

ABSTRACT
The mammalian intestinal epithelium establishes a selectively permeable barrier that supports nutrient absorption and prevents intrusion by noxious luminal substances and microbiota. The effectiveness and integrity of the barrier function are tightly regulated via well-controlled mechanisms. Long noncoding RNAs transcribed from ultraconserved regions (T-UCRs) control diverse cellular processes, but their roles in the regulation of gut permeability remain largely unknown. Here we report that the T-UCR uc.173 enhances intestinal epithelial barrier function by antagonizing microRNA 29b (miR-29b). Decreasing the levels of uc.173 by gene silencing led to dysfunction of the intestinal epithelial barrier in cultured cells and increased the vulnerability of the gut barrier to septic stress in mice. uc.173 specifically stimulated translation of the tight junction (TJ) claudin-1 (CLDN1) by associating with miR-29b rather than by binding directly to CLDN1 mRNA. uc.173 acted as a natural decoy RNA for miR-29b, which interacts with CLDN1 mRNA via the 3′ untranslated region and represses its translation. Ectopically expressed uc.173 abolished the association of miR-29b with CLDN1 mRNA and restored claudin-1 expression to normal levels in cells overexpressing miR-29b, thus rescuing the barrier function. These results highlight a novel function of uc.173 in controlling gut permeability and define a mechanism by which uc.173 stimulates claudin-1 translation, by decreasing the availability of miR-29b to CLDN1 mRNA.
KEYWORDS: T-UCRs, microRNA, posttranscriptional regulation, gut permeability, tight junction, septic stress
INTRODUCTION
The mammalian intestinal epithelial barrier depends on specialized structures named tight junctions (TJs) and adherens junctions (AJs) that protect the subepithelial tissue against a wide array of luminal noxious substances, allergens, and microbial pathogens (1, 2). TJs are located at the apical region of the epithelial lateral membrane and fence the paracellular space in the intestinal epithelium, which is selectively permeable to certain hydrophilic molecules, ions, and nutrients (1, 3). Functional TJ complexes in the epithelium primarily include TJ-transmembrane proteins, such as the claudin family of proteins, which interact directly with a cytosolic plaque of TJ-membrane-associated proteins, such as ZO-1, that bind to the cortical cytoskeleton (3, 4). Immediately below the TJs are the cadherin-rich AJs, which mediate strong cell-to-cell adhesion and also integrate distinct cellular signals to regulate epithelial paracellular permeability (5). Both TJ and AJ complexes are highly dynamic, and their constituent proteins undergo continuous remodeling and turnover (1, 6, 7). Maintenance of dynamic levels of TJ and AJ proteins is essential for the integrity of the epithelial barrier function, a process that is tightly controlled by numerous extracellular and intracellular factors (5, 8). Acute epithelial barrier dysfunction occurs commonly in various critical pathological conditions, leading to the translocation of luminal toxic substances and bacteria to the bloodstream and, in some instances, to multiple organ dysfunction syndrome and death (9), but the mechanisms responsible for this dysfunction are largely unknown.
The human genome transcribes a large number of noncoding RNAs (ncRNAs), such as microRNAs (miRNAs), endogenous small interfering RNAs (siRNAs), natural antisense transcripts (NATs), and long noncoding RNAs (lncRNAs), while protein-coding transcripts account for only a minority of the transcriptional output (10, 11). lncRNAs, defined as being at least 200 nucleotides long, have recently attracted much attention due to their abundant presence in the human genome, tissue-specific expression patterns in response to stress, and functional relevance in complex physiologic processes and pathological conditions (12, 13). lncRNAs can create complex regulatory networks by interacting with different molecular partners, such as transcription factors, histones or other chromatin-modifying proteins, and RNA-binding proteins (RBPs), to regulate gene expression at multiple levels (12, 14). Emerging evidence also shows that the interplay between lncRNAs and miRNAs controls gene regulatory programs involved in many aspects of cellular processes and human diseases (12, 15, 16). In this regard, lncRNAs can be primary miRNAs (pri-miRNAs) (15), act as molecular sponges for miRNAs (16), and regulate miRNA processing (17). Recently, lncRNA H19 was found to disrupt the intestinal epithelial barrier by serving as a precursor for miRNA 675 (miR-675) (18), whereas lncRNA SPRY4-IT1 enhanced intestinal epithelial barrier function by increasing the expression of TJs at the posttranscription level via interaction with RBP HuR (19, 20).
Ultraconserved regions (UCRs) are sequences located in both intra- and intergenic regions that are absolutely conserved (100%) among the orthologous regions of the human, rat, and mouse genomes (21). Although UCRs are actively transcribed in various tissues, more than half of all 481 known UCRs have no protein-coding potential. RNAs transcribed from these UCRs (T-UCRs) have been identified as a class of novel lncRNAs that regulate proliferation and apoptosis (22–24). The expression levels of T-UCRs are altered in response to stress conditions and pathologies. For example, the levels of cellular uc.349A, uc.352, uc.338, uc.8, uc.339, uc.63+, and uc.475 (22–28) increased dramatically in human cancers or after exposure to hypoxia. We have reported that 21 T-UCRs, including uc.173, are differentially expressed in the intestinal mucosae of fasted mice relative to expression in nonfasted mice and that increasing the levels of uc.173 promotes mucosal renewal of the intestine by inducing the degradation of pri-miR-195 (29). In this study, we investigate the role of uc.173 in the regulation of the intestinal epithelial barrier and present evidence that uc.173 stimulates the translation of TJ claudin-1 (CLDN1) through interaction with miR-29b, thus enhancing epithelial barrier function.
RESULTS
uc.173 silencing leads to intestinal epithelial barrier dysfunction in vitro.
To investigate the role of uc.173 in the regulation of the intestinal epithelial barrier function, we silenced the expression of uc.173 by transfecting human epithelial colorectal adenocarcinoma Caco-2 cells with locked nucleic acid (LNA)-modified anti-uc.173 oligonucleotides (anti-uc.173). As shown in Fig. 1A (left), levels of cellular uc.173 were dramatically lower in cells transfected with LNA-modified anti-uc.173 than in cells transfected with control oligonucleotides (Con-oligo). This effect of anti-uc.173–LNA was specific, as evidenced by the fact that it did not alter the abundance of T-UCR uc.346 (Fig. 1A, right). Decreasing the levels of uc.173 by anti-uc.173 transfection specifically inhibited expression of the TJ claudin-1 but failed to alter cellular levels of other TJs, such as claudin-3, occludin, JAM-1, and ZO-1, the AJ E-cadherin, and UBE2B protein (the product of the uc.173 host gene) (Fig. 1B). The levels of claudin-1 protein in a population of cells in which uc.173 was silenced decreased by ∼75% (n = 3) (P < 0.05) from those in cells transfected with control oligonucleotides. In accordance with this result, immunostaining also revealed that claudin-1 protein levels decreased remarkably after uc.173 silencing (Fig. 1C, top), although there were no differences in the extent of immunostaining of E-cadherin between anti-uc.173-transfected cells and cells transfected with control oligonucleotides (Fig. 1C, bottom).
FIG 1.

LNA-mediated uc.173 silencing inhibits claudin-1 expression and disrupts epithelial barrier function. (A) Levels of cellular uc.173 48 h after transfection with LNA-siRNA targeting uc.173 (anti-uc.173) or a control siRNA (Con-oligo) in Caco-2 cells. Values are relative to control levels and are means ± SEM from triplicate experiments. The asterisk indicates a significant difference (P < 0.05) from the Con-oligo result. (B) Representative immunoblots of tight junctions and an adherens junction in cells treated as described for panel A. Three experiments were performed, with similar results. (C) Distribution of claudin-1 and E-cadherin in cells treated as described for panel A. Forty-eight hours after transfection, cells were fixed, permeabilized, and incubated first with an antibody against claudin-1 or E-cadherin and then with FITC-conjugated anti-IgG. Original magnification, ×500. (D) Changes in epithelial barrier function, as indicated by changes in TEER (left) and FITC-dextran paracellular permeability (right), in cells treated as described for panel A. TEER assays were performed on 12-mm Transwell filters; paracellular permeability was assayed by adding the membrane-impermeant trace molecule FITC-dextran to the insert medium. Values are means ± SEM of data from six samples. Asterisks indicate significant differences (P < 0.05) from the Con-oligo results.
