

Abstract
Key points
This study investigates the impact of decreased fetal plasma glucose concentrations on the developing heart in late gestation, by subjecting pregnant ewes to a 50% global nutrient restriction.
Late gestation undernutrition (LGUN) decreased fetal plasma glucose concentrations whilst maintaining a normoxemic blood gas status.
LGUN increased the mRNA expression of IGF2 and IGF2R. Fetal plasma glucose concentrations, but not fetal blood pressure, were significantly correlated with IGF2 expression and the activation of CAMKII in the fetal right ventricle.
LGUN increased interstitial collagen deposition and altered the protein abundance of phospho‐PLB and phospho‐troponin I, regulators of cardiac contractility and relaxation.
This study shows that a decrease in fetal plasma glucose concentrations may play a role in the development of detrimental changes in the right ventricle in early life, highlighting CAMKII as a potential target for the development of intervention strategies.
Abstract
Exposure of the fetus to a range of environmental stressors, including maternal undernutrition, is associated with an increased risk of death from cardiovascular disease in adult life. This study aimed to determine the effect of maternal nutrient restriction in late gestation on the molecular mechanisms that regulate cardiac growth and development of the fetal heart. Maternal undernutrition resulted in a decrease in fetal glucose concentrations across late gestation, whilst fetal arterial remained unchanged between the control and late gestation undernutrition (LGUN) groups. There was evidence of an up‐regulation of IGF2/IGF2R signalling through the CAMKII pathway in the fetal right ventricle in the LGUN group, suggesting an increase in hypertrophic signalling. LGUN also resulted in an increased mRNA expression of COL1A, TIMP1 and TIMP3 in the right ventricle of the fetal heart. In addition, there was an inverse relationship between fetal glucose concentrations and COL1A expression. The presence of interstitial fibrosis in the heart of the LGUN group was confirmed through the quantification of picrosirius red‐stained sections of the right ventricle. We have therefore shown that maternal undernutrition in late gestation may drive the onset of myocardial remodelling in the fetal right ventricle and thus has negative implications for right ventricle function and cardiac health in later life.
Keywords: cardiac, heart, fetus, fibrosis, IGF2, signalling, undernutrition
Key points
This study investigates the impact of decreased fetal plasma glucose concentrations on the developing heart in late gestation, by subjecting pregnant ewes to a 50% global nutrient restriction.
Late gestation undernutrition (LGUN) decreased fetal plasma glucose concentrations whilst maintaining a normoxemic blood gas status.
LGUN increased the mRNA expression of IGF2 and IGF2R. Fetal plasma glucose concentrations, but not fetal blood pressure, were significantly correlated with IGF2 expression and the activation of CAMKII in the fetal right ventricle.
LGUN increased interstitial collagen deposition and altered the protein abundance of phospho‐PLB and phospho‐troponin I, regulators of cardiac contractility and relaxation.
This study shows that a decrease in fetal plasma glucose concentrations may play a role in the development of detrimental changes in the right ventricle in early life, highlighting CAMKII as a potential target for the development of intervention strategies.
Introduction
Epidemiological studies have shown that a suboptimal intrauterine environment is associated with an increased risk of cardiovascular disease in adult life (Barker et al. 1989; Barker, 2000). Inadequate fetal substrate supply as a consequence of maternal undernutrition, placental insufficiency or living at high altitude has detrimental long‐term consequences for cardiac health (McMillen & Robinson, 2005; Morrison, 2008; Giussani, 2016).
An ovine model of early‐onset placental insufficiency resulting in intrauterine growth restriction (IUGR) has been used to determine the impact of restricted oxygen and glucose supply on the development of the fetal heart in late gestation (Morrison et al. 2007; Wang et al. 2012b; Botting et al. 2014) and after birth (Wang et al. 2011). Placental insufficiency results in a delay in cardiomyocyte maturation, resulting in fewer but larger cardiomyocytes when normalized to fetal heart weight and increased left ventricular (LV) mass after birth in the absence of hypertension (Morrison et al. 2007; Wang et al. 2011; Botting et al. 2014).
During late gestation, cardiac growth is regulated by the insulin‐like growth factor (IGF) system (Botting et al. 2012). Physiological cardiac growth occurs via activation of the IGF1R/PI3K pathway and cardiomyocyte proliferation (first two‐thirds of gestation) and then predominantly by hypertrophy (increase in cardiomyocyte size) in late gestation (Burrell et al. 2003). However, in the growth‐restricted fetus and low birth weight (LBW) lamb, there is an increase in IGF2 and IGF2R gene and protein expression in the fetal heart (Wang et al. 2011). Interestingly, there is evidence that this is a consequence of epigenetic programming through increased histone acetylation in the IGF2/IGF2R promoter region, not methylation (Wang et al. 2011), and that increased IGF2R signalling may contribute to pathological cardiomyocyte and cardiac hypertrophy (Wang et al. 2015). Activation of IGF2R, a Gαq receptor, induces cardiac hypertrophy, through the activation of downstream effectors such as Ca2+–calmodulin‐dependent protein kinase II (CAMKII), protein kinase C (PKC)‐α and protein kinase A (PKA) (Chu et al. 2008, 2009; Wang et al. 2012b). Initially occurring as an adaptive response, the hypertrophy becomes detrimental to cardiac function if prolonged and is often associated with an up‐regulation of those molecules involved in myocardial fibrosis such as the two main isoforms of collagen in the heart collagen type 1 (COL1A) and collagen type 3 (COL3A) (Weber, 1989; Weber et al. 1993; Cleutjens et al. 1995). This interstitial accumulation of collagen and extracellular matrix dysregulation causes the myocardium to become increasingly less complaint (Khan & Sheppard, 2006) and is associated with systolic and diastolic dysfunction (Kitamura et al. 2001; Burlew & Weber, 2002).
Models of IUGR that involve placental insufficiency restrict both oxygen and glucose delivery to the fetus. There is a negative correlation between mean gestational and both the proportion of mononucleated cardiomyocytes and the size of binucleated cardiomyocytes (Morrison et al. 2007), suggesting that in the fetal IUGR heart, a low gestational may result in delayed cardiomyocyte maturation and an increase in cardiomyocyte size. Furthermore, these chronically hypoxemic fetuses have unchanged basal blood pressure in comparison with normoxemic controls (Danielson et al. 2005), indicating that the increase in cardiomyocyte size is independent of increased fetal blood pressure.
However, it has also been demonstrated that there blood pressure is increased in the nutrient‐restricted offspring in rodent models of total caloric restriction (Woodall et al. 1996) and low protein diet (Langley & Jackson, 1994) during pregnancy. As fetal blood pressure was not measured in these studies, we are unable to draw conclusions about the origins of hypertension in these models.
Experimental models of in utero programming of cardiovascular disease have generally focused on the impact of adverse intrauterine environments on the developmental changes within the left ventricle. Interestingly, right ventricular dysfunction predicts poorer cardiac outcomes in heart failure patients in the clinical setting (Polak et al. 1983) and pulmonary hypertension is associated with decreased right ventricular contractility (Segers et al. 2012). In postnatal life, the right ventricle pumps deoxygenated blood against the lesser pressure of the pulmonary circulation. However, during fetal life, when blood is oxygenated by the placenta, pulmonary resistance is high and blood flows through the ductus arteriosus and foramen ovale to maintain cerebral blood flow and brain growth. Thus, during gestation the right ventricle is the dominant ventricle and pumps blood against systemic arterial pressure.
