined by one‐way analysis of variance followed by Tukey's post‐hoc test. A P‐value less than 0·05 was considered statistically significant.
Results
AICAR and LOVA treatments, started after disease onset, cooperatively attenuate clinical and CNS diseases of EAE
Previously, we reported that combination of LOVA and AICAR attenuates EAE disease better than individual drug treatment with AICAR or statin in MBP‐immunized Lewis rats.43 Here, the therapeutic efficacies of LOVA and AICAR and their combination on the CNS disease of EAE were evaluated in MOG‐immunized C57BL/6 mice. The MOG‐immunized EAE mice were treated with vehicle (EAE), LOVA (1 mg/kg/day/i.p.; EAE + LOVA), AICAR (100 mg/kg/day/i.p.; EAE + AICAR), or LOVA + AICAR combination (EAE + LOVA + AICAR) at the onset of disease with a clinical score between 1 and 2 (day 14 post immunization) (Fig. 1). Similar to our previous study in rats,43 LOVA or AICAR treatments individually provided some efficacy against the clinical disease of EAE, but a greater degree of efficacy was provided by combination of these drugs (Fig. 2a).
Figure 2.
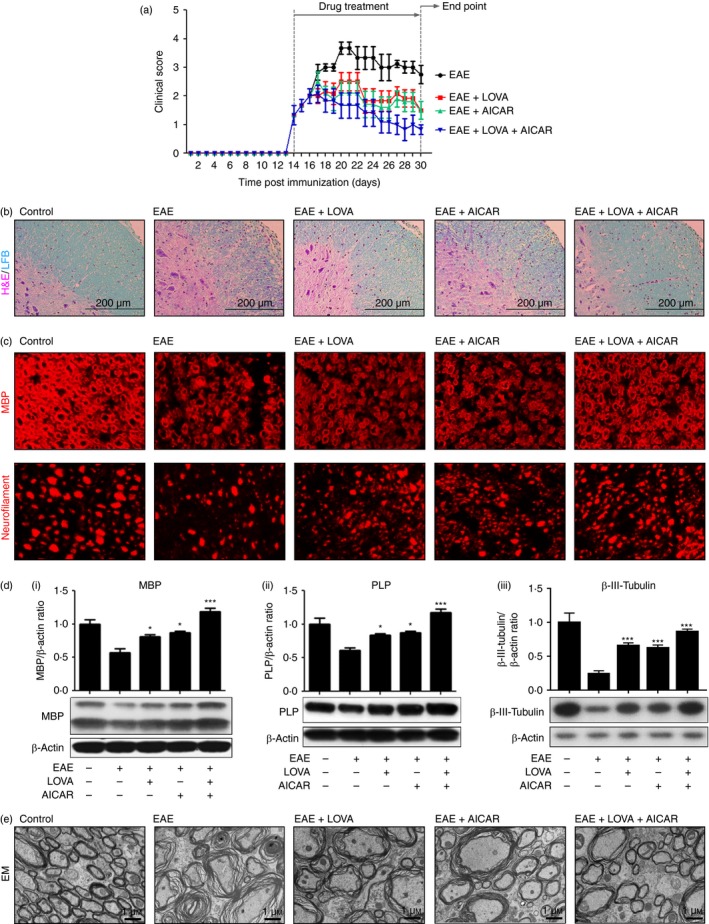
Therapeutic effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on clinical and central nervous system (CNS) diseases of experimental autoimmune encephalomyelitis (EAE). EAE was induced in female C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein (MOG) peptide. At the day of disease onset, the EAE mice were treated with LOVA (1 mg/kg/day/i.p.) or AICAR (100 mg/kg/day/i.p.) or combination of both drugs until day 30 (n = 8). Clinical disease of EAE of each mouse was analysed as described in the Materials and methods (a). At the day 30 post immunization the mice were killed and the CNS disease of EAE was analysed by H&E/LFB staining for the analysis of mononuclear inflammatory cells and myelin status (n = 4) (b), immunofluorescent staining for myelin basic protein (MBP) (myelin) and neurofilament (axons) (n = 4) (c), Western blot analysis for MBP (i) and proteolipid protein (PLP) (ii) and β‐III‐tubulin (iii) (n = 4) (d), and electron microscopy (EM) for spinal cords (n = 4) (d). In this study, control mice were also treated with pertussis toxin and complete Freund's adjuvant as vehicle. Each Western blot analysis was repeated three times and all blots were quantified and represented as bar graphs. Data represent mean ± standard error of mean of three independent experiments (six animals per group). *P < 0·05; ***P < 0·001; compared with the vehicle‐treated EAE groups. Panels (b), (c), and (e) are representative among the three independent experiments.
Next, spinal cord sections of control (treated with complete Freund's adjuvant without MOG peptide), EAE and drug‐treated EAE mice were subjected to Luxol fast blue–haematoxylin & eosin staining for analysis of myelin (blue)and mononuclear cell infiltration (magenta). Figure 2(b) shows that LOVA and AICAR treatment individually provided some protection against EAE‐induced demyelination and vascular immune cell infiltration, but combination treatment with these drugs provided greater efficacy. In addition, the observed greater efficacy of LOVA and AICAR combination, as compared with individual drugs, against demyelination [losses of MBPs in Fig. 2c,d(i) and PLP in Fig. 2d(ii)], axonal degeneration [loss of neurofilaments in Fig. 2c and β‐III‐tubulin in Fig. 2d(iii)], and loss of myelin integrity (splitting of the myelin sheath and loss of myelin in Fig. 2e) in the spinal cord of EAE animals compared with PTX‐treated control further indicate that LOVA and AICAR combination provides better protection against EAE‐induced demyelination than their individual treatments.
LOVA and AICAR treatments individually regulate RhoA/ROCK and AMPK activities in the spinal cords of EAE mice
Statins are known to attenuate de novo synthesis of isoprenoids, intermediates of the cholesterol de novo synthesis, and so inhibit RhoA/ROCK activity by inhibiting RhoA isoprenylation.35, 54 To evaluate the efficacy of LOVA on the inhibition of the RhoA/ROCK pathway in the spinal cord of EAE animals, we performed a ROCK activity assay using the spinal cord tissues of EAE animals (post‐immunization day 30). Figure 3(a) shows that the activity of ROCK was significantly increased in the spinal cords of EAE animals whereas treatment of EAE animals with LOVA, but not AICAR, significantly attenuated the ROCK activation. These data document a statin‐specific regulation of the RhoA/ROCK pathway under EAE conditions.
Figure 3.
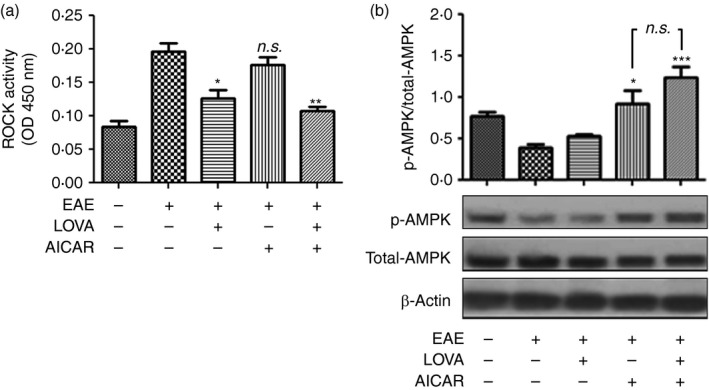
Effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on the activities of Rho‐associated kinase (ROCK) and AMP kinase (AMPK) in the spinal cords of experimental autoimmune encephalomyelitis (EAE) mice. Tissue lysates (n = 4) were extracted from spinal cords of the EAE mice treated with LOVA, AICAR, or LOVA and AICAR combination at the day 30 post‐immunization and analysed for activities of ROCK (a) and AMPK (b). AMPK activity was analysed by ratio of phospho‐AMPK (p‐AMPK) versus total AMPK following the Western blot analysis using antibody specific to phospho‐AMPK (Thr172) or pan‐AMPK (total AMPK). β‐Actin was used for internal loading control. Each analysis was repeated three times and the data are represented as a bar graph. Data represent mean ± standard error of mean of three independent experiments (six animals per group). *P < 0·05; **P < 0·01; ***P < 0·001; compared with the vehicle‐treated EAE groups. n.s. stands for not significant.