Importantly, uc.173 silencing disrupted epithelial barrier function in an in vitro model, as evidenced by a decrease in transepithelial electrical resistance (TEER) values (Fig. 1D, left) and an increase in the levels of paracellular flux of fluorescein isothiocyanate (FITC)-dextran (Fig. 1D, right). To exclude off-target effects, other LNA-modified anti-uc.173 oligonucleotides were tested; a similar repressive effect was observed on the expression of uc.173, as well as on claudin-1 expression levels and epithelial barrier function (data not shown). Transfection with either anti-uc.173 or control oligonucleotides did not affect cell viability, as examined by trypan blue staining (data not shown). These data indicate that the T-UCR uc.173 is essential for normal expression of the TJ claudin-1 and for the maintenance of epithelial barrier function in vitro.
uc.173 enhances the translation of claudin-1.
To examine the effect of increasing the levels of uc.173 on claudin-1 expression, Caco-2 cells were transfected with a plasmid vector expressing uc.173 under the control of the cytomegalovirus (CMV) promoter, as described in our previous study (29). As shown in Fig. 2A, the abundance of uc.173 increased substantially by 48 h after transfection, but that of uc.346 did not. Ectopically expressed uc.173 specifically stimulated the expression of claudin-1, with no effect on the expression levels of the claudin-3, occludin, JAM-1, ZO-1, E-cadherin, and UBE2B proteins (Fig. 2B).
FIG 2.

Ectopically expressed uc.173 enhances claudin-1 translation. (A) Levels of uc.173 (left) and uc.346 (right) in cells transfected with the uc.173 expression vector and measured 48 h later by RT-Q-PCR. Values are relative to those for the control (Vector) and are means ± SEM from triplicate experiments. O/E, overexpression. The asterisk indicates a significant difference (P < 0.05) from the result with the control vector. (B) Representative immunoblots of tight junctions and an adherens junction in cells treated as described for panel A. (C) Newly synthesized claudin-1 protein in cells treated as described for panel A. After cells were exposed to l-azidohomoalanine (AHA), cell lysates were incubated with the reaction buffer containing a biotin-alkyne reagent; the biotin-alkyne-azide-modified protein complex was pulled down by paramagnetic streptavidin-conjugated Dynabeads. (D) Distribution of CLDN1 mRNA in each gradient fraction prepared from polysomal profiles in cells treated as described for panel A. Nuclei were pelleted, and the resulting supernatants were fractionated through a 10-to-50% linear sucrose gradient. Total RNA was isolated from different fractions, and the level of CLDN1 or GAPDH mRNA in each fraction was measured and plotted as a percentage of the total level of CLDN1 or GAPDH mRNA, respectively, in each sample.
To investigate the mechanism underlying the stimulation of claudin-1 expression by uc.173, we examined the changes in CLDN1 mRNA levels and found that neither the silencing nor the overexpression of uc.173 altered the content of CLDN1 mRNA (see Fig. S1 in the supplemental material), suggesting that uc.173 might regulate claudin-1 expression at the translational level. To test this possibility, we examined the rate of CLDN1 mRNA translation by use of the Click-iT technology (see Materials and Methods) as described previously (18). The levels of newly synthesized claudin-1 protein in cells overexpressing uc.173 were significantly higher than those in cells transfected with a control vector (Fig. 2C). The stimulation of claudin-1 protein synthesis by uc.173 was specific, since there were no changes in the levels of nascent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) synthesis after transfection with the uc.173 expression vector.
To further investigate the role of uc.173 in the regulation of claudin-1 translation, we measured the relative distribution of CLDN1 mRNA in individual fractions from polyribosome gradients after ectopic overexpression of uc.173 as reported previously (30). Although increasing the levels of uc.173 did not affect global polysomal profiles (data not shown), the association of CLDN1 mRNA with actively translating fractions (fractions 10 to 12) increased in cells overexpressing uc.173, along with a reduction in the levels of CLDN1 mRNA in low-translating fractions (fractions 7 to 9) (Fig. 2D, left). In contrast, GAPDH mRNA, encoding a housekeeping protein, was distributed similarly in the two groups (Fig. 2D, right). These data reveal that uc.173 stimulates claudin-1 translation without affecting CLDN1 mRNA levels.
A reduction in uc.173 abundance increases the vulnerability of the gut barrier to pathological stress.
In an effort to define the in vivo function of uc.173 in regulating gut permeability, the levels of endogenous uc.173 in the intestinal mucosa were decreased by administering systemically the same LNA-modified anti-uc.173 oligonucleotides that were used in cultured intestinal epithelial cells (IECs). As reported in our previous study (29), treatment with anti-uc.173 for 4 consecutive days caused a sustained decrease in uc.173 levels in the small intestinal mucosa (Fig. 3A, left) but did not alter the levels of uc.346 (Fig. 3A, center) or U6 (Fig. 3A, right) RNA. The levels of mucosal uc.173 in the small intestine were ∼70% lower in mice treated with anti-uc.173 than in animals treated with control oligonucleotides. In contrast to the findings observed in vitro, decreasing the levels of uc.173 in the mucosa did not affect the basal level of gut permeability (Fig. 3B) in control mice (sham-treated groups), although the levels of claudin-1 protein decreased in anti-uc.173-treated mice (Fig. 3C).
FIG 3.

Inhibition of uc.173 impairs gut barrier function in mice subjected to CLP. (A) Levels of uc.173 (left), uc.346 (center), and U6 (right) RNAs in the small intestinal mucosae of mice injected intraperitoneally with LNA-modified anti-uc.173 or control oligonucleotides (Con-oligo) for 4 consecutive days. On day 5, total RNA was isolated for RT-Q-PCR analysis. Values are relative to those with Con-oligo and are means ± SEM of data from 5 animals. The asterisk indicates a significant difference (P < 0.05) from the Con-oligo result. (B) Gut permeability in sham-treated mice and in mice exposed to CLP for 24 h. FITC-dextran was given orally, and blood samples were collected 4 h later. The asterisk and plus sign indicate significant differences (P < 0.05) from sham-treated mice and from mice treated with Con-oligo and CLP, respectively. (C) Representative immunoblots of claudin-1, claudin-3, and β-catenin in the small intestinal mucosae of mice treated as described for panel B.
To determine if a reduction in the abundance of uc.173 in tissue affected the vulnerability of the gut barrier in critical pathological conditions, we subjected the mice to septic stress using the cecal ligation and puncture (CLP) model (31). Exposure to CLP for 24 h led to acute gut barrier dysfunction in both anti-uc.173-treated mice and mice treated with control oligonucleotides, as indicated by an increase in gut mucosal permeability to FITC-dextran. Of interest, however, the levels of increased gut permeability due to CLP in mice treated with anti-uc.173 were much higher than those in mice treated with control oligonucleotides (Fig. 3B). As expected, CLP stress decreased the levels of both claudin-1 and claudin-3 in the intestinal mucosa, but the inhibition of claudin-1 was amplified by decreasing the levels of uc.173 in anti-uc.173-treated mice (Fig. 3C). In fact, claudin-1 protein disappeared almost completely in anti-uc.173-treated mice subjected to CLP. On the other hand, there were no significant changes in claudin-3 reduction (Fig. 3C) or in histological features of the small intestinal mucosa between mice treated with anti-uc.173 and mice treated with control oligonucleotides after CLP (data not shown). Additionally, treatment with either anti-uc.173 or control oligonucleotides for 4 consecutive days did not result in diarrhea, loss of body weight, loss of hair, or decreased activity in mice (data not shown). These results show that decreasing the levels of uc.173 in the intestinal mucosa increases the vulnerability of the gut barrier to septic stress in mice.
uc.173 does not bind to CLDN1 mRNA but interacts directly with miR-29b.
