

Abstract
Hepatocellular carcinoma (HCC), is one of the most frequent human cancer and is characterized by a high mortality rate. The aggressiveness appears strictly related to the liver pathological background on which cancer develops. Inflammation and the consequent fibro/cirrhosis, derived from chronic injuries of several origins (viral, toxic and metabolic) and observable in almost all oncological patients, represents the most powerful risk factor for HCC and, at the same time, an important obstacle to the efficacy of systemic therapy. Multiple microenvironmental cues, indeed, play a pivotal role in the pathogenesis, evolution and recurrence of HCC as well as in the resistance to standard therapies observed in most of patients. The identification of altered pathways in cancer cells and of microenvironmental changes, strictly connected in pathogenic feedback loop, may permit to plan new therapeutic approaches targeting tumor cells and their permissive microenvironment, simultaneously.
Keywords: Hepatitis, hepatocyte nuclear factor 4 alpha (HNF4α), liver fibrosis, transforming growth factor beta (TGFβ), yes-associated protein (YAP)
Introduction
Hepatocellular carcinoma (HCC) represents one of the most frequent cause of cancer-related death worldwide, in spite of the increasing success in fighting different kinds of cancer.
The poor prognosis of HCC patients is largely attributed to tumor phenotypic and genetic heterogeneity, multifocal occurrence, high tumor recurrence rate after treatments and high predisposition to metastasize (1).
HCC almost always develops on the pathological background of a variety of chronic liver diseases, ranging from chronic viral hepatitis (hepatitis B and hepatitis C), hereditary disorders, alcoholic and chemical injury to metabolic syndromes, that contribute to its heterogeneity (2). All these chronic liver injuries are able to establish environmental changes that directly promote HCC onset, maintenance and progression.
Current therapeutic approaches include surgical resection, percutaneous radiofrequency ablation, trans-arterial chemoembolization and liver transplantation. Surgery and local treatments are applicable for patients with limited underlying liver disease. However, most patients present multifocal HCC and advanced-stage of liver disease or comorbidities at the time of diagnosis. Furthermore, HCC multifocal occurrence, due to intrahepatic metastatic spreading of primary tumor cells or to the development of multiple independent HCCs (3), is responsible for frequent early phase tumor recurrence following the described treatments and largely impacts on their outcome (4).
Liver transplantation represents the optimal therapy allowing to remove both tumor and underlying diseases. However, it is limited by the poor availability of liver donors and, consequently, by very long waiting lists, incompatible with the severity of the disease; moreover, not all patients are eligible for this treatment (5).
Therefore, systemic therapies are clearly needed to improve HCC patient’s prognosis. At this regard, the hard challenge is performing a therapy protocol taking into account the complex alterations that affect the entire organ and can largely interfere with most of the currently applied therapies making them ineffective (6).
Outline of the current molecular therapies of HCC
Similarly to many other tumors, the development of HCC is a multistep process involving the accumulation of genetic and epigenetic alterations in regulatory genes, resulting in the activation of oncogenes and in the inactivation/loss of tumor suppressor genes (7).
Extensive research over the past decade was focused on the identification of molecular pathways driving hepatocyte transformation, aberrantly activated or expressed, useful for early diagnosis and prognosis prediction. Recurrent focal amplifications or deletions involving genes that control oncogenic or tumor suppressing pathways (i.e., Wnt, MAPK, PI3K signaling and p53 pathway) were identified in primary HCCs (8).
Currently, a further effort was made to catalog HCCs in subclasses, based on different genetic and epigenetic alterations and clinical behavior, with the aim to develop tailored treatment strategies. Genomic studies have allowed to categorize HCCs into subsets with distinct molecular and clinical features. In particular, one subset is characterized by altered expression of genes mainly involved in cell proliferation (such as mTOR, IGF, Myc and Ras) and correlates with an aggressive tumor phenotype. Another subset is characterized by activation of the WNT/β-catenin and TGFβ signaling pathways (9), known to play a crucial role in liver development and liver cell differentiation, and found dysregulated in many human cancers. This subset is significantly associated to high risk of early tumor recurrence. A third subgroup is characterized by altered expression of genes involved in interferon signaling and inflammation (10). Moreover, increasing efforts enabled the identification of a number of HCC-related molecular biomarkers, including proteins and non-coding RNAs (particularly microRNAs), which role in transformation process is yet largely unknown, but which recognition could be important to further catalog HCCs and to plan diagnostic and therapeutic strategies (11).
At present, the targeted therapies represent the more effective treatment of HCC (12). In particular, the multi-kinase inhibitory sorafenib, was shown to improve the overall survival of HCC patients (13,14) with manageable toxicity and has become the standard systemic therapy. This molecule efficiently inhibits kinase receptors involved in the proliferative and stemness pathways found upregulated in a variety of human tumors (15). However, for patients in advanced-stage of HCC, unresponsive to sorafenib or with tumor recurrence, clinical options are yet very limited. In these cases, as well as in several other cancers unresponsive to the traditional chemotherapy, approaches of immunotherapy are promising (16). In particular, a targeted therapy of HCC with monoclonal antibodies able to induce an activation of the immune system and/or its escape from the inhibitory effects of tumor cells has been explored. The block of the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), which provides inhibitory signals to T cells, by means of a monoclonal antibody (tremelimumab), was found to lead to cytotoxic T-lymphocyte activation (17,18). The monoclonal antibody against the programmed cell death-1 (PD-1) receptor on activated T cells (nivolumab) was shown to impair its inactivating binding to the ligand PDL-1, expressed on HCC tumor cells, thus restoring T-cell-mediated antitumor response (19,20). The evaluation of efficacy of these immunotherapies in advanced-HCC patients in clinical trials (phase II and phase III) is still in progress.
Additional ongoing trials have been designed to test drugs in biomarker-based HCC subpopulations (21).
Lessons from phase III clinical trial
After the completion of a number of phase III clinical trials of patients with advanced HCC, appeared clear that targeted agents other than Sorafenib do not significantly improve the overall survival, and that combination therapies do not show significant advantage (22).
A part of the reason for the failure of these treatments resides in the HCC heterogeneity, together with an unsatisfactory classification, that reduces the efficacy of therapeutic protocols directed to a single target. Moreover, the liver toxicity of several compounds (a limiting feature in patients with pre-existing liver pathologies) and the marginal anti-tumor effects of tools utilized so far contributed to the disappointing results of phase III clinical trials (22).
The strong recurrence of the tumor after chemical/immune therapy or surgical eradication suggests a relevant role of liver progenitors or cancer stem cells (CSCs) in the reappearance of the disease. Whatever the origin of HCC, from mutations in normal stem cells or from events of reprogramming of differentiated hepatocytes, the presence of cancer cells with stemness features renders the therapies less effective (due to chemo-resistance, increased survival, resistance to hypoxia, immuno-tolerance, active drug-exporting systems observed in these cells) (23). CSC-targeted treatments would have potential high benefit at long-term ensuring the eradication of the cell subset responsible for the maintenance of tumor mass.
Notably, the acquisition or the maintenance of stemness features by pre-neoplastic and neoplastic cells can derive, other than from cell genetic/epigenetic modifications, also from tumor microenvironmental stimuli (e.g., extracellular matrix (ECM)-driven inputs, selective signals induced by treatments such as chemotherapy). Hence, specifically targeting these favorable tumor micro-environmental elements may increase the efficacy of therapies.