AICAR is a cell‐permeable activator of AMPK, a metabolic sensor that regulates cellular energy homeostasis as well as mitochondrial biogenesis.55 To evaluate the pharmacological efficacy of AICAR on the activation of AMPK in the spinal cords of EAE mice, AMPK activity in the spinal cords of EAE animals was accessed by Western blot analysis using antibody specific to phospho‐AMPK (Thr172; activated AMPK). As shown in Fig. 3(b) and reported previously by us,39 the activity of AMPK was decreased in spinal cords of EAE mice compared with control mice. AICAR treatment of EAE mice protected against the loss of AMPK activity in EAE animals but LOVA had no effect on the activity of AMPK. These data document that treatments with AICAR, but not LOVA, protect AMPK activity in the spinal cords of EAE animals.
LOVA and AICAR treatments cooperatively enhance the expression of transcription factors and regulators involved in mitochondrial and peroxisomal biogenesis in the spinal cord of EAE mice
PPARα and PPARγ play critical roles in regulation of peroxisomal and mitochondrial biogenesis.57, 58 Our laboratory and others have reported that statin treatment induces the expression and activities of PPARs35, 59, 60 through the attenuation of RhoA/ROCK activity.35, 54 Therefore, we next investigated the effects of LOVA and AICAR and their combination on the expression of PPARα and PPARγ. Figure 4(a) shows that the expression of PPARα and PPARγ in the spinal cords was decreased in EAE animals and LOVA treatment restored the decreased expression of PPARα and PPARγ. AICAR treatment had no effect on the expression of PPARα, but it partly restored the expression of PPARγ in EAE animals. These data indicate that LOVA efficiently protects against the EAE‐disease‐induced loss of both PPARα and PPARγ levels.
Figure 4.
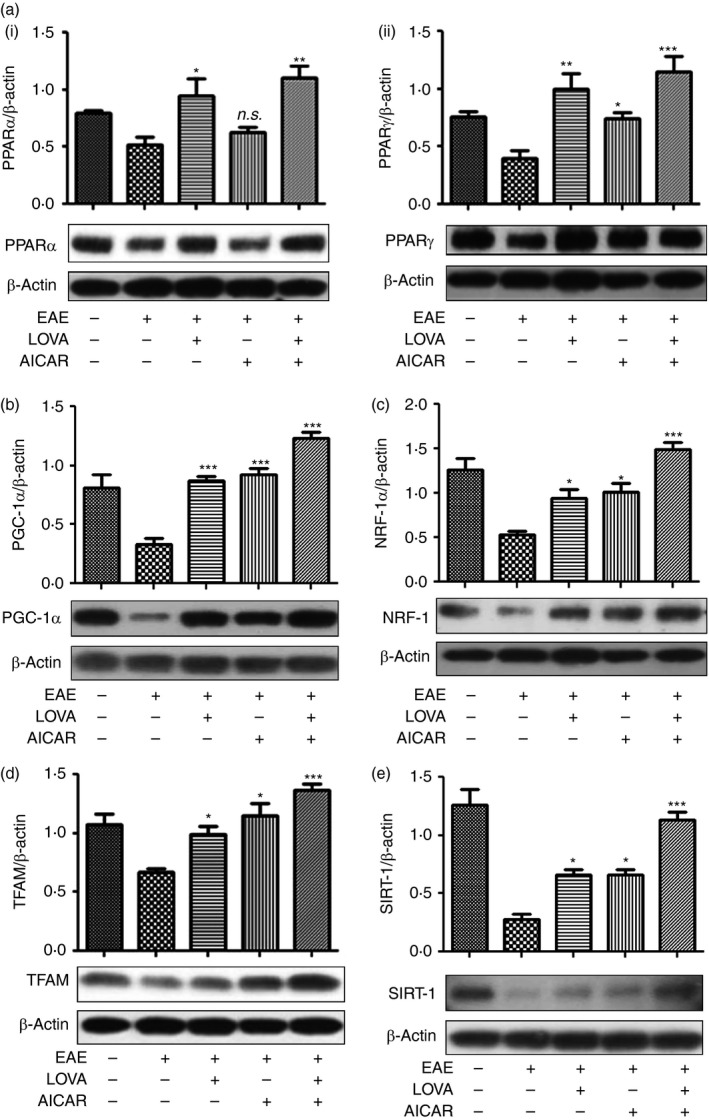
Effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on the expression of peroxisomal and mitochondrial biogenesis factors in the spinal cords of experimental autoimmune encephalomyelitis (EAE) mice. Tissue lysates (n = 4) were extracted from spinal cords of the EAE mice treated with LOVA, AICAR, or LOVA and AICAR in combination at day 30 post immunization and analysed for Western blot analysis for expressions of peroxisome proliferator‐activated receptor α (PPAR α) (a‐i), PPAR γ (a‐ii), peroxisome proliferator‐activated receptor‐γ coactivator‐1α (PGC‐1α) (b), nuclear respiratory factor 1 (NRF‐1) (c) mitochondrial transcription factor A (TFAM) (d), and sirtuin 1 (SIRT1) (e). β‐Actin was used for internal loading control. Data represent mean ± standard error of mean of three independent experiments (six animals per group). Each Western blot analysis was repeated three times and all blots were quantified and represented as bar graphs. *P < 0·05; **P < 0·01; ***P < 0·001; compared with the vehicle‐treated EAE groups. n.s. stands for not significant.
PPARγ coactivator‐1α (PGC‐1α) and its downstream factors, such as NRF1and NRF2, and TFAM, are known to play critical roles in mitochondrial biogenesis.55, 61 Figure 4(b–d) show that the expression of PGC‐1α, NRF‐1 and TFAM was significantly reduced in the spinal cords of EAE mice. Although treatment of EAE animals with LOVA or AICAR partly restored the loss of PGC‐1α, NRF‐1 and TFAM, treatment with their combination fully restored the losses of these transcription factors in EAE animals. AMPK and statin are known to activate NAD+‐dependent protein deacetylases (sirtuins or SIRT) for activation of PGC‐1α.62, 63 Figure 4(e) shows that expression of SIRT1 was significantly reduced in EAE spinal cords. The loss of SIRT1 in EAE spinal cords was partly restored by individual treatment with LOVA or AICAR, but fully restored by their combination treatment. These observations indicate that LOVA and AICAR combination optimizes the machinery for peroxisomal and mitochondrial biogenesis.
Lovastatin and AICAR treatments cooperatively protect against the loss of mitochondrial activity and functions in the spinal cords of EAE mice
There is a growing body of evidence that mitochondrial dysfunction contributes to the pathologies of various neurodegenerative disorders including MS.6, 64, 65 Accordingly, Fig. 5(a) shows that expressions of mitochondrial enzymes for OXPHOS were significantly reduced in EAE spinal cords. The loss of OXPHOS protein levels in the spinal cords of EAE animals was partly restored by individual treatment with LOVA or AICAR, but fully restored by treatment with their combination. In addition, spinal cords from EAE animals had decreased activities of complex I [Fig. 5b(i)], citrate synthase [TCA cycle activity; Fig. 5b(ii)], and low levels of ATP [Fig. 5b(iii)], whereas combination of LOVA and AICAR protected against these decreases. Mitochondrial dysfunction is associated with impaired anti‐oxidant defence resulting in oxidative insult.66, 67 Accordingly, Fig. 5(c) shows decreased levels of MnSOD. Individual drug treatment partly corrected this loss. The increased levels of MnSOD in EAE treated with combinations of LOVA and AICAR indicate optimized anti‐oxidant capacity of mitochondria. These data indicate that LOVA and AICAR combination treatment efficiently restored the disease‐induced mitochondrial dysfunction in the spinal cords of animals with EAE.
Figure 5.