To test if uc.173 activates claudin-1 translation through direct interaction with CLDN1 mRNA, we tagged uc.173 with MS2 hairpins (Fig. 4A) and coexpressed it in cells along with the chimeric protein MS2–glutathione S-transferase (GST). After affinity purification using glutathione-SH beads, the RNAs present in the ribonucleoprotein complexes were examined by real-time quantitative PCR (Q-PCR) analysis as reported previously (32). Cells were cotransfected with the pMS2 (control) or pMS2-uc.173 vector along with pMS2-GST, and the binding of uc.173 to CLDN1 mRNA and miRNAs was examined 24 h after transfection. As shown in Fig. 4B, uc.173 levels increased significantly. There were no differences in the levels of CLDN1 mRNA in materials pulled down by MS2-GST between cells transfected with pMS2-uc.173 and cells transfected with pMS2 (Fig. 4C), indicating that uc.173 does not bind directly to CLDN1 mRNA. Because miR-29b is highly expressed in IECs and plays an important role in maintaining intestinal epithelial homeostasis (33–35), we examined the interaction of uc.173 with miR-29b. Interestingly, miR-29b levels were enriched in materials pulled down from cells transfected with pMS2-uc.173 relative to levels from cells transfected with pMS2 (Fig. 4Ca). The interaction of uc.173 with miR-29b was specific, since increasing the amount of pMS2-uc.173 did not increase the level of miR-222 or lncRNA H19 in the pulldown materials (see Fig. S2 in the supplemental material). In addition, cotransfection with pMS2-GST and pMS2 or pMS2-uc.173 did not alter the steady-state levels of CLDN1 mRNA or miR-29b (Fig. 4Cb).
FIG 4.
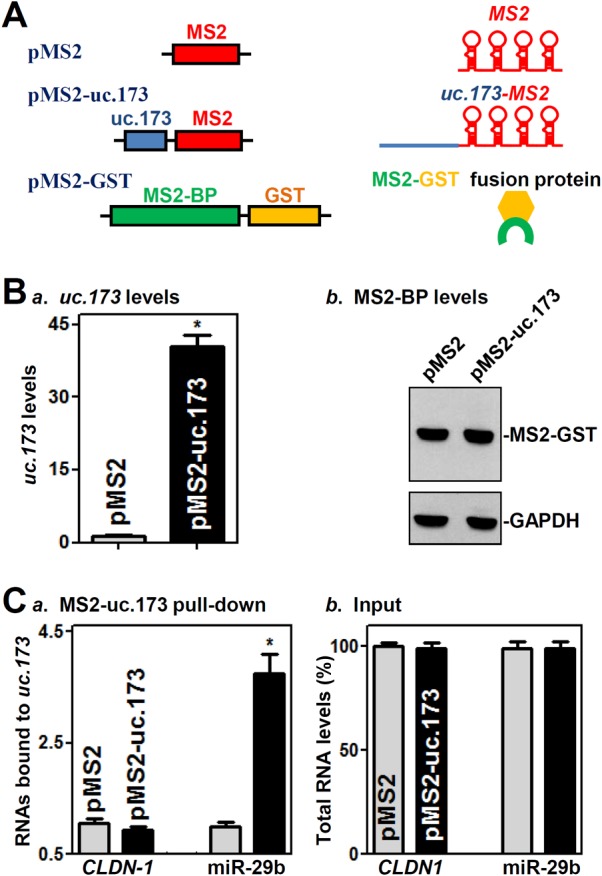
uc.173 interacts with miR-29b but not with CLDN1 mRNA. (A) Schematic of plasmids used for assays of CLDN1 mRNA and miR-29b pulldown. pMS2 and pMS2-uc.173 expressed MS2 and MS2-uc.173 RNAs, respectively, each containing 24 tandem MS2 hairpins; pMS2-GST expressed a fusion protein (MS2-GST) that can pull down MS2-containing RNA. (B) Levels of uc.173 (a) and MS2-BP (b) 24 h after cotransfection with pMS2-GST and the pMS2 or pMS2-uc.173 vector. Values are relative to those with the pMS2 vector and are means ± SEM from triplicate experiments. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with pMS2. (C) Binding of pMS2-uc.173 to RNAs. Shown are levels of CLDN1 mRNA and miR-29b in the materials pulled down by pMS2-uc.173, given relative to levels with pMS2 (a) and as percentages of total input RNAs (b). The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with pMS2.
Since uc.173 shows perfect complementarity with miR-29b at 8 nucleotides at its 3′ terminus (Fig. 5A), we investigated further the direct interaction between these two ncRNAs by using biotinylated miR-29b as described previously (19, 33). Twenty-four hours after transfection, miR-29b levels were significantly higher in cells transfected with biotin-labeled miR-29b than in cells transfected with a control scrambled oligomer (Fig. 5B, left), although the levels of U6 RNA did not differ between these two groups (Fig. 5B, right). As expected, uc.173 levels in samples pulled down using biotin-labeled miR-29b were much higher than those in samples pulled down using a biotin-labeled scrambled oligomer (Fig. 5Ca). Biotin-labeled miR-29b did not pull down other T-UCRs, such as uc.346 and uc.283; transfection with biotin-labeled miR-29b also failed to alter the levels of input RNAs (Fig. 5Cb). These results strongly suggest that uc.173 binds directly to miR-29b and can reduce the availability of miR-29b.
FIG 5.

Biotinylated miR-29b associates with uc.173. (A) Schematic of uc.173 depicting its complementarity with miR-29b. (B) Levels of biotinylated miR-29b (left) and U6 RNA (right) 24 h after transfection with biotinylated miR-29b as measured by Q-PCR analysis. Values are relative to those with the scrambled oligonucleotide and are means ± SEM of data from triplicate experiments. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the control scrambled oligomer. (C) Binding of biotinylated miR-29b to T-UCRs. Shown are levels of uc.173, uc.346, and uc.283 in the materials pulled down by biotin–miR-29b, given relative to the level with the scrambled oligomer (a) and as percentages of total input RNAs (b). The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the scrambled oligonucleotide.
miR-29 interacts with CLDN1 mRNA and represses its translation.
CLDN1 mRNA is a potential target of miR-29b, since there are two predicted binding sites for miR-29b within its 3′ untranslated region (3′ UTR) (Fig. 6A). Thus, we examined the association of miR-29b with CLDN1 mRNA by RNA pulldown assays using biotin-labeled miR-29b (Fig. 6Ba). Twenty-four hours after transfection with biotin-labeled miR-29b (Fig. 6Bb), CLDN1 mRNA was found to be enriched in materials pulled down by biotin–miR-29b but not in materials from cells transfected with a biotin-labeled control scrambled oligomer (Fig. 6Bc). On the other hand, biotin–miR-29b failed to pull down the mRNAs encoding E-cadherin and CUG-binding protein 1 (CUGBP1). To determine the functional consequence of miR-29b–CLDN1 mRNA association, we examined whether increasing the levels of miR-29b through transfection with its precursor (pre-miR-29b) reduced the expression of claudin-1. Transient transfection with pre-miR-29b dramatically increased miR-29b levels (Fig. 6C, left), although it did not alter U6 RNA levels (Fig. 6C, right). In contrast to the findings observed for cells overexpressing uc.173, increasing the levels of miR-29b by pre-miR-29b transfection specifically inhibited the expression of claudin-1 without affecting the expression levels of E-cadherin and β-catenin (Fig. 6D); CLDN1 mRNA levels decreased by ∼12% in cells transfected with pre-miR-29b, but this difference was not statistically significant. In turn, ectopically expressed miR-29b also resulted in epithelial barrier dysfunction, as revealed by a decrease in TEER values and an increase in paracellular permeability (see Fig. S3 in the supplemental material). Moreover, miR-29b overexpression inhibited claudin-1 expression predominantly by repressing its translation, since increasing the levels of miR-29b decreased the abundance of CLDN1 mRNA in actively translating fractions of polyribosome gradients and showed a significant shift to weakly translating fractions (fractions 4 and 5) (Fig. 6E, left). In contrast, transfection with pre-miR-29b did not alter the distribution of GAPDH mRNA (Fig. 6E, right) or global polysomal profiles (data not shown).