We have previously shown that maternal nutrient restriction during the rapid fetal growth phase in late gestation results in an increase in fetal arterial blood pressure (Edwards & McMillen, 2001). We hypothesize that exposure to maternal nutrient restriction will therefore result in both molecular and morphological changes within the right ventricle. Herein, we investigated the consequences of maternal nutrient restriction in late gestation on the molecular mechanisms that regulate the growth and development of the right ventricle.
Methods
All procedures were approved by the University of Adelaide Animal Ethics Committee and comply with the Australian code of practice for the care and use of animals for scientific purposes. All investigators understood and followed the ethical principles outlined by Grundy (2015) and the principles of the 3Rs, specifically the reduction of the use of animals in research (Russell & Burch, 1959).
Animals and surgery
Surgery was performed on pregnant Border‐Leicester cross Merino ewes (n = 14, sourced from Turretfield Research Centre, South Australia) between 110 and 113 days of gestation (term = 147 ± 3 days of gestation) to implant vascular catheters as previously described (Edwards & McMillen, 2001). Briefly, sodium thiopentane (1.25 g iv, Pentothal, Rhone Merieux, Pinkenba, Qld, Australia) was administered to induce general anaesthesia, which was maintained with 2.5‐4% halothane in oxygen (Fluothane, ICI, Melbourne, Vic., Australia). Vascular catheters were implanted into the maternal jugular vein, fetal jugular vein and carotid artery as well as the amniotic cavity as previously described (Muhlhausler et al. 2005). Ewes received an intramuscular injection of antibiotics (3.5 mL of Norocillin (150 mg mL−1 procaine penicillin and 112.5 mg mL−1 benzathine penicillin; Norbrook Laboratories Ltd., Gisborne, Australia) and 2 mL of 125 mg mL−1 dihydrostreptomycin in sterile saline (Sigma, St Louis, MO, USA) at the time of surgery. After surgery, ewes were administered xylazine (20 μg kg−1; im) for analgesia. Ewes were housed individually with a 12 h light–dark cycle and fed once daily at 11.00 h with water provided ad libitum. Ewes were allowed to recover from surgery for at least 4 days prior to experimentation.
Nutritional regime
Ewes were weighed once between 110 and 114 days gestation age (GA) and from 115 days GA ewes were fed either 20 g of Lucerne and 3 g oats per kg of body weight (Control, n = 6), or a 50% reduction, 10 g of Lucerne and 1.5 g oats per body weight [late gestation undernutrition (LGUN) group; n = 8). To account for the growing fetus, feed allowance was increased by 15% in both groups every 10 days until post‐mortem.
Blood sampling regime
Daily fetal blood gas as well as plasma glucose and cortisol concentrations have previously been reported for this cohort (Edwards & McMillen, 2001). Fetal arterial blood samples were collected every day for the first 4 days after surgery and then three times per week thereafter for the measurement of arterial , , pH and oxygen saturation temperature corrected to 39°C (ABL 520, Radiometer, Copenhagen, Denmark). Fetal arterial blood samples and maternal venous blood samples were also collected three times per week between 08.00 and 11.00 h. All details of glucose and cortisol assays and results for this cohort have been published previously (Edwards & McMillen, 2001). Briefly, plasma glucose concentrations were determined using hexokinase and glucose‐6‐phosphate dehydrogenase, measuring the formation of NADH photometrically at 340 nm (COBAS MIRA automated analysis system, Roche Diagnostic, Basle, Switzerland) and plasma cortisol concentrations were determined by radioimmunoassay using an Orion Diagnostica kit (Orion Diagnostica, Turku, Finland).
Fetal blood pressure measurements
The fetal carotid arterial catheter and amniotic catheters were connected to a MacLab data acquisition system via a MacLab 1050 displacement transducer and quad‐bridge amplifier (ADInstruments, Sydney, NSW, Australia). Arterial blood pressure, corrected for amniotic pressure, was measured using the MacLab Chart software on a Macintosh computer. Responses to blood pressure challenges have been previously reported (Edwards & McMillen, 2001).
Post‐mortem and tissue collection
At 144 or 145 days GA, ewes were humanely killed with an overdose of sodium pentobarbitone (Virbac, Peakhurst, NSW, Australia) and fetal sheep were delivered by hysterotomy, weighed and killed by decapitation. Fetal hearts were weighed, and right ventricles were excised and weighed separately. Samples of the right ventricle were both snap frozen in liquid nitrogen to be stored at −80°C for molecular analysis and fixed in 4% paraformaldehyde for histological analysis.
Quantification of mRNA transcripts within the right ventricle
Total RNA extraction
Total RNA was extracted from ∼50–60 mg of right ventricle tissue (Control, n = 6; LGUN, n = 8) using Qiagen QIAzol Lysis Reagent and Qiagen RNeasy purification columns (Qiagen Pty Ltd, Doncaster, Vic., Australia) (Soo et al. 2012; McGillick et al. 2013). All extracted RNA samples were checked for integrity by running them on an agarose gel stained with ethidium bromide. Total RNA was quantified by spectrophotometric measurements at 260 and 280 nm.
cDNA synthesis
The ratio of the optical density at 260 and 280 nm was used to calculate the correct dilutions of extracted RNA used for cDNA synthesis. The cDNA was synthesized according to the manufacturer's guidelines with a Superscript III First Strand Synthesis System (Invitrogen, Carlsbad, CA, USA), using 2 μg of total diluted RNA, random hexamers, dNTP, DTT and Superscript III in a final volume of 20 μL. A no template control (NTC) containing no RNA transcript and a no amplification control (NAC) containing no Superscript III were used to check for reagent contamination and genomic DNA contamination, respectively.
Quantitative real‐time RT‐PCR
All essential information regarding our procedure is included as per the MIQE guidelines (Bustin et al. 2009). The expression of genes regulating IGF signalling [IGF1, DQ152962; IGF1R, AY162434; IGF2, M89789; IGF2R, AF327649 (Gentili et al. 2009)], glucocorticoid signalling [glucocorticoid receptor (GR), NM_001114186.1; 11BHSD1, NM_001009395.1; 11BHSD2, NM_001009460.1 (McGillick et al. 2014)] and fibrosis (Table 1) were measured by quantitative RT‐PCR (qRT‐PCR). The expression of these target genes was determined in parallel with and normalized to three housekeeping genes: hypoxanthine phosphoribosyl transferase (HPRT; NM_001034035.1), tyrosine 3‐monooxygenase (YWAHZ; AY970970) and ribosomal protein P0 (RPPO; NM_001012682.1). These housekeeping genes were selected (based on their stability across samples in each treatment group) from a panel of eight candidate housekeeping genes using the geNorm component of the qBase relative quantification analysis software (Hellemans et al. 2007; Soo et al. 2012). Each sample was run in triplicate for each target and housekeeping gene and the mRNA amplification for each sample was determined using Fast SYBR Green Master Mix (Applied Biosystems) in a final volume of 6 μL on a ViiA7 Fast Real‐time PCR system (Applied Biosystems). Each well on the qRT‐PCR plate contained 1 μL of cDNA, 3 μL Fast SYBR Green Master Mix (2×), and 2 μL of forward and reverse primer mixed with differing amounts of H2O depending on the required final primer concentrations. NTCs for each primer set were included on each plate to check for non‐specific amplification. The threshold was set within the exponential growth phase of the amplification curve and the corresponding Ct values were obtained to quantify each reaction.
Table 1.
Primer sequences designed and validated for quantitative real‐time RT‐PCR analysis of fibrosis
| Gene | Primer sequence, 5′–3′ | Accession number |
|---|---|---|
| COL1A |
|
AF129287 |
| COL3A |
|
NM_001076831 |
| TIMP1 |
|
NM_001009319.2 |
| TIMP2 |
|
NM_001166186 |
| TIMP3 |
|
NM_001166187.1 |
FWD, forward; REV, reverse.
