

Summary
T‐cell immunoglobulin and mucin domain 3 (Tim‐3) is a surface receptor expressed by T helper type 1 (Th1) effector CD4 T cells, which are critical for defence against intracellular pathogens and have been implicated in autoimmune disease. Previous studies showed that Tim‐3 expression makes Th1 cells more susceptible to apoptosis and also marks functionally impaired T cells that arise due to chronic stimulation. However, other studies suggested that Tim‐3‐expressing Th1 cells do not always have these properties. To further define the relationship between Tim‐3 and Th1 cell function, we analysed the characteristics of Th1 cells that expressed Tim‐3 in response to brief stimulation in vitro or an acute viral infection in vivo. As expected, cultured CD4 T cells began expressing Tim‐3 during Th1 differentiation and secondary stimulation generated Tim‐3− and Tim‐3+ fractions that were separated and further analysed. When injected into naive mice, Tim‐3+ cells down‐regulated Tim‐3 and survived equally well compared with Tim‐3‐ cells. Further, Tim‐3− and Tim‐3+ Th1 cells had similar functional responses when transferred into naive mice that were subsequently infected with lymphocytic choriomeningitis virus (LCMV). Cultured Th1 cells that expressed Tim‐3 following T‐cell receptor stimulation had a greater capacity to express signature Th1 cytokines than their Tim‐3− counterparts and showed differential expression of genes that regulate CD4 T‐cell function. Consistent with these findings, Tim‐3+ Th1 cells generated in response to LCMV infection displayed augmented effector function relative to Tim‐3− cells. These results suggest that Tim‐3 expression by Th1 cells responding to acute stimulation can mark cells that are functionally competent and have an augmented ability to produce cytokines.
Keywords: CD4 cell, cytokines, inhibitory/activating receptors, T helper type 1, viral
Abbreviations
- B6
C57BL/6J
- FBS
fetal bovine serum
- IFN‐γ
interferon‐γ
- LCMV‐Arm
Armstrong strain of LCMV
- LCMV
lymphocytic choriomeningitis virus
- TCR
T‐cell receptor
- Th1 cells
Th1‐type effector CD4 T cells
- Tim
T‐cell immunoglobulin and mucin domain
- TNF
tumour necrosis factor
Introduction
Tim‐3 is a receptor expressed on the surface of immune cells that probably regulates both the innate and adaptive arms of the immune system.1, 2, 3 This molecule is a member of the T‐cell immunoglobulin and mucin domain (Tim) family, which includes three proteins in humans and four in mice.4, 5 A hallmark feature of the Tims is their extracellular region, which consists of an N‐terminal immunoglobulin variable region‐like domain and a glycosylated mucin‐like region. Tims also contain an intracellular cytoplasmic tail that varies in length between family members and can be phosphorylated by non‐receptor tyrosine kinases. Through their immunoglobulin variable region‐like domains, Tims are able to recognize the apoptotic cell marker phosphatidylserine and promote engulfment of apoptotic cells by phagocytes.6, 7, 8, 9 Apart from this role, biochemical studies have suggested that the Tims regulate the function of T cells and other immune cell types by tuning intracellular signalling in either a positive or negative manner.10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20
Tim‐3 was first discovered by a screen for molecules that are selectively expressed on the surface of T helper type 1 (Th1) effector CD4 T cells (Th1 cells),21 which are one of several effector cell types that can be formed by activated CD4 T cells during an immune response.22, 23 The generation of effector CD4 T cells is initiated by interaction between naive CD4 T cells and antigen‐presenting cells, which display peptide antigen–MHC II complexes that can engage the T‐cell receptor (TCR) and also provide ligands for co‐stimulatory receptors expressed by T cells.24, 25 CD4 T cells that receive these signals will proliferate and differentiate into effector cells as directed by cytokines in the local environment. For Th1 cells, differentiation is driven by the cytokines interleukin‐12 (IL‐12), IL‐27 and interferon‐γ (IFN‐γ),26, 27, 28 which induce activation of signal transducer and activator of transcription family transcription factors.29 These proteins, together with the transcription factor T‐bet and other transcriptional regulators, establish the gene expression programme required for Th1 cell formation.30 Th1 cells are critical for defence against pathogens that replicate within host cells, which include viruses and certain types of bacteria, fungi and protozoa. In addition, Th1 cell responses driven by recognition of self‐antigens probably contribute to the pathogenesis of autoimmune disorders such as type 1 diabetes and multiple sclerosis.31, 32, 33 Given these roles, it is important to elucidate pathways that regulate Th1 cell responses, which could unveil therapeutic approaches for manipulating their function.
Since its discovery, much effort has been made to understand how Tim‐3 influences Th1 cell function. The glycan‐binding protein Galectin‐9 can recognize carbohydrate chains attached to Tim‐3, and in vitro studies suggested that this interaction causes Tim‐3‐expressing Th1 cells to undergo apoptosis, so suppressing immune responses driven by these cells.34, 35 However, other in vitro studies showed that Galectin‐9 can induce apoptosis and augment cytokine production by CD4 T cells through Tim‐3‐independent pathways,36 whereas other reports suggested that Galectin‐9 influences CD4 T‐cell function through multiple pathways.37, 38, 39 Nonetheless, a number of in vivo studies using Tim‐3 antibodies or other agents thought to block Tim‐3–ligand interactions have provided support for the idea that Tim‐3 acts in some way to restrain Th1‐dependent immune responses.3
In addition to Th1 cells, Tim‐3 is expressed by functionally impaired, or exhausted, CD4 and CD8 T cells,40 which are generated during immune responses to chronic infections41, 42, 43, 44 or cancer.45, 46, 47 These findings established that Tim‐3 serves as a marker for exhausted T cells and also provided evidence that Tim‐3 acts together with other molecules to enforce the exhausted state. In contrast, Tim‐3 expression was associated with enhanced, rather than impaired, effector function by T cells from patients acutely infected with Mycobacterium tuberculosis.48 These latter findings suggest that Tim‐3 expression by T cells can be uncoupled from exhaustion and may, in some settings, be linked to augmented functionality.
In this study, we sought to further define the relationship between Tim‐3 expression and CD4 T‐cell function. To pursue this, we used in vitro and in vivo approaches to analyse the properties of Th1 cells that expressed Tim‐3 following acute, rather than chronic, stimulation. We found that Tim‐3+ Th1 cells generated in this manner were not more susceptible to apoptosis relative to those that did not express Tim‐3. In addition, Tim‐3+ Th1 cells generated by acute stimulation could mount robust responses to a viral infection, suggesting that these cells were not functionally impaired. Consistent with this conclusion, Th1 cells that expressed Tim‐3 had augmented, rather than impaired, functional responses to acute stimulation in vitro or in vivo relative to Tim‐3− cells. Hence, Tim‐3 expression by Th1 cells does not always correlate with increased apoptosis or functional impairment. In addition, these findings support the conclusion that Tim‐3 expression by acutely stimulated Th1 cells can be a marker for cells with enhanced effector function.
Materials and methods
Mice
Naive mice were housed in a specific pathogen‐free facility. For infection studies, mice were housed under animal biosafety level 2 conditions. Wild‐type C57BL/6J (B6) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). SMARTA mice49 expressing the Thy1.1 (CD90.1) variant of the surface protein Thy1 were provided by Dr Steven Varga (University of Iowa). All experimental procedures were approved by the University of Iowa Institutional Animal Care and Use Committee.
LCMV infection
The Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) was provided by Dr Steven Varga (University of Iowa) and was propagated using standard methods. Mice were injected intraperitoneally with 2 × 105 plaque‐forming units of LCMV‐Arm and analysed 8 days later.
Flow cytometric analysis
Splenocyte suspensions were generated by pushing the organ through a 70‐μm wire mesh followed by depletion of red blood cells using Pharm Lyse (BD Biosciences, San Jose, CA). Cells were resuspended in stain buffer [PBS containing 3% fetal bovine serum (FBS)] and incubated with anti‐mouse CD16/32 (eBioscience, San Diego, CA) to block non‐specific antibody binding. Cells were incubated with fluorochrome‐conjugated antibodies for 30 min on ice and washed twice with stain buffer. Flow cytometric analysis was performed using an LSR II (BD Biosciences) and collected data were analysed using flowjo (Tree Star, Ashland, OR). For all data analysis, debris and dead cells were excluded by gates drawn on plots of forward scatter area versus side scatter and cell doublets were excluded by gates drawn on plots of forward scatter area versus forward scatter width. Fluorochrome‐conjugated antibodies were purchased from BD Biosciences, eBioscience, BioLegend (San Diego, CA) or R&D Systems (Minneapolis, MN). Antibody clones used were: CD4 (RM4‐5), CD107a (1D4B), Granzyme B (GB11), IFN‐γ (XMG1.2), IL‐10 (JES5‐16E3), Ly6C (HK1.4), Tim‐3 (215008), Psgl‐1 (2PH1), T‐bet (4B10), Thy1.1 (OX‐7), Thy1.2 (53‐2.1) and tumour necrosis factor (TNF) (MP6‐XT22).
T‐cell culture medium
All cells were cultured in RPMI‐1640 medium supplemented with 10% FBS, 1% l‐glutamine, 50 units/ml penicillin, 50 μg/ml streptomycin and 50 μm β‐mercaptoethanol. FBS was obtained from Atlanta Biologicals (Flowery Branch, GA) and the medium and other additives were obtained from Life Technologies (Carlsbad, CA).