FIG 6.

miR-29b represses claudin-1 translation. (A) Schematic representation of CLDN1 mRNA depicting predicted target sites for miR-29b in its 3′ UTR. BS, predicted miR-29b-binding site. (B) Binding of biotinylated miR-29b to CLDN1 mRNA. (a) Schematic representation of biotinylated miR-29b; (b) miR-29b levels 24 h after transfection; (c) levels of mRNAs encoding claudin-1, E-cadherin (E-cad), and CUG-binding protein 1 (Cugbp1) in materials pulled down by biotin–miR-29b. Values are relative to that with the control scrambled oligomer and are means ± SEM of data from triplicate experiments. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the control scrambled oligomer. (C) Levels of miR-29b in cells transfected with pre-miR-29b for 48 h. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the control scrambled oligomer. (D) Representative immunoblots of claudin-1, E-cadherin, and β-catenin in cells treated as described for panel C. (E) Distribution of CLDN1 mRNA in each gradient fraction prepared from polysomal profiles in cells treated as described for panel C. The levels of CLDN1 or GAPDH mRNAs were measured and plotted as percentages of total CLDN1 or GAPDH mRNA levels in the samples, respectively.
To determine whether the repression of claudin-1 translation by miR-29b was mediated through the CLDN1 5′ UTR, coding region (CR), or 3′ UTR, fractions of the CLDN1 5′ UTR, CR, and 3′ UTR were subcloned into the Dual-Luciferase miRNA target expression vector pmirGLO to generate reporter constructs pmirGLO-CLDN1-5′UTR, pmirGLO-CLDN1-CR, and pmirGLO-CLDN1-3′UTR (Fig. 7A, schematic). As shown in Fig. 7A (right), miR-29b overexpression by transfection with pre-miR-29b selectively decreased the level of CLDN1-3′UTR luciferase reporter activity but did not inhibit the activity of the CLDN1-5′UTR or -CR reporter, indicating that miR-29b inhibits claudin-1 translation through the CLDN1 3′ UTR. To further characterize the specific binding site of miR-29b in the CLDN1 3′ UTR, we prepared various reporter constructs that expressed chimeric RNAs containing the luciferase and a partial CLDN1 3′ UTR with or without the potential binding site (Fig. 7B, schematic). Ectopic miR-29b overexpression decreased the levels of luciferase reporter gene activity when cells were transfected with the full-length 3′ UTR (3′UTR-FL) or with a construct containing the miR-29b binding site located at positions 2129 to 2151 (3′UTR-F3) but not when cells were transfected with 3′UTR-F1, -F2, -F4 (which also contains a predicted binding site), or -F5 (Fig. 7B, right).
FIG 7.

Deletion of the miR-29b binding site in the CLDN1 3′ UTR prevents miR-29b-mediated repression of claudin-1. (A) (Left) Schematic of CLDN1 5′ UTR, CR, and 3′ UTR luciferase (Luc) reporters. (Right) Levels of reporter activities after ectopic overexpression of miR-29b. BS, predicted miR-29b-binding site. Twenty-four hours after transfection with pre-miR-29b, cells were transfected with different CLDN1 luciferase reporter plasmids. The levels of firefly and Renilla luciferase activities were assayed 24 h later. The results were normalized to Renilla luciferase activities and are expressed as means ± SEM of data from triplicate experiments. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the scrambled oligomer. (B) Effects of 5′ deletions of the CLDN1 3′ UTR (as shown in the schematic) on its luciferase reporter activity in cells treated as described for panel A. Asterisks indicate significant differences (P < 0.05) from results for cells transfected with the scrambled oligomer. (C) Effect of point mutation of a specific miR-29b binding site (schematic) in CLDN1 3′UTR-F3 on its luciferase reporter activity in cells treated as described for panel A. WT, wild type. The asterisk indicates a significant difference (P < 0.05) from the result for cells transfected with the scrambled oligomer.
To gain a more detailed understanding of this regulatory paradigm, four nucleotides of CLDN1 3′UTR-F3, located at the F3 site of the CLDN1 3′ UTR, were mutated (Fig. 7C, schematic). As shown in Fig. 7C (right), repression of claudin-1 by miR-29b was completely prevented when this specific site from the CLDN1 3′ UTR was mutated. These results indicate that miR-29b interacts with CLDN1 mRNA via the specific binding site at positions 2129 to 2151, thereby repressing claudin-1 translation.
Elevation of uc.173 expression prevents miR-29b-induced epithelial barrier dysfunction.
To test the possibility that uc.173 stimulates claudin-1 translation by decreasing the association of miR-29b with CLDN1 mRNA, we cotransfected cells with pre-miR-29b and a uc.173 expression vector. As shown in Fig. 8A, increasing the levels of uc.173 did not affect induced miR-29b levels in cells cotransfected with pre-miR29b and the uc.173 expression vector. However, increased uc.173 levels abolished the binding of miR-29b to CLDN1 mRNA. There were no differences in the levels of CLDN1 mRNA in pulldown materials between cells transfected with biotin-labeled miR-29b and cells transfected with a biotin-labeled control scrambled oligomer after uc.173 overexpression (Fig. 8B). Cotransfection with biotin-labeled miR-29b and the uc.173 expression vector did not affect the total input of CLDN1 mRNA (see Fig. S4 in the supplemental material). In accordance with this effect, elevation of uc.173 expression also prevented the inhibition of claudin-1 expression (Fig. 8C) and restored epithelial barrier function (Fig. 8D) in cells overexpressing miR-29b. The levels of TEER and FITC-dextran flux in cells cotransfected with pre-miR-29b and the uc.173 expression vector were indistinguishable from those in cells transfected with the control scrambled oligomer. On the other hand, cotransfection with pre-miR-29b and the uc.173 expression vector did not alter the expression levels of occludin or E-cadherin protein. Taken together, these findings indicate that increasing uc.173 levels stimulates claudin-1 translation and enhances the barrier function, largely through the action of uc.173 as a molecular sponge for miR-29b to reduce its bioavailability to CLDN1 mRNA.
FIG 8.
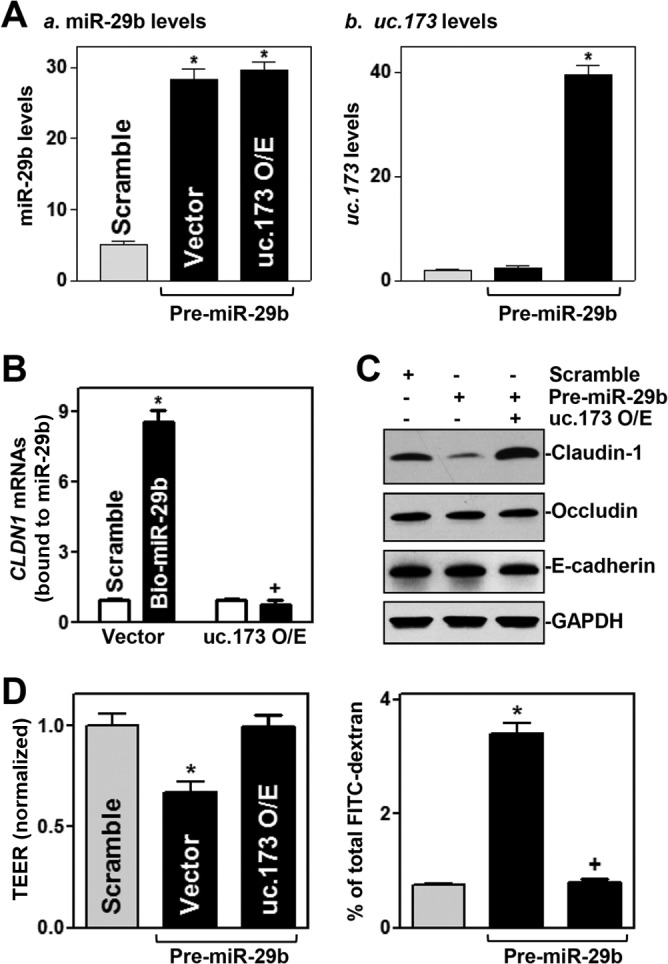
Ectopically expressed uc.173 prevents miR-29b-induced epithelial barrier dysfunction. (A) Levels of miR-29b (a) and uc.173 (b) 48 h after cells were either transfected with pre-miR-29b alone or cotransfected with pre-miR-29b and a uc.173 expression vector. O/E, overexpression. Values are relative to those with a scrambled control oligomer and are means ± SEM of data from triplicate experiments. Asterisks indicate significant differences (P < 0.05) from results for cells transfected with the scrambled oligomer. (B) Effect of increasing uc.173 levels on the association of miR-29b with CLDN1 mRNA. Twenty-four hours after cells were transfected with a uc.173 expression vector, they were transfected with biotin-labeled miR-29b (Bio-miR-29b). The levels of CLDN1 mRNA in the materials pulled down by miR-29b were examined 24 h thereafter. The asterisk and plus sign indicate significant differences (P < 0.05) from the results for cells transfected with a scrambled oligomer or for cells transfected with Bio-miR-29b and a control vector, respectively. (C) Representative immunoblots of claudin-1, occludin, and E-cadherin proteins in cells treated as described for panel A. (D) Changes in TEER (left) and FITC-dextran paracellular permeability (right) in cells treated as described for panel A. Values are means ± SEM of data from six samples. The asterisks and plus sign indicate significant differences (P < 0.05) from results for cells transfected with a scrambled oligomer or with pre-miR-29b alone, respectively.