Quantification of protein within the right ventricle
Protein extraction
Right ventricle tissue (∼100 mg; Control, n = 6; LGUN, n = 8) was sonicated (John Morris Scientific, SA, Australia) in lysis buffer consisting of 1 mL 100 mg−1 tissue of 1 mm Tris‐HCl (pH = 8, 5 m NaCl, 1% NP‐40, 1 mm sodium orthovanadate, 30 mm NaF, 10 mm sodium tetrapyrophosphate, 10 mm EDTA) and a protease inhibitor tablet (complete Mini; Roche). Samples were then centrifuged at 14,300 g and 4°C for 14 min (Eppendorf Centrifuge 5415, Crown Scientific, Vic., Australia). A Micro BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific Inc., Rockford, MA, USA) was used to determine the protein content of each sample. Bovine serum albumin (BSA; 2 mg mL−1 stock solution) was used to make a standard curve (Wang et al. 2012b; Lie et al. 2013; Botting et al. 2014).
Western blotting
Equal volumes of each sample were subjected to SDS‐PAGE (Lie et al. 2013, 2014; Wang et al. 2013). Proteins in each sample were then transferred onto a nitrocellulose membrane (Hybond ECL, GE Healthcare, NSW, Australia), which was subsequently stained with Ponceau S (0.5% Ponceau in 1% acetic acid) to determine the efficacy of the transfer. The membranes were briefly washed with 7% acetic acid to remove the Ponceau S and then subjected to three 5‐min washes in Tris‐buffered saline (TBS). The membranes were cut according to the size of the proteins and blocked in 5% BSA in TBS with 1% Tween (TBS‐T) for 1 h at room temperature. The membranes then underwent three 5‐min washes in TBS‐T and were incubated with their respective primary antibody: Troponin I (ab19615, Abcam, Cambridge, MA, USA), phospho‐Troponin I (#4004, Cell Signaling Technology, Danvers, MA, USA), phospho‐PLB (#8496, Cell Signaling Technology), CAMKII (#3362, Cell Signaling Technology), phospho‐CAMKII (ab22183, Abcam) and β‐actin [ATCB, horseradish peroxidase (HRP) conjugate, Cell Signaling Technology].
After incubation with the primary antibody, the blots were washed and incubated with the appropriate HRP‐labelled secondary IgG antibody for 1 h at room temperature. Enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) was used to detect the blots. The Western blot was imaged using ImageQuant LAS 4000 (GE Healthcare) and the protein abundance was quantified by densitometry using Image quant software (GE Healthcare).
Quantification of collagen deposition in the right ventricle
Fixed right ventricle samples (Control, n = 4; LGUN, n = 4) were embedded in paraffin and sectioned at 6 μm. Sections were then stained with Picrosirius Red and scanned at 40× magnification using a NanoZoomer‐XR (Hamamatsu, Japan) to produce whole slide images. The total area of Picrosirius Red staining was quantified as a percentage of total tissue area (excluding epicardial surface and intramyocardial vessels) using custom thresholds at 20× magnification (Visiopharm, Hoersholm, Denmark).
Statistical analyses
Data are presented as mean ± SEM and a probability of 5% (P ≤ 0.05) was considered significant for all analyses. To determine the effect of nutritional treatment (Control vs. LGUN) data were analysed using an unpaired t test using STATA (StataCorp, College Station, TX, USA). Linear regression was used to determine the relationship between mean fetal gestational plasma glucose as well as mean arterial blood pressure with mRNA and protein expression (GraphPad Prism version 7.03 for Windows, GraphPad Software, La Jolla, CA, USA). All genes and proteins studied were selected a priori based on their known biological role in cardiac development, growth and previously identified roles in the fetal heart (Morrison et al. 2007; Wang et al. 2011, 2012a, 2012b; Botting et al. 2012).
Results
Fetal outcome
Data regarding fetal weight at post‐mortem, daily blood gases as well as plasma glucose concentrations and blood pressure regulation have previously been reported (Edwards & McMillen, 2001). There was no effect of nutritional regime on fetal weight at 144–145 days gestation, or mean gestational arterial , and pH across late gestation; however, maternal undernutrition resulted in lower fetal plasma glucose concentrations (Fig. 1).
Figure 1. Fetal mean (A) and plasma glucose concentrations (B) across late gestation.
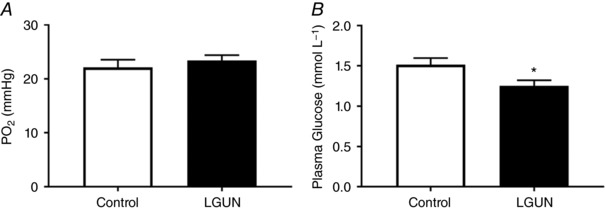
Data are expressed as mean ± SEM and analysed by unpaired t test. Control (n = 6), open bars; LGUN (n = 8), filled bars. * P < 0.05, effect of LGUN. Figure adapted, with permission, from Edwards & McMillen (2001).
Fetal mean arterial blood pressure was measured at two periods during late gestation (115–125 and 135–145 days GA) that were before and after the prepartum surge in fetal plasma cortisol concentration. At both periods, LGUN increased mean arterial blood pressure when compared to Controls (Edwards & McMillen, 2001).
Effect of LGUN on fetal heart development
Maternal undernutrition in late gestation had no effect on absolute or relative heart and ventricle weights (Table 2).
Table 2.
Morphometric cardiac measurements in fetuses from Control and LGUN groups
| Control (n = 6) | LGUN (n = 8) | |
|---|---|---|
| Body weight (kg) | 4.85 ± 0.21 | 4.37 ± 0.19 |
| Heart weight (g kg−1) | 29.83 ± 1.67 | 27.86 ± 1.10 |
| Heart weight: body weight (g kg−1) | 6.35 ± 0.16 | 6.15 ± 0.24 |
| Left ventricle weight (mg) | 15.71 ± 0.65 | 14.65 ± 0.67 |
| Left ventricle: heart weight (mg g−1) | 0.53 ± 0.02 | 0.53 ± 0.01 |
| Right ventricle weight (mg) | 7.85 ± 0.73 | 7.70 ± 0.49 |
| Right ventricle: heart weight (mg g−1) | 0.26 ± 0.01 | 0.23 ± 0.01 |
Values are mean ± SEM. P < 0.05 was considered significant. Data were analysed by an unpaired t test.
LGUN did not change the expression of glucocorticoid signalling molecules
LGUN had no effect on the mRNA expression of the glucocorticoid receptor (GR), 11BHSD1 or 11BHSD2 in the right ventricle (Table 3). There was no significant relationship between mean fetal cortisol concentrations in late gestation and the mRNA expression of GR, 11BHSD1 or 11BHSD2 (Table 3).
Table 3.
mRNA expression of glucocorticoid signalling molecules and their relationship with mean fetal cortisol concentrations in late gestation
| Control (n = 6) | LGUN (n = 8) | ||
|---|---|---|---|
| mRNA expression (MNE) | |||
| GR | 0.465 ± 0.036 | 0.482 ± 0.031 | |
| 11BHSD1 | 0.014 ± 0.002 | 0.020 ± 0.003 | |
| 11BHSD2 | 0.0006 ± 0.0002 | 0.0008 ± 0.0002 | |
| Relationship between mean fetal cortisol concentrations and mRNA expression (MNE) | |||
| Equation | r 2 | P | |
|---|---|---|---|
| GR | y = 0.0009x + 0.4931 | 0.0005 | NS |
| 11BHSD1 | y = −0.0001x + 0.0181 | 0.0024 | NS |
| 11BHSD2 | y = 9.0234E‐005x + 0.0003 | 0.2403 | NS |
Data were analysed by an unpaired t test. Values are mean ± SEM. P < 0.05. MNE, mean normalized expression; NS, not significant.