In vitro CD4 T‐cell differentiation and functional analysis
Spleen and lymph nodes were pooled and cell suspensions were prepared as described under flow cytometric analysis. Naive (CD62L+) CD4 T cells were isolated using the mouse CD4+ CD62L+ T Cell Isolation Kit II (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions and resuspended in T‐cell culture medium. To promote Th1 differentiation, medium was supplemented with 20 U/ml of IL‐2, 4 ng/ml of IL‐12 and 4 μg/ml of anti‐IL‐4. Both IL‐2 and IL‐12 were purchased from BD Biosciences; anti‐IL‐4 was purchased from eBioscience. Cells (5 × 105/well) were added to 24‐well plates coated with anti‐CD3 and anti‐CD28 (eBioscience). Wells were coated with antibodies by adding PBS containing anti‐CD3 and anti‐CD28 at 10 μg/ml followed by incubation and washing. Cells were cultured for 5 days at 37° in a 5% CO2 incubator. To elicit effector cytokine production after Th1 differentiation, cells were harvested, washed, resuspended in T‐cell medium supplemented with 20 U/ml of IL‐2, and plated onto wells coated with anti‐CD3. Cells were harvested 5–24 hr later for RNA isolation, collection of supernatant for ELISA, or intracellular cytokine analysis.
For intracellular staining of cytokines or Granzyme B, Th1 cells were incubated on wells coated with anti‐CD3 for 12 hr at 37° in a 5% CO2 incubator, after which GolgiPlug or GolgiStop (BD Biosciences) was added followed by a 5‐hr incubation at 37° in a 5% CO2 incubator. Cells were harvested, washed with stain buffer and stained for cell surface markers. After washing, cells were resuspended in Cytofix/Cytoperm (BD Biosciences) and incubated for 15 min at room temperature. Cells were washed with Perm/Wash (BD Biosciences) and resuspended in Perm/Wash containing fluorochrome‐conjugated anti‐cytokine or anti‐Granzyme B antibodies. Cells were incubated for 20 min at 4°, washed with Perm/Wash and resuspended in stain buffer for analysis. For intracellular staining of T‐bet, cells were cultured and processed as described above except cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience).
Microarray analysis
CD4 T cells were isolated and differentiated under Th1 conditions, harvested and restimulated on wells coated with anti‐CD3 for 6 hr. Cells were harvested and incubated with fluorochrome‐conjugated antibodies specific for CD4, Thy1.2 and Tim‐3. After washing, CD4+ Thy.12+ cells were sorted into Tim‐3− and Tim‐3+ fractions using a BD FACSAria (BD Bioscience). Recovered cells were resuspended in Trizol and stored at −80°. Cell samples from three independent sort purifications were collected and RNA was purified by standard methods. RNA concentrations were determined using a NanoDrop 1000 spectrophotometer and the integrity and purity of RNA was assessed using an Agilent 2100 bioanalyzer. Twenty‐five nanograms of RNA was converted to single primer isothermally amplified cDNA using the WT‐Ovation Pico RNA Amplification System, v1 (NuGEN Technologies, San Carlos, CA). Amplified cDNA was passed through a Qiagen MinElute Reaction Cleanup column (Qiagen, Valencia, CA) according to modifications from NuGEN; 3·3 µg of single primer isothermally amplified DNA was used to generate ST‐cDNA using the WT‐Ovation Exon Module v1 (NuGEN Technologies) and was passed through a Qiagen MinElute Reaction Cleanup column. Then, 5 µg of purified ST‐cDNA was fragmented (average fragment size = 85 bases) and biotin‐labelled using the FL‐Ovation cDNA Biotin Module, v2 (NuGEN Technologies). Biotin‐labelled cDNA was mixed with Affymetrix (Santa Clara, CA) eukaryotic hybridization buffer, placed onto Affymetrix Mouse Genome 430 2.0 arrays and incubated at 45° for 18 hr with rotation in an Affymetrix Model 640 Genechip Hybridization Oven. Arrays were washed and stained with streptavidin‐phycoerythrin (Molecular Probes, Eugene OR), and signals were amplified with anti‐streptavidin antibody (Vector Laboratories, Burlingcame, CA) using the Affymetrix Model 450 Fluidics Station. Arrays were scanned with the Affymetrix Model 3000 scanner and data were collected using the genechip operating software (GCOS) v1.4. Data were analysed using Partek Genomics Suite (Partek, Inc, St Louis, MO). Significant differences in gene expression were defined as showing a greater than twofold difference and a P value < 0·05. webgestalt 50 was used for Gene Ontology analysis.
Quantitative RT‐PCR analysis
Cells were resuspended in Trizol (Thermo Fisher Scientific, Waltham, MA) and RNA purified according to the manufacturer's instructions. Superscript III (Life Technologies) and random hexamer primers were used to synthesize cDNA. Quantitative real‐time PCR was performed using SYBR Green PCR Master Mix (Life Technologies) and the Model 7900HT detection system (Applied Biosystems) to obtain cycle threshold (Ct) values for target and internal reference (Hprt) cDNAs. Target cDNA levels were normalized to Hprt cDNA levels using the equation 2−[∆Ct] where ΔCt is defined as Cttarget − CtHprt.51 PCR primers used are listed in the Supplementary material (Table S1).
Adoptive transfer of SMARTA CD4 T cells
For adoptive transfer of naive cells, peripheral blood was collected from SMARTA mice and depleted of red blood cells using VitaLyse (BioE, St Paul, MN). Numbers of SMARTA cells in samples were calculated by multiplying total cell counts by the frequencies of SMARTA cells (defined as CD4+ Thy1.1+ and Vβ8.3 TCR+) as determined by flow cytometry. For adoptive transfer of Th1 cells, SMARTA cells were isolated from spleens and differentiated under Th1 conditions for two consecutive rounds. Dead cells were removed using the Dead Cell Removal Kit (Miltenyi Biotec) according to the manufacturer's instructions. Cells were resuspended in stain buffer and incubated for 20 min on ice with phycoerythrin‐conjugated anti‐Tim‐3. Cells were washed and separated into Tim‐3− and Tim‐3+ fractions using Anti‐phycoerythrin Microbeads (Miltenyi Biotec) according to the manufacturer's instructions. Cells were suspended in saline and injected intravenously into host mice: 2 × 106 cells were injected into mice that were left uninfected and 7·5 × 105 cells were injected into mice that were infected with LCMV‐Arm 16 hr later. To analyse responses by naive SMARTA cells, 1 × 104 cells were injected intravenously into host mice, which were infected with LCMV‐Arm 16 hr later.
Ex vivo stimulation and functional analysis of SMARTA cells
Splenocytes were resuspended in T‐cell medium containing LCMV GP61–80 peptide (1 μm) and either GolgiPlug or GolgiStop and then incubated for 5 hr at 37° in a 5% CO2 incubator. Cells were harvested, washed and stained for cell surface markers. After washing, cells were suspended in Cytofix/Cytoperm (BD Biosciences) and incubated for 15 min at room temperature. Cells were then washed with Perm/Wash (BD Biosciences) and resuspended in Perm/Wash containing fluorochrome‐conjugated anti‐Granzyme B, anti‐IFN‐γ, or anti‐TNF. Cells were incubated for 20 min at 4°, washed with Perm/Wash and resuspended in stain buffer for flow cytometric analysis. For intracellular staining of T‐bet, cells were cultured and processed as described above except cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience). For CD107a staining, anti‐CD107a was added to cells along with LCMV GP61–80 peptide and GolgiStop when cultures were initiated, and then cells were incubated for 5 hr at 37° in a 5% CO2 incubator. Cells were then harvested, washed and stained for cell surface markers.
Statistical analysis
Data were analysed using the unpaired, two‐tailed Student's t‐test within prism (GraphPad Software, La Jolla, CA).
Results
Tim‐3+ Th1 cells generated in vitro have augmented effector function
To analyse Tim‐3 mRNA and protein expression during Th1 differentiation in vitro, naive CD4 T cells from B6 mice were cultured under the appropriate conditions and samples of cells were harvested on days 2, 3 and 5 of culture. A portion of cells was analysed immediately and the remainder were restimulated overnight through the TCR using an antibody specific for the CD3ε subunit and then analysed. Expression of Tim‐3 mRNA and protein was measured by quantitative RT‐PCR (qRT‐PCR) and flow cytometry, respectively (Fig. 1a,b). Tim‐3 mRNA levels in cells harvested on day 2 were similar to the baseline values of naive cells and increased only slightly in response to restimulation. Also, cells harvested on day 2 did not express Tim‐3 protein either before or after restimulation. On day 3, the levels of Tim‐3 mRNA and the frequencies of Tim‐3+ cells were near baseline, but both increased in response to restimulation. For cells harvested on day 5, the levels of Tim‐3 mRNA and the frequencies of Tim‐3+ cells were substantially higher relative to naive cells and both greatly increased when cells were restimulated. Hence, as shown previously,21, 52 these data confirm that CD4 T cells begin expressing Tim‐3 upon Th1 differentiation. These results also show that Tim‐3 expression is sharply up‐regulated by differentiated cells in response to secondary TCR stimulation.
Figure 1.
