

ABSTRACT
Cleavage and polyadenylation specificity factor 6 (CPSF6) is a human protein that binds HIV-1 capsid and mediates nuclear transport and integration targeting of HIV-1 preintegration complexes. Truncation of the protein at its C-terminal nuclear-targeting arginine/serine-rich (RS) domain produces a protein, CPSF6-358, that potently inhibits HIV-1 infection by targeting the capsid and inhibiting nuclear entry. To understand the molecular mechanism behind this restriction, the interaction between CPSF6-358 and HIV-1 capsid was characterized using in vitro and in vivo assays. Purified CPSF6-358 protein formed oligomers and bound in vitro-assembled wild-type (WT) capsid protein (CA) tubes, but not CA tubes containing a mutation in the putative binding site of CPSF6. Intriguingly, binding of CPSF6-358 oligomers to WT CA tubes physically disrupted the tubular assemblies into small fragments. Furthermore, fixed- and live-cell imaging showed that stably expressed CPSF6-358 forms cytoplasmic puncta upon WT HIV-1 infection and leads to capsid permeabilization. These events did not occur when the HIV-1 capsid contained a mutation known to prevent CPSF6 binding, nor did they occur in the presence of a small-molecule inhibitor of capsid binding to CPSF6-358. Together, our in vitro biochemical and transmission electron microscopy data and in vivo intracellular imaging results provide the first direct evidence for an oligomeric nature of CPSF6-358 and suggest a plausible mechanism for restriction of HIV-1 infection by CPSF6-358.
IMPORTANCE After entry into cells, the HIV-1 capsid, which contains the viral genome, interacts with numerous host cell factors to facilitate crucial events required for replication, including uncoating. One such host cell factor, called CPSF6, is predominantly located in the cell nucleus and interacts with HIV-1 capsid. The interaction between CA and CPSF6 is critical during HIV-1 replication in vivo. Truncation of CPSF6 leads to its localization to the cell cytoplasm and inhibition of HIV-1 infection. Here, we determined that truncated CPSF6 protein forms large higher-order complexes that bind directly to HIV-1 capsid, leading to its disruption. Truncated CPSF6 expression in cells leads to premature capsid uncoating that is detrimental to HIV-1 infection. Our study provides the first direct evidence for an oligomeric nature of truncated CPSF6 and insights into the highly regulated process of HIV-1 capsid uncoating.
KEYWORDS: CPSF6, HIV, TEM, capsid, imaging, restriction
INTRODUCTION
The HIV-1 capsid comprises multiple copies of the capsid (CA) protein assembled into hexamers and pentamers (1–3), which further assemble into a conical protein shell that encapsulates the viral RNA genome. Following HIV-1 entry into cells, the viral capsid interacts with numerous host cell factors that facilitate uncoating and downstream viral events, including reverse transcription, nuclear entry, and integration site targeting. The surface of the capsid acts as a docking platform for many host cell factors, which either promote infection or enable virus restriction by innate immune responses (4, 5). While the structure of the capsid has been well characterized (3, 6, 7), much less is known about the detailed structure and function of capsid-binding host factors and their interactions with HIV-1 capsid. In addition, some host factors, including TRIM5α, TRIMCyp, and MxB, must oligomerize to functionally interact with the assembled viral capsid (8–11). As such, capsid pattern recognition by host factor oligomers is emerging as a key feature and determinant of viral restriction by these factors. For most capsid-binding host proteins, including cleavage and polyadenylation specificity factor 6 (CPSF6), how virus-host protein-protein interactions contribute to viral replication or restriction remains unclear.
CPSF6 has been suggested to regulate HIV-1 nuclear entry and integration site targeting through interaction with HIV-1 capsid (12, 13). CPSF6 is a pre-mRNA processing protein that dynamically shuttles between the nucleus and the cytoplasm, with a C-terminal nuclear-targeting arginine/serine-rich (RS) domain (14–17). Truncation of CPSF6 at amino acid 358 (CPSF6-358), which removes the RS domain, leads to an exclusively cytoplasmic localization of the protein and potent inhibition of HIV-1 infection (18, 19). The antiviral activity of CPSF6-358 depends on its direct docking to the HIV-1 capsid at a stage after reverse transcription and before nuclear entry (19), but the mechanism of this inhibition is unclear. The crystal structures of cross-linked CA hexamer in complex with a CPSF6-358 peptide (residues 314 to 322) reveal a hydrophobic binding pocket encompassing the intermolecular interface between the N-terminal domain (NTD) and the C-terminal domain (CTD), in which the peptide is anchored via a phenylalanine-glycine (FG) motif (15, 20). This binding pocket is shared among CPSF6, NUP153, and the small-molecule compounds PF-74 and BI-2 (15, 20). Single point mutations in the pocket are sufficient to diminish binding of CPSF6-358 and to rescue infectivity (15, 18, 19, 21). Importantly, several HIV-1 capsid mutations (A105T, N74D, N57A, A77V, R132K/L136M, M66F, Q67A, K70A, and T107A) have been highly informative for understanding the role of CPSF6 as an HIV-1 cofactor (15, 22). Of particular interest, the HIV-1 CA mutation N74D abolishes CPSF6-358 binding and its antiviral activity (22, 23). While the structural detail of the specific interaction between CA hexamer and the linear binding epitope of CPSF6 is available (12, 20), the mechanistic basis upon which CPSF6-358 potently restricts HIV-1 infection is not known. Understanding the molecular mechanism by which CPSF6-358 restricts HIV requires both a fully assembled HIV-1 capsid and purified full-length CPSF6-358 protein, which have been a challenge to obtain.
In vivo studies of HIV-1 capsid and its interactions with host factors has also been difficult, hampered by the fragile and dynamic nature of the capsid (24). As HIV-1 CA itself cannot be directly labeled in functional virions, indirect approaches to image capsid in infected cells have been developed, including antibody staining (25), staining of viral RNA after capsid permeabilization (26), labeling of capsid-binding oligomeric cyclophilin A (CypA-DsRed) (27), or labeling with a cleavable fluorescent reporter encoded within gag (28). In addition, microscopy assays for imaging HIV-1 nucleic acids and other factors that are expected to be present in reverse transcriptase (RT) complexes and/or preintegration complexes early after infection have been developed. These assays have applied fluorescent nucleotides (25), fluorescent integrase (IN) (29), fluorescent RNA-binding proteins (30), and staining of modified viral RNA (26) or DNA (31). While expression of restrictive CPSF6-358 has been studied in cells (18, 21, 32), its localization with and effect on HIV-1 complexes after infection have not been visualized.