LGUN increases IGF2R‐related signalling molecules
Insulin‐like growth factor signalling has previously been implicated in normal fetal cardiac growth as well as in hypertrophic signalling in the fetal heart on the background of reduced substrate supply. LGUN had no effect on cardiac mRNA expression of IGF1 and IGF1R (Fig. 2 A, C) but resulted in an increase in cardiac mRNA expression of IGF2 and IGF2R (Fig. 2 B, D).
Figure 2. Normalized mRNA expression of IGF1 (A), IGF2 (B), IGF1R (C) and IGF2R (D).
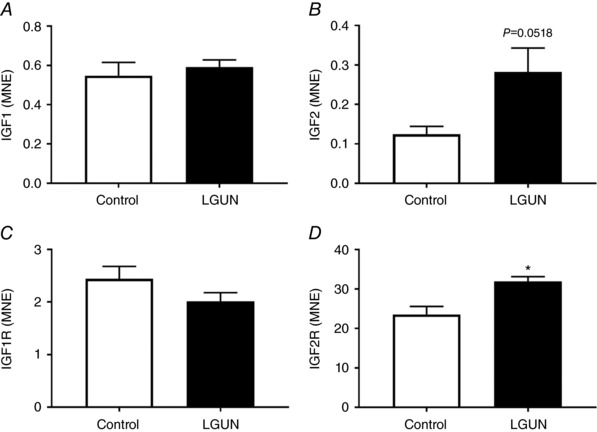
Data were analysed by unpaired t test. Data are expressed as mean ± SEM. Control, unfilled bars (n = 6); LGUN, filled bars (n = 8). MNE, mean normalized expression;* P ≤ 0.05.
There was no significant relationship between fetal mean arterial pressure and the mRNA expression of IGF2 (Fig. 3 A, C) or IGF2R (Fig. 3 B, D) at either age range measured.
Figure 3. Relationship between fetal mean arterial pressure recorded at 115–125 and 135–145 days GA with mRNA expression of IGF2 (A, C) and IGF2R (B, D).
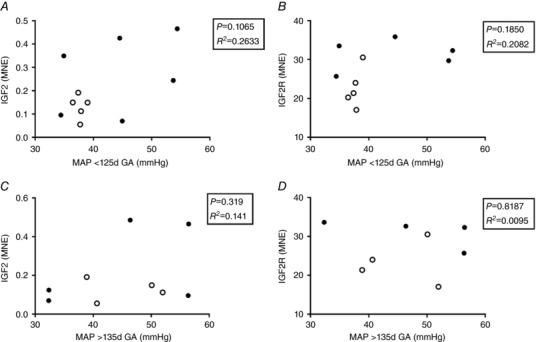
Each circle represents an individual data point. Control, open circles; LGUN, filled circles; MAP, mean arterial pressure; MNE, mean normalized expression; GA, gestational age.
The relationship between mean fetal plasma glucose concentrations and IGF2 mRNA was significantly different between the Control and LGUN groups (Fig. 4 A). The slopes of the lines of best fit for each treatment group were significantly different (P = 0.032). There was an inverse relationship between fetal cardiac IGF2 mRNA expression (y) and fetal glucose concentrations (x) within the LGUN group (P < 0.05, R 2 = 0.574, y = −1.212x + 1.94) but there was no significant relationship within the Control group. The relationship between IGF2R mRNA expression and fetal mean plasma glucose concentrations was not significant for either treatment group (Fig. 4 B).
Figure 4. Relationship between mean fetal plasma glucose concentration and the mRNA expression of IGF2 (A) and IGF2R (B).
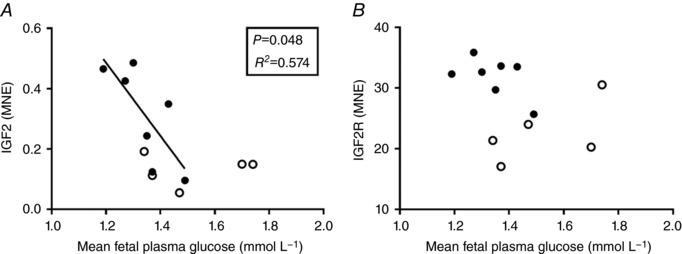
Each circle represents an individual data point. Control, open circles (n = 5); LGUN, filled circles (n = 7); MNE, mean normalized expression.
CAMKII activation
The protein abundance of phosphorylated CAMKII was higher in the right ventricle in the LGUN compared to the Control group (Fig. 5 A). The phosphorylation of CAMKII was positively correlated with both IGF2 (4 E) and IGF2R (Fig. 5 F). There was no difference in the ratio of phosphorylated PKA to total PKA abundance between the Control and LGUN groups (6.54 ± 0.31 vs. 5.84 ± 0.43 au).
Figure 5. Protein abundance of phosphorylated CAMKII (A) is increased as a result of LGUN and positively correlated to the mRNA expression of both IGF2 (E) and IGF2R (F).
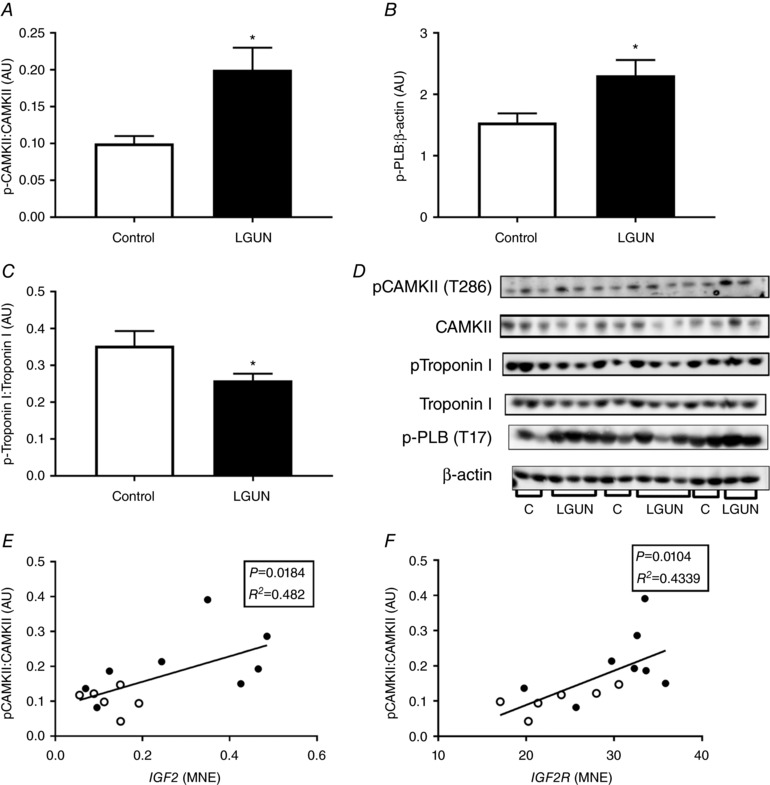
Protein abundance of phospho‐PLB (B) and Troponin I (C) was increased and decreased, respectively, as a result of LGUN. Values are mean ± SEM. *Significantly different from control fetuses (P < 0.05). AU, arbitrary units. Control, open bars/circles (n = 6); LGUN, filled bars/circles (n = 8); MNE, mean normalized expression.
Markers of cardiac contractility
The protein abundance of phospho‐PLB was higher (Fig. 5 B) and phosphorylated Troponin I was lower (Fig. 5 C) in the right ventricles of the LGUN compared to the Control group.