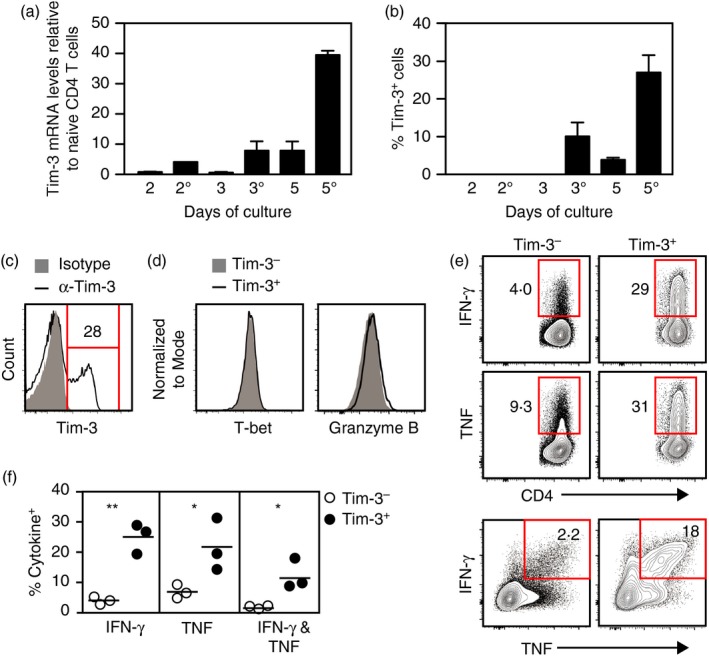
Tim‐3+ T helper type 1 (Th1) cells generated by acute stimulation in vitro have enhanced effector function. (a,b) CD4 T cells were cultured under Th1 conditions, harvested and analysed. Cells were harvested on days 2, 3 and 5 of culture (denoted as 2, 3 and 5). Additionally, cells taken at these time‐points were washed, restimulated overnight on plates coated with anti‐CD3 antibody and harvested (denoted as 2°, 3° and 5°). (a) Tim‐3 mRNA expression levels. Tim‐3 mRNA levels were normalized to those detected in naive CD4 T cells, which were set at 1. (b) Frequencies of cells expressing Tim‐3 on their surface as determined by flow cytometry. (c–f) Cells were cultured under Th1 conditions for 5 days, harvested, and restimulated overnight on plates coated with anti‐CD3 antibody. (c) Surface expression of Tim‐3. Isotype denotes staining with a non‐specific isotype‐matched antibody. (d) T‐bet and Granzyme B expression by Tim‐3− and Tim‐3+ cells. (e) Interferon‐γ (IFN‐γ) and tumour necrosis factor (TNF) expression by Tim‐3− and Tim‐3+ cells. (f) Frequencies of IFN‐γ‐ and TNF‐expressing cells within Tim‐3− and Tim‐3+ populations as calculated from data represented in (e). For (a) and (b), bars represent the mean and standard error of values from two independent experiments. Data shown in (c), (d) and (e) are representative of results from at least two independent experiments. Circles in (f) each represent a result obtained from an independent experiment (n = 3). *P < 0·05; **P < 0·01. [Colour figure can be viewed at http://wileyonlinelibrary.com]
To characterize the functional properties of Tim‐3+ Th1 cells, naive CD4 T cells were differentiated in culture, recovered and restimulated through the TCR to elicit expression of Tim‐3 and the Th1 cytokines IFN‐γ and TNF. Tim‐3− and Tim‐3+ cells were detected after TCR restimulation as expected (Fig. 1c). These populations expressed the transcription factor T‐bet and Granzyme B at similar levels (Fig. 1d), indicating that both had undergone Th1 differentiation. However, the Tim‐3+ fraction contained significantly higher frequencies of cells producing IFN‐γ or TNF, and more Tim‐3+ cells expressed both IFN‐γ and TNF simultaneously (Fig. 1e,f). These results show that Tim‐3 expression by Th1 cells generated in vitro correlates with an augmented ability to produce Th1 cytokines.
Tim‐3− and Tim‐3+ Th1 cells show distinct gene expression patterns
We next asked whether Tim‐3− and Tim‐3+ Th1 cells show differences in global gene expression. To explore this, CD4 T cells were differentiated in culture, restimulated with anti‐CD3 and sort‐purified based on Tim‐3 expression to isolate RNA that was used for microarray analysis. This identified 128 genes that were differentially expressed between Tim‐3− and Tim‐3+ cells (Fig. 2a and see Supplementary material, Table S2), with roughly equal numbers being expressed at higher or lower levels in Tim‐3+ cells relative to Tim‐3− cells. Gene Ontology analysis of this data set revealed significant enrichment for genes associated with immune responses (data not shown). Among differentially expressed genes were those encoding cell surface proteins, secreted molecules and transcription factors with roles in the immune system (Fig. 2b–d).
Figure 2.
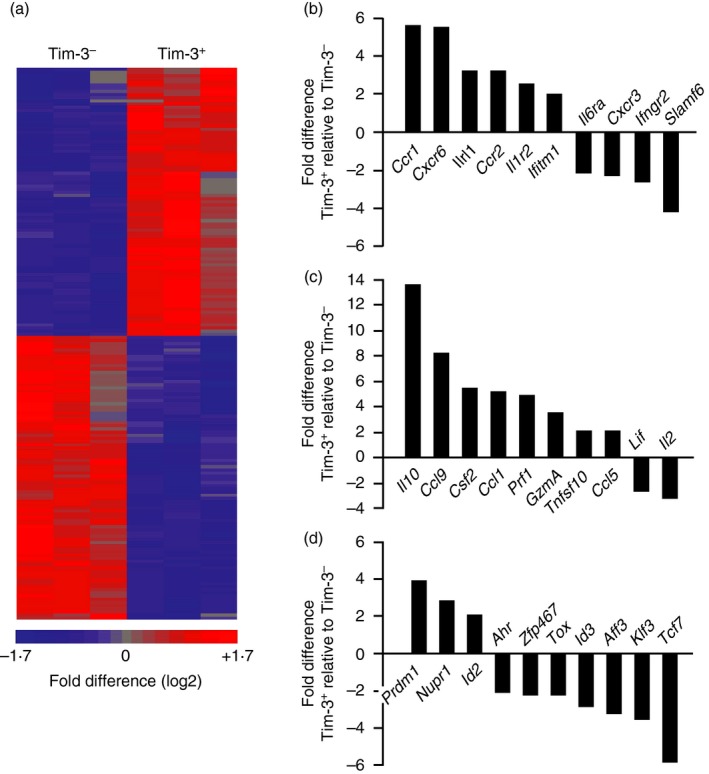
Tim‐3− and Tim‐3+ T hekper type 1 (Th1) cells show distinct gene expression patterns. CD4 T cells were differentiated under Th1 conditions, harvested, and restimulated for 6 hr on plates coated with anti‐CD3 antibody. Tim‐3− and Tim‐3+ CD4 T cells were sort‐purified and used to isolate RNA for microarray analysis. (a) Heat map generated by unbiased clustering of data sets from Tim‐3− and Tim‐3+ cells. Each column represents data obtained from an independent experiment (n = 3 total). Significant differences in the expression of 128 genes are shown. (b–d) Differential expression of genes encoding (b) cell surface proteins and receptors, (c) secreted factors and (d) transcription factors. [Colour figure can be viewed at http://wileyonlinelibrary.com]
We performed qRT‐PCR analysis to verify some of the differences in gene expression identified by microarray analysis. Among the differentially expressed genes encoding cell surface proteins, Tim‐3 mRNA levels were significantly higher in Tim‐3+ cells as expected, whereas those encoding the surface proteins IFN‐γR2 and SLAMF6 were expressed at lower levels (see Supplementary material, Fig. S1a). Analysis of genes in the group encoding secreted factors confirmed that mRNAs encoding IL‐10, CCL9 and Perforin were all expressed at significantly higher levels in Tim‐3+ cells and that IL‐2 RNA was significantly reduced (see Supplementary material, Fig. S1b). Increased expression of IL‐10 was also confirmed at the protein level (see Supplementary material, Fig. S1c). Analysis of genes encoding transcription factors showed that mRNAs encoding B lymphocyte‐induced maturation protein 1 (BLIMP‐1; PRDM1) and inhibitor of differentiation 2 protein (ID2) were expressed at significantly higher levels in Tim‐3+ cells, whereas those encoding ID3 and T‐cell factor 7 (TCF7) were expressed at significantly lower levels (see Supplementary material, Fig. S1d). Together, these data confirm that genes with key roles in the immune system are expressed at different levels in Tim‐3− and Tim‐3+ Th1 cells, supporting the conclusion that Tim‐3+ Th1 cells are functionally distinct from those that are Tim‐3−.
Tim‐3+ Th1 cells persist following transfer into naive hosts
Cell culture studies showed that Tim‐3+ Th1 cells undergo apoptosis when exposed to Galectin‐9, implying that Tim‐3 marks cells destined for apoptotic cell death.34, 36 To explore the link between Tim‐3 and apoptosis in vivo, we asked whether Tim‐3− and Tim‐3+ Th1 cells showed differences in survival following adoptive transfer into host mice. To carry out these studies, we used CD4 T cells from SMARTA TCR‐transgenic mice49 expressing the Thy1.1 variant of the surface protein Thy1. SMARTA CD4 T cells (SMARTA cells) express a TCR specific for the GP61–80 peptide from LCMV, which can be used to restimulate cells ex vivo. Thy1.1 expression allowed SMARTA cells to be distinguished from host CD4 T cells during analysis (see Fig. 4b). SMARTA cells were isolated and differentiated under Th1 conditions for two successive rounds, which gave rise to roughly equal proportions of Tim‐3− and Tim‐3+ cells (Fig. 3a) that expressed similar levels of T‐bet and Granzyme B (Fig. 3b,c). When restimulated in vitro through the TCR using anti‐CD3, Tim‐3+ SMARTA cells generated higher frequencies of IFN‐γ and TNF‐producing cells compared with Tim‐3− cells (Fig. 3d–f), again showing that Tim‐3 can mark cells with a greater capacity to produce Th1 cytokines.
Figure 4.