Altogether, the features of the HCC described so far underline how in the treatment of this tumor it is necessary to consider several aspects, including genetic and epigenetic alterations of each HCC, the persistence and the role of CSCs and, non-last, the tumor environment that favors both the development and the progression of the cancer and the reduction of current therapy efficacy. Therefore, new therapeutic approaches should aim to combine antitumor efficacy with “normalizing” effects on the environment where HCC develops with the final goal to reduce both cell-autonomous and non-cell autonomous oncogenic stimuli, to render more accessible to the drugs the transformed cells and, ultimately, to reduce cancer recurrence after treatment.
Now, we will analyze the cellular, chemical and physical aspects of the microenvironment influencing the crosstalk between cells and tumor niche to indicate possible therapeutic targets and tools.
Role of microenvironmental cues in HCC pathogenesis
HCC development is commonly associated with pre-existing chronic liver diseases representing high risk factors (viral, alcoholic, toxic and metabolic liver hepatitis). Whatever the etiology, the tumor pathogenesis is invariably related to inflammation process, high rate of hepatocyte death and regenerative proliferation and activation of hepatic stellate cells (HSCs) into fibril-producing cells. All these conditions, leading to liver fibrosis, are highly predisposing to tumor onset and favor tumor progression and metastasization (intraorgan and/or at distance).
Therefore, it is now clear that HCC development is strictly dependent on environmental cues able to induce and maintain biological changes in tumor cells and in their tissue niche. The main cellular components of HCC microenvironment are stromal cells [including carcinoma-associated fibroblasts (CAFs)], endothelial cells, HSCs and immune cells [including tumor-associated macrophages (TAMs)]; all of them are influenced by the molecular interplay (modulated via growth factors, inflammatory cytokines, ECM proteins and cell-cell contacts) with the other ones (24).
In this section, we will discuss the main micro-environmental changes triggered by chronic liver damage that on one hand is a source of hepatocyte proliferative stimuli and, on the other one, induces progressive stromal alteration. Taking these changes into account will allow the acquisition of better knowledge of the mechanisms involved in tumor occurrence and the set-up of more effective therapeutic approaches.
Liver inflammation as driving force of liver cell transformation
HCC is strictly related to the inflammation process (25). The recruitment of inflammatory cells in the chronically injured tissue, indeed, is crucial for the microenvironment changes driving to fibrosis and HCC insurgence.
Infected or in various ways injured parenchymal cells secrete a variety of cytokines, including interleukin-1β (IL-1β), tumor necrosis factor α (TNFα), transforming growth factor β (TGFβ) and interleukin-6 (IL-6), responsible for the recruitment and the activation of other cell types. Among these cells, fibrogenic cells (mainly HSCs) are activated; this leads to the production of an excessive amount of ECM proteins that, in turn, altering stroma composition and stiffness, are responsible for further activation of HSCs and recruitment of inflammatory cells, effectively establishing a positive pathogenic loop (Figure 1).
Figure 1.
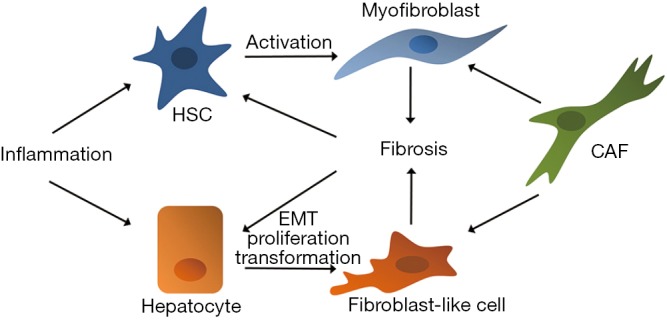
Cell-cell and cell-stroma interplay sustaining fibrosis and inducing hepatocyte transformation. HSC, hepatic stellate cell; CAF, cancer activated fibroblast; EMT, epithelial-to-mesenchymal transition.
IL-1β is an inflammatory cytokine released from infiltrating macrophages during liver chronic inflammation and contributing to the establishment of fibrosis (26).
TNFα, produced by inflammatory and endothelial cells, induces hepatocyte survival and resistance to apoptotic stimuli, so having an important role in promoting HCC (27).
IL-6, mainly released by resident liver macrophages (Kupffer cells) early after hepatocyte damage or by hepatitis virus infected hepatocytes (28,29), has been found at high serum level in chronic liver diseases and correlates with an increased risk to develop HCC (30). This well-known inflammatory cytokine activates and maintains liver inflammation as well as tumor-associated signaling pathways, leading to hepatitis progression, hepatocyte continuous proliferation, cirrhosis and ultimately HCC. When tumor has arisen, IL-6 is secreted by infiltrating TAMs and promotes the expansion of HCC progenitor/CSCs; its high plasma level, in fact, correlates with tumor recurrence, metastasis and poor prognosis (31,32).
Among the cytokines secreted by inflammatory cells, the TGFβ represents one of the most relevant pro-fibrogenic molecules, responsible for HSCs activation and consequently synthesis and secretion of high amount of ECM proteins (33). Moreover, the cytokine induces the differentiation and function of CAFs (34).
The establishment of an inflammatory response is a common aspect of chronic viral hepatitis, the most frequent risk factor of HCC. About three-quarters of HCCs worldwide are ascribed to chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections (35). HCV infection, in particular, is one of the main causes of chronic liver disease, in most Western countries, leading to liver fibrosis, cirrhosis and HCC (36). HCV has been strongly implicated in HCC pathogenesis because of its role in the alteration of ROS and other signaling pathways and in modulation of immune responses and apoptosis (36,37). On the other hand, infection creates an inflammatory environment, elevating IL-1β levels and altering TGFβ signaling (38). The continuous inflammation in infected liver cells, in fact, induces alternating cycles of cell elimination by apoptosis and tissue regeneration contributing to the long-term establishment of fibrosis.
Over the last years, a combination of direct-acting antiviral drugs (DAAs) has been very effective in eradicating HCV and inhibiting continuous inflammation in the liver. However, treating HCV infection is not sufficient to ameliorate existing liver diseases, such as advanced cirrhosis, and to protect against HCC development (39). Also, the incidence of HCC after HCV clearance is still very high in those patients and complicated by concomitant risk factors (e.g., HBV co-infection or metabolic syndromes) suggesting that the viral infection can leave a “mark” in liver cells or in liver microenvironment favoring the establishment/promotion of liver cancer. Importantly, wide range analysis of gene expression in HCV-infected livers at early stage of cirrhosis, unveiled a 186-gene signature that was able to predict cirrhosis progression and HCC insurgence. Marker genes that defined the signature were enriched in inflammation signals, including the NF-kB pathway and IL-6 signaling (40).
Fibrosis as tumor promoting background
Fibrosis represents an invariable evolution of chronic inflammatory processes, and its pathological endpoint, cirrhosis, a high-risk factor for the occurrence of HCCs.
Fibrosis is characterized by a deregulated remodeling of ECM, consisting in an increased deposition and a reduced turnover of its components. ECM accumulation not only causes deeply changes in liver architecture and function but can also create a physical barrier to drug delivery. The main fibrogenic cells are HSCs while the contribution of parenchymal cells (hepatocytes and cholangiocytes undergoing epithelial-to-mesenchymal transition, EMT), of endothelial cells (undergoing endothelial-to-mesenchymal transition) and of bone marrow stem cells, which has also been described (41), appears to be at lesser extent.
HSCs secrete ECM proteins, metalloproteinases (MMPs) and their tissue specific inhibitors (i.e., TIMPs), together with a lot of cytokines, including platelet-derived growth factors (PDGFs), fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF) and TGFβ. All of these contribute to sustain and increase the fibrotic process and, when the hepatocyte transformation has occurred, to positively promote the proliferation and survival of tumor cells (42).