Right ventricular fibrosis
There was an increase in the deposition of interstitial collagen in the right ventricle in the LGUN group (Fig. 7). The increased collagen deposition was associated with an increase in the mRNA expression of COL1A as well as increases in TIMP‐1 and TIMP‐3 but not in COL3A or TIMP‐2 (Fig. 6). COL1A mRNA expression was negatively correlated with the mean fetal plasma glucose concentrations across late gestation (Fig. 6 C). There was also a higher COL1A:COL3A ratio in the LGUN group (Control, 2.242 ± 0.278; LGUN, 3.309 ± 0.228; P < 0.05).
Figure 7. Interstitial collagen deposition (A) determined by picrosirius red staining in Control (B) and LGUN fetuses (C).
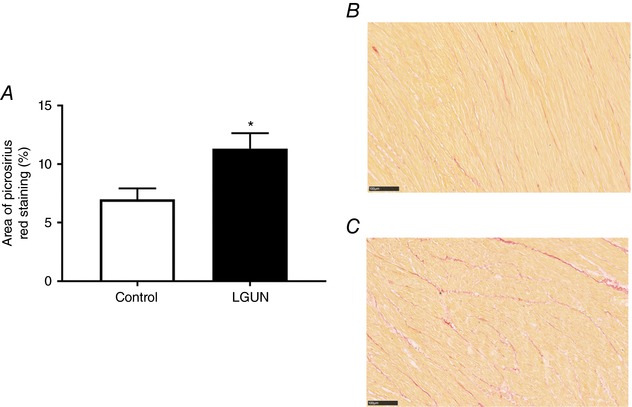
Stained slides scanned for whole slide images using a Nanozoomer at 40× magnification. Scale bar on representative images = 100 μm. Data analysed by unpaired t test. Data presented as mean ± SEM. P < 0.05 was considered significant. Control, unfilled bars (n = 4); LGUN, filled bars (n = 4). [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 6. Normalized cardiac mRNA expression of COL1A (A), COL3A1 (B), TIMP‐1(D), TIMP‐2 (E) and TIMP‐3 (F).
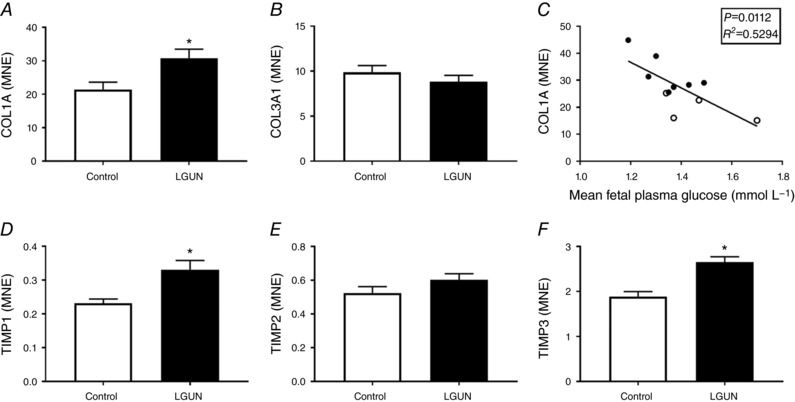
Relationship between cardiac COL1A mRNA expression and mean fetal plasma concentrations in late gestation (C). Values are mean ± SEM. Control, open bars/circles (n = 6); LGUN, filled bars/circles (n = 8); MNE, mean normalized expression. *Significantly different from control fetuses (P < 0.05).
Discussion
Cardiovascular health in adult life is related to nutritional status in utero, with a range of studies showing that there is up to a 50% greater risk of cardiovascular disease in adult life if an individual was IUGR (Barker et al. 1989, 2002, 2005; Fall et al. 1995; McMillen & Robinson, 2005). LBW is a proxy for low substrate supply in fetal life and has been correlated with poorer cardiovascular outcomes in postnatal life (Barker et al. 1989, 2000). LBW as a result of IUGR is most commonly caused by either placental insufficiency, where fetal nutrient and oxygen supply are each restricted, or by maternal undernutrition, where fetal nutrient supply alone is decreased. We have previously used a model of placental insufficiency to demonstrate that there is increased hypertrophic signalling in the left ventricle in the heart of the chronically hypoxaemic fetus and that there is an increase in relative left ventricular mass after birth. The majority of studies investigating the effects of decreased fetal nutrient supply on cardiac development have focused on left ventricle development. Although the left ventricle is dominant in postnatal life, pumping oxygenated blood into systemic circulation, cardiac output and stroke volumes are equal in the left and right ventricle in the fetus (Hamill et al. 2011). There have been relatively few studies focusing on the impact of IUGR on the growth and development of the right ventricle of the fetal heart.
Unlike models of early‐onset placental insufficiency and moderate maternal undernutrition (15% maternal global nutrient restriction; Hawkins et al. 2000), LGUN resulted in increased fetal mean arterial blood pressure at both 115–125 and 135–145 days GA. This is consistent with rodent models of fetal protein (Woodall et al. 1996) and global nutrient restriction (Langley & Jackson, 1994) in which offspring had increased blood pressure after birth. The increase in postnatal blood pressure in the rodent model of nutrient restriction is due to increased glucocorticoid signalling or sensitivity because blocking maternal corticosterone biosynthesis in rats exposed to a low‐protein diet during gestation prevented the increase in postnatal blood pressure. Although LGUN did not increase fetal plasma cortisol concentrations throughout late gestation, mean arterial blood pressure was positively correlated with fetal plasma cortisol concentrations between 135 and 145 days of gestation, but not from 115 to 125 days of gestation (Edwards & McMillen, 2001).
It has been demonstrated that increased systolic pressure in late gestation causes cardiomyocyte hypertrophy in the right ventricle (Barbera et al. 2000). However, in the present study, there was no change in the absolute or relative weight of the right ventricle, suggesting that either there was no hypertrophy of cardiomyocytes in fetal life or there was an increase in cardiomyocyte size balanced by a decrease in cardiomyocyte number. Future studies would be required to answer this question because we were not able to determine cardiomyocyte size as cardiomyocytes were not isolated during tissue collection and alternative methods for calculating cardiomyocyte size from fixed tissue samples do not account for tissue shrinkage in paraformaldehyde‐fixed and paraffin‐embedded samples (Bruel & Nyengaard, 2005).
Cortisol can increase both cell cycle entry of cardiomyocytes (Giraud et al. 2006), ventricle thickness and weight (Reini et al. 2008) of the developing heart in the absence of hypertension. Activation of the glucocorticoid receptor (GR) by cortisol is regulated by the enzymatic activity of both 11BHSD1 and 11BHSD2. Both GR and 11BHSD1 are abundantly expressed in cardiomyocytes and blood vessels within the fetal heart, whereas 11BHSD2 is localized more predominantly in blood vessels than cardiomyocytes (Reini et al. 2006), demonstrating that cortisol has access to the developing heart. However, although LGUN increased maternal plasma cortisol concentrations for the first 10 days of the nutritional regime; fetal plasma cortisol concentrations were not increased. Furthermore, we observed no change in the cardiac gene expression of GR, 11BHSD1 or 11BHSD2 in the fetal heart, suggesting that systemic cortisol was not having a local effect within the right ventricle.