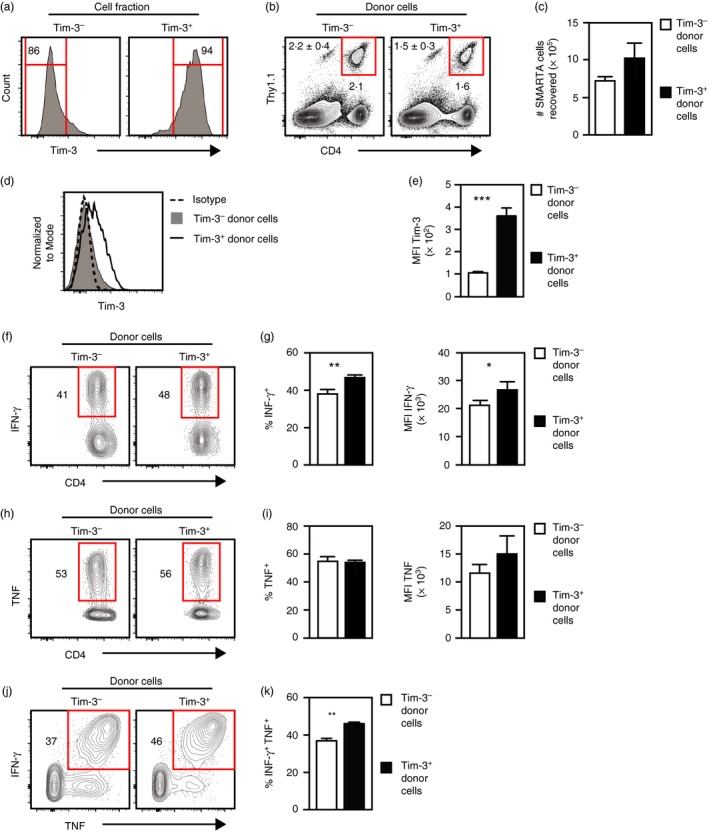
Tim‐3− and Tim‐3+ Th1 cells show similar persistence and functionality following adoptive transfer into naive mice. SMARTA cells were differentiated under T helper type 1 (Th1) conditions and separated into Tim‐3− and Tim‐3+ fractions, which were then injected into naive host mice. After 7 days, spleens were harvested for analysis. (a) Tim‐3 expression by cells immediately following separation into Tim‐3− and Tim‐3+ fractions. (b) Detection of SMARTA cells (CD4+ Thy1.1+) in spleen cell suspensions. Shown at the upper left are the mean and standard error of cell frequencies as calculated from the data represented in the panel. (c) Total numbers of SMARTA cells recovered as calculated from data represented in (b). (d) Tim‐3 expression by recovered SMARTA cells. Isotype denotes staining with a non‐specific isotype‐matched antibody. (e) Median fluorescence intensities (MFI) of Tim‐3 expression by the indicated cell populations. (f) Interferon‐γ (IFN‐γ) expression by SMARTA cells following ex vivo stimulation with GP 61–80 peptide. (g) Frequencies and MFI of IFN‐γ‐expressing cells as calculated from data represented in (f). (h) Tumour necrosis factor (TNF) expression by SMARTA cells following ex vivo stimulation with GP 61–80 peptide. (i) Frequencies and MFI of TNF‐expressing cells as calculated from data represented in (h). (j) Coexpression of IFN‐γ and TNF by SMARTA cells following ex vivo stimulation with GP 61–80 peptide. (k) Frequencies of SMARTA cells coexpressing IFN‐γ and TNF as determined from data represented in (j). All histograms are representative of data from two independent experiments involving a combined total of five mice per group. Bar graphs represent the mean and standard error of values pooled from two independent experiments involving a combined total of five mice per group.*P < ·05; **P < ·01; ***P < ·001. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
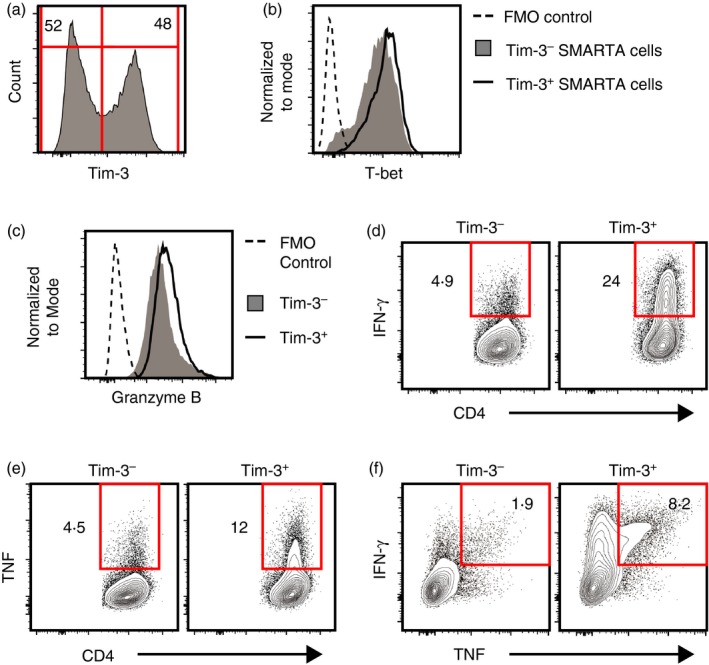
Differentiation of SMARTA CD4 T cells into Tim‐3− and Tim‐3+ T helper type 1 (Th1) cells. SMARTA CD4 T cells were differentiated under Th1 conditions for 5 days, washed and then cultured under Th1 conditions for another 5 days. Cells were then harvested and analysed. (a) Surface Tim‐3 expression by recovered cells. (b–f) Expression of T‐bet (b), Granzyme B (c), interferon‐γ (IFN‐γ) (d), tumour necrosis factor (TNF) (e), and IFN‐γ and TNF (f) by Tim‐3− and Tim‐3+ cells. For (b) and (c), FMO control represents data from cells stained with all antibodies except those specific for T‐bet or Granzyme B. All data shown are representative of results from at least two independent experiments. [Colour figure can be viewed at http://wileyonlinelibrary.com]
SMARTA Th1 cells were separated into Tim‐3− and Tim‐3+ subsets (Fig. 4a). Immediately after separation, each population was injected into different host mice and spleens were harvested for analysis 7 days later. Similar frequencies and total numbers of SMARTA cells were detected in mice that received either Tim‐3− or Tim‐3+ cells (Fig. 4b,c). In addition, Tim‐3 expression was down‐regulated by SMARTA cells that were Tim‐3+ at the time of injection, although these cells still expressed more Tim‐3 relative to those that were Tim‐3− before injection (Fig. 4d,e).
We also evaluated functional responses by recovered SMARTA cells by culturing splenocytes in the presence of GP61–80 peptide to stimulate cytokine production. Control cultures confirmed that cytokine expression required the addition of the peptide (data not shown). SMARTA cells from hosts that received Tim‐3+ cells generated higher frequencies of IFN‐γ‐producing cells compared with those from mice that received Tim‐3− cells, and had greater median fluorescence intensities (MFI) for IFN‐γ staining, indicating these cells expressed more IFN‐γ on a per cell basis (Fig. 4f,g). SMARTA cells from mice that received either Tim‐3− or Tim‐3+ cells generated similar frequencies of TNF‐producing cells and no differences in the MFI for TNF staining were observed (Fig. 4h,i). However, cells from hosts that received Tim‐3+ cells generated higher frequencies of cells that produced both IFN‐γ and TNF (Fig. 4j,k).
To investigate if Tim‐3 expression affected cell persistence or function after an extended time period, we injected Tim‐3− and Tim‐3+ Th1 cells into naive mice and then waited 23 days (rather than 7 days) before harvesting spleens for analysis. Similar frequencies and numbers of SMARTA cells were present in mice that received either Tim‐3− or Tim‐3+ cells (see Supplementary material, Fig. S2a,b). As expected, Tim‐3 expression was down‐regulated by SMARTA cells that were Tim‐3+ before injection, although these still expressed more Tim‐3 relative to cells that were Tim‐3− before injection (see Supplementary material, Fig. S2c,d). When stimulated with GP61–80 peptide, SMARTA cells from hosts injected with Tim‐3+ cells produced higher frequencies of IFN‐γ‐producing cells that had greater MFI for IFN‐γ staining relative to those from mice that received Tim‐3− cells (see Supplementary material, Fig. S2e,f). The frequencies and MFI of TNF‐producing cells were similar between SMARTA cells from hosts that initially received Tim‐3− or Tim‐3+ cells (see Supplementary material, Fig. S2g,h), but those from mice injected with Tim‐3+ cells produced greater frequencies of cells expressing both IFN‐γ and TNF (see Supplementary material, Fig. S2i,j). Taken together, these data show that there was no difference in survival between Tim‐3− and Tim‐3+ Th1 cells following adoptive transfer into naive mice. These data also show that, although Tim‐3+ cells down‐regulated Tim‐3 in the absence of stimulation, these cells still maintained a greater ability to express Th1 cytokines compared with Tim‐3− cells.
Th1 cells responding to infection generate Tim‐3+ effectors with augmented function
We next determined how Tim‐3− and Tim‐3+ Th1 cells responded to an acute stimulation following adoptive transfer. SMARTA CD4 T cells were differentiated under Th1 conditions for two consecutive rounds, separated into Tim‐3− and Tim‐3+ populations and injected into host mice. The next day, host mice were challenged with LCMV‐Arm to induce an acute infection and spleens were harvested for analysis 8 days later. Similar frequencies and numbers of SMARTA cells were detected in infected mice given either Tim‐3− or Tim‐3+ cells, again indicating no difference in survival between the two populations (Fig. 5a,b). Additionally, both populations of transferred cells generated similar frequencies of Tim‐3‐expressing cells (Fig. 5c,d). Hence, Tim‐3− or Tim‐3+ Th1 cells persist to a similar extent in LCMV‐infected mice and both populations can give rise to Tim‐3‐expressing cells.
Figure 5.