Fibrotic microenvironment, moreover, is characterized by increased stiffness of the entire organ that, while affects liver cell proliferation, differentiation state, functionality (43) and sensitivity to the drugs, further promotes activation of quiescent HSCs, establishing a positive feedback loop, responsible for the formation of pathological scar tissue and then cirrhosis (44) (Figure 1).
While the late stages of liver fibrosis and cirrhosis are irreversible, the early stages of fibrosis and, possibly, of cirrhosis are to some extent reversible (45). Thus, the development of strategies targeting liver fibrosis could stop the progression of disease.
Finding new molecular targets
Inflammation and fibrosis are processes largely investigated and most of the molecular mechanisms have been clarified. The dissection of molecular pathways driving to and/or activated by these pathological situations and directly involved in HCC initiation and development, permits to identify molecular targets for innovative therapeutic protocols.
Hippo/yes-associated protein (YAP) pathway
Hippo pathway is a molecular signaling starting from several extracellular signals, including diffusible molecules and mechanical stimuli, all parameters strongly altered in fibrotic livers. Hippo signaling consists of a specific and evolutionary conserved set of kinases, such as Hippo/MSTs and LATS1/2 (46), largely converging on YAP, a co-transcriptional factor responsible for a wide modulation of gene expression. Activation of Hippo pathway results in YAP inhibitory phosphorylation and degradation and, ultimately, in the inactivation of the YAP-dependent transcriptional activity (46). The Hippo/YAP signaling in the liver controls development, mass homeostasis, ECM composition and elasticity, hepatocyte proliferation and differentiation, as well as the maintenance of stem/precursor compartments. Its dysregulation, by inactivation of Hippo kinases and/or activation of YAP, has been observed in several human cancers and has been experimentally demonstrated to have a significant impact on cancer development, both by eliciting microenvironment changes triggering fibrosis and promoting tumor onset (47). YAP, in fact, can influence the chemical and physical composition of the tumor microenvironment through the functional regulation of different cell types. This function is mainly accomplished through the regulation of HSCs, where YAP induces the secretion of ECM and pro-angiogenetic proteins (48). YAP function is required for the establishment and maintenance of CAFs (49). Moreover, YAP stimulates the production of chemoattractants for the recruitment of T cell suppressing myeloid-derived cells by carcinoma cells (50). YAP hyperactivation can promote tumorigenesis through the stimulation of proliferation, reduction of cell death and expansion of stem cell/precursor populations (51,52). At the same time, the reactivation of YAP in differentiated hepatocytes (where it is normally inactive) can induce their de-differentiation (52) and the acquisition of mesenchymal traits, thus triggering cell migration and invasion and conferring chemo-resistance (53). Diethylnitrosamine (DENA)-induced liver tumorigenesis in rat models showed an early accumulation of YAP in pre-neoplastic foci and in oval cells, promoting their tumorigenic and metastatic properties (54), thus indicating YAP as a sensitive marker of pre-neoplastic stages.
Accordingly with the role of YAP in HCC development, its increased expression and nuclear localization have been observed in more than 85% of adult HCC tumor samples (55), and in 62% of patients with HBV-associated HCC after surgical liver resection (56). Furthermore, its overexpression has been significantly associated with a poor prognosis of HCC patients (tumor recurrence, shorter overall survival and lower disease-free survival after treatment) resulting an independent prognostic marker for HCC (56,57).
Other than genetic alterations of Hippo pathway components, microenvironmental cues can control YAP subcellular localization and activation. Mechanical inputs from ECM, such as matrix stiffness and cytoskeletal tension, represent a crucial trigger for nuclear localization and transcriptional activation of YAP (58). When resident liver stem cells are cultured on stiff ECM, YAP is retained in the nucleus in the active form, whereas it is inhibited and re-localized in the cytoplasm when the same cells are cultured on a soft ECM (43). Interestingly, YAP subcellular localization correlates with differentiation state of liver cells, such as of several other cell types (43,58). Moreover, culturing HSCs on soft substrates inhibits YAP signaling and cell activation that, instead, occurs when HSCs are cultured in high stiffness (48).
In vivo, the altered stiffness of the inflamed/fibrotic liver can represent a major trigger of YAP activation for hepatocytes (as well as for HSCs), that therefore go towards de-differentiation and proliferation. The same de-differentiative and proliferative stimuli can be exerted by rigid stroma also on transformed hepatocytes so influencing tumor progression. These findings, while explain why chronic liver diseases can increase the risk of HCC, support the need to take into account microenvironment changes in the design of new strategy for tumor eradication. Since YAP activation, as said above, may influence and be influenced by the composition and the chemical/physical characteristics of the tumor microenvironment, the targeting of this pathway could represent an efficient approach to “normalize” both tumor cells and tumor niche.
Targeting of YAP
The described role of YAP in the pathogenesis of HCC suggests YAP as a possible therapeutic target for the treatment of HCC. Several observations in animal models demonstrated the efficacy of YAP silencing in the prevention and regression of liver fibrosis and in the reduction of experimentally induced liver cancers; this suggests that genetic or pharmacological interference of YAP expression/activity could provide therapeutic benefits in humans.
In the last few years, different approaches to inhibit YAP oncogenic function have been proposed. A pharmacological inhibitor of YAP activity, verteporfin, has been recently identified by a drug library screening for the ability to interfere with YAP-dependent transcription (59). It is a small molecule able to selectively bind YAP and, by a conformational change, to impairs its interaction with its transcriptional factor TEAD. TEAD factor, in fact, is essential in mediating YAP-dependent gene expression leading to cell proliferation, oncogenic transformation and EMT (59,60).
Verteporfin appears an ideal drug for selective inhibition of YAP-induced tumorigenesis. Its experimental administration in rats and mice, in fact, has been shown to inhibit the YAP oncogenic activity (54,59) without affecting liver size and safety of wild-type animals (59). Moreover, it has been shown that verteporfin interferes with the high stiffness-dependent HSC activation in vitro and ameliorates the CCl4-induced liver fibrosis in vivo (48,61). Notably, phase I/II clinical trials in pancreatic cancer reported a safe utilization of the drug (62), suggesting a feasible application to HCC treatment.
Approaches using small peptides to inhibit YAP function through competition with TEAD interaction have been also described. The use of cyclic peptides mimicking the binding of YAP to TEAD significantly reduced HCC growth rate in mice (63). Moreover, a small peptide consisting in the inhibitory domain of the vestigial-like protein 4, a natural antagonist of YAP in TEAD binding, has been successfully assessed in the suppression of gastric cancers in vivo (64).
In addition, an engineered TEAD-dominant negative protein, lacking its DNA-binding domain, has been shown to suppress YAP-mediated transactivation and to prevent YAP-driven tumorigenesis (59). Notably, these pre-clinical studies have also demonstrated that the TEAD-dominant negative can inhibit liver tumorigenesis caused by YAP overexpression without compromising normal liver homeostasis and physiology (59); this suggests that YAP pathway targeting can ensure high selectivity for cancer cells, compared to conventional therapies.
Direct targeting of YAP has been performed also by using small interfering RNA encapsulated into lipid nanoparticles (siRNA-LNPs); this treatment reduces tumor cell proliferation, restores hepatocyte differentiation and zonal gene expression, eventually causing tumor regression in mice models of advanced HCC (65). The only mild and reversible side effects observed in treated animals encourage clinical application.
Altogether, these results render YAP pathway a promising target for molecular therapy of HCC.
TGFβ signaling
The pleiotropic TGFβ cytokine has emerged as a pivotal player in HCC pathogenesis and progression, arranging microenvironment and cell autonomous changes, both favorable for tumor onset and growth (66).