IGF2R signalling induces cardiac hypertrophy through a plethora of downstream effectors such as CAMKII, PKC‐α and p44/42 MAP kinase (ERK) (Chu et al. 2008; Wang et al. 2012a). Suboptimal intrauterine conditions can result in epigenetic changes to the IGF2/H19 and IGF2R genes (Li et al. 2010; Zhang et al. 2010a). In a sheep model of early‐onset IUGR, in which the fetus is chronically hypoxaemic in late gestation, there is increased IGF2/IGF2R‐mediated hypertrophic signalling in the left ventricle and this persists into postnatal life where LBW lambs also have relatively larger left ventricles due to increased CAMKII signalling (Wang et al. 2012b, 2015). In a model of maternal nutrient restriction from early to mid‐gestation in sheep, ventricular enlargement and thickening were evident along with increased expression of IGF1R and IGF2R in mid‐gestation (Dong et al. 2005). LGUN had no effect on either absolute or relative heart or ventricle weights, which is consistent with an early‐onset model of placental insufficiency in which increased cardiomyocyte size in late gestation was not accompanied by increased ventricular weight (Morrison et al. 2007). Increased IGF2R mRNA expression and downstream CAMKII activation without increased expression of IGF1 or IGF1R suggests that LGUN may cause pathological hypertrophic signalling (Chu et al. 2008). Indeed, CAMKII activation is involved in both hypertrophy and heart failure (Boknik et al. 2001; Zhang et al. 2005). Although not entirely similar to the LGUN‐induced increase in mean arterial pressure of the present study, transverse aortic constriction in mice resulting in pressure overload and cardiac hypertrophy also increased CAMKII activation (Colomer et al. 2003). Increased activation of CAMKII has also been reported in spontaneously hypertensive rats and as a result of increased electrical field stimulation (Zhang et al. 2010b). Further to this and similar to our results, pressure overload increased phosphorylation of PLB at the CAMKII phosphorylation site (Thr 17) (Zhang et al. 2003). The activation of CAMKII in the present study along with the decreased phosphorylation of Troponin I are consistent with the molecular changes observed in the left ventricle of LBW lambs (Wang et al. 2015).
The current study provides evidence that LGUN may alter contractility in the right ventricle. When un‐phosphorylated, PLB acts to inhibit sarcoplasmic reticulum Ca2+‐ATPase (SERC2A) thereby limiting calcium (Ca2+) uptake by the sarcoplasmic reticulum. However, when phosphorylated either by CAMKII or PKA, the inhibition on SERC2A by PLB is relieved. In the present study, LGUN increased the phosphorylation of PLB. This suggests that LGUN may increase the rate of relaxation (due to decreased cystostolic Ca2+) of the right ventricle through the activation of CAMKII and subsequent phosphorylation of PLB. It should be noted that PLB may also be phosphorylated by PKA. However, in the present study this does not appear to be the case as LGUN did not increase either total PKA or phosphorylated PKA levels. Interestingly, PKA is the downstream mediator of increased IGF‐2R hypertrophic signalling in the left ventricle when Leu27IGF‐2 (specific for IGF‐2R activation) was infused into the left circumflex coronary artery of late‐gestation fetal sheep (Wang et al. 2012b).
A more moderate (30%) maternal nutrient restriction in the non‐human primate causes signs of both left and right ventricle dysfunction in the offspring, including a decreased ejection fraction, wall shortening and increased chamber volume as assessed using magnetic resonance imaging (Kuo et al. 2017a, b ). Interestingly, these two partner studies highlighted that right ventricular function was more severely impacted by maternal nutrient restriction than left ventricular function. Consistent with pathological hypertrophy and cardiac dysfunction (Weber et al. 1993), we have shown that LGUN increased interstitial collagen deposition in the right ventricle and this was accompanied by increased mRNA expression of COL1A, TIMP1 and TIMP3. Notably, the stiffness of collagen type 1 (measured as COL1A) is 30 times greater than that of a cardiomyocyte (MacKenna et al. 1997), suggesting that the right ventricles of LGUN fetuses may be much stiffer than those of controls. Furthermore, the expression of COL1A was inversely correlated with mean fetal plasma glucose concentrations. Interestingly, there was no difference in COL3A mRNA expression between the Control and LGUN groups; however, the increase in COL1A:COL3A in the LGUN group suggests decreased cardiac contractility and compliance (Wei et al. 1999). Furthermore, despite the previously reported sex effect, our results are consistent with an increased prevalence of myocardial fibrosis in the hearts of IUGR male non‐human primates (Muralimanoharan et al. 2017). These data along with the changes to contractility proteins (PLB and Troponin I) in the present study may explain the emergence of dysfunction in the right ventricle after birth in the IUGR non‐human primate (Kuo et al. 2017a).
In summary, we have shown that on a background of a normoxemic blood gas status, reduced plasma glucose concentrations may activate the IGF2/IGF2R signalling pathway within the right ventricle of the fetal heart. Activation of the IGF2R pathway in the fetal heart as a result of a reduced fetal substrate supply is well described. However, to the best of our knowledge this study is the first to implicate CAMKII as a downstream mediator of the detrimental effects of reduced fetal glucose supply in the developing fetal heart. Furthermore, LGUN increases interstitial collagen deposition and alters the abundance of contractility proteins in the right ventricle. This may predispose the right ventricle to altered contractility and dysfunction in postnatal life. However, the lack of both fetal and maternal in vivo or ex vivo cardiac functional data is a key limitation of the present study. Indeed, maternal cardiovascular conditions during pregnancy, such as gestational hypertension, have the potential to impact offspring cardiovascular parameters, with offspring born to hypertensive mothers having higher blood pressure (Geelhoed et al. 2010). As such, maternal haemodynamic changes during the LGUN regime may influence fetal cardiac development. Future studies would aim to measure maternal blood pressure during the LGUN regime as well as fetal functional cardiac characteristics either by magnetic resonance imaging, ultrasound or ex vivo Langendorf.
Placental insufficiency is associated with a decrease in oxygen and glucose supply to the fetus. The results in the current study suggest that a decrease in fetal plasma glucose may play a role in up‐regulating IGF2R/CAMKII signalling and fibrosis in the absence of fetal hypoxaemia. It is therefore possible that decreases in fetal glucose and oxygen have separate but additive impacts on the developing heart and hence each contribute to an increased risk of cardiovascular disease in adult life.
Additional information
Competing interests
The authors have no conflicts of interest.
Author contributions
Conception or design of the work: JRTD, ICM, JM. Acquisition, analysis or interpretation of data for the work: JRTD, ICM, JM. Drafting the work or revising it critically for important intellectual content: JRTD, ICM, JM. Final approval of the version to be published: JRTD, ICM, JM. Agreement to be accountable for all aspects of the work: JRTD, ICM, JM
Funding
The animal component of the work was funded by an NHMRC Program Grant (ICM). JRTD was funded by an Australian Government Research Training Program (RTP) scholarship. JLM was funded by an NHMRC Career Development Fellowship (APP1066916) and an Australian Research Council Future Fellowship (Level 3; FT170100431).
Acknowledgements
We acknowledge the contribution of Lisa Edwards to this study and the assistance of Anne Jurisevic in performing the surgical procedures, and providing expert post‐surgical care of the ewe and her fetus. We thank Stacey Holman for assistance with Western blotting.
Biographies
Jack R.T. Darby is a PhD student in the Early Origins of Adult Health Research Group in the School of Pharmacy and Medical Sciences at the University of South Australia, Adelaide, Australia. His research is focused on understanding the mechanisms by which IUGR offspring are at an increased risk of developing cardiovascular disease in adult life.

Janna L. Morrison is Head of the Early Origins of Adult Health Research Group in the School of Pharmacy and Medical Sciences at the University of South Australia, Adelaide, Australia. Professor Morrison has been funded as a fellow by the Heart Foundation (2004‐2013), NHMRC Career Development Fellow (2014‐2017), and is currently funded by an Australian Research Council Future Fellowship. Her work focuses on examining the link between low birth weight and heart disease in adulthood. She is a fellow of the Cardiovascular Section of the American Physiological Society (2015).