To gain insight into how CPSF6-358 restricts HIV-1 infection, we purified CPSF6-358 from a mammalian expression system using albumin as a secretion signal fusion tag. CPSF6-358 purified as dimers and higher-order oligomers and was found to bind and physically disrupt tubular HIV-1 CA assemblies in vitro. This disruption of CA tubes was dependent on the oligomeric state of the protein, with more drastic disruption in the presence of higher-order CPSF6-358 oligomers. In addition, imaging of cells expressing fluorescently tagged CPSF6-358 showed the formation of higher-order CPSF6-358 complexes upon infection with wild-type (WT) HIV-1, but not with N74D HIV-1 or in the presence of the competitive inhibitor PF-74. Furthermore, expression of CPSF6-358 destabilized HIV-1 capsids in cells, as visualized by viral RNA staining. Together, our data provide the first direct evidence of oligomerization of CPSF6-358, suggesting a mechanism for its restriction of HIV-1 infection.
RESULTS
CPSF6-358 is purified as dimers and higher-order oligomers.
Detailed analysis of the direct interaction between HIV-1 capsid and CPSF6-358 has historically been hindered by the inability to express and purify high-quality and high-quantity yields of CPSF6-358 protein. To overcome this challenge, we attached a His6-albumin fusion tag to the N terminus of CPSF6-358 (His6-albumin–CPSF6-358) and expressed the protein in mammalian cells. The albumin-fused protein was robustly expressed (Fig. 1A) and subsequently purified using Ni-nitrilotriacetic acid (NTA) resin followed by a Superdex 200 26/60 gel filtration column. Two broad peaks were observed in the gel filtration profile of the tagged protein (Fig. 1B, P1 and P2), both of which corresponded to His6-albumin–CPSF6-358, as confirmed by Western blotting with anti-His and anti-CPSF6 antibodies (Fig. 1A). The presence of two peaks suggests that the purified fusion protein may adopt different oligomeric states. Size exclusion chromatography coupled with in-line multiangle light scattering (SEC-MALS) further showed that the P1 protein contained primarily large oligomers with molecular masses around 2.1 MDa, whereas the P2 sample was a mixture of oligomeric states, with the majority around 260 kDa and 148 kDa (Fig. 2A, red curve) and a small fraction similar to P1, around 2 MDa. Negative-stain electron microscopy (EM) revealed this to be the case, with the P1 peak showing linear strings of varying protein oligomer lengths (Fig. 1C, left), while P2 contained a complex mixture of oligomeric species that were mostly shorter and thinner than those observed in P1 (Fig. 1C, right).
FIG 1.

Purification of CPSF6-358 with an albumin tag from the mammalian secretory expression system. (A) SDS-PAGE and Western blot analysis of His6-albumin–CPSF6-358 expression and purification. Samples taken from untransfected cells (U), transfected cells (T), the flowthrough (FT) and elution (E) from Ni-NTA resin, and peaks (P1 and P2) from the Superdex 200 26/60 column (shown in panel B) were stained with Coomassie blue (top) or processed with anti-His (middle) or anti-CPSF6 (bottom) antibody, following Western blotting. (B) Gel filtration profile of the protein eluted from the Superdex 200 26/60 column. The two His6-albumin–CPSF6-358 peaks are labeled P1 and P2. (C) Representative EM images of negatively stained His6-albumin–CPSF6-358 samples from fractions P1 (left) and P2 (right), as shown in panel B. Scale bars, 100 nm.
FIG 2.

Characterization of CPSF6-358 oligomerization states. (A) SEC-MALS analysis of His6-albumin–CPSF6-358 samples from P1 (black) and P2 (red) samples, shown in Fig. 1; the estimated molecular mass of the monomeric form of the protein should be 110 kDa. (B) Superdex 200 gel filtration of CPSF6-358 after TEV cleavage of the His6-albumin tag of P2 (top) with an EM image of the purified CPSF6-358 fraction from the position of the peak indicated by the arrow (inset) and SDS-PAGE of the corresponding peaks, stained with Coomassie blue (bottom). (C) Analytical ultracentrifugation analysis of His6-albumin–CPSF6-358 from P1 (blue), P2 (black), and CPSF6-358 (red) at 1.0 mg/ml. The expected oligomeric state for each peak is indicated.
To evaluate whether the albumin tag affects the oligomeric state of CPSF6-358, we incubated the P1 and P2 samples with TEV protease to remove His6-albumin and purified untagged CPSF6-358 protein. Gel filtration analysis of the digested P1 protein revealed the continued presence of tagged protein (data not shown), likely because access to the TEV protease cleavage site was limited by the higher-order assembly of the P1 protein. Furthermore, partially cleaved CPSF6-358 in P1 remained bound in large oligomers and could not be separated from uncleaved protein (data not shown). In contrast, removal of the His6-albumin fusion tag was very efficient for the P2 fraction (Fig. 2B), resulting in a broad peak of purified CPSF6-358 (Fig. 2B, peak I) comprising multiple oligomeric species; the morphology of the purified CPSF6-358 at the peak position is shown in Fig. 2B, inset. To determine the exact oligomeric species present in this sample, in addition to the undigested P1 and P2 samples, sedimentation velocity scans were recorded for a series of dilutions starting at 1.0 mg/ml (Table 1). Before incubation with TEV protease, P1 contained small amounts of monomers and dimers; however, the vast majority of the sample was soluble large oligomers (Fig. 2C, blue). P2, on the other hand, primarily contained a mixture of monomers and dimers, with a small fraction of large oligomers (Fig. 2C, black). The proportion of dimers in P2 increased with increasing sample concentrations (data not shown), suggesting an equilibrium between the two species. After tag removal from the P2 sample, CPSF6-358 appeared to be mostly dimers, with a small amount of large oligomers also present (Fig. 2C, red). Together, these data indicate that in vitro-purified CPSF6-358 forms dimers and higher-order oligomers.