DISCUSSION
Claudin-1 is a member of the claudin family of TJs. Claudins seal epithelial cells together in the intestinal epithelium in a way that prevents even small molecules from leaking between cells (1, 36), but the exact mechanisms that control cellular claudin-1 abundance are largely unknown. In this study, we show that the T-UCR uc.173 is a novel regulator of claudin-1 expression and that increasing the levels of uc.173 stimulates claudin-1 translation without affecting total CLDN1 mRNA levels. As shown above, uc.173 did not bind to CLDN1 mRNA but acted as a decoy RNA for miR-29b, which represses claudin-1 translation via direct interaction with the CLDN1 3′ UTR. The present study also highlights the importance of uc.173-mediated claudin-1 expression in the regulation of gut permeability, since decreased levels of claudin-1 due to uc.173 silencing not only resulted in epithelial barrier dysfunction in vitro but also increased the vulnerability of the gut barrier to septic stress in mice. Because of the striking evolutionary retention of T-UCRs in the human genome, these findings provide a strong rationale and a fundamental basis for developing new therapeutic strategies directed at uc.173 and/or its target, miR-29b, in order to preserve the integrity of gut barrier function in stressful environments, especially in surgical intensive care patients supported with total parenteral nutrition.
T-UCRs are highly expressed in the epithelium of the intestinal mucosa, and their levels change remarkably after food starvation, during inflammation, and in cancers (23, 29, 37). T-UCR expression profiles are also dysregulated in human Barrett's esophagus (38). However, our understanding of the biological functions of T-UCRs and their involvement in pathology in the gut mucosa is limited. Our present results constitute the first demonstration that uc.173 is essential for maintaining the normal function of the intestinal epithelial barrier by enhancing claudin-1 expression. In cultured IECs, uc.173 silencing specifically decreased the levels of cellular claudin-1 protein, since it did not affect the expression levels of other TJs, such as claudin-3, occludin, JAM-1, and ZO-1, nor that of the AJ E-cadherin. Decreased levels of claudin-1 in uc.173-silenced cells were associated with epithelial barrier dysfunction in an in vitro model. In keeping with these results, the degree of CLP-induced gut permeability in mice treated with LNA-modified anti-uc.173 was much higher than that observed in mice treated with a control oligomer. Moreover, ectopically expressed uc.173 enhanced nascent claudin-1 protein synthesis and increased the levels of CLDN1 mRNA associated with actively translating fractions of polyribosomes. In support of our present findings, uc.173 enhances the growth of the small intestinal mucosa in mice (29) and inhibits lead-induced neuronal apoptosis in brain tissue (39). In contrast, the T-UCR uc.261 was found to decrease TJ expression and repress TJ assembly, thus damaging intestinal epithelial barrier function (37). Clearly, more studies are needed to fully investigate the exact roles and mechanisms of different T-UCRs in the regulation of epithelial barrier function under various pathophysiological conditions.
In this study, we also observed that uc.173 interacted directly with miR-29b via the complementary sites located at its 3′ end, suggesting the possibility that uc.173 can act as a molecular sponge for miR-29b so as to reduce the availability of miR-29b to CLDN1 mRNA. As shown in Fig. S2 in the supplemental material, MS2-tagged uc.173 specifically pulled down miR-29b in IECs but did not bind to miR-222 or lncRNA H19. Furthermore, miR-29b was associated only with uc.173, not with uc.346 or uc.283, as measured by RNA pulldown assays using biotin-labeled miR-29b (Fig. 5C). In keeping with our present results, several T-UCRs have been shown to regulate gene expression by interacting with given miRNAs but not directly targeting mRNAs (23). For example, uc.339 functions as a natural decoy RNA for three miRNAs—miR-339-3p, miR-663-3p, and miR-95-5p—in lung cancer cells (26), while uc.8+ entraps miR-596 and reduces the availability of this miRNA to MMP9 mRNA in bladder tissues (25). In addition, T-UCRs can also regulate miRNA activity at another level. It has been reported that uc.283+ controls miRNA processing by preventing pri-miRNA cleavage by Drosha (17). We have recently demonstrated that uc.173 interferes with miRNA stability by binding to pri-miR-195 via sequence complementarity in IECs (29). It is likely that uc.173 affects gene regulatory programs governing intestinal epithelial homeostasis and barrier function predominantly by functioning as a decoy RNA, as for miR-29b, and/or by modulating miRNA degradation, as for miR-195.
Our present results also indicate that miR-29b represses claudin-1 translation by interacting directly with the 3′ UTR of CLDN1 mRNA, but not with its 5′ UTR or CR, in IECs. Studies using various ectopic luciferase reporters bearing partial transcripts spanning the CLDN1 3′ UTR with or without the miR-29b binding site further show that a specific sequence at positions 2129 to 2151 in 3′-UTR-F3 was the predominant functional site through which miR-29b interacted with claudin-1 and repressed its translation, whereas another predicted site, located in 3′-UTR-F4, exhibited a lesser effect. These findings are consistent with results from others, who demonstrate that miR-29b commonly interacts with the 3′ UTRs of its target transcripts, such as bcr-abl1, gata3, pdpn, col1a1, col3a1, col5a1, eln, Cdk2, dnmt3a, and dnmt3b mRNAs, thus destabilizing mRNAs and/or repressing their translation (20, 33, 40–44). In some instances, miR-29b also binds to the CRs of target mRNAs for its regulatory actions. For example, miR-29b destabilizes Men1 mRNA through a single binding site in the CR of Men1 mRNA rather than in its 3′ UTR (34). In a similar manner, miR-322/503 represses Smurf2 mRNA translation by interacting with the Smurf2 CR (45), and miR-222 inhibits Cdk4 mRNA translation through interaction with the Cdk4 CR (46).
Finally, our findings show that increasing uc.173 levels in cells overexpressing miR-29b rescued claudin-1 expression and restored epithelial barrier function by reducing the association of miR-29b with CLDN1 mRNA. miR-29b is a potent repressor of intestinal mucosal regeneration, and elevation of cellular miR-29b levels causes mucosal atrophy and compromises the integrity of the intestinal epithelium in mice (20, 33, 35). Our present studies provide additional evidence that uc.173-mediated repression of miR-29b also contributes to the stimulation of intestinal mucosal growth after uc.173 overexpression. On the other hand, miR-195 represses IEC migration over the wounded area after injury by targeting Stim1 mRNA, thus impairing the repair of the intestinal epithelium (47). Therefore, inhibition of miR-195 activity by uc.173 (29) also plays a role in the uc.173-mediated protective effect on the epithelial barrier. Because the basal level of mucosal uc.173 is relatively high in the intestine and its levels in tissue decrease dramatically in stressful environments, enhancement of epithelial barrier function and mucosal regeneration by uc.173 through antagonism of miR-29b and miR-195 is crucial for homeostasis of the intestinal epithelium, representing a novel therapeutic target for patients with gut barrier dysfunction and mucosal atrophy or hyperplasia.