Edited by: Kim Barrett & Jeffrey Ardell
Linked articles This article is highlighted by a Perspective by Clarke & Nathanielsz. To read this Perspective, visit https://doi.org/10.1113/JP276234.
This is an Editor's Choice article from the 15 June 2018 issue.
References
- Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ & Thornburg KL (2000). Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol 279, R1157–1164. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Eriksson JG, Forsen T & Osmond C (2002). Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol 31, 1235–1239. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Forsen TJ, Kajantie E & Eriksson JG (2005). Trajectories of growth among children who have coronary events as adults. N Engl J Med 353, 1802–1809. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B & Simmonds SJ (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580. [DOI] [PubMed] [Google Scholar]
- Barker DJP ( 2000). In utero programming of cardiovascular disease. Theriogenology 53, 555–574. [DOI] [PubMed] [Google Scholar]
- Boknik P, Heinroth‐Hoffmann I, Kirchhefer U, Knapp J, Linck B, Luss H, Muller T, Schmitz W, Brodde O & Neumann J (2001). Enhanced protein phosphorylation in hypertensive hypertrophy. Cardiovasc Res 51, 717–728. [DOI] [PubMed] [Google Scholar]
- Botting KJ, McMillen IC, Forbes H, Nyengaard JR & Morrison JL (2014). Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia‐responsive genes. J Am Heart Assoc 3, e000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botting KJ, Wang KC, Padhee M, McMillen IC, Summers‐Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA & Morrison JL (2012). Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin Exp Pharmacol Physiol 39, 814–823. [DOI] [PubMed] [Google Scholar]
- Bruel A & Nyengaard JR (2005). Design‐based stereological estimation of the total number of cardiac myocytes in histological sections. Basic Res Cardiol 100, 311–319. [DOI] [PubMed] [Google Scholar]
- Burlew BS & Weber KT (2002). Cardiac fibrosis as a cause of diastolic dysfunction. Herz 27, 92–98. [DOI] [PubMed] [Google Scholar]
- Burrell JH, Boyn AM, Kumarasamy V, Hsieh A, Head SI & Lumbers ER (2003). Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat Rec A Disc Mol Cell Evol Biol 274, 952–961. [DOI] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J & Wittwer CT (2009). The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem 55, 611–622. [DOI] [PubMed] [Google Scholar]
- Chu CH, Huang CY, Lu MC, Lin JA, Tsai FJ, Tsai CH, Chu CY, Kuo WH, Chen LM & Chen LY (2009). Enhancement of AG1024‐induced H9c2 cardiomyoblast cell apoptosis via the interaction of IGF2R with Galpha proteins and its downstream PKA and PLC‐β modulators by IGF‐II. Chin J Physiol 52, 31–37. [DOI] [PubMed] [Google Scholar]
- Chu CH, Tzang BS, Chen LM, Kuo CH, Cheng YC, Chen LY, Tsai FJ, Tsai CH, Kuo WW & Huang CY (2008). IGF‐II/mannose‐6‐phosphate receptor signaling induced cell hypertrophy and atrial natriuretic peptide/BNP expression via Galphaq interaction and protein kinase C‐α /CaMKII activation in H9c2 cardiomyoblast cells. J Endocrinol 197, 381–390. [DOI] [PubMed] [Google Scholar]
- Cleutjens JP, Verluyten MJ, Smiths JF & Daemen MJ (1995). Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 147, 325–338. [PMC free article] [PubMed] [Google Scholar]
- Colomer JM, Mao L, Rockman HA & Means AR (2003). Pressure overload selectively up‐regulates Ca2+/calmodulin‐dependent protein kinase II in vivo . Mol Endocrinol 17, 183–192. [DOI] [PubMed] [Google Scholar]
- Danielson L, McMillen IC, Dyer JL & Morrison JL (2005). Restriction of placental growth results in greater hypotensive response to α‐adrenergic blockade in fetal sheep during late gestation. J Physiol 563, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Ford SP, Fang CX, Nijland MJ, Nathanielsz PW & Ren J (2005). Maternal nutrient restriction during early to mid gestation up‐regulates cardiac insulin‐like growth factor (IGF) receptors associated with enlarged ventricular size in fetal sheep. Growth Horm IGF Res 15, 291–299. [DOI] [PubMed] [Google Scholar]
- Edwards LJ & McMillen IC (2001). Maternal undernutrition increases arterial blood pressure in the sheep fetus during late gestation. J Physiol 533, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fall CH, Vijayakumar M, Barker DJ, Osmond C & Duggleby S (1995). Weight in infancy and prevalence of coronary heart disease in adult life. BMJ 310, 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geelhoed JJ, Fraser A, Tilling K, Benfield L, Davey Smith G, Sattar N, Nelson SM & Lawlor DA (2010). Preeclampsia and gestational hypertension are associated with childhood blood pressure independently of family adiposity measures: the Avon Longitudinal Study of Parents and Children. Circulation 122, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili S, Morrison JL & McMillen IC (2009). Intrauterine growth restriction and differential patterns of hepatic growth and expression of IGF1, PCK2, and HSDL1 mRNA in the sheep fetus in late gestation. Biol Reprod 80, 1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud GD, Louey S, Jonker S, Schultz J & Thornburg KL (2006). Cortisol stimulates cell cycle activity in the cardiomyocyte of the sheep fetus. Endocrinology 147, 3643–3649. [DOI] [PubMed] [Google Scholar]
- Giussani DA (2016). The fetal brain sparing response to hypoxia: physiological mechanisms. J Physiol 594, 1215–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill N, Yeo L, Romero R, Hassan SS, Myers SA, Mittal P, Kusanovic JP, Balasubramaniam M, Chaiworapongsa T, Vaisbuch E, Espinoza J, Gotsch F, Goncalves LF & Lee W (2011). Fetal cardiac ventricular volume, cardiac output, and ejection fraction determined with 4‐dimensional ultrasound using spatiotemporal image correlation and virtual organ computer‐aided analysis. Am J Obstet Gynecol 205, 76.e1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins P, Steyn C, Ozaki T, Saito T, Noakes DE & Hanson MA (2000). Effect of maternal undernutrition in early gestation on ovine fetal blood pressure and cardiovascular reflexes. Am J Physiol Regul Integr Comp Physiol 279, R340–348. [DOI] [PubMed] [Google Scholar]
- Hellemans J, Mortier G, De Paepe A, Speleman F & Vandesompele J (2007). qBase relative quantification framework and software for management and automated analysis of real‐time quantitative PCR data. Genome Biol 8, R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R & Sheppard R (2006). Fibrosis in heart disease: understanding the role of transforming growth factor‐β in cardiomyopathy, valvular disease and arrhythmia. Immunology 118, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M, Shimizu M, Ino H, Okeie K, Yamaguchi M, Funjno N, Mabuchi H & Nakanishi I (2001). Collagen remodeling and cardiac dysfunction in patients with hypertrophic cardiomyopathy: the significance of type III and VI collagens. Clin Cardiol 24, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AH, Li C, Huber HF, Schwab M, Nathanielsz PW & Clarke GD (2017a). Maternal nutrient restriction during pregnancy and lactation leads to impaired right ventricular function in young adult baboons. J Physiol 595, 4245–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AH, Li C, Li J, Huber HF, Nathanielsz PW & Clarke GD (2017b). Cardiac remodelling in a baboon model of intrauterine growth restriction mimics accelerated ageing. J Physiol 595, 1093–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley SC & Jackson AA (1994). Increased systolic blood pressure in adult rats induced by fetal exposure to maternal low protein diets. Clin Sci 86, 217–222; discussion 121. [DOI] [PubMed] [Google Scholar]