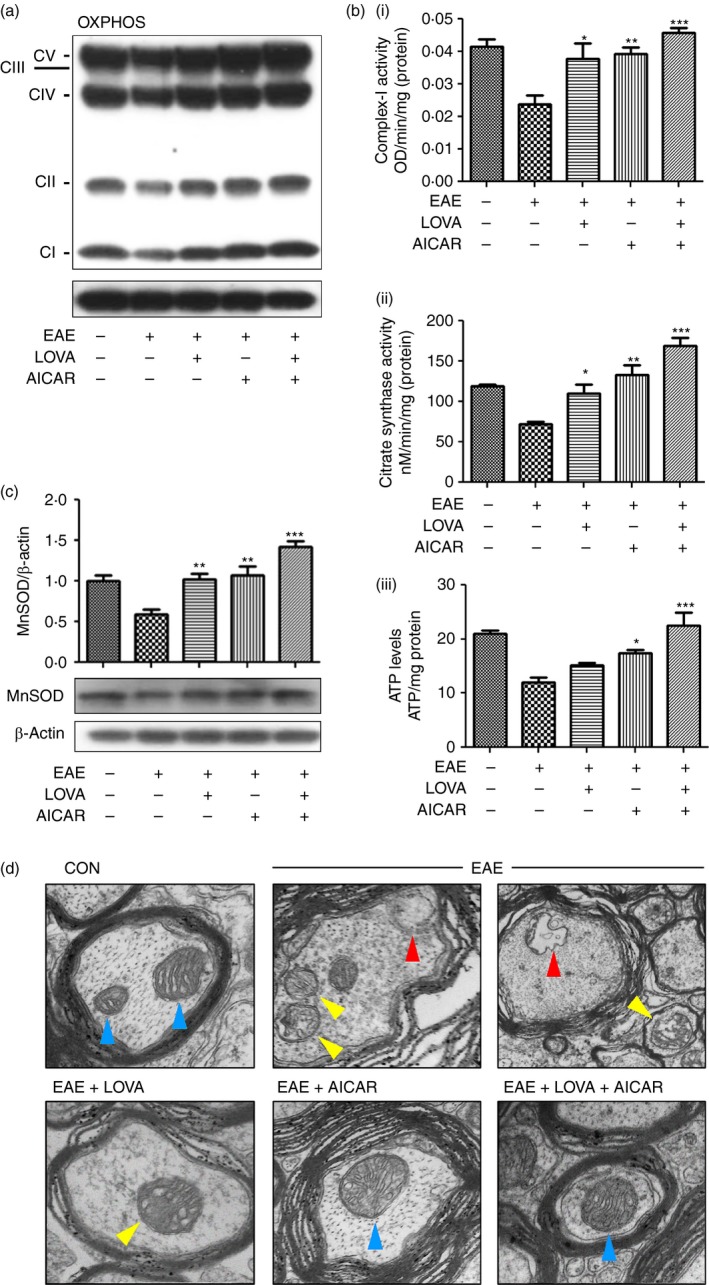
Effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on the function and structure of mitochondria in the spinal cords of experimental autoimmune encephalomyelitis (EAE) mice. Tissue lysates (n = 4) were extracted from spinal cords of the EAE mice treated with LOVA, AICAR, or LOVA and AICAR combination at the day 30 post immunization and analysed for expressions of mitochondrial oxidative phosphorylation (OXPHOS) (a), enzyme activities for mitochondrial complex‐I (b‐i) and citrate synthase (b‐ii), tissue levels of ATP (b‐iii), expression of mitochondrial manganese superoxide dismutase (MnSOD) (c). In OXPHOS Western blot analysis, CI–CVI stand for NUDFB8/Complex‐I NADH dehydrogenase 1β8 (CI), SDHB/complex‐II succinate ubiquinone oxidoreductase (CII), UQCRC2/complex‐III Ubiquinol cytochrome c oxidoreductase (CIII), MTCO1/complex‐IV cytochrome c oxidase subunit 1 (CIV), and ATP5A/complex V ATP synthase 5A (CV). β‐Actin was used for internal loading control. Data represent mean ± standard error of mean of three independent experiments (six animals per group). Each analysis was repeated three times and represented as a bar graph. Panels (a) and (d) are representative among 12 images from four mice. *P < 0·05; **P < 0·01; ***P < 0·001; compared with the vehicle‐treated EAE groups. Effect of LOVA, AICAR or LOVA + AICAR combination on EAE‐induced alteration in mitochondrial structure was analysed by electron microscopy. Blue arrowheads represent the healthy mitochondrial structure. Yellow arrowheads represent damaged mitochondrial structure with cristae disarrangement and partial cristolysis. Red arrowheads represent mitochondrial lysis. In this study, control mice were also treated with pertussis toxin and complete Freund's adjuvant as vehicle. Panels (a) and (d) are representative among the three independent experiments.
Mitochondrial dysfunction has been implicated in the degeneration of axons under the demyelinating disease conditions of MS and EAE,68 so next we investigated mitochondrial structure in axons of spinal cords of EAE animals by electron microscopy. Figure 5(d) shows that control mice had densely packed myelin and healthy mitochondrial cristae and structure (blue arrowheads) in the spinal cord. However, EAE mice showed loosened myelin sheets and damaged mitochondrial structure as evidenced by cristae disintegration, cristolysis (yellow arrowheads) and mitochondrial lysis (red arrowheads). LOVA‐treated EAE mice showed improved myelin and mitochondrial structures with some degree of cristae disintegration (yellow arrowhead). AICAR‐treated EAE mice showed improved cristae arrangement in axonal mitochondria (blue arrowhead) but some degree of loosened myelin was still observed. EAE mice treated with the LOVA and AICAR combination showed improved myelin and mitochondrial structures, which is comparable to control mice. These data indicate that the LOVA and AICAR combination cooperatively optimizes the myelin structure as well as mitochondrial structure and function and hence neuroprotection.
LOVA and AICAR treatments cooperatively increase the expression levels of peroxisomal proteins in EAE
Recent studies have reported decreased peroxisomal proteins and thus functions in the CNS of EAE and MS.8, 69 Therefore, we examined the efficacies of LOVA, AICAR and their combination on the loss of peroxisomes in the spinal cords of EAE mice. Figure 6(a,b) shows that, consistent with our previous observations,8 EAE mice had decreased expression of PMP70 and catalase. The observed decreased expression of PMP70, a peroxisomal membrane protein, reflects altered peroxisomal number and function. Accordingly, we also observed that EAE mice showed decreased number of peroxisomes in the spinal cords (punctuated orange dots in Fig. 6c). The EAE‐induced loss of peroxisomes was partly restored by LOVA or AICAR but fully restored by LOVA and AICAR in combination. Catalase, a major antioxidant in peroxisomes, is responsible for detoxification of H2O2 and hence altered catalase expression reflects altered antioxidant capacity of peroxisomes.70, 71 Treatments of EAE mice with LOVA or AICAR provided partial degrees of protection against the loss of peroxisomes in EAE mice. In addition, treatment of EAE mice with LOVA and AICAR in combination fully normalized the expressions of both PMP70 and catalase. These observations indicate that LOVA and AICAR‐mediated mechanisms protect against the loss of peroxisomal proteins and peroxisomes in the spinal cords of EAE mice.
Figure 6.