MATERIALS AND METHODS
Cell culture and animals.
Caco-2 cells were purchased from the American Type Culture Collection (Manassas, VA) and were maintained under standard culture conditions (48). Tissue culture medium and fetal bovine serum were purchased from Invitrogen (Carlsbad, CA), and biochemicals were from Sigma (St. Louis, MO). The antibodies recognizing claudin-1, claudin-3, occludin, JAM-1, ZO-1, E-cadherin, β-catenin, UBE2B, and GAPDH were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and BD Biosciences (Sparks, MD). The secondary antibody conjugated to horseradish peroxidase was from Sigma. LNA-modified anti-uc.173 oligonucleotides and control LNA-scrambled oligonucleotides were custom-generated by Exiqon (Vedbæk, Denmark). The pre-miR miRNA precursor of miRNA-29b (pre-miR-195) was purchased from Ambion (Austin, TX). Biotin-labeled miRNA-29b was custom-made by Dharmacon (Lafayette, CO).
C57BL/6J mice (male and female, 6 to 9 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and were housed in a pathogen-free animal facility at the Baltimore VA Medical Center. All animal experiments were conducted in accordance with NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine and the Baltimore VA hospital. In studies of LNA-mediated uc.173 silencing, mice were injected intraperitoneally (i.p.) with LNA–anti-uc.173 (500 μg/100 g of body weight/day) for 4 consecutive days, whereas control mice were injected with an equal dose of control LNA-scrambled oligonucleotides as described previously (29). On day 5, a 4-cm segment taken from the middle of the small intestine was removed, and the mucosa was scraped with a glass slide for various measurements.
Plasmid construction.
An expression vector containing a 284-bp fragment flanking the human uc.173 locus under the control of the pCMV promoter was constructed as described previously (29). The chimeric firefly luciferase reporter constructs containing the full-length CLDN1 5′ UTR, CR, or different 3′ UTR fragments were subcloned into the Dual-Luciferase miRNA target expression vector pmirGLO (Promega, Madison, WI) to generate pmirGLO-Luc-CLDN1-5′UTR, pmirGLO-Luc-CLDN1-CR, and pmirGLO-CLDN1-3′UTR as described previously (49). DNA sequencing and enzyme digestion were used to confirm the sequence and orientation of the fragment in the luciferase reporter. Transient transfections were performed using the Lipofectamine reagent as recommended by the manufacturer (Invitrogen) (30). Luciferase activity was examined using the Dual-Luciferase assay system, and the levels of firefly luciferase activity were normalized to the levels of Renilla luciferase activity and were further compared with the levels of luciferase mRNA in every experiment. All the primer sequences used to generate these constructs are given in Table S1 in the supplemental material. Both the pcDNA-MS2 and pcDNA-MS2-GST plasmids have been described previously (32), and full-length uc.173 was inserted into pcDNA-MS2 at the XhoI site as described previously (7, 47).
RT-PCR and Q-PCR analysis.
Total RNA was isolated by using an RNeasy minikit (Qiagen, Valencia, CA), and reverse transcription (RT) and PCR amplification reactions were performed as described previously (50). The levels of the GAPDH PCR product were examined to monitor the evenness of RNA input in RT-PCR samples. Real-time quantitative PCR (Q-PCR) analysis was conducted using 7500 Fast Real-Time PCR systems with specific primers, probes, and software (Applied Biosystems, Foster City, CA). For miRNA studies, the levels of miRNA-29b were also quantified by Q-PCR by using a TaqMan microRNA assay; the small nuclear RNA (snRNA) U6 was used as an endogenous control.
Western blot analysis.
Whole-cell lysates were prepared using 2% SDS, sonicated, and centrifuged (12,000 rpm) at 4°C for 15 min. The supernatants were boiled for 5 min and were size-fractionated by SDS-PAGE (7.5% acrylamide). After proteins were transferred to nitrocellulose filters, the blots were incubated with primary antibodies recognizing claudin-1 or other TJs; following incubations with secondary antibodies, immunocomplexes were developed by using chemiluminescence.
Analysis of newly translated protein.
De novo synthesis of nascent proteins was detected by a Click-iT protein analysis detection kit (Life Technologies, Grand Island, NY) according to the manufacturer's instructions, with minor modifications as described previously (18). Briefly, cells were first incubated in a methionine-free medium and then exposed to l-azidohomoalanine (AHA). After the cell lysates were mixed with the reaction buffer containing a biotin-alkyne reagent and CuSO4 for 20 min, the biotin-alkyne-azide-modified protein complex was pulled down using paramagnetic streptavidin-conjugated Dynabeads. The pulldown material was resolved by 10% SDS-PAGE and was analyzed by Western immunoblot analysis using antibodies that recognized claudin-1 or GAPDH protein.
Polysome analysis was performed as described previously (51). Briefly, cells at ∼70% confluence were incubated for 15 min in 0.1 mg/ml cycloheximide, lifted by scraping in 1 ml of polysome extraction buffer, and lysed on ice for 10 min. Nuclei were pelleted, and the resulting supernatant was fractionated through a 10-to-50% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. Fractions were eluted with a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm using a UV-6 detector (ISCO, Louisville, KY). After the RNA in each fraction was extracted, the level of each individual mRNA was quantified by Q-PCR in each of the fractions.
Biotin-labeled RNA pulldown assays.
Biotin-labeled pri-miR-29b was transfected, and 24 h later, whole-cell lysates were collected, mixed with streptavidin-coupled Dynal beads, and incubated on a rotator overnight (52). After the beads were washed thoroughly, the bead-bound RNA was isolated and subjected to RT, followed by Q-PCR analysis. Input RNA was extracted and served as a control.
Measurements of epithelial barrier function in vitro.
Epithelial barrier function in vitro was examined by paracellular tracer flux assays using the 12-well Transwell plate (surface area, 1.12 cm2) as described previously (19). FITC-dextran (70 kDa; Sigma), a membrane-impermeant molecule, served as the paracellular tracer and was added to the apical bathing wells. The basal bathing well had no added tracers and contained the same flux assay medium as that in the apical compartment. All flux assays were performed at 37°C, and the basal medium was collected at different times after the addition of FITC-dextran. The concentration of FITC-dextran in the basal medium was determined using a fluorescence plate reader with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. TEER was measured with an epithelial volt/ohm meter under open-circuit conditions (World Precision Instruments, Sarasota, FL) as described previously (53), and the TEER values of all monolayers were normalized to those of control monolayers in the same experiment.
Surgical procedures.
Mice were anesthetized with pentobarbital (Nembutal; 5.5 mg/100 g of body weight, given i.p.), and CLP was performed as described previously (31). The distal portion of the cecum (1 cm) was ligated with 5-0 silk sutures. The ligated cecum was then punctured with a 25-gauge needle and was slightly compressed with an applicator until a small amount of stool appeared. In sham-operated animals, the cecum was manipulated, but without ligation and puncture, and was placed back in the peritoneum. The incision was closed using a 2-layer procedure: 5-0 silk sutures on the muscle layer and the skin, respectively. Mice received 1 ml of saline i.p. for fluid resuscitation at the time of closure and 0.1 mg buprenorphine (Buprenex)/100 g of body weight subcutaneously (s.c.) 4 times at 12-h intervals to minimize distress.
Gut permeability in vivo was determined by examining the appearance in blood of FITC-dextran administered by gavage as described previously (54). Briefly, mice were gavaged with FITC-dextran at a dose of 60 mg/100 g of body weight 4 h before harvest. Blood samples were collected by cardiac puncture. The concentration of FITC-dextran in serum was determined using a fluorescence plate reader as described above.