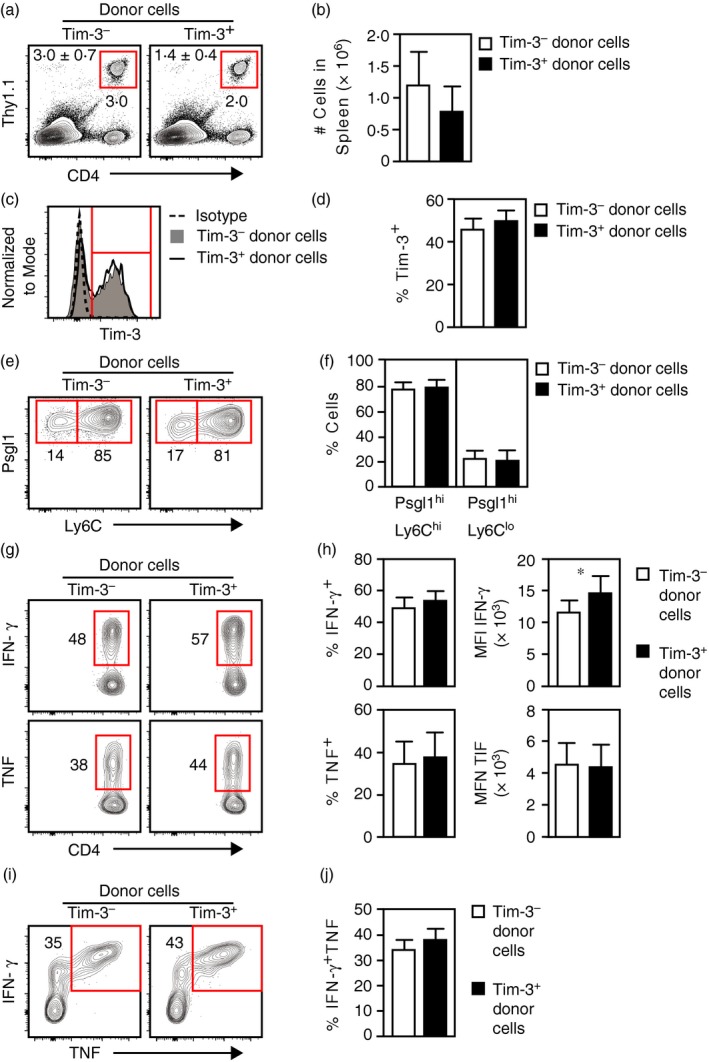
Adoptively transferred Tim‐3− and Tim‐3+ T helper type 1 (Th1) cells both give rise to Tim‐3‐expressing effector cells in response to infection with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm). SMARTA CD4 T cells were differentiated under Th1 conditions, separated into Tim‐3− and Tim‐3+ fractions and injected into naive host mice. The next day, hosts were infected with LCMV‐Arm and spleens were harvested for analysis 8 days later. (a) Detection of SMARTA cells (CD4+ Thy1.1+). Shown at the upper left are the mean and standard error of cell frequencies as calculated from data represented in the panel. (b) Total numbers of SMARTA cells recovered as calculated from the data represented in (a). (c) Tim‐3 expression by SMARTA cells. Isotype denotes staining with a non‐specific isotype‐matched antibody. (d) Frequencies of Tim‐3‐expressing SMARTA cells as determined from data represented in (c). (e) Expression of Psgl1 and Ly6C by SMARTA cells. (f) Frequencies of Psgl1hi Ly6Chi and Psglhi Ly6Clo SMARTA cells as determined from data represented in (e). (g) Interferon‐γ (IFN‐γ) and tumour necrosis factor (TNF) expression by SMARTA cells following ex vivo stimulation with GP 61–80 peptide. (h) Frequencies and median fluorescence intensities (MFI) of IFN‐γ‐ and TNF‐expressing SMARTA cells as determined from data represented in (g). (i) Coexpression of IFN‐γ and TNF by SMARTA cells following ex vivo stimulation with GP 61–80 peptide. (j) Frequencies of SMARTA cells coexpressing IFN‐γ and TNF as determined from data represented in (i). All histograms are representative of data obtained from two independent experiments involving a combined total of eight (for Tim‐3− donor cells) or six (for Tim‐3+ donor cells) mice per group. Bar graphs represent the mean and standard error of values pooled from two independent experiments involving a combined total of eight (for Tim‐3− donor cells) or six (for Tim‐3+ donor cells) mice per group. *P < 0·05. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Studies by others have shown that CD4 T cells responding to LCMV‐Arm infection generate Psgl1+ Ly6C+ and Psgl1+ Ly6C− subsets of cells, with the former containing primarily effector Th1 cells and the latter enriched for memory CD4 T‐cell precursors.53, 54 Tim‐3− and Tim‐3+ donor cells generated similar frequencies of Psgl1+ Ly6C+ and Psgl1+ Ly6C− cells (Fig. 5e,f). Also, ex vivo GP61–80 peptide stimulation of splenocytes from mice given Tim‐3− or Tim‐3+ cells generated similar frequencies of IFN‐γ‐ and TNF‐producing cells and the MFI for staining were similar between the two groups (Fig. 5g,h). However, SMARTA cells from mice given Tim‐3+ cells generated significantly greater MFI for IFN‐γ staining. Regardless of the donor cell type, the frequencies of SMARTA cells able to produce both TNF and IFN‐γ simultaneously were similar (Fig. 5i,j). We also analysed CD107a expression to measure degranulation by Th1 cells in response to peptide stimulation. SMARTA cells from mice given either Tim‐3− or Tim‐3+ cells generated similar frequencies of CD107a+ cells, but the MFI of cells from mice injected with Tim‐3+ cells were significantly higher (see Supplementary material, Fig. S3a,b). Hence, Tim‐3− and Tim‐3+ SMARTA Th1 cells mount similar responses to an acute infection and both generate the expected effector cell subsets.
We next asked if Tim‐3+ Th1 cells generated in response to LCMV‐Arm infection showed augmented effector function, which would be predicted from our in vitro studies. SMARTA cells in LCMV‐infected mice that initially received Tim‐3+ Th1 cells were subdivided based on Tim‐3 expression and cytokine production was analysed (Fig. 6a,b). Significantly greater frequencies of IFN‐γ‐ and TNF‐producing cells were detected in the Tim‐3+ fraction of cells relative to the Tim‐3− subset. Also, the MFI for IFN‐γ and TNF staining were significantly higher for Tim‐3+ cells compared with those that were Tim‐3−. Analysis of CD107a expression produced the same pattern of results (see Supplementary material, Fig. S4a,b). The frequencies of Tim‐3+ cells that expressed IFN‐γ together with TNF were also significantly elevated (Fig. 6c,d). Similar results were obtained from analysis of SMARTA cells in host mice that received Tim‐3− Th1 cells before LCMV infection (Fig. 6e‐h; see Supplementary material, Fig. S4c,d). So, the Tim‐3+ Th1 cells generated in response to an acute infection in vivo have enhanced effector function relative to their Tim‐3− counterparts.
Figure 6.
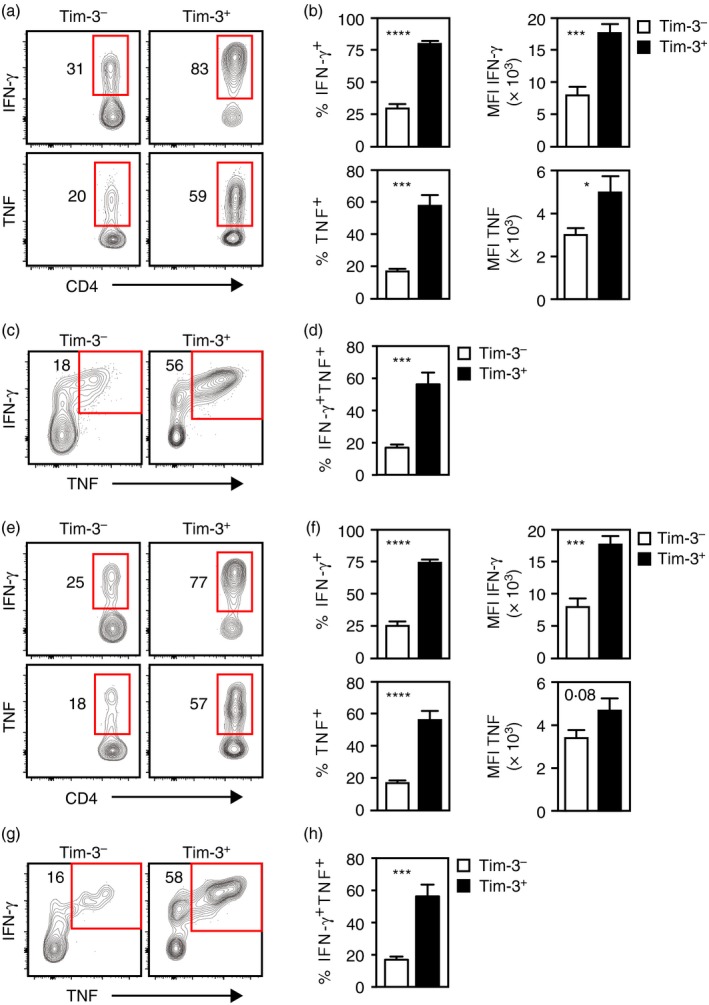
Adoptively transferred T helper type 1 (Th1) cells responding to Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) infection generate Tim‐3+ cells with enhanced effector function. Tim‐3+ (a–d) or Tim‐3− (e–h) SMARTA Th1 cells generated in vitro were injected into host mice that were infected with LCMV‐Arm on the next day. Spleens were harvested 8 days later and SMARTA cells were stimulated ex vivo with GP 61–80 peptide. Stimulated cells were subdivided into Tim‐3− and Tim‐3+ subsets as indicated by the histogram labels and legends. (a) Interferon‐γ (IFN‐γ) and tumour necrosis factor (TNF) expression by ex vivo stimulated SMARTA cells from hosts that initially received Tim‐3+ donor cells. (b) Frequencies of IFN‐γ‐ and TNF‐producing SMARTA cells and their median fluorescence intensities (MFI) as determined from data represented in (a). (c) Coexpression of IFN‐γ and TNF by ex vivo stimulated SMARTA cells from hosts that initially received Tim‐3+ donor cells. (d) Frequencies of cells coexpressing IFN‐γ and TNF as determined from data represented in (c). (e) IFN‐γ and TNF expression by ex vivo stimulated SMARTA cells from hosts that initially received Tim‐3− donor cells. (f) Frequencies of IFN‐γ‐ and TNF‐producing SMARTA cells and their MFI as determined from data represented in (e). (g) Coexpression of IFN‐γ and TNF by ex vivo stimulated SMARTA cells from hosts that initially received Tim‐3− donor cells. (h) Frequencies of cells coexpressing IFN‐γ and TNF as determined from data represented in (g). All histograms are representative of two independent experiments involving a combined total of six (for Tim‐3+ donor cells) or eight (for Tim‐3− donor cells) mice per group. Bar graphs represent the mean and standard error of values from two independent experiments involving a combined total of eight (for Tim‐3− donor cells) or six (for Tim‐3+ donor cells) mice per group. *P < 0·05; ***P < 0·001; ****P < 0·0001. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Naive CD4 T cells responding to acute infection generate Tim‐3+ Th1 cells with enhanced effector function
The experiments described above involved adoptive transfer of Th1 cells generated in vitro, but left unknown whether Tim− and Tim‐3+ Th1 cells generated in vivo from naive cells have similar properties. To address this, naive SMARTA cells were injected into host mice, which were then infected with LCMV‐Arm. Spleens from infected mice were harvested for analysis 8 days after infection. SMARTA cells were present at a relatively low frequency (~1·6% of the total cells; Fig. 7a) and ~45% were Tim‐3+ (Fig. 7b,c). As observed in vitro, Tim‐3− and Tim‐3+ SMARTA cells generated in response to LCMV infection expressed similar levels of T‐bet and Granzyme B (Fig. 7d,e), indicating that both subsets had differentiated into Th1 cells. Analysis of cytokine production in response to ex vivo stimulation with GP61–80 peptide showed that the Tim‐3+ subset of SMARTA cells contained significantly higher frequencies of IFN‐γ‐ and TNF‐producing cells relative to the Tim‐3− fraction (Fig. 7f,g). Also, the MFI generated by staining for IFN‐γ or TNF were significantly higher for the Tim‐3+ subset (Fig. 7g). Consistent with these results, the Tim‐3+ fraction contained greater frequencies of cells that produced both IFN‐γ and TNF (Fig. 7h). Also, the Tim‐3+ subset contained significantly higher frequencies of CD107a+ cells (Fig. 7i). These results demonstrate that Tim‐3+ Th1 cells generated in vivo in response to an acute infection have augmented Th1 effector function.