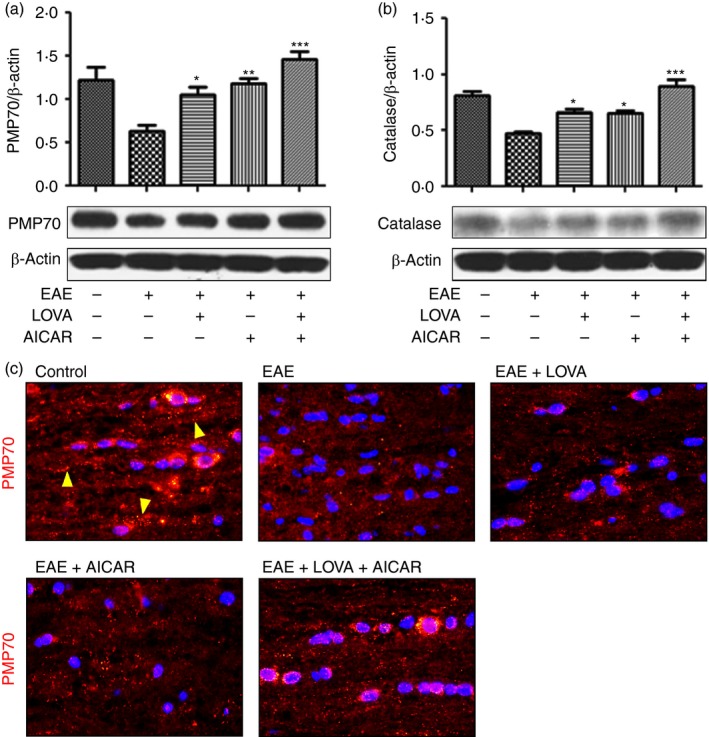
Effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on the expression of peroxisomal proteins in the spinal cords of (experimental autoimmune encephalomyelitis) (EAE) mice. Tissue lysates (n = 4) were extracted from spinal cords of the EAE mice treated with LOVA, AICAR or LOVA and AICAR combination at the day 30 post immunization and analysed for Western blot analysis for expressions of 70 000 MW peroxisomal membrane protein (PMP70) (a) and catalase (b). β‐Actin was used for internal loading control. Each Western analysis was repeated three times and all blots were quantified and represented as bar graph. Data represents mean ± standard error of mean of three independent experiments (six animals per group). *P < 0·05; **P < 0·01; ***P < 0·001; compared with the vehicle‐treated EAE groups. Next, PMP70 expression in peroxisomes was analysed by immunofluorescent staining of the spinal cord section (c). Panel (c) is the representative among the three independent experiments. The punctuated red/orange dots represent intact peroxisomes (yellow arrowheads).
AICAR and lovastatin treatments, started after peak of the disease, repairs mitochondrial and peroxisomal defects and attenuates clinical and CNS disease of EAE
To assess anti‐inflammatory and immunomodulatory roles of LOVA and AICAR combination, mononuclear cells were isolated from the CNS of control and EAE mice and EAE mice treated with LOVA and/or AICAR, and then MOG peptide induced production of CD4+ T‐cell‐associated cytokines (IFN‐γ for Th1, IL‐4 for Th2, IL‐17 for Th17 and IL‐10 for regulatory T cells) were analysed at 20 days after immunization. Figure 7(a,b) shows that treatment of EAE mice with LOVA and AICAR at the day of disease onset (day 14) cooperatively attenuated the release of pro‐inflammatory cytokines (e.g. IFN‐γ and IL‐17) from the CNS mononuclear cells. In addition, AICAR treatment of EAE mice also restored the decreased release of anti‐inflammatory cytokines (e.g. IL‐4 and IL‐10) from these cells (Fig. 7c,d). These data suggest that the observed improvement of neurological and biochemical parameters by LOVA and AICAR combination may result from the anti‐inflammatory and immuno‐modulatory activities of these drugs.
Figure 7.
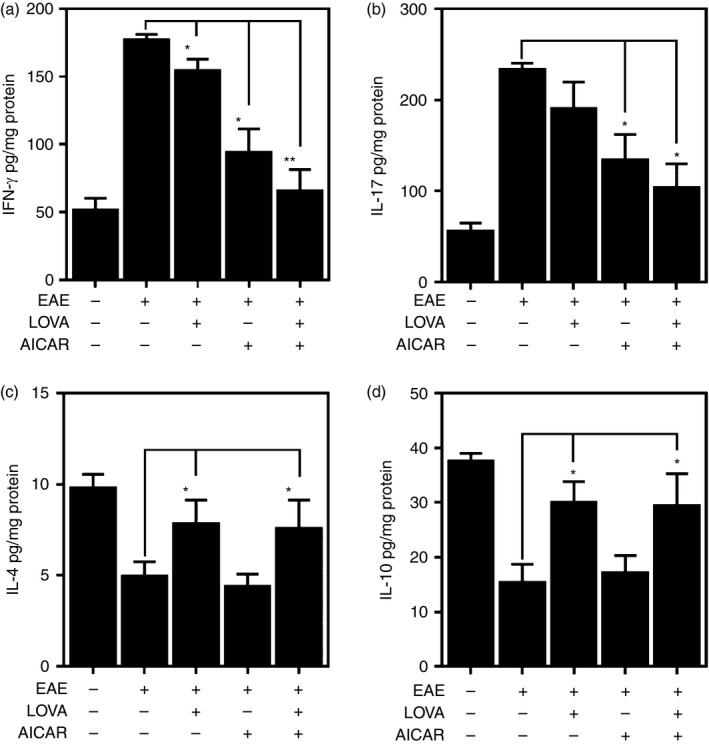
Release of pro‐inflammatory and anti‐inflammatory cytokines by central nervous system (CNS) mononuclear cells isolated from experimental autoimmune encephalomyelitis (EAE) mice treated with lovastatin (LOVA), 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) or LOVA and AICAR combination. EAE was induced in female C57BL/6 mice by immunization with myelin oligodendrocyte glycoprotein (MOG) peptide. At the day of disease onset, the EAE mice were treated with LOVA or AICAR or a combination of both drugs until day 30. Mononuclear cells were isolated from spinal cords and brains from these mice (n = 4) and stimulated with MOG peptide and release of interferon (IFN). Data represent mean ± standard error mean (n = 4 animals). *P < 0·05; **P < 0·01; compared to EAE groups.
To define the role of LOVA and AICAR combination in the processes of neurorepair, the drug treatments were initiated after the peak of EAE disease (Figs 1 and 8a). At the peak of disease, the spinal cords of EAE mice exhibited loosened myelin sheets in electron microscopy analyses [Fig. 8b(i)] as well as loss of axons (β‐III‐tubulin) and [Fig. 8b(ii)]. Moreover, the spinal cords of EAE mice at the peak of disease had decreased expression of mitochondrial (e.g. OXPHOS and MnSOD) and peroxisomal protein (e.g. PEX14), indicating the loss of mitochondria and peroxisomes. Treatment of these animals with LOVA and AICAR individually slightly attenuated the severity of clinical EAE disease (without any statistical significance) but their combination significantly attenuated clinical EAE disease (Fig. 8a). Accordingly, LOVA or AICAR treatment had relatively little effect, if any, on the decreased expression of axonal (β‐tubulin), myelin (MBP), mitochondrial (OXPHOS and MnSOD) and peroxisomal (e.g. PMP70 and PEX14) proteins in the spinal cords of EAE mice. On the other hand, these combination treatments normalized the expression levels of these proteins [Fig. 8c(i)]. These data indicate that LOVA and AICAR combination treatments are not only effective in protection of mitochondria, peroxisomes and axons from inflammatory insults, but also effectively promote repair of damaged mitochondria and peroxisomes, and hence myelin and axonal protection and repair.
Figure 8.