Statistical analysis.
All values are expressed as means ± standard errors of the means (SEM). An unpaired, two-tailed Student t test was used where indicated, and a P value of <0.05 was considered significant. In assessing multiple groups, one-way analysis of variance (ANOVA) was utilized with Tukey's post hoc test (55). The statistical software used was SPSS 17.1.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Merit Review Awards (to Jian-Ying Wang and J. N. Rao) from the U.S. Department of Veterans Affairs, grants from the National Institutes of Health (DK57819, DK61972, and DK68491, to Jian-Ying Wang), and funding from the National Institute on Aging Intramural Research Program, NIH (to M. Gorospe). Jian-Ying Wang is a Senior Research Career Scientist, Biomedical Laboratory Research & Development Service, U.S. Department of Veterans Affairs.
Jun-Yao Wang and Y.-H. Cui performed most experiments and summarized the data. L. Xiao and H. K. Chung performed in vivo experiments and biotin pulldown assays. Y. Zhang and J. N. Rao performed experiments with the miR-29b promoter. M. Gorospe contributed to the experimental design and data analysis. Jian-Ying Wang designed the experiments, analyzed the data, prepared figures, and drafted the manuscript.
We declare that we have no competing interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00010-18.
REFERENCES
- 1.Turner JR. 2009. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 2.Yang H, Rao JN, Wang JY. 2014. Posttranscriptional regulation of intestinal epithelial tight junction barrier by RNA-binding proteins and microRNAs. Tissue Barriers 2:e28320. doi: 10.4161/tisb.28320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higashi T, Miller AL. 2017. Tricellular junctions: how to build junctions at the TRICkiest points of epithelial cells. Mol Biol Cell 28:2023–2034. doi: 10.1091/mbc.E16-10-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furuse M, Izumi Y, Oda Y, Higashi T, Iwamoto N. 2014. Molecular organization of tricellular tight junctions. Tissue Barriers 2:e28960. doi: 10.4161/tisb.28960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malinova TS, Huveneers S. 2018. Sensing of cytoskeletal forces by asymmetric adherens junctions. Trends Cell Biol 28:328–341. doi: 10.1016/j.tcb.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Xiao L, Rao JN, Zou T, Liu L, Bellavance E, Gorospe M, Wang JY. 2008. JunD represses transcription and translation of the tight junction protein zona occludens-1 modulating intestinal epithelial barrier function. Mol Biol Cell 19:3701–3712. doi: 10.1091/mbc.E08-02-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu TX, Wang PY, Rao JN, Zou T, Liu L, Xiao L, Gorospe M, Wang JY. 2011. Chk2-dependent HuR phosphorylation regulates occludin mRNA translation and epithelial barrier function. Nucleic Acids Res 39:8472–8487. doi: 10.1093/nar/gkr567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatt T, Rizvi A, Batta SP, Kataria S, Jamora C. 2013. Signaling and mechanical roles of E-cadherin. Cell Commun Adhes 20:189–199. doi: 10.3109/15419061.2013.854778. [DOI] [PubMed] [Google Scholar]
- 9.Carter SR, Zahs A, Palmer JL, Wang L, Ramirez L, Gamelli RL, Kovacs EJ. 2013. Intestinal barrier disruption as a cause of mortality in combined radiation and burn injury. Shock 40:281–289. doi: 10.1097/SHK.0b013e3182a2c5b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang KC, Chang HY. 2011. Molecular mechanisms of long noncoding RNAs. Mol Cell 43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulitsky I, Bartel DP. 2013. lincRNAs: genomics, evolution, and mechanisms. Cell 154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Batista PJ, Chang HY. 2013. Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 14.Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. 2010. Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58. doi: 10.1016/j.cell.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W. 2012. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 14:659–665. doi: 10.1038/ncb2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Y, Wu F, Zhou J, Yan L, Jurczak MJ, Lee HY, Yang L, Mueller M, Zhou XB, Dandolo L, Szendroedi J, Roden M, Flannery C, Taylor H, Carmichael GG, Shulman GI, Huang Y. 2014. The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res 42:13799–13811. doi: 10.1093/nar/gku1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liz J, Portela A, Soler M, Gómez A, Ling H, Michlewski G, Calin GA, Guil S, Esteller M. 2014. Regulation of pri-miRNA processing by a long noncoding RNA transcribed from an ultraconserved region. Mol Cell 55:138–147. doi: 10.1016/j.molcel.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Zou T, Jaladanki SK, Liu L, Xiao L, Chung HK, Wang JY, Xu Y, Gorospe M, Wang JY. 2016. H19 long noncoding RNA regulates intestinal epithelial barrier function via microRNA 675 by interacting with RNA-binding protein HuR. Mol Cell Biol 36:1332–1341. doi: 10.1128/MCB.01030-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao L, Rao JN, Cao S, Liu L, Chung HK, Zhang Y, Zhang J, Liu Y, Gorospe M, Wang JY. 2016. Long noncoding RNA SPRY4-IT1 regulates intestinal epithelial barrier function by modulating the expression levels of tight junction proteins. Mol Biol Cell 27:617–626. doi: 10.1091/mbc.E15-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang JY, Xiao L, Wang JY. 2017. Posttranscriptional regulation of intestinal epithelial integrity by noncoding RNAs. Wiley Interdiscip Rev RNA 8:1399. doi: 10.1002/wrna.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. 2004. Ultraconserved elements in the human genome. Science 304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 22.Calin GA, Liu CG, Ferracin M, Hyslop T, Spizzo R, Sevignani C, Fabbri M, Cimmino A, Lee EJ, Wojcik SE, Shimizu M, Tili E, Rossi S, Taccioli C, Pichiorri F, Liu X, Zupo S, Herlea V, Gramantieri L, Lanza G, Alder H, Rassenti L, Volinia S, Schmittgen TD, Kipps TJ, Negrini M, Croce CM. 2007. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 12:215–229. doi: 10.1016/j.ccr.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 23.Ling H, Vincent K, Pichler M, Fodde R, Berindan-Neagoe I, Slack FJ, Calin GA. 2015. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 34:5003–5011. doi: 10.1038/onc.2014.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braconi C, Valeri N, Kogure T, Gasparini P, Huang N, Nuovo GJ, Terracciano L, Croce CM, Patel T. 2011. Expression and functional role of a transcribed noncoding RNA with an ultraconserved element in hepatocellular carcinoma. Proc Natl Acad Sci U S A 108:786–791. doi: 10.1073/pnas.1011098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olivieri M, Ferro M, Terreri S, Durso M, Romanelli A, Avitabile C, De Cobelli O, Messere A, Bruzzese D, Vannini I, Marinelli L, Novellino E, Zhang W, Incoronato M, Ilardi G, Staibano S, Marra L, Franco R, Perdonà S, Terracciano D, Czerniak B, Liguori GL, Colonna V, Fabbri M, Febbraio F, Calin GA, Cimmino A. 2016. Long noncoding RNA containing ultraconserved genomic region 8 promotes bladder cancer tumorigenesis. Oncotarget 7:20636–20654. doi: 10.18632/oncotarget.7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vannini I, Wise PM, Challagundla KB, Plousiou M, Raffini M, Bandini E, Fanini F, Paliaga G, Crawford M, Ferracin M, Ivan C, Fabris L, Davuluri RV, Guo Z, Cortez MA, Zhang X, Chen L, Zhang S, Fernandez-Cymering C, Han L, Carloni S, Salvi S, Ling H, Murtadha M, Neviani P, Gitlitz BJ, Laird-Offringa IA, Nana-Sinkam P, Negrini M, Liang H, Amadori D, Cimmino A, Fabbri M, Calin GA. 2017. Transcribed ultraconserved region 339 promotes carcinogenesis by modulating tumor suppressor microRNAs. Nat Commun 8:1801. doi: 10.1038/s41467-017-01562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sekino Y, Sakamoto N, Goto K, Honma R, Shigematsu Y, Sentani K, Oue N, Teishima J, Matsubara A, Yasui W. 2017. Transcribed ultraconserved region Uc.63+ promotes resistance to docetaxel through regulation of androgen receptor signaling in prostate cancer. Oncotarget 8:94259–94270. doi: 10.18632/oncotarget.21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferdin J, Nishida N, Wu X, Nicoloso MS, Shah MY, Devlin C, Ling H, Shimizu M, Kumar K, Cortez MA, Ferracin M, Bi Y, Yang D, Czerniak B, Zhang W, Schmittgen TD, Voorhoeve MP, Reginato MJ, Negrini M, Davuluri RV, Kunej T, Ivan M, Calin GA. 2013. HINCUTs in cancer: hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ 20:1675–1687. doi: 10.1038/cdd.