The TGFβ signaling pathway involves the binding of the cytokine to a serine-threonine kinase type II receptor (TGFβ RII) that recruits and phosphorylates a type I receptor (TGFβ RI). The TGFβ RI subsequently phosphorylates downstream effectors, typically Smad transcription factors (Smad-dependent pathway). Moreover, TGFβ can signal in a Smad-independent manner, integrating pathways starting from a variety of extracellular signals and adhesion molecules, thus inducing a complex modulation of gene expression (67).
TGFβ signaling is involved in all stages of HCC progression, from initial liver injury and inflammation through fibro/cirrhosis to tumor initiation, development and metastasis.
TGFβ has cytostatic, apoptotic and aging effects in hepatocytes during embryogenesis, controls mass homeostasis and suppresses tumor development at early stages in adults (68). On the contrary, high level of the cytokine or an upregulation of its pathway promotes liver inflammation, fibrosis, activation of HCSs and, when the transformation process had occurred, the apoptosis resistance and proliferation of transformed hepatocytes (68). Moreover, TGFβ is a major inducer on EMT, thus promoting progression and metastasis of primary tumors (69).
TGFβ is synthesized and secreted by inflammatory cells and, while stimulates the activation of HSCs into fibrogenic cells, it is produced and secreted by these. Moreover, hyperactivation of its signaling in tumor cells can derive from autocrine loop, other than from paracrine mechanisms (70,71).
Tumor-secreted TGFβ may reduce antitumor immune responses by increasing the quantity of immunosuppressive cells, such as regulatory T cells (72,73), and may regulate the production/secretion of growth factors and cytokines by CAFs, thus contributing to tumor cell proliferation and invasion, and to neoangiogenesis (34).
Accordingly with its tumor promoting role, the TGFβ signaling was found constitutively activated in HCC cell lines (74,75). Moreover, high serum levels of the cytokine were found in advanced-stage HCC patients and correlated with tumor vascularization, metastasis formation, reduced survival and poor prognosis (75-78).
Targeting of TGFβ signaling
As described above, the presence of TGFβ signaling is not restricted to late stages of HCC but it is observed throughout the development of HCC, working at different levels during HCC pathogenesis in an autocrine/paracrine manner. It can modulate both cancer cell function and microenvironment changes, and its inhibition can affect different stages of HCC onset and progression and offer a wide therapeutic application.
Specific TGFβ-receptor inhibitors have been developed and utilized in preclinical models to test their efficacy as anti-fibrotic and anti-cancer tools. The in vitro characterization of these inhibitors showed anti-EMT effects with re-expression of E-cadherin (79). In vivo, it has been observed a reduced fibro-cirrhotic process, inhibition of neoangiogenesis due to a block in the VEGF production (71) and a reduced migration and invasion of HCC cell lines (79). In particular, the orally active TGFβ RI/II kinase dual inhibitor LY2109761, has been proven effective in the reduction of vascular invasion (79) and in the inhibition of tumor growth and metastasis in xenograft model of HCC (80). Another TGFβ-receptor inhibitor, LY2157299 monohydrate (Galunisertib) (81), is now under clinical evaluation in phase II clinical trials and suggests improved clinical outcome in patients with HCC (82).
EMT-driving signaling pathways
EMT is a trans-differentiation process involving a deep reprogramming of the cellular transcriptional profile, leading to the loss of polarity and cell-cell contacts and the acquisition of a mesenchymal phenotype and migratory capacity. EMT is dynamically regulated in physiological background where it is reversible [in mesenchymal-to-epithelial transition (MET)] (83).
The EMT/MET dynamics characterizes physiological situations, such as organogenesis during embryonic development, wound healing and tissue regeneration in the adult, but it also drives pathological states, including fibrosis and epithelial tumor progression (84).
In the liver, several tissue conditions and diffusible molecules can induce EMT program. Major role in its activation is played by the mechano-stimuli starting from high rigidity of the fibrotic stroma and by the cytokines, primarily TGFβ, influencing tumor microenvironment and tumor cells.
While the contribution to the fibrogenesis of normal hepatocytes induced in EMT is still debated (85), the relevance of EMT in transformed hepatocytes during tumor progression is well established. In particular, cells lose epithelial differentiation and possibly acquire self-renewal ability and stemness gene expression. The occurrence of an EMT program in a tumor cell induces the acquisition of more aggressive properties, including motility and invasivity, increased resistance to apoptotic stimuli and chemoresistance (86).
Various master factors, mainly acting as transcriptional repressors of epithelial genes, were shown to be sufficient to trigger and orchestrate EMT. In particular, Snail is sufficient to induce EMT in vitro in many epithelial cells, and its over-expression in vivo correlates with drug resistance, metastasis, high risk for recurrence and poor prognosis (87-89). Snail has been shown to negatively regulate a plethora of genes ranging from adhesion molecules (mainly E-cadherin), pro-apoptotic genes (e.g., Bid and p53), cytoskeletal molecules (e.g., cytokeratins) to master genes of epithelial/hepatocytes differentiation (i.e., HNF4α and HNF1α, see below) through the direct binding to their promoters and the subsequent transcriptional repression (90-93).
Furthermore, Snail, through the transcriptional repression of miRNAs can upregulate mesenchymal markers, survival genes, genes involved in cell movement and invasion (e.g., MMPs) and stemness genes (94).
EMT transcriptional master regulators appear sufficient to reprogram differentiated cell towards an alternative identity thanks to their ability to deeply impact the chromatin state by directly recruiting chromatin modifiers to specific genes. Recent evidence revealed that Snail repressive activity, pivotal in EMT onset and maintenance, depends on upregulation and recruitment on the promoters of target genes of repressive chromatin modifiers. In particular, Snail recruits the histone demethylase LSD1 (95) and the member of Polycomb group proteins EZH2 (93) and up-regulates DNA methyl transferases by downregulating their inhibitory microRNA (96). Notably, Snail/EZH2 complex requires the enrolment of the long non-coding RNA HOTAIR, previously indicated by in vivo studies as an excellent predictor of metastasis and tumor recurrence in HCC (97,98). Accordingly, recent evidence revealed that genome-wide epigenetic modifications are required for the execution of EMT program (99) and are extensively involved in the development of HCC (100).
Targeting of EMT program
Strategies to counteract EMT in tumor progression were largely investigated. One kind of therapeutic approach consists in an epigenetic cancer therapy targeting chromatin regulators involved in EMT. Different classes of drugs, including histone deacetylase inhibitors, DNA methyltransferase inhibitors or demethylating agents (i.e., 5-azacytidine) have been utilized both in HCC cell lines and in xenograft models showing reduced cell proliferation and survival, and inhibition of tumor growth (101). Furthermore, highly specific EZH2 and LSD1 inhibitors have been developed and currently utilized in clinical trials for cancer of different origins (102,103). This strategy has not yet applied in vivo to HCC.
Others therapeutic approaches aimed at directly interfering with the functional role of Snail can be pursued, for example, by using Snail dominant-negative mutants or the expression of Snail repressors able to drive genetic and epigenetic reprogramming of tumor cells. One of these master tumor suppressors will be described in the next section.
Targeting of oncogenic function of lncRNAs such as HOTAIR, already prognostic biomarker for different kind of cancers (104), can also represent a promising strategy for EMT impairment in HCC (105).
Unbiased molecular tools: the case of hepatocyte nuclear factor 4α (HNF4α)
A major challenge in cancer research is the identification of therapeutic strategies able to selectively target cancer cells without affecting normal cells.