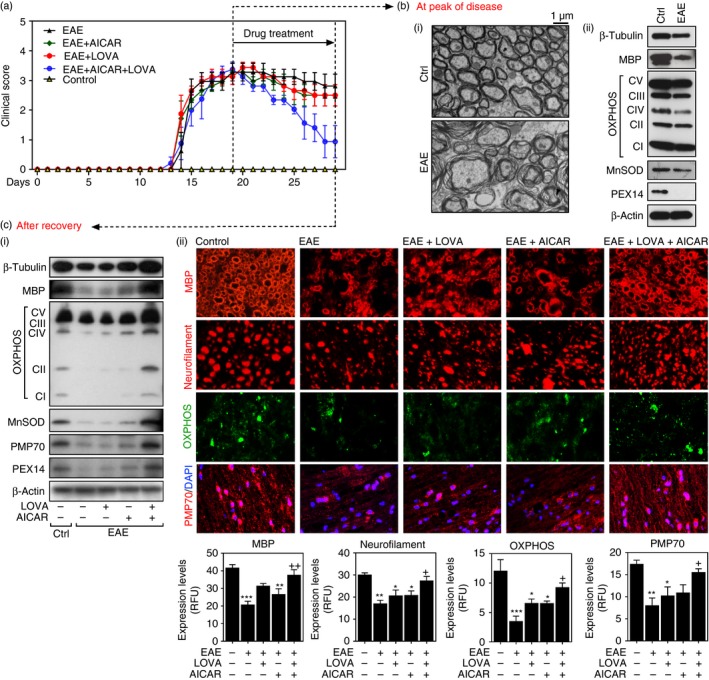
Neurorepair effects of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and their combination on clinical and central nervous system (CNS) disease of experimental autoimmune encephalomyelitis (EAE). EAE was induced in female C57BL/6 mice by immunization with MOG peptide. At the peak of disease (day 19 post immunization), the EAE mice were treated with lovastatin (LOVA; 1 mg/kg/day/i.p.) or AICAR (100 mg/kg/day/i.p.) or a combination of both drugs until day 29 (n = 12 for control and EAE groups and n = 8 for AICAR, LOVA and AICAR+LOVA treated groups). Clinical disease of EAE of each mouse was analysed as described under Materials and methods (a). At the peak of the disease, the control and EAE mice (n = 4) were killed and myelin structure in the spinal cord was analysed by electron microscopy (b‐i) and the spinal cord expressions of β‐III‐tubulin (for axons), MBP (for myelin), OXPHOS and MnSOD (for mitochondria), PEX14 and (for peroxisomes) were analysed by Western blot analysis (b‐ii). Panel (b) is the representative among the three independent experiments. For Western analysis, the spinal cord samples (n = 4) were homogenized and equal amounts of proteins were pooled. The levels of β‐actin were used for internal loading control for Western blot analysis. At day 29 post immunization, the control and EAE mice and EAE mice treated with LOVA, AICAR or LOVA + AICAR combination (n = 8 per each group) were killed and the spinal cord expression of β‐III‐tubulin, MBP, OXPHOS, MnSOD, 70 000 MW peroxisomal membrane protein (PMP70) and PEX14 was analysed by Western blot analysis (c‐i). For Western blot analysis, the spinal cord samples (n = 4) were homogenized and equal amounts of proteins were pooled. Status of myelin (MBP/coronal sections), axons (neurofilament/coronal sections), mitochondria (OXPHOS/coronal sections), and peroxisomes (PMP70/sagittal sections) was also analysed by immunofluorescent staining of spinal cord sections (c‐ii). DAPI was used for the staining of nuclei. The bar graphs represent immunofluorescence quantified using imagepro plus. Data represent mean ± standard error of mean (n = 4 animals). *P < 0·05; **P < 0·01; ***P < 0·001; compared with the vehicle‐treated control groups. + P < 0·05; ++ P < 0·01; compared with the vehicle‐treated EAE groups.
LOVA or AICAR or their combination also protect mitochondria and peroxisomes in in vitro culture of MO3.13 human OL‐like cells
We previously reported that cytokine‐induced OL death involves mitochondrial and peroxisomal dysfunctions.72, 73 Accordingly, treatment of differentiated MO3.13 human OLs51 with pro‐inflammatory cytokines (IL‐17; 25 ng/ml, TNF‐α; 10 ng/ml, and IL‐1β; 10 ng/ml) induced cell death as observed in cell viability (MTT) assays and increased caspase‐3 cleavage (caspase‐3 activation) (Fig. 9a). The cytokine‐induced cell death was partly attenuated by individual treatment with LOVA (10 μm) or AICAR (200 μm) but their combination treatment further reduced cell death, indicating the efficacy of LOVA/AICAR combination in the protection of OLs from inflammatory cytotoxicity. Pro‐inflammatory cytokine treatment of MO3.13 OLs also resulted in induction of RhoA activation and treatment of these cells with LOVA, but not AICAR, restored the increased cytokine‐induced RhoA activation [Fig. 9b(i)]. On the other hand, LOVA has no effect on cellular AMPK activity while AICAR treatment induced AMPK activation in a dose‐dependent manner [Fig. 9b(ii)]. Pro‐inflammatory cytokine treatment of MO3.13 cells resulted in a decreased ratio of mitochondrial versus chromosomal DNAs (an index of mitochondrial copy number), and the decreased ratio was partly restored by LOVA treatment but fully restored by AICAR treatment (Fig. 9c), indicating an efficacy of AICAR in increasing the mitochondrial copy number. Pro‐inflammatory cytokine treatment of MO3.13 OLs also resulted in decreased expression of transcription factors for mitochondrial biogenesis (e.g. PGC‐1α and NRF‐1) [Fig. 9d(i)] and mitochondrial respiratory complexes (OXPHOS) [Fig. 9d(ii)] and decreased mitochondrial membrane potential [Fig. 9d(iii)] and cellular ATP levels [Fig. 9d(iii)], and LOVA and AICAR treatment cooperatively restored these activities to the control levels or above control levels. These data indicate the cooperative actions of LOVA and AICAR in restoration of defective mitochondrial function under inflammatory conditions.
Figure 9.
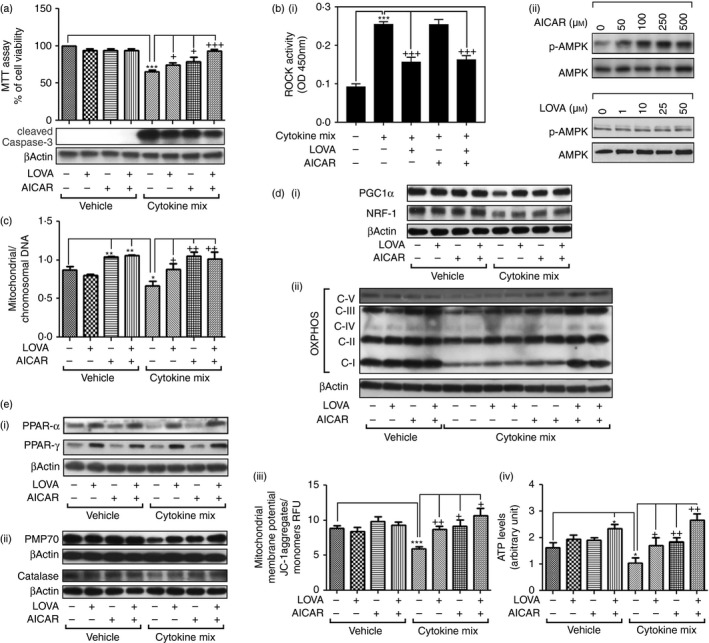
Effect of lovastatin (LOVA) and 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) combination on inflammatory oligodendrocyte (OL) death and mitochondrial and peroxisomal biogenesis in cell culture model. Effects of LOVA (10 μm), AICAR (200 μm), or their combination on pro‐inflammatory cytokines interleukin‐17 (IL‐17; 25 ng/ml), tumour necrosis factor‐α (TNF‐α; 10 ng/ml), and IL‐1β (10 ng/ml for 24 hr) induced loss of cell viability (MTT assay) and caspase‐3 activation (cleaved caspase‐3) were analysed in differentiated MO3.13 oligodendrocyte‐like cells (a). Effects of LOVA and AICAR on pro‐inflammatory cytokine‐induced ROCK activation (b‐i) and AMPK activation (b‐ii) were analysed as described in Materials and methods. Effect of LOVA and AICAR combination on pro‐inflammatory cytokine‐induced losses of mitochondrial copy number (ratio of mitochondrial DNA versus chromosomal DNA) (c), transcription factors for mitochondrial biogenesis (PGC1α, and NRF‐1) (d‐i), expressions of OXPHOS (d‐ii), mitochondrial membrane potential (d‐iii), cellular ATP levels (d‐iv), PPAR expression (e‐i), peroxisomal proteins [70 000 MW peroxisomal membrane protein (PMP70) and catalase] (e‐ii) were analysed as described in Materials and methods. Each analysis was repeated three times and represented as a bar graph.