TABLE 1.
Estimated molecular masses of the CPSF6-358 proteins from the c(s) analysisa
| Protein | Monomer mass (kDa)b | Concn (mg/ml) | Major species |
f/f0d | |||
|---|---|---|---|---|---|---|---|
| Peak 1 |
Peak 2 |
||||||
| Mass (kDa) | Sedimentation coefficient (S) | Mass (kDa) | Sedimentation coefficient (S) | ||||
| H6–albumin–CPSF6-358 P1 | 110.0 | 1.0 | 56.4 | 5.4 | 117.9 | 8.8 | 0.98c |
| 0.5 | 98.4 | 5.0 | 217.3 | 8.4 | 1.55 | ||
| 0.25 | 125.1 | 5.4 | 275.2 | 9.2 | 1.66 | ||
| H6–albumin–CPSF6-358 P2 | 110.0 | 1.0 | 88.1 | 4.9 | 141.4 | 6.7 | 1.46 |
| 0.5 | 88.7 | 5.0 | 152.3 | 7.1 | 1.45 | ||
| 0.25 | 102.7 | 5.1 | 216.8 | 8.3 | 1.56 | ||
| CPSF6-358 | 37.8 | 1.0 | 89.0 | 4.3 | 1.68 | ||
| 0.5 | 89.6 | 4.3 | 1.68 | ||||
| 0.25 | 65.2 | 4.2 | 1.40 | ||||
For each sample concentration, the signal-weighted sedimentation coefficient and the estimated molecular mass of each species are shown, together with the best-fit frictional ratio for the distribution.
Samples also include an amount of aggregated material.
Frictional ratios of <1.0 are not possible and do not actually have any physical meaning. This value reflects the software's attempt to fit such a diverse range of species.
Frictional ratio parameter.
CPSF6-358 binds and disrupts WT CA tubular assemblies.
A number of studies have shown that CPSF6-358 is a potent inhibitor of HIV-1 infection and prevents HIV-1 nuclear entry and integration by targeting the viral capsid (18, 19, 23, 33). To understand whether CPSF6-358 alters the capsid upon binding, we incubated preassembled WT HIV-1 CA tubes, generated in vitro from purified CA protein (3), with either His6-albumin–CPSF6-358 (P1 or P2) or untagged CPSF6-358 from P2. In all cases, cosedimentation of the CPSF6-358 proteins with the CA tubes was observed (Fig. 3A, B, and M, left). In contrast, binding of CPSF6-358 proteins to tubes assembled with N74D HIV-1 CA, a mutation previously shown to abolish CPSF6-358 binding and restriction (19), was negligible (Fig. 3A and B). Binding of assembled CA tubes by untagged CPSF6-358 was more efficient than that by the tagged protein, as almost all of the untagged CPSF6-358 came down with CA tubes, whereas only about 50% of the tagged protein cosedimented with CA (Fig. 3A and B). Quantitative analysis of CPSF6-358 binding was performed by measuring the molar ratio of CA-bound CPSF6-358 over a range of CPSF6-358 concentrations. Dose-dependent binding was observed for both tagged CPSF6-358 (P1 and P2) and untagged CPSF6-358 (Fig. 3M, left).
FIG 3.

CPSF6-358 binds and disrupts WT CA tubular assemblies. (A) SDS-PAGE of WT and N74D CA assemblies, following incubation with His6-albumin–CPSF6-358, from P1 or P2 and centrifugation. The gel was Coomassie blue stained, with supernatant (s) and pellet (p) samples indicated. (B) SDS-PAGE of WT and N74D CA assemblies following incubation with untagged CPSF6-358 and centrifugation. (C to H) Representative negative-stain EM micrographs of the samples in panel A. (C to E) WT CA tubular assemblies alone (C) or with 30 μM P1 (D) or 30 μM P2 (E) His6-albumin–CPSF6-358. (F to H) CA N74D alone (F) or with 30 μM P1 (G) or 30 μM P2 (H) His6-albumin–CPSF6-358. The arrows indicate the capsid fragments. (I to L) Representative negative-stain EM micrographs of the samples in panel B. Shown are WT CA tubular assemblies alone (I) or with 30 μM CPSF6-358 (J) and CA N74D tubular assemblies alone (K) or with 30 μM CPSF6-358 (L). Scale bars, 100 nm. (M) Dose-dependent effect of CPSF6-358 on CA tubes. Shown is binding of P1 (blue), P2 (black), and CPSF6-358 (red) to assembled WT CA tubes (left). The effects of P1 (blue), P2 (black), and CPSF6-358 (red) binding on the average length of tubes (middle) and on the number of remaining initial tubular assemblies (right) were measured. The error bars indicate the standard deviation of the values.
To investigate the effect of CPSF6-358 binding on CA tubular assemblies, we examined the samples from our CPSF6-358/CA binding assays using transmission EM (TEM) (Fig. 3C to L). Remarkably, TEM micrographs showed a drastic structural disruption of WT CA tubes upon incubation with either His6-albumin–CPSF6-358 (P1 or P2) (Fig. 3D and E) or untagged CPSF6-358 (Fig. 3J), whereas N74D CA tubes remained intact (Fig. 3G to H and L). Still, significant differences in the morphology of the breakdown products was evident in the presence of P1 protein compared to P2 protein. Binding of the large P1 oligomers resulted in dissolution of tubes and an appearance of distinct curved capsid remnants (Fig. 3D, arrows), whereas P2 protein and untagged CPSF6-358 derived from P2, consisting of mostly dimers, broke the tubes into short segments and a mixture of cones and spheres, respectively (Fig. 3E and J). In all cases, CPSF6-358 densities around the CA tube fragments apparently remained bound to the surfaces of CA tube fragments (Fig. 3D, E, and J). Intriguingly, the amount of pelletable capsid did not change upon tube breakdown (Fig. 3A and B), suggesting that the predominant effect of CPSF6-358 is fragmentation without dissociation into soluble proteins. Quantitative analysis of CA tube fragmentation by CPSF6-358, by measuring tube numbers and lengths at different concentrations of CPSF6-358, revealed a dose-dependent reduction in both the number of long, unfragmented tubular assemblies and the lengths of fragmentation products as the CPSF6-358 concentration increased (Fig. 3M, middle and right).