Figure 7.
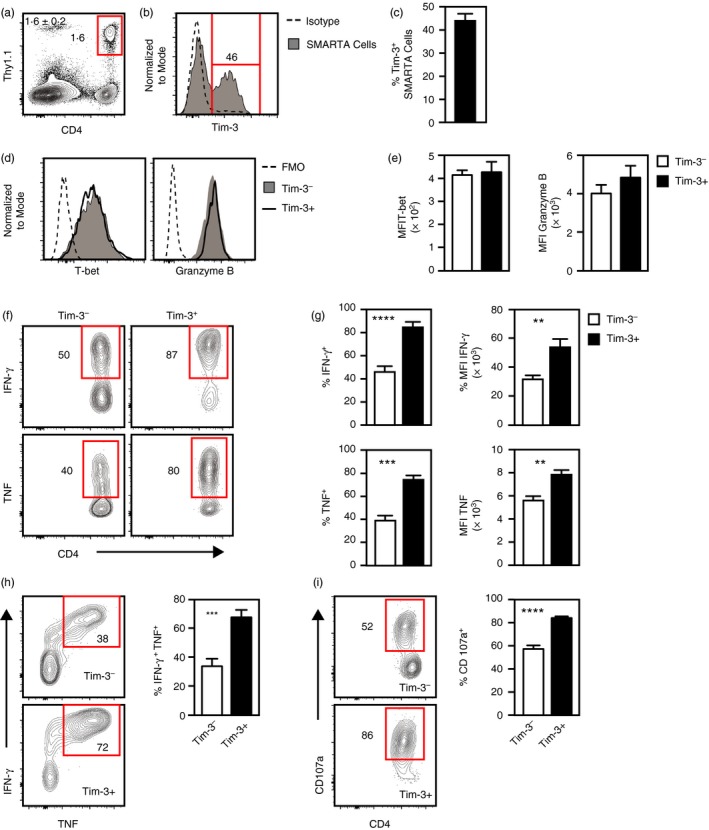
Tim‐3+ T helper type 1 (Th1) cells generated in vivo in response to acute lymphocytic choriomeningitis virus (LCMV) infection have augmented effector function. Naive SMARTA CD4 T cells were injected into naive host mice, which were infected with the Armstrong strain of LCMV (LCMV‐Arm) on the next day. 8 days later, spleens were harvested and analysed. (a) Detection of SMARTA cells (CD4+ Thy1.1+). Shown at the upper left are the mean and standard error of cell frequencies as calculated from data represented in the panel. (b) Tim‐3 expression by SMARTA cells. Isotype denotes staining with a non‐specific isotype‐matched antibody. (c) Frequencies of Tim‐3+ SMARTA cells as calculated from data represented in (b). (d) T‐bet and Granzyme B expression by Tim‐3− and Tim‐3+ SMARTA cells. FMO control represents data from cells stained with all antibodies except those specific for T‐bet or Granzyme B. (e) Summary of median fluorescent intensities (MFI) for T‐bet and Granzyme B staining as determined from data represented in (d). (f) Interferon‐γ (IFN‐γ) and tumour necrosis factor (TNF) expression by Tim‐3− and Tim‐3+ SMARTA cells stimulated ex vivo with GP 61–80 peptide. (g) Frequencies and MFI of IFN‐γ‐ and TNF‐expressing cells as determined from data represented in (e). (h) Coexpression of IFN‐γ and TNF by Tim‐3− and Tim‐3+ SMARTA cells stimulated ex vivo with GP 61–80 peptide. (i) Expression of CD107a by Tim‐3− and Tim‐3+ SMARTA cells stimulated ex vivo with GP 61–80 peptide. All histograms are representative of two independent experiments involving a combined total of seven mice. All bar graphs represent the mean and standard error of values pooled from two independent experiments involving a combined total of seven mice. **P < 0·01; ***P < 0·001; ****P < 0·0001. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Discussion
We used in vitro and in vivo models to define the characteristics of Tim‐3+ Th1 cells responding to acute stimulation. Consistent with previous studies,21, 52 CD4 T cells began expressing Tim‐3 as they differentiated into Th1 effector cells. We further noted that Tim‐3 expression by Th1 cells was strongly up‐regulated by secondary TCR stimulation and so was coordinated with the induction of effector function. Following adoptive transfer into naive mice, Tim‐3− and Tim‐3+ Th1 cells were maintained at similar numbers and both mounted robust responses to acute LCMV infection. Hence, under these conditions, Tim‐3 expression did not promote apoptosis of Th1 cells or suppress their function. In contrast to the idea that Tim‐3 exclusively marks exhausted CD4 T cells, we found that Tim‐3+ Th1 T cells generated in vitro were not functionally impaired, but rather displayed enhanced effector function relative to Tim‐3− cells and, as discussed below, seemed more strongly polarized toward a Th1 phenotype based on gene expression analysis. Consistent with these results, Tim‐3+ Th1 cells generated in vivo in response to acute LCMV infection also showed greater effector function compared with Tim‐3− cells. Combined, our data indicate that Tim‐3‐expressing Th1 cells are not necessarily prone to apoptosis or functionally disabled and, in some settings, have augmented effector function.
As expected, we found that CD4 T cells undergoing Th1 differentiation begin expressing Tim‐3 and our time–course analysis suggested that Tim‐3 mRNA and protein expression becomes increasingly permissive as Th1 differentiation progresses. Previous studies showed that Tim‐3 expression is regulated by the Th1 transcription factor T‐bet.52 Interestingly, our data show that Tim‐3− and Tim‐3+ Th1 cells generated in vitro or in vivo express T‐bet at similar levels, suggesting that T‐bet alone does not dictate Tim‐3 expression levels. Consistent with this idea, additional mechanisms have been implicated in the regulation of Tim‐3 expression, including IL‐27‐induced activation of the transcription factor NFIL3 and DNA methylation.55, 56 Further, Tim‐3 mRNA and protein levels were increased when Th1 cells were restimulated through TCR, suggesting that Tim‐3 expression is regulated by transcription factors activated because of TCR signalling. Additionally, our global gene expression analysis suggested that Tim‐3+ Th1 cells are distinct from their Tim‐3− counterparts and are more differentiated, which could facilitate Tim‐3 expression. Evidence supporting the conclusion that Tim‐3+ cells are more polarized toward a Th1 phenotype includes increased expression of mRNAs encoding the proteins BLIMP‐1, ID2, CCR2, CXCR6, Granzyme A and Perforin and decreased expression of mRNAs encoding the proteins TCF‐1/TCF‐7, ID3, IL‐2, IFN‐γR2, and IL‐6Rα, which are features associated with highly differentiated Th1 cells.54, 57, 58, 59, 60, 61 The idea that Tim‐3+ Th1 cells are more differentiated is also supported by our data showing that Tim‐3+ Th1 cells have a greater capacity to produce IFN‐γ and TNF and to degranulate in response to TCR stimulation. Hence, in some circumstances Tim‐3 expression may mark highly differentiated Th1 cells.
Following adoptive transfer into naive mice, Tim‐3+ cells down‐regulated Tim‐3, suggesting that some form of stimulation is required to sustain surface Tim‐3 expression in vivo. In accord with this, studies by others have shown that T cells responding to acute infections48, 62 or exposed to chronic antigen and cytokine stimulation41, 42, 43, 44, 45, 46, 47 express high levels of Tim‐3. This conclusion is also supported by our data showing that Tim‐3 is expressed by adoptively transferred naive CD4 T cells or Th1 cells responding to LCMV infection. These findings are in line with the idea that Tim‐3 expression is regulated in part by TCR‐induced signalling pathways as suggested by our in vitro studies.
The finding that Tim‐3− and Tim‐3+ Th1 cells persisted equally well following transfer into naive hosts argues that the latter are not more susceptible to Galectin‐9‐dependent (or independent) pathways for apoptosis as was observed in cell culture studies.21 In addition to Galectin‐9, Tim‐3 can also recognize phosphatidylserine and HMGB1 as ligands,9, 63 but whether these factors induce apoptosis or have some other impact on T‐cell function has not been established. Although Galectin‐9 appears to be constitutively expressed in several organs including the spleen,64, 65 it is possible that the levels of Galectin‐9 expressed in naive mice are not sufficient to induce apoptosis through Tim‐3. Studies by others have shown that Galectin‐9 expression is up‐regulated by inflammatory cytokines or viral infections.66, 67, 68, 69, 70, 71, 72, 73, 74 Therefore, we also tested whether Tim‐3+ Th1 cells were prone toward apoptosis in the context of acute LCMV infection, which induces a robust inflammatory response. Again, we found no evidence for Tim‐3+ Th1 cells being more sensitive to apoptosis compared with Tim‐3− cells. Further, our data showed that Tim‐3− and Tim‐3+ Th1 cells mount similar effector responses to LCMV infection, suggesting that Tim‐3+ Th1 cells are not more susceptible to other potential mechanisms of functional suppression compared with Tim‐3− cells. This conclusion contrasts with that idea that Tim‐3 expression invariably enforces inhibition on Th1 cells, as suggested by studies using agents to block Tim‐3–ligand interactions.3 Hence, in some contexts Tim‐3 expression alone may not be sufficient to suppress or restrain Th1 cell function.