A possible approach for HCC treatment that can be conceived is the genetic and epigenetic cell reprogramming through the delivery of master transcription factors.
HNFs are transcription factors that, cooperatively, are responsible for the maintenance of differentiation and metabolic functions of hepatocytes (106). In particular, HNF4α was shown to maintain hepatocyte identity by activating epithelial/hepatocyte genes (107,108) and by repressing the expression of mesenchymal genes (109). The high number of target genes revealed by genome-scale studies (110) demonstrated that HNF4α is a wide-range regulator of liver gene expression.
In human liver, HNF4α expression was found to be reduced in fibro-cirrhotic tissues and decreased in tumor tissue compared to the non-tumoral one of the same patients (111,112). Reduction/loss of HNF4α was found in about 70% of HCC, also not associated with hepatitis virus infection (111). The impaired HNF4α expression correlates with poor differentiation, metabolic defects and altered liver architecture, with metastatic potential of neoplastic cells and, ultimately, with an unfavorable outcome in HCC patients (111).
Several liver injuries target HNF4α (113) and lead to significant changes in its expression or DNA binding (114-117).
In this context, it has been reported that genes sensitive to HNF4α knock-down significantly overlap genes affected by inflammatory cytokines (118), supporting the hypothesis that HNF4α is involved in regulating the inflammatory response in the liver.
A recent report showed that transient inhibition of HNF4α activates an inflammatory feedback loop triggering and sustaining hepatocellular transformation (119). In particular, in this study, HNF4α was found to repress the IL-6/STAT3 inflammatory pathway. Interestingly, perturbation of this circuit has been also observed in human HCC tissues at different stages of HCC pathogenesis (earlystage and advanced stage), supporting a functional role of HNF4α in the inflammation-related HCC.
Loss of hepatocyte differentiation and reduced levels of HNF4α have been observed also upon pathological stimuli from microenvironment (i.e., ECM high stiffness, reminiscent of a liver fibro-cirrhotic state; presence of TGFβ) (43,120,121) and viral infection (122,123) and associated to several tumor-related properties such as chemoresistance, strong proliferative response and EMT (112,122,124) (Figure 2).
Figure 2.
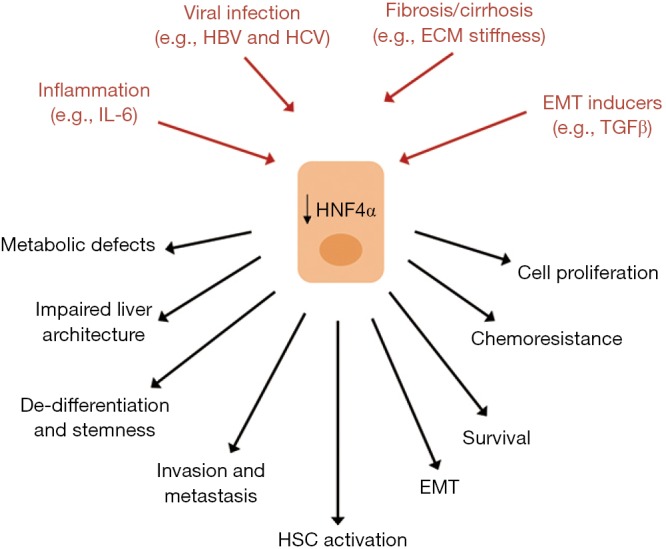
Deregulation of HNF4α in hepatocytes during HCC pathogenesis. Signals from microenvironment responsible for a reduced/loss of HNF4α expression/activity were shown and correlated to tumor-associated outcomes. See text for references. HNF4α, hepatocyte nuclear factor 4α; HSC, hepatic stellate cells; HCC, hepatocellular carcinoma; EMT, epithelial-to-mesenchymal transition; IL-6, interleukin 6; HBV, hepatitis B virus; HCV, hepatitis C virus; ECM, extracellular matrix; TGFβ, transforming growth factor β.
The pivotal role of HNF4α protein as tumor suppressor suggested the design of therapeutic approaches based on the restoration of its expression and function in HCC.
Several evidences supported the use of HNF4α as “drug” for HCC treatment. The advantage of this kind of approach resides first of all in the possibility to target tumor cells independently of their origin (transformed hepatocytes in EMT or liver CSCs). Secondly, the wide-range tumor suppressing activity of HNF4α allows the simultaneous targeting of several cellular tumor-related properties (Figure 3). In particular, extensive in vivo studies demonstrated that HNF4α restores hepatocyte differentiation and induces MET in HCC cell lines (109), controls the epigenetic state of differentiated hepatocytes through the down-regulation of DNA methyltransferases (96), interferes with the expression of EMT-master genes through their direct transcriptional repression (109) and with the expression of stemness markers through the upregulation of the epithelial miRNAs 200s and 34a (94).
Figure 3.
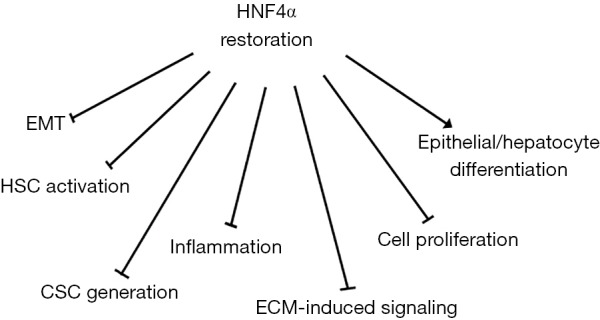
Potential tumor suppressor effects of HNF4α restoration/delivery in HCC. See text for references. HNF4α, hepatocyte nuclear factor 4α; HCC, hepatocellular carcinoma; HSC, hepatic stellate cells; EMT, epithelial-to-mesenchymal transition; ECM, extracellular matrix; CSC, cancer stem cells.
Finally, the restoration of HNF4 expression/activity can “normalize” tumor microenvironment ameliorating hepatic fibrosis in rats (125) and reducing hepatic inflammation (116).
Accordingly with these observations, HNF4α delivery in pre-clinical models resulted in the impairment of HCC occurrence and in the reduced growth of established tumors (112,126,127).
However, recent data showed that the HNF4α capability to restore a more differentiated phenotype and a less aggressive behavior in transformed hepatocyte could be impaired in the presence of TGFβ, thus suggesting that the therapeutic use of HNF4α gene delivery could be limited by a TGFβ-containing tumor environment. In particular, TGFβ signaling appeared responsible of several post-translational modifications able to interfere with the binding of HNF4α to its target genes, including mesenchymal genes normally suppressed by HNF4α (120). In this context, efforts are required to identify the inactivating post-translational modifications induced by TGFβ on HNF4α and consequently, to engineer mutants insensitive to the inactivating action of the cytokine.
Interestingly, in the last few years other members of HNF family have been described as tumor suppressors in HCC and their restoration produced successful results in terms of treatment of fibrosis and cancer in pre-clinical studies (128). Focusing also on these proteins as tools for molecular therapy of HCC can be a promising challenge for the future.
Conclusions
As highlighted from recent literature and outlined in this review, the complexity of cellular and microenvironmental networks influencing efficacy of current systemic molecular therapies needs to be taken into account for a successful management of HCC. Further characterization of cell-cell and cell-stroma interplay in livers with HCC will contribute to the design of novel and more efficient therapeutic approaches.