We also examined whether CPSF6-358 binds individual cross-linked CA hexamers carrying 4 mutations on the CA protein (A14C/E45C/W184A/M185A), using size exclusion chromatography. The elution profiles of mixtures of CA hexamers and His6-albumin–CPSF6-358 or CPSF6-358 coincided precisely with their individual profiles (Fig. 4A to C) without any shift in the peak positions, as would be expected in the case of complex formation. Consistent with this observation, SDS-PAGE gels did not show coelution of CA hexamers and CPSF6-358, with or without the albumin tag (Fig. 4D). These results demonstrate that CPSF6-358 has a much lower capacity to bind to CA hexamers than to assembled CA tubes, implying that higher-order assemblies of CA are required for efficient CPSF6-358 interaction.
FIG 4.

Binding of CPSF6-358 with 14C/45C/W184A/M185A hexamer. (A to C) Gel filtration (Superdex 200) profile of CA hexamer with His6-albumin–CPSF6-358 from P1 (A) or P2 (B) or with untagged CPSF6-358 (C). Red, CA hexamer alone; blue, CPSF6-358 proteins alone; black, mixtures. (D) SDS-PAGE analysis of fractions in panels A to C.
HIV-1 infection induces higher-order complexes of CPSF6-358 in cells.
Our in vitro data suggest that multiple copies of CPSF6-358 bind HIV-1 capsid and that binding induces capsid disassembly. To visualize the interaction of CPSF6-358 with HIV-1 in vivo, we engineered HeLa cells to stably express fluorescently tagged CPSF6-358 and then infected the cells with HIV-1. Since direct labeling of CA is not ideal, as any tags on the protein may impact capsid stability and/or interactions, we labeled the contents of the capsid by generating a fluorescently tagged integrase (IN). Specifically, the fluorescent protein mRuby3 (34) was introduced between Vpr and IN in a previously described fusion construct (35) and was expressed during HIV-1 production in trans (Fig. 5A). The labeled IN was determined to be functional, as it was able to rescue the infectivity of HIV-1 containing an IN active-site mutation, D116N (36) (Fig. 5B). mRuby3-IN allowed visualization of virus particles prior to (Fig. 5C) and after (Fig. 5D) infection of HeLa cells and was colocalized with HIV-1 RNA after the infection of cells (Fig. 5D), suggesting that it is present within capsids and reverse transcription-preintegration complexes. For CPSF6-358, the fluorescent protein enhanced green fluorescent protein (eGFP) or iRFP670 was fused to the C terminus and stably expressed in HeLa cells. Similar to what has previously been shown (18), expression of CPSF6-358–eGFP or CPSF6-358–iRFP670 potently restricted infection by WT HIV-1, but not N74D HIV-1 (data not shown).
FIG 5.

Generation of fluorescently labeled HIV-1. (A) Schematic design of the Vpr-mRuby3-IN construct used to label HIV-1 particles in trans. (B) Specific infectivity (luciferase per nanogram of p24) was measured for D116N HIV-1 complemented in trans with no plasmid, Vpr-RT, Vpr-IN, or Vpr-mRuby3-IN. (C) TIRF image of WT HIV-1 labeled with Vpr-mRuby3-IN. (D) Confocal image of HeLa cells synchronously infected with WT HIV-1 labeled with Vpr-mRuby3-IN and 5-ethynl uridine and fixed 30 min postinfection.
In uninfected HeLa cells expressing CPSF6-358–eGFP, CPSF6-358–eGFP expression was relatively uniform throughout the cell cytoplasm and nucleus (Fig. 6A). In contrast, upon WT HIV-1 infection of HeLa cells expressing CPSF6-358–eGFP, distinct green puncta were observed in the cytoplasm (Fig. 6A). However, formation of CPSF6-358–eGFP puncta was not observed after infection with N74D HIV-1 (Fig. 6A). To determine if HIV-1 capsid binding is necessary for formation of the puncta, infections were performed with WT HIV-1 in the presence of PF-74 or with another HIV-1 CA mutant that does not bind to CPSF6, A77V (22). Like N74D HIV-1, formation of CPSF6-358–eGFP puncta was not observed despite the presence of mRuby-IN-containing complexes within the cells (Fig. 6B). Similar results were observed in cells expressing CPSF6-358–iRFP670 or HIV-1 labeled with Vpr-red fluorescent protein (RFP) tag-IN (data not shown). These results suggest that an increase in the local concentration of CPSF6-358 upon HIV-1 capsid binding likely leads to formation of higher-order complexes of CPSF6-358.
FIG 6.

WT HIV-1 infection induces formation of CPSF6-358 higher-order complexes in HeLa cells. (A) Confocal images of HeLa cells stably expressing CPSF6-358–eGFP before or 30 min after infection with WT HIV-1 or N74D HIV-1. (B) CPSF6-358–eGFP puncta and mRuby-IN particles were quantified per cell (n ≥ 25 z-stacks) at 30 min postinfection with WT HIV-1 in the presence or absence of 10 μM PF-74, N74D HIV-1, or A77V HIV-1. The asterisks denote comparisons with P values of <0.05. (C) HeLa cells stably expressing CPSF6-358–eGFP were treated (open symbols) or not (solid symbols) with 2 μM CsA and synchronously infected with WT HIV-1 or N74D HIV-1. The number of CPSF6-358–eGFP puncta per field of view was determined. The error bars represent standard error of the mean (SEM). *, P < 0.05; **, P < 0.005; ***, P < 0.001.
CsA treatment leads to faster formation of CPSF6-358 higher-order complexes.