The PPARs are transcription factors for expression of peroxisomal proteins and hence peroxisomal biogenesis. Figure 9e(i) shows that treatment of MO3.13 OLs with pro‐inflammatory cytokines results in decreased expression of PPARα but had no effect on PPARγ expression. LOVA treatment increased PPARα and PPARγ expression under normal and pro‐inflammatory cytokine conditions [Fig. 9e(i)]. On the other hand, AICAR treatment had no effect on the expression of PPARα or PPARγ. However, AICAR and LOVA combination treatment increased the expressions of peroxisomal protein (PMP70 and catalase) under pro‐inflammatory cytokine conditions [Fig. 9e(ii)]. These data indicate that LOVA‐induced PPARα/γ expression and AICAR‐induced AMPK pathways protect against defective expression of peroxisomal proteins under inflammatory conditions in in vitro cell culture model and in in vivo EAE model. Overall, these data indicate that LOVA and AICAR combination cooperatively protects and repairs mitochondrial and peroxisomal functions under inflammatory conditions of autoimmune disease and so ameliorates CNS disease of EAE.
Discussion
The present day US Food and Drug Administration‐approved immunomodulatory drugs have limited efficacies against the CNS disease as the disease progression continues despite the treatments.2 Recent studies underscore the importance of mitochondrial and peroxisomal dysfunctions in inflammation‐induced CNS pathology of MS and EAE.3, 4, 5, 6, 8 Therefore, an effective therapy for MS should include immunomodulatory as well as neuroprotective activities. This study reports that, in addition to immunomodulatory activities, LOVA and AICAR combination treatment targets peroxisomal and mitochondrial mechanisms for protection of myelin and axons and hence clinical disease of EAE.
Mitochondrial dysfunction is now well recognized to play a pivotal role in inflammation‐induced loss of myelin and axonal degeneration in EAE/MS.3, 4, 5, 6 In addition, peroxisomes also play an indispensable role in the metabolism of myelin lipids and maintenance of cellular redox states.9 Peroxisomes and mitochondria have a significant degree of metabolic and redox (ROS) cross‐talk and mutual dependence, and so these organelles are referred to as the ‘big brother and little sister’ relationship.27 EAE and MS involve loss and dysfunction of both mitochondria and peroxisomes.8, 35, 68, 74 While functional mitochondria are required for cellular survival, the loss and dysfunction of peroxisomes are reported to lead to mitochondrial dysfunction and demyelination, and hence to neurodegeneration.10, 11, 12, 13, 14, 15, 16, 17 Peroxisomes and mitochondria play critical roles in the metabolism of lipids and ROS and in ATP production and hence cell survival. Genetic loss of different peroxisomal proteins is known to cause 18 distinct neurological diseases.75 In addition, peroxisomes are also responsible for catabolism and turnover of various bioactive lipids and fatty acids, such as arachidonic acid metabolites (e.g. prostaglandins and leukotrienes), which are known to participate in various cellular pathological activities. Mitochondria and peroxisomes are responsible for generation and degradation of ROS.76 In mitochondria, MnSOD detoxifies superoxide anion () to hydrogen peroxide (H2O2) and catalase degrades it to water (H2O) and oxygen (O2) in peroxisomes. Moreover, mitochondria and peroxisomes also share transcription factors for their biogenesis. The observed cooperative expressions of mitochondrial and peroxisomal proteins by PPAR activators (fibrates)77 or by over‐expression of PGC‐1α 78 further confirms the biogenetic link between mitochondria and peroxisomes. Therefore, for efficient protection against inflammatory damage to myelin and axons, functional and structural protection of mitochondria and peroxisomes and myelin/axons are important.
Our laboratory was the first to report pleiotropic effects of statin on neuroinflammation and its therapeutic value for EAE28, 29, 30, 31 as well as efficacy in relapsing–remitting MS patients.32 The potential of statin as a possible candidate drug for MS has also been recently highlighted by a recent placebo‐controlled double blind study of 140 patients with progressive MS.37 Statins inhibit HMG‐CoA reductase and so inhibit de novo synthesis of isoprenoids (intermediates of cholesterol biosynthesis). We reported that depletion of isoprenoids, especially geranylgeranyl‐pyrophosphate (GGPP), with statin treatment, while maintaining normal cholesterol homeostasis, promotes differentiation and protection of oligodendrocytes by inhibiting RhoA/ROCK pathways and hence induction of PPARs and peroxisomal functions.34 In this study, we also observed that LOVA treatment efficiently restored EAE‐induced increase in RhoA/ROCK activity (Fig. 3a) as well as EAE or pro‐inflammatory cytokine‐induced decrease in expressions of PPARα and PPARγ (Figs 4a and 9e). However, LOVA alone had limited efficacy in normalization of other transcription factors required for mitochondrial/peroxisomal biogenesis, such as PGC‐1α, NRF‐1 and TFAM (Fig. 4b–d). Accordingly, LOVA treatment showed limited efficacy against the CNS and clinical diseases of EAE (Fig. 2), so suggesting a requirement of drugs targeting both PPARs and PGC‐1α signalling pathways for optimal protections of peroxisomes and mitochondria in EAE/MS.
AMPK is an energy sensor, which governs various cellular processes for maintaining the cellular energy and redox homeostasis. AMPK also plays a key role in PGC‐1α‐mediated regulation of mitochondrial/peroxisomal biogenesis.46, 79 We previously reported that AMPK activators (e.g. metformin and AICAR) alleviate EAE disease severity by modulating pro‐inflammatory and immune responses and/or by elevating glial expression of neurotrophic factors.40, 41, 42, 43, 44 However, AICAR treatment and hence activation of AMPK in the spinal cords of EAE mice (Fig. 3b) had limited efficacy on EAE‐induced deficits in PPARα/PPARγ expression and this correlated with partial protection against the clinical and CNS diseases of EAE (Fig. 2). Similarly, AICAR treatment of differentiated MO3.13 cells had no obvious effect on PPARα/PPARγ expressions under normal and pro‐inflammatory cytokine conditions [Fig. 9e(i)]. These data indicate that LOVA and AICAR provide neuroprotective mechanisms through modulation of different mechanisms (RhoA/ROCk versus AMPK) and that each pathway provides limited neuroprotection against EAE disease with partial efficacies on mitochondrial and peroxisomal dysfunctions (Figs 5 and 6). Individual treatments showed limited efficacies, but we observed that combination of LOVA and AICAR cooperatively restored the EAE‐induced decreases in PGC‐1α and SIRT‐1 and their downstream transcription factors (NRF‐1 and TFAM) (Fig. 4) as well as mitochondrial function (Fig. 5). Accordingly, LOVA and AICAR combination provided greater protection, compared with individual treatments, against the EAE‐induced dysfunction and disintegration of mitochondria and peroxisomes (Figs 5 and 6) and hence improved protection against the CNS and clinical diseases of EAE (Figs 2a and 8a).
Various laboratories including ours have reported on the immunomodulatory roles of LOVA28, 49 and AICAR.40, 80 Potentially, the observed therapeutic efficacy of LOVA and AICAR combination on the protection of mitochondrial and peroxisomal defects could result from the anti‐inflammatory and immunomodulatory effects of these drugs. Indeed, we observed that LOVA and AICAR treatments cooperatively attenuate the expression of pro‐inflammatory cytokines (e.g. IFN‐γ and IL‐17) in the spinal cords of EAE mice (Fig. 7a,b). In addition, AICAR treatment also restored the expression of anti‐inflammatory cytokines (e.g. IL‐4 and IL‐10), which were decreased in the spinal cords of EAE mice (Fig. 7c,d). To define the role of LOVA and AICAR combination in the processes of neurorepair, the EAE mice were treated with LOVA and AICAR after the peak of EAE disease when the myelin/axons and mitochondria/peroxisomes were already damaged (Fig. 8b). LOVA and AICAR individually had relatively little effect on clinical disease and related CNS pathology in EAE but their combination treatment significantly attenuated clinical disease of EAE as well as myelin, axonal, mitochondrial and peroxisomal defects (Fig. 8a,c). Moreover, LOVA and AICAR treatment cooperatively protects against inflammatory OL damage in in vitro cell culture model by restoring the pro‐inflammatory cytokine‐induced abnormalities in mitochondria and peroxisomes as well as transcription factors associated with their biogenesis (e.g. PGC‐1α, NRF‐1, PPARα, and PPARγ) with normalized levels of ATP (Fig. 9). These data indicate that LOVA and AICAR combination is not only effective in the modulation of pro‐inflammatory autoimmune responses, but also in protection and repair of disease‐associated mitochondria and peroxisomes and myelin/axon abnormalities and hence the CNS disease of EAE.