Our studies showed that Th1 cells expressing Tim‐3 produce more effector cytokines in response to stimulation (relative to Tim‐3− cells) and may be more highly differentiated. Other studies demonstrated that exhausted T cells generated in response to chronic stimulation express high levels of Tim‐341, 42, 43, 44, 45, 46, 47 and provided evidence that Tim‐3 enforces the functional impairment of T‐cell exhaustion. Yet, in patients actively infected with M. tuberculosis, Tim‐3 expression marks CD4 and CD8 T cells with a greater propensity to produce IFN‐γ, TNF and other effector molecules.48 Likewise, tumour‐infiltrating CD8 T cells expressing Tim‐3 can have increased effector function compared with their Tim‐3− counterparts.75, 76 Similar to these findings, our data demonstrated that, after acute stimulation in vitro or in response to acute LCMV infection, Tim‐3 expression by CD4 T cells was associated with enhanced effector function. These findings suggest that Tim‐3 expression is not restricted to exhausted T cells and does not invariably promote functional impairment. One explanation for the differences between our findings and those involving exhausted T cells may be that the impact of Tim‐3 on T‐cell function is context‐dependent and varies due to factors that are specific to acute or chronic stimulation. Nonetheless, our data support the conclusion that Tim‐3 is expressed by CD4 T cells with augmented function that are generated in response to acute stimulation.
Tim‐3 has long been considered to be a negative regulator of T‐cell function. Although not directly addressed by the results presented here, our data at least support the idea that Tim‐3 can augment, rather than suppress, CD4 T‐cell activation. This conclusion is consistent with studies demonstrating that Tim‐3 enhances functional responses by CD8 T cells, mast cells and transformed dendritic cells.12, 62, 77, 78, 79 In addition, molecular studies have provided evidence that Tim‐3 can augment activation of signal transduction events induced by T‐cell receptor engagement18 and can increase the activity of the mTORC1 signalling pathway.77 Hence, accumulating data suggest that, at least under some circumstances, Tim‐3 can positively regulate immune cell responses.
Seminal studies of Tim‐3 function demonstrated that treatment with anti‐Tim‐3 antibodies can reverse T‐cell exhaustion caused by chronic antigen stimulation.41, 44, 45 These findings were interpreted to signify that anti‐Tim‐3 antibodies interfered with interactions between Tim‐3 and its ligands, so relieving Tim‐3‐mediated negative regulation. In light of evidence that Tim‐3 can function as a positive regulator, an alternative interpretation is that anti‐Tim‐3 antibody treatment results in cross‐linking of Tim‐3, so inducing downstream signalling pathways that stimulate T‐cell function. Such a mechanism would also explain data showing that in vitro stimulation of T cells in the presence of anti‐Tim‐3 antibodies promotes proliferation, differentiation and effector function.41, 48, 77
Disclosures
The authors declare no conflicts of interest.
Supporting information
Figure S1. Analysis of differential gene expression between Tim‐3− and Tim‐3+ T helper type 1 cells.
Figure S2. Persistence and functionality of Tim‐3− and Tim‐3+ T helper type 1 cells 23 days after transfer into naive mice.
Figure S3. Degranulation, as measured by CD107a expression, of Tim‐3− and Tim‐3+ T helper type 1 cells following exposure to Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) infection.
Figure S4. Increased degranulation, as measured by CD107a expression, of Tim‐3+ cells generated by adoptively‐transferred T helper type 1 cells following exposure to Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) infection.
Table S1. Genes expressed at significantly different levels in Tim‐3+ T helper type 1 cells relative to Tim‐3− Th1 cells.
Table S2. Oligonucleotide primers used for quantitative real‐time PCR analysis.
Acknowledgements
JVG performed the experiments; JVG and JDC designed the study, analysed the data and wrote the manuscript. We thank Renee Goodfellow for technical assistance and Diana F. Colgan for critical reading of the manuscript. Research reported in this publication was supported by the National Institute of Allergy and Infectious Disease (NIAID) of the National Institutes of Health (NIH) under award numbers R01 AI054821 and R01 AI093737 (to JDC). JVG was supported in part by the NIAID of the NIH under award number T32 AI007485. Some data presented herein were obtained at the Flow Cytometry Facility, a Carver College of Medicine and Holden Comprehensive Cancer Center core research facility at the University of Iowa funded through user fees and financial support from the Carver College of Medicine, the Holden Comprehensive Cancer Center, and the Iowa City Veteran's Administration Medical Center. The Flow Cytometry Facility is also supported by the National Cancer Institute (NCI) of the NIH under award number P30 CA086862 and by the National Center for Research Resources of the NIH under award number 1S10 RR027219. Some data presented herein were obtained at the Iowa Institute of Human Genetics Genomics Division at the University of Iowa, which is supported in part by the NCI of the NIH under award number P30 CA086862.
References
- 1. Han G, Chen G, Shen B, Li Y. Tim‐3: an activation marker and activation limiter of innate immune cells. Front Immunol 2013; 4:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim‐3 functions in antimicrobial and tumor immunity. Trends Immunol 2011; 32:345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gorman JV, Colgan JD. Regulation of T cell responses by the receptor molecule Tim‐3. Immunol Res 2014; 59:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kane LP. T cell Ig and mucin domain proteins and immunity. J Immunol 2010; 184:2743–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 2010; 235:172–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature 2007; 450:435–9. [DOI] [PubMed] [Google Scholar]
- 7. Kobayashi N, Karisola P, Pena‐Cruz V, Dorfman DM, Jinushi M, Umetsu SE et al TIM‐1 and TIM‐4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 2007; 27:927–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M et al Tim‐3 mediates phagocytosis of apoptotic cells and cross‐presentation. Blood 2009; 113:3821–30. [DOI] [PubMed] [Google Scholar]
- 9. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH et al T cell/transmembrane, Ig, and mucin‐3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 2010; 184:1918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Souza AJ, Oriss TB, O'Malley KJ, Ray A, Kane LP. T cell Ig and mucin 1 (TIM‐1) is expressed on in vivo‐activated T cells and provides a costimulatory signal for T cell activation. Proc Natl Acad Sci USA 2005; 102:17113–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim‐3 is phosphorylated upon stimulation by its ligand galectin‐9. Biochem Biophys Res Commun 2006; 351:571–6. [DOI] [PubMed] [Google Scholar]
- 12. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C et al Promotion of tissue inflammation by the immune receptor Tim‐3 expressed on innate immune cells. Science 2007; 318:1141–3. [DOI] [PubMed] [Google Scholar]
- 13. Binne LL, Scott ML, Rennert PD. Human TIM‐1 associates with the TCR complex and up‐regulates T cell activation signals. J Immunol 2007; 178:4342–50. [DOI] [PubMed] [Google Scholar]
- 14. de Souza AJ, Oak JS, Jordanhazy R, DeKruyff RH, Fruman DA, Kane LP. T cell Ig and mucin domain‐1‐mediated T cell activation requires recruitment and activation of phosphoinositide 3‐kinase. J Immunol 2008; 180:6518–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rodriguez‐Manzanet R, Meyers JH, Balasubramanian S, Slavik J, Kassam N, Dardalhon V et al TIM‐4 expressed on APCs induces T cell expansion and survival. J Immunol 2008; 180:4706–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mariat C, Degauque N, Balasubramanian S, Kenny J, DeKruyff RH, Umetsu DT et al Tim‐1 signaling substitutes for conventional signal 1 and requires costimulation to induce T cell proliferation. J Immunol 2009; 182:1379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Curtiss ML, Hostager BS, Stepniak E, Singh M, Manhica N, Knisz J et al Fyn binds to and phosphorylates T cell immunoglobulin and mucin domain‐1 (Tim‐1). Mol Immunol 2011; 48:1424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA et al Phosphotyrosine‐dependent coupling of Tim‐3 to T‐cell receptor signaling pathways. Mol Cell Biol 2011; 31:3963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clayton KL, Haaland MS, Douglas‐Vail MB, Mujib S, Chew GM, Ndhlovu LC et al T cell Ig and mucin domain‐containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol 2014; 192:782–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomkowicz B, Walsh E, Cotty A, Verona R, Sabins N, Kaplan F et al TIM‐3 suppresses anti‐CD3/CD28‐induced TCR activation and IL‐2 expression through the NFAT signaling pathway. PLoS ONE 2015; 10:e0140694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T et al Th1‐specific cell surface protein Tim‐3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002; 415:536–41. [DOI] [PubMed] [Google Scholar]
- 22. Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell‐mediated effector immunity. J Allergy Clin Immunol 2015; 135:626–35. [DOI] [PubMed] [Google Scholar]
- 23. DuPage M, Bluestone JA. Harnessing the plasticity of CD4+ T cells to treat immune‐mediated disease. Nat Rev Immunol 2016; 16:149–63. [DOI] [PubMed] [Google Scholar]
- 24. Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol 2014; 15:798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL‐12 produced by Listeria‐induced macrophages. Science 1993; 260:547–9. [DOI] [PubMed] [Google Scholar]
- 27. Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW et al Cutting edge: role of IL‐27/WSX‐1 signaling for induction of T‐bet through activation of STAT1 during initial Th1 commitment. J Immunol 2003; 170:4886–90. [DOI] [PubMed] [Google Scholar]
- 28. Macatonia SE, Hsieh CS, Murphy KM, O'Garra A. Dendritic cells and macrophages are required for Th1 development of CD4+ T cells from αβ TCR transgenic mice: IL‐12 substitution for macrophages to stimulate IFN‐γ production is IFN‐γ‐dependent. Int Immunol 1993; 5:1119–28. [DOI] [PubMed] [Google Scholar]