CPSF6-358–eGFP puncta did not always form simultaneously after synchronized infection with WT HIV-1, leading us to hypothesize that another host factor could prevent immediate access of CPSF6-358 to intracellular capsid. As CypA is known to bind to HIV-1 capsid (37), disruption of CypA-capsid interaction was performed by treatment of cells with cyclosporine (CsA). We observed that treatment of cells with CsA resulted in greater numbers of CPSF6-358–eGFP puncta than in untreated cells (Fig. 6C). Quantification of mRuby3-IN particles and CPSF6-358–eGFP puncta showed no difference in the number of IN-containing complexes in cells under each treatment condition (data not shown). However, the number of CPSF6-358–eGFP higher-order complexes was significantly higher 30 min postinfection in the presence of CsA (Fig. 6C). Live-cell imaging similarly showed that CPSF6-358–eGFP puncta formed more rapidly and increased in number in CsA-treated cells after infection with WT HIV-1 (data not shown), but not after N74D HIV-1 infection. These results suggest that CypA may shield capsid from access and binding by CPSF6-358.
CPSF6-358 higher-order complexes associate with HIV-1 particles and lead to permeabilization of HIV-1 capsid.
To determine whether CPSF6-358–eGFP higher-order complexes associate with HIV-1 complexes in cells, high-speed live-cell imaging was performed to visualize early formation of CPSF6-358–eGFP complexes after synchronized infection with WT HIV-1. eGFP puncta formed as early as 10 min postinfection with WT HIV-1 in HeLa cells stably expressing CPSF6-358–eGFP, increasing in number up to 30 min postinfection (Fig. 7A; see Movie S1 in the supplemental material). Formation of the CPSF6-358–eGFP complexes often occurred at the location of mRuby3-IN signal (Fig. 7A and B), suggesting that they may form around capsids. However, over time, the mRuby3 signal often separated from all or some of the CPSF6-358–eGFP signal (Fig. 7; see Movie S1 in the supplemental material). CPSF6-358–eGFP puncta were not always colocalized with mRuby3-IN signal, possibly due to capsid uncoating prior to the start of imaging or because mRuby3-IN packaging did not occur in all virions during virus production.
FIG 7.
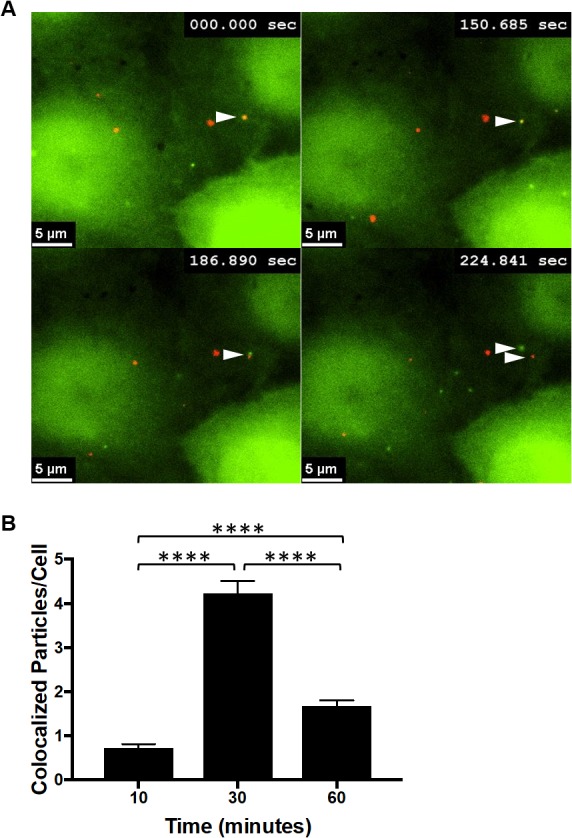
Dynamic interactions occur between CPSF6-358 and WT HIV-1 particles. (A) Images were obtained by live-cell frustrated TIRF imaging 10 min after synchronized infection with WT HIV-1 of HeLa cells stably expressing CPSF6-358–eGFP. The arrowheads indicate initial colocalization of CPSF6-358–eGFP (green) with mRuby3-IN (red) and then separation approximately 3 min later. (B) eGFP and mRuby3 colocalized particles were quantified at 10, 30, and 60 min postinfection. The error bars represent SEM. ****, P < 0.0001.
As mRuby3-IN was shown to generally separate from CPSF6-358–eGFP between 30 and 60 min postinfection and CPSF6-358 can disrupt CA tubes in vitro, we hypothesized that CPSF6-358–eGFP may promote more rapid capsid uncoating. To test this hypothesis, a capsid permeabilization assay was performed on HeLa cells with or without CPSF6-358–eGFP expression after WT HIV-1 infection. Previously, labeling of HIV-1 RNA with 5-ethynyl uridine (EU) during virus production could be detected by staining with an EU-specific dye only when the viral core was permeabilized, approximately 30 to 45 min postinfection of HeLa cells, consistent with capsid uncoating using other assays (26). In this study, peak WT HIV-1 RNA staining in HeLa cells occurred at 30 min postinfection, whereas peak staining in cells expressing CPSF6-358–eGFP occurred at 20 min postinfection (Fig. 8A). Viral RNA staining of N74D HIV-1 did not differ in HeLa cells with or without CPSF6-358–eGFP (Fig. 8B). Collectively, these results suggest that CPSF6-358 oligomerizes around WT HIV-1 capsid in cells and may lead to premature capsid opening.
FIG 8.
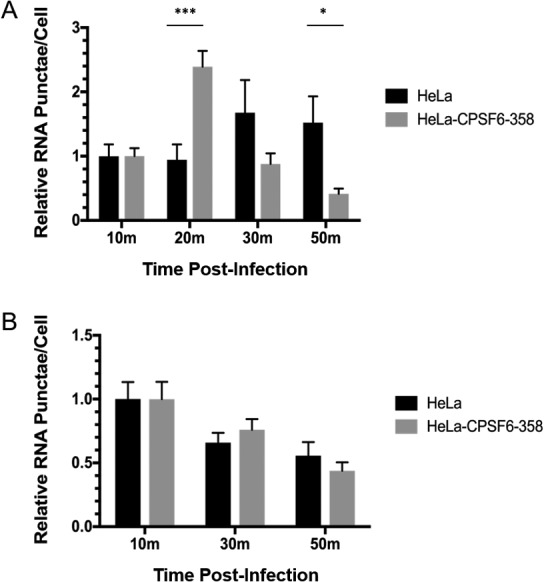
Capsid permeabilization of WT HIV-1 occurs more quickly in HeLa cells expressing CPSF6-358–eGFP. HeLa cells and HeLa cells expressing CPSF6-358–eGFP were infected with WT HIV-1 (A) or N74D HIV-1 (B) and stained for viral RNA at different times. The error bars represent SEM of two (WT) or one (N74D) independent experiment. *, P < 0.05; ***, P < 0.001.