- Li CC, Maloney CA, Cropley JE & Suter CM (2010). Epigenetic programming by maternal nutrition: shaping future generations. Epigenomics 2, 539–549. [DOI] [PubMed] [Google Scholar]
- Lie S, Hui M, McMillen IC, Muhlhausler BS, Posterino GS, Dunn SL, Wang KC, Botting KJ & Morrison JL (2014). Exposure to rosiglitazone, a PPAR‐gamma agonist, in late gestation reduces the abundance of factors regulating cardiac metabolism and cardiomyocyte size in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 306, R429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie S, Sim SM, McMillen IC, Williams‐Wyss O, MacLaughlin SM, Kleemann DO, Walker SK, Roberts CT & Morrison JL (2013). Maternal undernutrition around the time of conception and embryo number each impact on the abundance of key regulators of cardiac growth and metabolism in the fetal sheep heart. J Dev Orig Health Disease 4, 377–390. [DOI] [PubMed] [Google Scholar]
- MacKenna DA, Vaplon SM & McCulloch AD (1997). Microstructural model of perimysial collagen fibers for resting myocardial mechanics during ventricular filling. Am J Physiol 273, H1576–1586. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Morrison JL, McMillen IC & Orgeig S (2014). Intrafetal glucose infusion alters glucocorticoid signalling and reduces surfactant protein mRNA expression in the lung of the late gestation sheep fetus. Am J Physiol Regul Integr Comp Physiol 307, R538–545. [DOI] [PubMed] [Google Scholar]
- McGillick EV, Orgeig S, McMillen IC & Morrison JL (2013). The fetal sheep lung does not respond to cortisol infusion during the late canalicular phase of development. Physiol Rep 1, e00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillen IC & Robinson JS (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85, 571–633. [DOI] [PubMed] [Google Scholar]
- Morrison JL ( 2008). Sheep models of intrauterine growth restriction: fetal adaptations and consequences. Clin Exp Pharmacol Physiol 35, 730–743. [DOI] [PubMed] [Google Scholar]
- Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL & McMillen IC (2007). Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 293, R306–313. [DOI] [PubMed] [Google Scholar]
- Muhlhausler BS, Adam CL, Marrocco EM, Findlay PA, Roberts CT, McFarlane JR, Kauter KG & McMillen IC (2005). Impact of glucose infusion on the structural and functional characteristics of adipose tissue and on hypothalamic gene expression for appetite regulatory neuropeptides in the sheep fetus during late gestation. J Physiol 565, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralimanoharan S, Li C, Nakayasu ES, Casey CP, Metz TO, Nathanielsz PW & Maloyan A (2017). Sexual dimorphism in the fetal cardiac response to maternal nutrient restriction. J Mol Cell Cardiol 108, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak JF, Holman BL, Wynne J & Colucci WS (1983). Right ventricular ejection fraction: an indicator of increased mortality in patients with congestive heart failure associated with coronary artery disease. J Am Coll Cardiol 2, 217–224. [DOI] [PubMed] [Google Scholar]
- Reini SA, Dutta G, Wood CE & Keller‐Wood M (2008). Cardiac corticosteroid receptors mediate the enlargement of the ovine fetal heart induced by chronic increases in maternal cortisol. J Endocrinol 198, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reini SA, Wood CE, Jensen E & Keller‐Wood M (2006). Increased maternal cortisol in late‐gestation ewes decreases fetal cardiac expression of 11beta‐HSD2 mRNA and the ratio of AT1 to AT2 receptor mRNA. Am J Physiol Regul Integr Comp Physiol 291, R1708‐1716. [DOI] [PubMed] [Google Scholar]
- Russell WMS & Burch RL (1959). The principles of humane experimental technique. Methuen, London. [Google Scholar]
- Segers VF, Brutsaert DL & De Keulenaer GW (2012). Pulmonary hypertension and right heart failure in heart failure with preserved left ventricular ejection fraction: pathophysiology and natural history. Curr Opin Cardiol 27, 273–280. [DOI] [PubMed] [Google Scholar]
- Soo PS, Hiscock J, Botting KJ, Roberts CT, Davey AK & Morrison JL (2012). Maternal undernutrition reduces P‐glycoprotein in guinea pig placenta and developing brain in late gestation. Reprod Toxicol 33, 374–381. [DOI] [PubMed] [Google Scholar]
- Wang KC, Botting KJ, Padhee M, Zhang S, McMillen IC, Suter CM, Brooks DA & Morrison JL (2012a). Early origins of heart disease: low birth weight and the role of the insulin‐like growth factor system in cardiac hypertrophy. Clin Exp Pharmacol Physiol 39, 958–964. [DOI] [PubMed] [Google Scholar]
- Wang KC, Brooks DA, Thornburg KL & Morrison JL (2012b). Activation of IGF‐2R stimulates cardiomyocyte hypertrophy in the late gestation sheep fetus. J Physiol 590, 5425–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Lim CH, McMillen IC, Duffield JA, Brooks DA & Morrison JL (2013). Alteration of cardiac glucose metabolism in association to low birth weight: experimental evidence in lambs with left ventricular hypertrophy. Metabolism 62, 1662–1672. [DOI] [PubMed] [Google Scholar]
- Wang KC, Tosh DN, Zhang S, McMillen IC, Duffield JA, Brooks DA & Morrison JL (2015). IGF‐2R‐Galphaq signaling and cardiac hypertrophy in the low‐birth‐weight lamb. Am J Physiol Regul Integr Comp Physiol 308, R627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KC, Zhang L, McMillen IC, Botting KJ, Duffield JA, Zhang S, Suter CM, Brooks DA & Morrison JL (2011). Fetal growth restriction and the programming of heart growth and cardiac insulin‐like growth factor 2 expression in the lamb. J Physiol 589, 4709–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber KT (1989). Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13, 1637–1652. [DOI] [PubMed] [Google Scholar]
- Weber KT, Brilla CG & Janicki JS (1993). Myocardial fibrosis: functional significance and regulatory factors. Cardiovasc Res 27, 341–348. [DOI] [PubMed] [Google Scholar]
- Wei S, Chow LT, Shum IO, Qin L & Sanderson JE (1999). Left and right ventricular collagen type I/III ratios and remodeling post‐myocardial infarction. J Card Fail 5, 117–126. [DOI] [PubMed] [Google Scholar]
- Woodall SM, Johnston BM, Breier BH & Gluckman PD (1996). Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatr Res 40, 438–443. [DOI] [PubMed] [Google Scholar]
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr. , Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ & Anderson ME (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 11, 409–417. [DOI] [PubMed] [Google Scholar]
- Zhang S, Rattanatray L, MacLaughlin SM, Cropley JE, Suter CM, Molloy L, Kleemann D, Walker SK, Muhlhausler BS, Morrison JL & McMillen IC (2010a). Periconceptional undernutrition in normal and overweight ewes leads to increased adrenal growth and epigenetic changes in adrenal IGF2/H19 gene in offspring. FASEB J 24, 2772–2782. [DOI] [PubMed] [Google Scholar]
- Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr. , Bers DM & Brown JH (2003). The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 92, 912–919. [DOI] [PubMed] [Google Scholar]
- Zhang W, Qi F, Chen DQ, Xiao WY, Wang J & Zhu WZ (2010b). Ca2+/calmodulin‐dependent protein kinase IIδ orchestrates G‐protein‐coupled receptor and electric field stimulation‐induced cardiomyocyte hypertrophy. Clin Exp Pharmacol Physiol 37, 795–802. [DOI] [PubMed] [Google Scholar]