- 29. Villarino AV, Kanno Y, Ferdinand JR, O'Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol 2015; 194:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oestreich KJ, Weinmann AS. Transcriptional mechanisms that regulate T helper 1 cell differentiation. Curr Opin Immunol 2012; 24:191–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev 2012; 248:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015; 74:5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol 2016; 183:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ et al The Tim‐3 ligand galectin‐9 negatively regulates T helper type 1 immunity. Nat Immunol 2005; 6:1245–52. [DOI] [PubMed] [Google Scholar]
- 35. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A et al Bat3 promotes T cell responses and autoimmunity by repressing Tim‐3‐mediated cell death and exhaustion. Nat Med 2012; 18:1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Su EW, Bi S, Kane LP. Galectin‐9 regulates T helper cell function independently of Tim‐3. Glycobiology 2011; 21:1258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vaitaitis GM, Wagner DH Jr. Galectin‐9 controls CD40 signaling through a Tim‐3 independent mechanism and redirects the cytokine profile of pathogenic T cells in autoimmunity. PLoS ONE 2012; 7:e38708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Madireddi S, Eun SY, Lee SW, Nemcovicova I, Mehta AK, Zajonc DM et al Galectin‐9 controls the therapeutic activity of 4‐1BB‐targeting antibodies. J Exp Med 2014; 211:1433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C et al Galectin‐9‐CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity 2014; 41:270–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR et al Tim‐3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV‐1 infection. J Exp Med 2008; 205:2763–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Golden‐Mason L, Palmer BE, Kassam N, Townshend‐Bulson L, Livingston S, McMahon BJ et al Negative immune regulator Tim‐3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol 2009; 83:9122–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kassu A, Marcus RA, D'Souza MB, Kelly‐McKnight EA, Golden‐Mason L, Akkina R et al Regulation of virus‐specific CD4+ T cell function by multiple costimulatory receptors during chronic HIV infection. J Immunol 2010; 185:3007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K et al Cooperation of Tim‐3 and PD‐1 in CD8 T‐cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA 2010; 107:14733–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med 2010; 207:2187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C et al Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen‐specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 2010; 207:2175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ et al IL‐12 upregulates TIM‐3 expression and induces T cell exhaustion in patients with follicular B cell non‐Hodgkin lymphoma. J Clin Invest 2012; 122:1271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qiu Y, Chen J, Liao H, Zhang Y, Wang H, Li S et al Tim‐3‐expressing CD4+ and CD8+ T cells in human tuberculosis (TB) exhibit polarized effector memory phenotypes and stronger anti‐TB effector functions. PLoS Pathog 2012; 8:e1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Oxenius A, Bachmann MF, Zinkernagel RM, Hengartner H. Virus‐specific MHC‐class II‐restricted TCR‐transgenic mice: effects on humoral and cellular immune responses after viral infection. Eur J Immunol 1998; 28:390–400. [DOI] [PubMed] [Google Scholar]
- 50. Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 2005; 33:W741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2–ΔΔCT method. Methods 2001; 25:402–8. [DOI] [PubMed] [Google Scholar]
- 52. Anderson AC, Lord GM, Dardalhon V, Lee DH, Sabatos‐Peyton CA, Glimcher LH et al T‐bet, a Th1 transcription factor regulates the expression of Tim‐3. Eur J Immunol 2010; 40:859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA et al Differential expression of Ly6C and T‐bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity 2011; 35:633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS et al Distinct memory CD4+ T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity 2013; 38:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G et al An IL‐27/NFIL3 signalling axis drives Tim‐3 and IL‐10 expression and T‐cell dysfunction. Nat Commun 2015; 6:6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chou FC, Kuo CC, Chen HY, Chen HH, Sytwu HK. DNA demethylation of the TIM‐3 promoter is critical for its stable expression on T cells. Genes Immun 2016; 17:179–86. [DOI] [PubMed] [Google Scholar]
- 57. Bach EA, Szabo SJ, Dighe AS, Ashkenazi A, Aguet M, Murphy KM et al Ligand‐induced autoregulation of IFN‐γ receptor β chain expression in T helper cell subsets. Science 1995; 270:1215–8. [DOI] [PubMed] [Google Scholar]
- 58. Pernis A, Gupta S, Gollob KJ, Garfein E, Coffman RL, Schindler C et al Lack of interferon γ receptor β chain and the prevention of interferon γ signaling in TH1 cells. Science 1995; 269:245–7. [DOI] [PubMed] [Google Scholar]
- 59. Hwang ES, Hong JH, Glimcher LH. IL‐2 production in developing Th1 cells is regulated by heterodimerization of RelA and T‐bet and requires T‐bet serine residue 508. J Exp Med 2005; 202:1289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al Bcl6 and Blimp‐1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009; 325:1006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F et al LEF‐1 and TCF‐1 orchestrate TFH differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol 2015; 16:980–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gorman JV, Starbeck‐Miller G, Pham NL, Traver GL, Rothman PB, Harty JT et al Tim‐3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. J Immunol 2014; 192:3133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka‐Akita H et al Tumor‐infiltrating DCs suppress nucleic acid‐mediated innate immune responses through interactions between the receptor TIM‐3 and the alarmin HMGB1. Nat Immunol 2012; 13:832–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wada J, Kanwar YS. Identification and characterization of galectin‐9, a novel β‐galactoside‐binding mammalian lectin. J Biol Chem 1997; 272:6078–86. [DOI] [PubMed] [Google Scholar]
- 65. Matsumoto R, Matsumoto H, Seki M, Hata M, Asano Y, Kanegasaki S et al Human ecalectin, a variant of human galectin‐9, is a novel eosinophil chemoattractant produced by T lymphocytes. J Biol Chem 1998; 273:16976–84. [DOI] [PubMed] [Google Scholar]
- 66. Imaizumi T, Kumagai M, Sasaki N, Kurotaki H, Mori F, Seki M et al Interferon‐γ stimulates the expression of galectin‐9 in cultured human endothelial cells. J Leukoc Biol 2002; 72:486–91. [PubMed] [Google Scholar]
- 67. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X et al Tim‐3/galectin‐9 signaling pathway mediates T‐cell dysfunction and predicts poor prognosis in patients with hepatitis B virus‐associated hepatocellular carcinoma. Hepatology 2012; 56:1342–51. [DOI] [PubMed] [Google Scholar]
- 68. Gieseke F, Kruchen A, Tzaribachev N, Bentzien F, Dominici M, Muller I. Proinflammatory stimuli induce galectin‐9 in human mesenchymal stromal cells to suppress T‐cell proliferation. Eur J Immunol 2013; 43:2741–9. [DOI] [PubMed] [Google Scholar]
- 69. Mengshol JA, Golden‐Mason L, Arikawa T, Smith M, Niki T, McWilliams R et al A crucial role for Kupffer cell‐derived galectin‐9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE 2010; 5:e9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Reddy PB, Sehrawat S, Suryawanshi A, Rajasagi NK, Mulik S, Hirashima M et al Influence of Galectin‐9/Tim‐3 interaction on herpes simplex virus‐1 latency. J Immunol 2011; 187:5745–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Steelman AJ, Smith R 3rd, Welsh CJ, Li J. Galectin‐9 protein is up‐regulated in astrocytes by tumor necrosis factor and promotes encephalitogenic T‐cell apoptosis. J Biol Chem 2013; 288:23776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McSharry BP, Forbes SK, Cao JZ, Avdic S, Machala EA, Gottlieb DJ et al Human cytomegalovirus upregulates expression of the lectin galectin 9 via induction of β interferon. J Virol 2014; 88:10990–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tandon R, Chew GM, Byron MM, Borrow P, Niki T, Hirashima M et al Galectin‐9 is rapidly released during acute HIV‐1 infection and remains sustained at high levels despite viral suppression even in elite controllers. AIDS Res Hum Retroviruses 2014; 30:654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hsu YL, Wang MY, Ho LJ, Huang CY, Lai JH. Up‐regulation of galectin‐9 induces cell migration in human dendritic cells infected with dengue virus. J Cell Mol Med 2015; 19:1065–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E et al PD‐1 identifies the patient‐specific CD8+ tumor‐reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124:2246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM et al Apoptosis of tumor infiltrating effector TIM‐3+ CD8+ T cells in colon cancer. Sci Rep 2015; 5:15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sabins NC, Chornoguz O, Leander K, Kaplan F, Carter R, Kinder M et al TIM‐3 engagement promotes effector memory T cell differentiation of human antigen‐specific CD8 T cells by activating mTORC1. J Immunol 2017; 199:4091–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakae S, Iikura M, Suto H, Akiba H, Umetsu DT, Dekruyff RH et al TIM‐1 and TIM‐3 enhancement of Th2 cytokine production by mast cells. Blood 2007; 110:2565–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD et al Tim‐3 enhances FcεRI‐proximal signaling to modulate mast cell activation. J Exp Med 2015; 212:2289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of differential gene expression between Tim‐3− and Tim‐3+ T helper type 1 cells.
Figure S2. Persistence and functionality of Tim‐3− and Tim‐3+ T helper type 1 cells 23 days after transfer into naive mice.
Figure S3. Degranulation, as measured by CD107a expression, of Tim‐3− and Tim‐3+ T helper type 1 cells following exposure to Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) infection.
Figure S4. Increased degranulation, as measured by CD107a expression, of Tim‐3+ cells generated by adoptively‐transferred T helper type 1 cells following exposure to Armstrong strain of lymphocytic choriomeningitis virus (LCMV‐Arm) infection.
Table S1. Genes expressed at significantly different levels in Tim‐3+ T helper type 1 cells relative to Tim‐3− Th1 cells.
Table S2. Oligonucleotide primers used for quantitative real‐time PCR analysis.