DISCUSSION
CPSF6-358 restricts HIV-1 infection through interaction with capsid and affects nuclear entry and integration of viral DNA (19). Here, we have shown the first direct evidence of the oligomeric nature of CPSF6-358. Binding of CPSF6-358 to WT CA tubes results in disruption of the tube assemblies into fragments, with the higher oligomerization form of CPSF6-358 being more effective, suggesting that it might enhance the capsid pattern-sensing ability and facilitate early capsid dissociation. Another C-terminally truncated CPSF6, CPSF6-375, was reported to promote rapid capsid disassembly and inhibition of viral cDNA synthesis (38). Previously, the N74D CA mutant was shown to escape CPSF6-358 restriction in cells and to bind less efficiently to CA tubes (18, 39). Our in vitro and in vivo experiments both showed that N74D capsid assemblies fail to interact with CPSF6-358, presumably evading the nuclear import restriction (18).
CPSF6 residues 314 to 322, which constitute the minimal binding epitope of CPSF6 for the CA protein, have been reported to have a low binding affinity (100 μM) for soluble, disulfide-stabilized CA hexamer (20). Consistent with these results, our data indicate that the CA hexamer interacts very weakly with CPSF6-358. In contrast, preassembled CA tubes have a stronger interaction with CPSF6-358, as we could detect substantial binding to CA tubular assemblies in pelleting assays. Since both CA assembly and CPSF6-358 oligomerization are required for efficient interaction, CPSF6-358 may have an intrinsic capsid lattice-sensing ability, similar to other capsid-interacting restriction factors, such as TRIM5α (1, 2, 40, 41). CPSF6-358 oligomers achieve both recognition and disruption of viral capsid assemblies in vitro. Apparent higher-order complexes were also observed in the cytoplasm of cells expressing CPSF6-358–eGFP after WT HIV-1 infection, but not after N74D HIV-1 infection or in the presence of the competitive inhibitor PF-74. The formation of higher-order CPSF6-358 oligomers correlated with HIV-1 capsid disruption, which may contribute to the antiviral function of the restriction factor.
Uncoating of the HIV-1 capsid is highly regulated and plays a critical role during early postentry stages of infection (33). However, the exact timing of uncoating, or disassembly of the capsid lattice and its physical separation from the genome, is still poorly defined but likely occurs in a multistep fashion as it travels toward the cell nucleus (26). Our results show that WT HIV-1 capsid permeabilization occurs more rapidly in the presence of fluorescently labeled CPSF6-358, suggesting that CPSF6-358–eGFP may promote more rapid capsid uncoating. Capsid uncoating has been shown to be linked to viral DNA synthesis (42, 43). However, the kinetics and completion of reverse transcription are not affected by expression of CPSF6-358 in cells (18). Although CPSF6-358 leads to disruption of HIV-1 capsid, it is possible that it can also protect viral nucleic acids from the detrimental effects of premature uncoating. The host cell factor CypA also interacts directly with HIV-1 capsid and modulates capsid uncoating and viral infectivity in certain cell types (37, 44). Treatment of HeLa cells with CsA leads to more rapid formation of higher-order CPSF6-358–eGFP complexes after infection with WT HIV-1. Previously, we showed that CsA blocks the infectivity of CPSF6-358-resistant N74D HIV-1 in HeLa cells prior to or at reverse transcription (23). Recently, CsA has been shown to prevent higher-order complexes of full-length CPSF6 in the nuclei of HIV-infected cells and to prevent capsid use of nucleoporins, suggesting that CypA shields the N74 pocket from interaction with multiple host factors, including CPSF6 (V. KewalRamani, personal communication). These combined results suggest that CypA influences capsid uncoating in the context of CPSF6-358, perhaps by shielding WT capsid from access and binding to CPSF6-358.
Overall, we demonstrated that the host protein CPSF6-358 can form oligomers both in vivo and in vitro and can interact with and disrupt HIV-1 capsid tubes in vitro and HIV-1 capsids in vivo. Full-length endogenous CPSF6 is largely expressed in the nucleus and likely interacts with capsid after most of it has been dissociated. When enriched in the cytoplasm, such as during protein synthesis, CPSF6 could form dimers and oligomers, targeting HIV-1 capsid for dissociation either as an antiviral effect or as part of the normal capsid-uncoating process. CypA binding to capsid may be a mechanism to prevent premature binding of capsid to CPSF6 in the cytoplasm, which could lead to innate immune responses targeting cytoplasmic viral DNA. Better understanding of the complex and intertwined processes of HIV-1 capsid uncoating, reverse transcription, and nuclear entry will aid in the development of novel therapeutic targets to inhibit infection.
MATERIALS AND METHODS
Plasmid and protein expression.
The cDNA encoding CPSF6 1 to 358 was amplified and cloned into pcDNA3.1(+) mammalian expression vectors (Thermo Fisher), which were modified to encode a 6×His tag at the N terminus, followed by albumin as a secretion signal protein, using EcoRI and XhoI sites [modified pcDNA3.1(+) with a His6-albumin tag was a gift from Troy Krzysiak, University of Pittsburgh] and with a TEV cleavage site immediately following the albumin sequence.
The protein was expressed in a suspension-adapted HEK293 cell line (Expi 293F, a human embryonic kidney cell line; Thermo Fisher). Cells growing in flasks were transfected at a density of 2.5 × 106/ml using ExpiFectamine 293 (Thermo Fisher) according to the manufacturer's instructions. Following transfection, the cells were grown at 37°C with shaking at 125 rpm in 8% CO2 and 80% humidity for 2 days. The conditioned media, containing secreted protein, were harvested on day 2 by centrifugation at 5,000 rpm for 30 min. The clarified media were pooled and used for purification.
Protein purification.