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao L, Wu J, Wang JY, Chung HK, Kalakonda S, Rao JN, Gorospe M, Wang JY. 2018. Long noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of microRNA 195. Gastroenterology 154:599–611. doi: 10.1053/j.gastro.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK, Wu J, Donahue JM, Gorospe M, Wang JY. 2015. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell 26:1797–1810. doi: 10.1091/mbc.E14-11-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW III, Bland KI, Chaudry IH. 2005. Cecal ligation and puncture. Shock 1:52–57. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 32.Yoon JH, Srikantan S, Gorospe M. 2012. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods 58:81–87. doi: 10.1016/j.ymeth.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao L, Rao JN, Zou T, Liu L, Cao S, Martindale JL, Su W, Chung HK, Gorospe M, Wang JY. 2013. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 24:3038–3046. doi: 10.1091/mbc.E13-05-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ouyang M, Su W, Xiao L, Rao JN, Jiang L, Li Y, Turner DJ, Gorospe M, Wang JY. 2015. Modulation by miR-29b of intestinal epithelium homoeostasis through the repression of menin translation. Biochem J 465:315–323. doi: 10.1042/BJ20141028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Chen G, Wang J, Zou T, Liu L, Xiao L, Chung HK, Rao JN, Wang JY. 2016. Posttranscriptional regulation of Wnt-coreceptor LRP6 and RNA-binding protein HuR by miR-29b in intestinal epithelial cells. Biochem J 473:1641–1649. doi: 10.1042/BCJ20160057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schneeberger EE, Lynch RD. 2004. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 37.Qian XX, Peng JC, Xu AT, Zhao D, Qiao YQ, Wang TR, Shen J, Ran ZH. 2016. Noncoding transcribed ultraconserved region (T-UCR) uc.261 participates in intestinal mucosa barrier damage in Crohn's disease. Inflamm Bowel Dis 22:2840–2852. doi: 10.1097/MIB.0000000000000945. [DOI] [PubMed] [Google Scholar]
- 38.Fassan M, Dall'Olmo L, Galasso M, Braconi C, Pizzi M, Realdon S, Volinia S, Valeri N, Gasparini P, Baffa R, Souza RF, Vicentini C, D'Angelo E, Bornschein J, Nuovo GJ, Zaninotto G, Croce CM, Rugge M. 2014. Transcribed ultraconserved noncoding RNAs (T-UCR) are involved in Barrett's esophagus carcinogenesis. Oncotarget 5:7162–7171. doi: 10.18632/oncotarget.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nan A, Zhou X, Chen L, Liu M, Zhang N, Zhang L, Luo Y, Liu Z, Dai L, Jiang Y. 2016. A transcribed ultraconserved noncoding RNA uc.173 is a key molecule for the inhibition of lead-induced neuronal apoptosis. Oncotarget 7:112–124. doi: 10.18632/oncotarget.6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cortez MA, Nicoloso MS, Shimizu M, Rossi S, Gopisetty G, Molina JR, Carlotti C Jr, Tirapelli D, Neder L, Brassesco MS, Scrideli CA, Tone LG, Georgescu MM, Zhang W, Puduvalli V, Calin GA. 2010. miR-29b and miR-125a regulate podoplanin and suppress invasion in glioblastoma. Genes Chromosomes Cancer 49:981–990. doi: 10.1002/gcc.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang X, Schwind S, Yu B, Santhanam R, Wang H, Hoellerbauer P, Mims A, Klisovic R, Walker AR, Chan KK, Blum W, Perrotti D, Byrd JC, Bloomfield CD, Caligiuri MA, Lee RJ, Garzon R, Muthusamy N, Lee LJ, Marcucci G. 2013. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res 19:2355–2367. doi: 10.1158/1078-0432.CCR-12-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melo SA, Kalluri R. 2013. miR-29b moulds the tumour microenvironment to repress metastasis. Nat Cell Biol 15:139–140. doi: 10.1038/ncb2684. [DOI] [PubMed] [Google Scholar]
- 43.Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, Sung JJ, Lan HY. 2011. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol 22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, Janssen J, Koppe C, Knolle P, Castoldi M, Tacke F, Trautwein C, Luedde T. 2011. MicroRNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53:209–218. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 45.Cao S, Xiao L, Rao JN, Zou T, Liu L, Zhang D, Turner DJ, Gorospe M, Wang JY. 2014. Inhibition of Smurf2 translation by miR-322/503 modulates TGF-β/Smad2 signaling and intestinal epithelial homeostasis. Mol Biol Cell 25:1234–1243. doi: 10.1091/mbc.E13-09-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao L, Cui YH, Rao JN, Zou T, Liu L, Smith A, Turner DJ, Gorospe M, Wang JY. 2011. Regulation of cyclin-dependent kinase 4 translation through CUG-binding protein 1 and microRNA-222 by polyamines. Mol Biol Cell 22:3055–3069. doi: 10.1091/mbc.E11-01-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhuang R, Rao JN, Zou T, Liu L, Xiao L, Cao S, Hansraj NZ, Gorospe M, Wang JY. 2013. miR-195 competes with HuR to modulate stim1 mRNA stability and regulate cell migration. Nucleic Acids Res 41:7905–7919. doi: 10.1093/nar/gkt565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cui YH, Xiao L, Rao JN, Zou T, Liu L, Chen Y, Turner DJ, Gorospe M, Wang JY. 2012. miR-503 represses CUG-binding protein 1 translation by recruiting CUGBP1 mRNA to processing bodies. Mol Biol Cell 23:151–162. doi: 10.1091/mbc.E11-05-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Zhang Y, Xiao L, Yu TX, Li JZ, Rao JN, Turner DJ, Gorospe M, Wang JY. 2017. Cooperative repression of insulin-like growth factor type 2 receptor translation by microRNA 195 and RNA-binding protein CUGBP1. Mol Cell Biol 37:e00225-. doi: 10.1128/MCB.00225-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, Christodoulou-Vafeiadou E, Rao JN, Zou T, Xiao L, Chung HK, Yang H, Gorospe M, Kontoyiannis D, Wang JY. 2014. RNA-binding protein HuR promotes growth of small intestinal mucosa by activating the Wnt signaling pathway. Mol Biol Cell 25:3308–3318. doi: 10.1091/mbc.E14-03-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Zhuang R, Xiao L, Chung HK, Luo J, Turner DJ, Rao JN, Gorospe M, Wang JY. 2017. HuR enhances early restitution of the intestinal epithelium by increasing Cdc42 translation. Mol Cell Biol 37:e00574-16. doi: 10.1128/MCB.00574-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung HK, Chen Y, Rao JN, Liu L, Xiao L, Turner DJ, Yang P, Gorospe M, Wang JY. 2015. Transgenic expression of miR-222 disrupts intestinal epithelial regeneration by targeting multiple genes including Frizzled-7. Mol Med 21:676–687. doi: 10.2119/molmed.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu TX, Rao JN, Zou T, Liu L, Xiao L, Ouyang M, Cao S, Gorospe M, Wang JY. 2013. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol Biol Cell 24:85–99. doi: 10.1091/mbc.E12-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. 2001. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harter JL. 1960. Critical values for Duncan's new multiple range tests. Biometrics 16:671–685. doi: 10.2307/2527770. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.