This study documents that combination of the inhibition of the RhoA/ROCK signalling pathway by LOVA and the activation of AMPK‐mediated signalling pathway by AICAR attenuates the immune/inflammatory disease of EAE, provides protection against CNS disease, and promotes neurorepair in the EAE disease model. Treatment with statin was reported to provide efficacy in patients with relapsing–remitting MS32 and in patients with progressive MS.37 The studies described here support the rationale for a future clinical study for supplementation of AMPK activator to statin therapy to improve the efficacy of statin observed with MS patients.32, 37
Disclosures
The authors declare that they have no conflict of interest in the publication.
Acknowledgements
We acknowledge Ms Joyce Bryan for her help in procurement of animals and supplies. Drs Devadoss J. Samuvel, Seungho Choi and Nishant Saxena performed the experiments. Drs Inderjit Singh, Avtar K. Singh and Jeseong Won designed and wrote the paper. This work was supported in part by the U.S. Department of Veterans Affairs (BX002829) and National Institutes of Health (NS037766).
Contributor Information
Inderjit Singh, Email: singhi@musc.edu.
Jeseong Won, Email: wonj@musc.edu.
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2002; 359:1221–31. [DOI] [PubMed] [Google Scholar]
- 2. Michel L, Larochelle C, Prat A. Update on treatments in multiple sclerosis. Presse Med 2015; 44:e137–51. [DOI] [PubMed] [Google Scholar]
- 3. Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008; 131:1722–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zambonin JL, Zhao C, Ohno N, Campbell GR, Engeham S, Ziabreva I et al Increased mitochondrial content in remyelinated axons: implications for multiple sclerosis. Brain 2011; 134:1901–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nikic I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM et al A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 2011; 17:495–9. [DOI] [PubMed] [Google Scholar]
- 6. Dutta R, McDonough J, Yin X, Peterson J, Chang A, Torres T et al Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann Neurol 2006; 59:478–89. [DOI] [PubMed] [Google Scholar]
- 7. Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS et al Mitochondrial changes within axons in multiple sclerosis. Brain 2009; 132:1161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh I, Paintlia AS, Khan M, Stanislaus R, Paintlia MK, Haq E et al Impaired peroxisomal function in the central nervous system with inflammatory disease of experimental autoimmune encephalomyelitis animals and protection by lovastatin treatment. Brain Res 2004; 1022:1–11. [DOI] [PubMed] [Google Scholar]
- 9. Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol 2015; 3:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Faust PL, Kaye EM, Powers JM. Myelin lesions associated with lysosomal and peroxisomal disorders. Expert Rev Neurother 2010; 10:1449–66. [DOI] [PubMed] [Google Scholar]
- 11. Fourcade S, Lopez‐Erauskin J, Ruiz M, Ferrer I, Pujol A. Mitochondrial dysfunction and oxidative damage cooperatively fuel axonal degeneration in X‐linked adrenoleukodystrophy. Biochimie 2014; 98:143–9. [DOI] [PubMed] [Google Scholar]
- 12. Wanders RJ. Peroxisomes in human health and disease: metabolic pathways, metabolite transport, interplay with other organelles and signal transduction. Subcell Biochem 2013; 69:23–44. [DOI] [PubMed] [Google Scholar]
- 13. Baumgart E, Vanhorebeek I, Grabenbauer M, Borgers M, Declercq PE, Fahimi HD et al Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model for Zellweger syndrome (PEX5 knockout mouse). Am J Pathol 2001; 159:1477–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dirkx R, Vanhorebeek I, Martens K, Schad A, Grabenbauer M, Fahimi D et al Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology 2005; 41:868–78. [DOI] [PubMed] [Google Scholar]
- 15. McGuinness MC, Lu JF, Zhang HP, Dong GX, Heinzer AK, Watkins PA et al Role of ALDP (ABCD1) and mitochondria in X‐linked adrenoleukodystrophy. Mol Cell Biol 2003; 23:744–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer‐Charlier N, Broccoli V et al Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late‐onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 2005; 14:3565–77. [DOI] [PubMed] [Google Scholar]
- 17. Baarine M, Beeson C, Singh A, Singh I. ABCD1 deletion‐induced mitochondrial dysfunction is corrected by SAHA: implication for adrenoleukodystrophy. J Neurochem 2014; 133:380–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Delille HK, Alves R, Schrader M. Biogenesis of peroxisomes and mitochondria: linked by division. Histochem Cell Biol 2009; 131:441–6. [DOI] [PubMed] [Google Scholar]
- 19. Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold‐inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998; 92:829–39. [DOI] [PubMed] [Google Scholar]
- 20. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V et al Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 1999; 98:115–24. [DOI] [PubMed] [Google Scholar]
- 21. Vega RB, Huss JM, Kelly DP. The coactivator PGC‐1 cooperates with peroxisome proliferator‐activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 2000; 20:1868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kobayashi S, Tanaka A, Fujiki Y. Fis1, DLP1, and Pex11p coordinately regulate peroxisome morphogenesis. Exp Cell Res 2007; 313:1675–86. [DOI] [PubMed] [Google Scholar]
- 23. Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 2005; 16:5077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Santel A, Frank S. Shaping mitochondria: the complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life 2008; 60:448–55. [DOI] [PubMed] [Google Scholar]
- 25. Ryter SW, Lee SJ, Smith A, Choi AM. Autophagy in vascular disease. Proc Am Thorac Soc 2010; 7:40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martinet W, Knaapen MW, Kockx MM, De Meyer GR. Autophagy in cardiovascular disease. Trends Mol Med 2007; 13:482–91. [DOI] [PubMed] [Google Scholar]
- 27. Schrader M, Yoon Y. Mitochondria and peroxisomes: are the ‘big brother’ and the ‘little sister’ closer than assumed? BioEssays 2007; 29:1105–14. [DOI] [PubMed] [Google Scholar]
- 28. Pahan K, Sheikh FG, Namboodiri AM, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest 1997; 100:2671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stanislaus R, Gilg AG, Singh AK, Singh I. Immunomodulation of experimental autoimmune encephalomyelitis in the Lewis rats by Lovastatin. Neurosci Lett 2002; 333:167–70. [DOI] [PubMed] [Google Scholar]
- 30. Stanislaus R, Pahan K, Singh AK, Singh I. Amelioration of experimental allergic encephalomyelitis in Lewis rats by lovastatin. Neurosci Lett 1999; 269:71–4. [DOI] [PubMed] [Google Scholar]
- 31. Stanislaus R, Singh AK, Singh I. Lovastatin treatment decreases mononuclear cell infiltration into the CNS of Lewis rats with experimental allergic encephalomyelitis. J Neurosci Res 2001; 66:155–62. [DOI] [PubMed] [Google Scholar]
- 32. Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic‐Plese S et al Oral simvastatin treatment in relapsing–remitting multiple sclerosis. Lancet 2004; 363:1607–8. [DOI] [PubMed] [Google Scholar]
- 33. Paintlia AS, Paintlia MK, Hollis BW, Singh AK, Singh I. Interference with RhoA‐ROCK signaling mechanism in autoreactive CD4+ T cells enhances the bioavailability of 1,25‐dihydroxyvitamin D3 in experimental autoimmune encephalomyelitis. Am J Pathol 2012; 181:993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Paintlia AS, Paintlia MK, Singh AK, Orak JK, Singh I. Activation of PPAR‐γ and PTEN cascade participates in lovastatin‐mediated accelerated differentiation of oligodendrocyte progenitor cells. Glia 2010; 58:1669–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paintlia AS, Paintlia MK, Singh AK, Singh I. Modulation of Rho‐Rock signaling pathway protects oligodendrocytes against cytokine toxicity via PPAR‐α‐dependent mechanism. Glia 2013; 61:1500–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paintlia AS, Paintlia MK, Singh AK, Singh I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol Pharmacol 2008; 73:1381–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K et al Effect of high‐dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS‐STAT): a randomised, placebo‐controlled, phase 2 trial. Lancet 2014; 383:2213–21. [DOI] [PubMed] [Google Scholar]