Purification of His-tagged proteins from the conditioned media was performed by adding 5 mM CaCl2 and 1 mM NiCl2 to the media, followed by centrifugation at 12,000 × g for 30 min. The supernatant was collected and applied to Ni Sepharose high-performance resin (GE Healthcare) that had been equilibrated with 0.1 M sodium phosphate buffer, pH 7.5, 250 mM NaCl, 5% glycerol, and 1 mM dithiothreitol (DTT). The sample was incubated for 2 h with the resin at 4°C prior to a 20 mM imidazole wash step and elution with 500 mM imidazole. The elution was then applied to a Hi-Load Superdex 200 26/60 column (GE Healthcare) in a buffer containing 0.1 M sodium phosphate buffer, pH 7.9, 150 mM NaCl, 5% glycerol, and 1 mM DTT. Fractions containing the target protein were collected and concentrated to around 30 mg/ml using Amicon concentrators (Millipore, Billerica, MA, USA), flash-frozen with liquid N2, and stored at −80°C.
SDS-PAGE and Western blot analysis.
Equal volumes of each pooled conditioned medium sample and each fraction from the column were mixed with 4× NuPAGE LDS sample buffer (Thermo Fisher) supplemented with 10 mM DTT and loaded onto a 4 to 12% Bis-Tris NuPAGE gel (Thermo Fisher), alongside a protein molecular mass marker (Bluestain protein ladder; Gold Biotechnology, USA). The gels were run at 100 V for 15 min and then 150 V for 40 min in NuPAGE MES SDS running buffer (Thermo Fisher), and the proteins were subsequently transferred onto polyvinylidene difluoride (PVDF) membranes using iBlot transfer stacks (Thermo Fisher). The membranes were blocked at ambient temperature for 1 h in bovine serum albumin (BSA) blocking buffers, followed by overnight incubation with mouse anti-His antibody (H1029; Sigma) or rabbit anti-CPSF6 antibody (EPR12898; Abcam) at 4°C and then for an additional hour with monoclonal anti-rabbit or anti-mouse immunoglobulin-alkaline phosphatase antibody at ambient temperature. Between antibody incubations, the membranes were washed three times with Tris-buffered saline (TBS) containing 0.1% Tween 20, and finally, the membranes were developed with BCIP (5-bromo-4-chloro-3-indolylphosphate)-Nitro Blue Tetrazolium (NBT) color development substrate (Promega, USA) to enable visualization of protein bands. Each experiment was carried out at least three times.
SEC-MALS and analytical ultracentrifugation.
The molecular masses of CPSF6-358 proteins were determined using an analytical Superdex 200 column with in-line multiangle light scattering (Heleos; Wyatt Technology), variable-wavelength UV (1100 series; Agilent Technology), and refractive index detectors (Optilab rEX; Wyatt Technology). Typically, 100 μl of the protein solution at 3.5 mg/ml was injected into the column equilibrated with 0.1 M sodium phosphate buffer, pH 7.9, containing 150 mM NaCl, 5% glycerol, and 1 mM DTT at a flow rate of 0.5 ml/min at room temperature. The ASTRA program (version 6.1; Wyatt Technology) was used for light scattering data analysis.
For characterization of the protein samples, analytical ultracentrifugation (AUC) sedimentation velocity scans were recorded for a 2-fold dilution series of each sample, starting from 1.0 mg/ml. All the AUC experiments were performed at 40,000 rpm, using a Beckman XL-I analytical ultracentrifuge with an An-50Ti rotor at 20°C. Data were recorded using the absorbance (at 280 nm) optical detection system. The density and viscosity of the buffer were measured experimentally using an Anton Paar DMA 5000M densitometer equipped with a Lovis 200ME viscometer module. The partial specific volumes of the protein constructs were calculated from the amino acid sequences using SEDFIT (45). The data were processed using SEDFIT, fitting to the sedimentation coefficient distribution [c(s)] model.
Capsid-binding assay.
Tubular assemblies of HIV-1 CA were prepared at 80 μM (2 mg/ml) in 1 M NaCl and 50 mM Tris-HCl (pH 8.0) buffer at 37°C for 1 h. For the binding assays, the binding buffer was the same as the stock buffer for CPSF6-358 proteins, which contained 0.1 M sodium phosphate buffer, pH 7.9, 150 mM NaCl, 5% glycerol, and 1 mM DTT. Briefly, different concentrations of CPSF6-358 were added to preassembled CA tubes, and for the control samples, the same amount of the CPSF6-358 protein stock buffer was added to make the final CA concentrations all the same. The CA concentration was slightly reduced to 64 μM in the binding assays. The reaction mixtures were incubated on a rocking platform at room temperature for 1 h with gentle mixing at 10-min intervals. At the end of incubation, 5-μl samples were withdrawn from the reaction mixtures and immediately used for EM analysis. The remaining samples were pelleted at 20,000 × g with an Eppendorf 5417R centrifuge for 30 min, and supernatants and pellets (resuspended in the same volume) were mixed with 4× LDS loading buffer for gel analysis. Supernatant and pellet samples, without boiling, were loaded on SDS-4 to 12% PAGE gels and stained with Coomassie blue. Each experiment was performed at least three times.
Soluble CA hexamers (A14C/E45C/W184A/M185A; CA concentration, 80 μM), His6-albumin–CPSF6-358 proteins (P1 or P2; 30 μM) or CPSF6-358 (120 μM), and their mixtures at the same protein concentrations were loaded onto a Superdex 200 10/300GL column (GE Healthcare) separately. The elution profiles were compared to monitor whether the complex was formed.
To determine the binding ratio of CPSF6-358:CA, the SDS-PAGE gels were scanned using an Epson 4990 scanner. The integrated intensities of CA and CPSF6-358 protein bands were measured using the Image J 1.40 program (NIH). The molar ratios were calculated according to the formula (CPSF6-358 intensity/CPSF6-358 molecular mass)/(CA intensity/CA molecular mass) and calibrated using the input ratios as standards.
TEM analysis.
The morphologies of different variants of CA assemblies and CA–CPSF6-358 complexes were characterized by TEM. Samples were stained with fresh 2% uranyl formate, deposited onto 400-mesh carbon-coated copper grids, and dried for 30 min. TEM images were acquired on a Tecnai T12 transmission electron microscope at 120 kV.
Virus production.