Acknowledgments
None
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Waller LP, Deshpande V, Pyrsopoulos N. Hepatocellular carcinoma: A comprehensive review. World J Hepatol 2015;7:2648-63. 10.4254/wjh.v7.i26.2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-76. 10.1053/j.gastro.2007.04.061 [DOI] [PubMed] [Google Scholar]
- 3.Feo F, Pascale RM. Multifocal hepatocellular carcinoma: intrahepatic metastasis or multicentric carcinogenesis? Ann Transl Med 2015;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poon RT, Fan ST, Ng IO, et al. Different risk factors and prognosis for early and late intrahepatic recurrence after resection of hepatocellular carcinoma. Cancer 2000;89:500-7. [DOI] [PubMed] [Google Scholar]
- 5.Belghiti J, Fuks D. Liver resection and transplantation in hepatocellular carcinoma. Liver Cancer 2012;1:71-82. 10.1159/000342403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomaa AI, Waked I. Recent advances in multidisciplinary management of hepatocellular carcinoma. World J Hepatol 2015;7:673-87. 10.4254/wjh.v7.i4.673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanda M, Sugimoto H, Kodera Y. Genetic and epigenetic aspects of initiation and progression of hepatocellular carcinoma. World J Gastroenterol 2015;21:10584-97. 10.3748/wjg.v21.i37.10584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang K, Lim HY, Shi S, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013;58:706-17. 10.1002/hep.26402 [DOI] [PubMed] [Google Scholar]
- 9.Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385-92. 10.1158/0008-5472.CAN-09-1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Llovet JM, Villanueva A, Lachenmayer A, et al. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:436. 10.1038/nrclinonc.2015.121 [DOI] [PubMed] [Google Scholar]
- 11.Chauhan R, Lahiri N. Tissue- and Serum-Associated Biomarkers of Hepatocellular Carcinoma. Biomark Cancer 2016;8:37-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomaa A WI. Management of advanced hepatocellular carcinoma: review of current and potential therapies. Hepatoma Res 2017;3:112-22. 10.20517/2394-5079.2017.03 [DOI] [Google Scholar]
- 13.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90. 10.1056/NEJMoa0708857 [DOI] [PubMed] [Google Scholar]
- 14.Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009;10:25-34. 10.1016/S1470-2045(08)70285-7 [DOI] [PubMed] [Google Scholar]
- 15.Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64:7099-109. 10.1158/0008-5472.CAN-04-1443 [DOI] [PubMed] [Google Scholar]
- 16.Ma W, Chen X, Yuan Y. T-cell-associated immunotherapy: a promising strategy for the treatment of hepatocellular carcinoma. Immunotherapy 2017;9:523-5. 10.2217/imt-2017-0053 [DOI] [PubMed] [Google Scholar]
- 17.Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81-8. 10.1016/j.jhep.2013.02.022 [DOI] [PubMed] [Google Scholar]
- 18.Duffy AG, Ulahannan SV, Makorova-Rusher O, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545-51. 10.1016/j.jhep.2016.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol 2007;19:813-24. 10.1093/intimm/dxm057 [DOI] [PubMed] [Google Scholar]
- 20.Brahmer JR, Hammers H, Lipson EJ. Nivolumab: targeting PD-1 to bolster antitumor immunity. Future Oncol 2015;11:1307-26. 10.2217/fon.15.52 [DOI] [PubMed] [Google Scholar]
- 21.Desai JR, Ochoa S, Prins PA, et al. Systemic therapy for advanced hepatocellular carcinoma: an update. J Gastrointest Oncol 2017;8:243-55. 10.21037/jgo.2017.02.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res 2014;20:2072-9. 10.1158/1078-0432.CCR-13-0547 [DOI] [PubMed] [Google Scholar]
- 23.Albini A, Bruno A, Gallo C, et al. Cancer stem cells and the tumor microenvironment: interplay in tumor heterogeneity. Connect Tissue Res 2015;56:414-25. 10.3109/03008207.2015.1066780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hernandez-Gea V, Toffanin S, Friedman SL, et al. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013;144:512-27. 10.1053/j.gastro.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berasain C, Castillo J, Perugorria MJ, et al. Inflammation and liver cancer: new molecular links. Ann N Y Acad Sci 2009;1155:206-21. 10.1111/j.1749-6632.2009.03704.x [DOI] [PubMed] [Google Scholar]
- 26.Kolb M, Margetts PJ, Anthony DC, et al. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 2001;107:1529-36. 10.1172/JCI12568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knight B, Yeoh GC, Husk KL, et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med 2000;192:1809-18. 10.1084/jem.192.12.1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quetier I, Brezillon N, Duriez M, et al. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J Hepatol 2013;59:285-91. 10.1016/j.jhep.2013.03.021 [DOI] [PubMed] [Google Scholar]
- 29.Chang TS, Wu YC, Chi CC, et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res 2015;21:201-10. 10.1158/1078-0432.CCR-13-3274 [DOI] [PubMed] [Google Scholar]
- 30.Aleksandrova K, Boeing H, Nothlings U, et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014;60:858-71. 10.1002/hep.27016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wan S, Zhao E, Kryczek I, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014;147:1393-404. 10.1053/j.gastro.2014.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang CQ, Sun HT, Gao XM, et al. Interleukin-6 enhances cancer stemness and promotes metastasis of hepatocellular carcinoma via up-regulating osteopontin expression. Am J Cancer Res 2016;6:1873-89. [PMC free article] [PubMed] [Google Scholar]
- 33.Evans RA, Tian YC, Steadman R, et al. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp Cell Res 2003;282:90-100. 10.1016/S0014-4827(02)00015-0 [DOI] [PubMed] [Google Scholar]
- 34.Calon A, Tauriello DV, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 2014;25:15-22. 10.1016/j.semcancer.2013.12.008 [DOI] [PubMed] [Google Scholar]
- 35.Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol 2010;7:448-58. 10.1038/nrgastro.2010.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 2006;25:3834-47. 10.1038/sj.onc.1209562 [DOI] [PubMed] [Google Scholar]
- 37.Koike K. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J Gastroenterol Hepatol 2007;22 Suppl 1:S108-11. 10.1111/j.1440-1746.2006.04669.x [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki K, Murata M, Yoshida K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology 2007;46:48-57. 10.1002/hep.21672 [DOI] [PubMed] [Google Scholar]
- 39.Lee YA, Friedman SL. Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antiviral Res 2014;107:23-30. 10.1016/j.antiviral.2014.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoshida Y, Villanueva A, Sangiovanni A, et al. Prognostic gene expression signature for patients with hepatitis C-related early-stage cirrhosis. Gastroenterology 2013;144:1024-30. 10.1053/j.gastro.2013.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brenner DA, Kisseleva T, Scholten D, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair 2012;5:S17. 10.1186/1755-1536-5-S1-S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elpek GO. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J Gastroenterol 2014;20:7260-76. 10.3748/wjg.v20.i23.7260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cozzolino AM, Noce V, Battistelli C, et al. Modulating the Substrate Stiffness to Manipulate Differentiation of Resident Liver Stem Cells and to Improve the Differentiation State of Hepatocytes. Stem Cells Int 2016;2016:5481493. [DOI] [PMC free article] [PubMed]
- 44.Zhubanchaliyev A, Temirbekuly A, Kongrtay K, et al. Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front Pharmacol 2016;7:462. 10.3389/fphar.2016.00462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X, Xu J, Brenner DA, et al. Reversibility of Liver Fibrosis and Inactivation of Fibrogenic Myofibroblasts. Curr Pathobiol Rep 2013;1:209-14. 10.1007/s40139-013-0018-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu FX, Zhao B, Guan KL. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015;163:811-28. 10.1016/j.cell.2015.10.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016;29:783-803. 10.1016/j.ccell.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mannaerts I, Leite SB, Verhulst S, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol 2015;63:679-88. 10.1016/j.jhep.2015.04.011 [DOI] [PubMed] [Google Scholar]