- 38. Pihl‐Jensen G, Tsakiri A, Frederiksen JL. Statin treatment in multiple sclerosis: a systematic review and meta‐analysis. CNS Drugs 2015; 29:277–91. [DOI] [PubMed] [Google Scholar]
- 39. Nath N, Khan M, Rattan R, Mangalam A, Makkar RS, de Meester C et al Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem Biophys Res Commun 2009; 386:16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5‐aminoimidazole‐4‐carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol 2005; 175:566–74. [DOI] [PubMed] [Google Scholar]
- 41. Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol 2009; 182:8005–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paintlia AS, Paintlia MK, Mohan S, Singh AK, Singh I. AMP‐activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am J Pathol 2013; 183:526–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5‐aminoimidazole‐4‐carboxamide‐1‐β‐d‐ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol 2006; 169:1012–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prasad R, Giri S, Nath N, Singh I, Singh AK. 5‐aminoimidazole‐4‐carboxamide‐1‐β‐4‐ribofuranoside attenuates experimental autoimmune encephalomyelitis via modulation of endothelial‐monocyte interaction. J Neurosci Res 2006; 84:614–25. [DOI] [PubMed] [Google Scholar]
- 45. Paintlia AS, Mohan S, Singh I. Combinatorial effect of metformin and lovastatin impedes T‐cell autoimmunity and neurodegeneration in experimental autoimmune encephalomyelitis. J Clin Cell Immunol 2013; 4:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scarpulla RC. Metabolic control of mitochondrial biogenesis through the PGC‐1 family regulatory network. Biochim Biophys Acta 2011; 1813:1269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ota H, Eto M, Kano MR, Kahyo T, Setou M, Ogawa S et al Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler Thromb Vasc Biol 2010; 30:2205–11. [DOI] [PubMed] [Google Scholar]
- 48. Lei J, Gu X, Ye Z, Shi J, Zheng X. Antiaging effects of simvastatin on vascular endothelial cells. Clin Appl Thromb Hemost 2014; 20:212–8. [DOI] [PubMed] [Google Scholar]
- 49. Nath N, Giri S, Prasad R, Singh AK, Singh I. Potential targets of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitor for multiple sclerosis therapy. J Immunol 2004; 172:1273–86. [DOI] [PubMed] [Google Scholar]
- 50. Buntinx M, Vanderlocht J, Hellings N, Vandenabeele F, Lambrichts I, Raus J et al Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: morphology and oligodendrocyte‐specific gene expression. J Neurocytol 2003; 32:25–38. [DOI] [PubMed] [Google Scholar]
- 51. Won JS, Kim J, Paintlia MK, Singh I, Singh AK. Role of endogenous psychosine accumulation in oligodendrocyte differentiation and survival: implication for Krabbe disease. Brain Res 2013; 1508:44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Baarine M, Beeson C, Singh A, Singh I. ABCD1 deletion‐induced mitochondrial dysfunction is corrected by SAHA: implication for adrenoleukodystrophy. J Neurochem 2015; 133:380–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Won JS, Kim J, Annamalai B, Shunmugavel A, Singh I, Singh AK. Protective role of S‐nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J Alzheimers Dis 2013; 34:621–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nohria A, Prsic A, Liu PY, Okamoto R, Creager MA, Selwyn A et al Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis 2009; 205:517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem 2010; 47:69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 2009; 89:1025–78. [DOI] [PubMed] [Google Scholar]
- 57. Mello T, Materozzi M, Galli A. PPARs and mitochondrial metabolism: from NAFLD to HCC. PPAR Res 2016; 2016:7403230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab 2014; 19:380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Landrier JF, Thomas C, Grober J, Duez H, Percevault F, Souidi M et al Statin induction of liver fatty acid‐binding protein (L‐FABP) gene expression is peroxisome proliferator‐activated receptor‐α‐dependent. J Biol Chem 2004; 279:45512–8. [DOI] [PubMed] [Google Scholar]
- 60. Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K et al Statins activate peroxisome proliferator‐activated receptor γ through extracellular signal‐regulated kinase 1/2 and p38 mitogen‐activated protein kinase‐dependent cyclooxygenase‐2 expression in macrophages. Circ Res 2007; 100:1442–51. [DOI] [PubMed] [Google Scholar]
- 61. Austin S, St‐Pierre J. PGC1α and mitochondrial metabolism – emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 2012; 125:4963–71. [DOI] [PubMed] [Google Scholar]
- 62. Du G, Song Y, Zhang T, Ma L, Bian N, Chen X et al Simvastatin attenuates TNFα‐induced apoptosis in endothelial progenitor cells via the upregulation of SIRT1. Int J Mol Med 2014; 34:177–82. [DOI] [PubMed] [Google Scholar]
- 63. Jing H, Yao J, Liu X, Fan H, Zhang F, Li Z et al Fish‐oil emulsion (ω‐3 polyunsaturated fatty acids) attenuates acute lung injury induced by intestinal ischemia‐reperfusion through adenosine 5′‐monophosphate‐activated protein kinase‐sirtuin1 pathway. J Surg Res 2014; 187:252–61. [DOI] [PubMed] [Google Scholar]
- 64. Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis 2013; 51:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 1990; 54:823–7. [DOI] [PubMed] [Google Scholar]
- 66. Bowling AC, Schulz JB, Brown RH Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem 1993; 61:2322–5. [DOI] [PubMed] [Google Scholar]
- 67. Emamgholipour S, Hossein‐Nezhad A, Sahraian MA, Askarisadr F, Ansari M. Evidence for possible role of melatonin in reducing oxidative stress in multiple sclerosis through its effect on SIRT1 and antioxidant enzymes. Life Sci 2016; 145:34–41. [DOI] [PubMed] [Google Scholar]
- 68. Mahad D, Lassmann H, Turnbull D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol 2008; 34:577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gray E, Rice C, Hares K, Redondo J, Kemp K, Williams M et al Reductions in neuronal peroxisomes in multiple sclerosis grey matter. Mult Scler 2014; 20:651–9. [DOI] [PubMed] [Google Scholar]
- 70. Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS‐metabolism: implications for human disease. Biochim Biophys Acta 2012; 1822:1363–73. [DOI] [PubMed] [Google Scholar]
- 71. Guy J, Ellis EA, Hope GM, Rao NA. Antioxidant enzyme suppression of demyelination in experimental optic neuritis. Curr Eye Res 1989; 8:467–77. [DOI] [PubMed] [Google Scholar]
- 72. Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin‐17 and tumor necrosis factor‐α enhances oxidative stress‐mediated oligodendrocyte apoptosis. J Neurochem 2011; 116:508–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Paintlia MK, Paintlia AS, Khan M, Singh I, Singh AK. Modulation of peroxisome proliferator‐activated receptor‐α activity by N‐acetyl cysteine attenuates inhibition of oligodendrocyte development in lipopolysaccharide stimulated mixed glial cultures. J Neurochem 2008; 105:956–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med 2014; 20:179–87. [DOI] [PubMed] [Google Scholar]
- 75. Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta 2006; 1763:1733–48. [DOI] [PubMed] [Google Scholar]
- 76. Lismont C, Nordgren M, Van Veldhoven PP, Fransen M. Redox interplay between mitochondria and peroxisomes. Front Cell Dev Biol 2015; 3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Monsalve FA, Pyarasani RD, Delgado‐Lopez F, Moore‐Carrasco R. Peroxisome proliferator‐activated receptor targets for the treatment of metabolic diseases. Mediators Inflamm 2013; 2013:549627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huang TY, Zheng D, Houmard JA, Brault JJ, Hickner RC, Cortright RN. Overexpression of PGC‐1α increases peroxisomal and mitochondrial fatty acid oxidation in human primary myotubes. Am J Physiol Endocrinol Metab 2017; 312:E253–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 2013; 12:465–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ayasolla KR, Singh AK, Singh I. 5‐aminoimidazole‐4‐carboxamide‐1‐β‐4‐ribofuranoside (AICAR) attenuates the expression of LPS‐ and Aβ peptide‐induced inflammatory mediators in astroglia. J Neuroinflammation 2005; 2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]