- 49.Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013;15:637-46. 10.1038/ncb2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G, Lu X, Dey P, et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov 2016;6:80-95. 10.1158/2159-8290.CD-15-0224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Camargo FD, Gokhale S, Johnnidis JB, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol 2007;17:2054-60. 10.1016/j.cub.2007.10.039 [DOI] [PubMed] [Google Scholar]
- 52.Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324-38. 10.1016/j.cell.2014.03.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huo X, Zhang Q, Liu AM, et al. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol Rep 2013;29:840-6. 10.3892/or.2012.2176 [DOI] [PubMed] [Google Scholar]
- 54.Perra A, Kowalik MA, Ghiso E, et al. YAP activation is an early event and a potential therapeutic target in liver cancer development. J Hepatol 2014;61:1088-96. 10.1016/j.jhep.2014.06.033 [DOI] [PubMed] [Google Scholar]
- 55.Li H, Wolfe A, Septer S, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int 2012;32:38-47. 10.1111/j.1478-3231.2011.02646.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu MZ, Yao TJ, Lee NP, et al. Yes-associated protein is an independent prognostic marker in hepatocellular carcinoma. Cancer 2009;115:4576-85. 10.1002/cncr.24495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin C, Hu Z, Lei B, et al. Overexpression of Yes-associated protein and its association with clinicopathological features of hepatocellular carcinoma: A meta-analysis. Liver Int 2017;37:1675-81. 10.1111/liv.13428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474:179-83. 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- 59.Liu-Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 2012;26:1300-5. 10.1101/gad.192856.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962-71. 10.1101/gad.1664408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin K, Pritchett J, Llewellyn J, et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun 2016;7:12502. 10.1038/ncomms12502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huggett MT, Jermyn M, Gillams A, et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer 2014;110:1698-704. 10.1038/bjc.2014.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou Z, Hu T, Xu Z, et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J 2015;29:724-32. 10.1096/fj.14-262980 [DOI] [PubMed] [Google Scholar]
- 64.Jiao S, Wang H, Shi Z, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014;25:166-80. 10.1016/j.ccr.2014.01.010 [DOI] [PubMed] [Google Scholar]
- 65.Fitamant J, Kottakis F, Benhamouche S, et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep 2015. [Epub ahead of print]. 10.1016/j.celrep.2015.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fabregat I, Moreno-Caceres J, Sanchez A, et al. TGF-beta signalling and liver disease. FEBS J 2016;283:2219-32. 10.1111/febs.13665 [DOI] [PubMed] [Google Scholar]
- 67.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003;425:577-84. 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- 68.Fabregat I, Fernando J, Mainez J, et al. TGF-beta signaling in cancer treatment. Curr Pharm Des 2014;20:2934-47. 10.2174/13816128113199990591 [DOI] [PubMed] [Google Scholar]
- 69.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005;24:5764-74. 10.1038/sj.onc.1208927 [DOI] [PubMed] [Google Scholar]
- 70.Liu Y, Liu H, Meyer C, et al. Transforming growth factor-beta (TGF-beta)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J Biol Chem 2013;288:30708-19. 10.1074/jbc.M113.478685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazzocca A, Fransvea E, Dituri F, et al. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010;51:523-34. 10.1002/hep.23285 [DOI] [PubMed] [Google Scholar]
- 72.Han Y, Guo Q, Zhang M, et al. CD69+ CD4+ CD25- T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane-bound TGF-beta 1. J Immunol 2009;182:111-20. 10.4049/jimmunol.182.1.111 [DOI] [PubMed] [Google Scholar]
- 73.Wang Y, Liu T, Tang W, et al. Hepatocellular Carcinoma Cells Induce Regulatory T Cells and Lead to Poor Prognosis via Production of Transforming Growth Factor-beta1. Cell Physiol Biochem 2016;38:306-18. 10.1159/000438631 [DOI] [PubMed] [Google Scholar]
- 74.Matsuzaki K, Date M, Furukawa F, et al. Autocrine stimulatory mechanism by transforming growth factor beta in human hepatocellular carcinoma. Cancer Res 2000;60:1394-402. [PubMed] [Google Scholar]
- 75.Lee D, Chung YH, Kim JA, et al. Transforming growth factor beta 1 overexpression is closely related to invasiveness of hepatocellular carcinoma. Oncology 2012;82:11-8. 10.1159/000335605 [DOI] [PubMed] [Google Scholar]
- 76.Shirai Y, Kawata S, Tamura S, et al. Plasma transforming growth factor-beta 1 in patients with hepatocellular carcinoma. Comparison with chronic liver diseases. Cancer 1994;73:2275-9. [DOI] [PubMed] [Google Scholar]
- 77.Ito N, Kawata S, Tamura S, et al. Positive correlation of plasma transforming growth factor-beta 1 levels with tumor vascularity in hepatocellular carcinoma. Cancer Lett 1995;89:45-8. 10.1016/0304-3835(95)90156-6 [DOI] [PubMed] [Google Scholar]
- 78.Okumoto K, Hattori E, Tamura K, et al. Possible contribution of circulating transforming growth factor-beta1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver Int 2004;24:21-8. 10.1111/j.1478-3231.2004.00882.x [DOI] [PubMed] [Google Scholar]
- 79.Fransvea E, Angelotti U, Antonaci S, et al. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008;47:1557-66. 10.1002/hep.22201 [DOI] [PubMed] [Google Scholar]
- 80.Fransvea E, Mazzocca A, Santamato A, et al. Kinase activation profile associated with TGF-beta-dependent migration of HCC cells: a preclinical study. Cancer Chemother Pharmacol 2011;68:79-86. 10.1007/s00280-010-1459-x [DOI] [PubMed] [Google Scholar]
- 81.Herbertz S, Sawyer JS, Stauber AJ, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther 2015;9:4479-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Faivre SJ, Santoro A, Gane E, et al. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J Clin Oncol 2016;34:4070. [Google Scholar]
- 83.Cicchini C, Amicone L, Alonzi T, et al. Molecular mechanisms controlling the phenotype and the EMT/MET dynamics of hepatocyte. Liver Int 2015;35:302-10. 10.1111/liv.12577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thiery JP, Acloque H, Huang RY, et al. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139:871-90. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 85.Taura K, Iwaisako K, Hatano E, et al. Controversies over the Epithelial-to-Mesenchymal Transition in Liver Fibrosis. J Clin Med 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. J Clin Med 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miyoshi A, Kitajima Y, Kido S, et al. Snail accelerates cancer invasion by upregulating MMP expression and is associated with poor prognosis of hepatocellular carcinoma. Br J Cancer 2005;92:252-8. 10.1038/sj.bjc.6602266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woo HY, Min AL, Choi JY, et al. Clinicopathologic significance of the expression of Snail in hepatocellular carcinoma. Korean J Hepatol 2011;17:12-8. 10.3350/kjhep.2011.17.1.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang Y, Shi J, Chai K, et al. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets 2013;13:963-72. 10.2174/15680096113136660102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kaufhold S, Bonavida B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Res 2014;33:62. 10.1186/s13046-014-0062-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005;132:3151-61. 10.1242/dev.01907 [DOI] [PubMed] [Google Scholar]
- 92.Cicchini C, Filippini D, Coen S, et al. Snail controls differentiation of hepatocytes by repressing HNF4alpha expression. J Cell Physiol 2006;209:230-8. 10.1002/jcp.20730 [DOI] [PubMed] [Google Scholar]
- 93.Battistelli C, Cicchini C, Santangelo L, et al. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017;36:942-55. 10.1038/onc.2016.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garibaldi F, Cicchini C, Conigliaro A, et al. An epistatic mini-circuitry between the transcription factors Snail and HNF4alpha controls liver stem cell and hepatocyte features exhorting opposite regulation on stemness-inhibiting microRNAs. Cell Death Differ 2012;19:937-46. 10.1038/cdd.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin T, Ponn A, Hu X, et al. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010;29:4896-904. 10.1038/onc.2010.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cicchini C, de Nonno V, Battistelli C, et al. Epigenetic control of EMT/MET dynamics: HNF4alpha impacts DNMT3s through miRs-29. Biochim Biophys Acta 2015;1849:919-29. [DOI] [PMC free article] [PubMed]
- 97.Yang Z, Zhou L, Wu LM, et al. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol 2011;18:1243-50. 10.1245/s10434-011-1581-y [DOI] [PubMed] [Google Scholar]
- 98.Gao JZ, Li J, Du JL, et al. Long non-coding RNA HOTAIR is a marker for hepatocellular carcinoma progression and tumor recurrence. Oncol Lett 2016;11:1791-8. 10.3892/ol.2016.4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sun L, Fang J. Epigenetic regulation of epithelial-mesenchymal transition. Cell Mol Life Sci 2016;73:4493-515. 10.1007/s00018-016-2303-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Amicone L, Citarella F, Cicchini C. Epigenetic regulation in hepatocellular carcinoma requires long noncoding RNAs. Biomed Res Int 2015;2015:473942. [DOI] [PMC free article] [PubMed]
- 101.Ma L, Chua MS, Andrisani O, et al. Epigenetics in hepatocellular carcinoma: an update and future therapy perspectives. World J Gastroenterol 2014;20:333-45. 10.3748/wjg.v20.i2.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yan KS, Lin CY, Liao TW, et al. EZH2 in Cancer Progression and Potential Application in Cancer Therapy: A Friend or Foe? Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maiques-Diaz A, Somervaille TC. LSD1: biologic roles and therapeutic targeting. Epigenomics 2016;8:1103-16. 10.2217/epi-2016-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hajjari M, Salavaty A. HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol Med 2015;12:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu L, Zhang L, Zheng S. Role of the long non-coding RNA HOTAIR in hepatocellular carcinoma. Oncol Lett 2017;14:1233-9. 10.3892/ol.2017.6312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kyrmizi I, Hatzis P, Katrakili N, et al. Plasticity and expanding complexity of the hepatic transcription factor network during liver development. Genes Dev 2006;20:2293-305. 10.1101/gad.390906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Parviz F, Matullo C, Garrison WD, et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat Genet 2003;34:292-6. 10.1038/ng1175 [DOI] [PubMed] [Google Scholar]
- 108.Fang B, Mane-Padros D, Bolotin E, et al. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res 2012;40:5343-56. 10.1093/nar/gks190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Santangelo L, Marchetti A, Cicchini C, et al. The stable repression of mesenchymal program is required for hepatocyte identity: a novel role for hepatocyte nuclear factor 4alpha. Hepatology 2011;53:2063-74. 10.1002/hep.24280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science 2004;303:1378-81. 10.1126/science.1089769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lazarevich NL, Shavochkina DA, Fleishman DI, et al. Deregulation of hepatocyte nuclear factor 4 (HNF4)as a marker of epithelial tumors progression. Exp Oncol 2010;32:167-71. [PubMed] [Google Scholar]
- 112.Ning BF, Ding J, Yin C, et al. Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res 2010;70:7640-51. 10.1158/0008-5472.CAN-10-0824 [DOI] [PubMed] [Google Scholar]
- 113.Babeu JP, Boudreau F. Hepatocyte nuclear factor 4-alpha involvement in liver and intestinal inflammatory networks. World J Gastroenterol 2014;20:22-30. 10.3748/wjg.v20.i1.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li X, Salisbury-Rowswell J, Murdock AD, et al. Hepatocyte nuclear factor 4 response to injury involves a rapid decrease in DNA binding and transactivation via a JAK2 signal transduction pathway. Biochem J 2002;368:203-11. 10.1042/bj20020233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chahar S, Gandhi V, Yu S, et al. Chromatin profiling reveals regulatory network shifts and a protective role for hepatocyte nuclear factor 4alpha during colitis. Mol Cell Biol 2014;34:3291-304. 10.1128/MCB.00349-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Li M, Tang Y, Wu L, et al. The hepatocyte-specific HNF4alpha/miR-122 pathway contributes to iron overload-mediated hepatic inflammation. Blood 2017;130:1041-51. 10.1182/blood-2016-12-755967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang Z, Burke PA. Effects of hepatocyte nuclear factor-4alpha on the regulation of the hepatic acute phase response. J Mol Biol 2007;371:323-35. 10.1016/j.jmb.2007.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang Z, Bishop EP, Burke PA. Expression profile analysis of the inflammatory response regulated by hepatocyte nuclear factor 4alpha. BMC Genomics 2011;12:128. 10.1186/1471-2164-12-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hatziapostolou M, Polytarchou C, Aggelidou E, et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011;147:1233-47. 10.1016/j.cell.2011.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cozzolino AM, Alonzi T, Santangelo L, et al. TGFbeta overrides HNF4alpha tumor suppressing activity through GSK3beta inactivation: implication for hepatocellular carcinoma gene therapy. J Hepatol 2013;58:65-72. 10.1016/j.jhep.2012.08.023 [DOI] [PubMed] [Google Scholar]
- 121.de Lucas S, Lopez-Alcorocho JM, Bartolome J, et al. Nitric oxide and TGF-beta1 inhibit HNF-4alpha function in HEPG2 cells. Biochem Biophys Res Commun 2004;321:688-94. 10.1016/j.bbrc.2004.07.025 [DOI] [PubMed] [Google Scholar]
- 122.Wu Q, Liu HO, Liu YD, et al. Decreased expression of hepatocyte nuclear factor 4alpha (Hnf4alpha)/microRNA-122 (miR-122) axis in hepatitis B virus-associated hepatocellular carcinoma enhances potential oncogenic GALNT10 protein activity. J Biol Chem 2015;290:1170-85. 10.1074/jbc.M114.601203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Vallianou I, Dafou D, Vassilaki N, et al. Hepatitis C virus suppresses Hepatocyte Nuclear Factor 4 alpha, a key regulator of hepatocellular carcinoma. Int J Biochem Cell Biol 2016;78:315-26. 10.1016/j.biocel.2016.07.027 [DOI] [PubMed] [Google Scholar]
- 124.Bonzo JA, Ferry CH, Matsubara T, et al. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J Biol Chem 2012;287:7345-56. 10.1074/jbc.M111.334599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yue HY, Yin C, Hou JL, et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut 2010;59:236-46. 10.1136/gut.2008.174904 [DOI] [PubMed] [Google Scholar]
- 126.Yin C, Lin Y, Zhang X, et al. Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology 2008;48:1528-39. 10.1002/hep.22510 [DOI] [PubMed] [Google Scholar]
- 127.Lazarevich NL, Cheremnova OA, Varga EV, et al. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 2004;39:1038-47. 10.1002/hep.20155 [DOI] [PubMed] [Google Scholar]
- 128.Marchetti A, Bisceglia F, Cozzolino AM, et al. New Tools for Molecular Therapy of Hepatocellular Carcinoma. Diseases 2015;3:325-40. 10.3390/diseases3040325 [DOI] [PMC free article] [PubMed] [Google Scholar]