HEK293T cells (a human embryonic kidney cell line; ATCC) were routinely tested for mycoplasma (MycoAlert detection kit;, Lonza) and transfected with (i) WT or mutant pNLdE-luc (18); (ii) pVpr-mRuby3-IN, pVpr-IN (35), or pVpr-RT (35); and (iii) pCMV-VSV-G plasmids at a 5:5:1 ratio using Lipofectamine 2000 (Thermo Fisher Scientific). After 48 h, the cell supernatant was collected, filtered through a 30-mm diameter sterile syringe filter with a 0.45-µm-pore-size hydrophilic polyethersulfone (PES) membrane (Millipore), and stored at −80°C. For the capsid permeabilization assay, mRuby3-IN-labeled HIV-1 was produced in the presence of EU, as previously described (26).
Luciferase assay.
HeLa cells cells (a human cervical adenocarcinoma cell line; ATCC) were routinely tested for mycoplasma (MycoAlert detection kit;, Lonza) and plated in 24-well plates in Dulbecco's modified Eagle medium (DMEM) (Thermo Fisher Scientific) at 37°C and 5% CO2. The next day, HIV-1 was added to the wells in duplicate. Forty-eight hours postinfection, the cells were lysed in Glo Lysis buffer (Promega) and added to luciferase assay substrate (Promega). Luminescence was measured using a 1450 MicroBeta TriLux machine (PerkinElmer).
Synchronized infection and fixation of cells.
HeLa cells stably expressing CPSF6-358–eGFP (deposited in Addgene; no. 110693) or CPSF6-358-iRFP670 (deposited in Addgene; no. 110694) were seeded in MatTek dishes overnight in Fluorobrite medium (Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS) (Atlanta Biologicals) and penicillin, streptomycin, and glutamine (Thermo Fisher Scientific) at 37°C and 5% CO2. Prior to imaging, the cells were incubated with DMEM with or without 2 μM CsA (Sigma-Aldrich) or 10 μM PF-74 (Sigma) for 1 h. Afterward, the cells were chilled at 4°C for 10 min. Virus was added to the microwell above the coverslip in the center of the MatTek dish and incubated at 37°C for 10 min. Before imaging, the cells were washed three times with medium to remove unbound virus. For fixation, the cells were washed with phosphate-buffered saline (PBS) and fixed with fresh 2% paraformaldehyde for 15 min. The cells were washed with PBS and stained with Hoechst 33342 (Sigma) for 5 min. After washing off the Hoechst stain with PBS, Gelvatol mounting medium (Sigma-Aldrich) was added to the dish, and a coverslip was added on top of the cells.
Capsid permeabilization assay.
HeLa cells and HeLa cells stably expressing CPSF6-358–eGFP were synchronously infected with EU-labeled HIV-1 (20 ng of p24, as determined by enzyme-linked immunosorbent assay [ELISA]; XpressBio). The cells were fixed 10 to 50 min postinfection, permeabilized, stained for viral RNA (EU-AF647 Click-iT; Invitrogen) and cell nuclei (Hoechst 33342), and mounted with coverslips.
Confocal and live-cell imaging.
Fixed-cell images were collected with a Nikon A1 scanning confocal microscope. Z-stack images were collected at 0.5-μm steps up to 10 μm in 3 colors (408 nm, 488 nm, and 561 nm). For live-cell experiments, MatTek dishes containing cells were placed on a STX heat stage (Tokai Hit, Inc.) to maintain 37°C and 5% CO2. A Nikon Ti live-cell microscope was used to collect frustrated total internal-reflection fluorescence (TIRF) images using a Prime 95B sCMOS camera (Photometrics, Inc.) with an LBX-4C illumination module (Oxxius, Inc.). Images were collected at up to 3 frames per second with two colors (488 nm and 561 nm).
Quantification of puncta and virus particles.
Nikon Elements 5.0 was used to quantify the CPSF6-358 puncta during HIV-1 infection. Ten or more z-stacks (0.5-μm spacing) of confocal images were collected for each time point. In brief, a 3-dimensional (3D) nucleus mask was made based on Hoechst staining. Both CPSF6-358 puncta and HIV-1 particles were detected using the 3D spot detection function. The number of CPSF6-358 puncta was determined by subtraction of the initial CPSF6-358 puncta from the signals within the nucleus mask. RNA puncta were enumerated with Imaris software. Grubbs' extreme Studentized deviate test was used to identify and exclude statistical outliers.
Statistical analysis.
Statistical significance was determined by two-sided unpaired Student's t test using Prism (GraphPad). P values of <0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Troy Krzysiak for the pcDNA3.1(+) plasmid encoding the albumin fusion tag, John Kappes for the Vpr-IN construct, Michael Lin for the mRuby3 construct, Frances J. D. Alvarez for technical assistance, and Teresa Brosenitsch for reading the manuscript.
This work was supported by National Institutes of Health P50 grant GM082251 (P.Z., Z.A., and S.C.W.), National Institutes of Health T32 training grant AI065380 (D.K.F.), and UK Wellcome Trust Investigator Award 206422/Z/17/Z (P.Z.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00368-18.
REFERENCES
- 1.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 2.Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365. doi: 10.1038/nature09976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu PT, Schenk M, Walker VP, Dempsey PW, Kanchanapoomi M, Wheelwright M, Vazirnia A, Zhang X, Steinmeyer A, Zugel U, Hollis BW, Cheng G, Modlin RL. 2009. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One 4:e5810. doi: 10.1371/journal.pone.0005810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu C, Perilla JR, Ning J, Lu M, Hou G, Ramalho R, Himes BA, Zhao G, Bedwell GJ, Byeon I-J, Ahn J, Gronenborn AM, Prevelige PE, Rousso I, Aiken C, Polenova T, Schulten K, Zhang P. 2016. Cyclophilin A stabilizes the HIV-1 capsid through a novel non-canonical binding site. Nat Commun 7:10714. doi: 10.1038/ncomms10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Wu W, Liu F, Yao M. 2009. Boundedness and convergence of online gradient method with penalty for feedforward neural networks. IEEE transactions on neural networks 20:1050–1054. doi: 10.1109/TNN.2009.2020848. [DOI] [PubMed] [Google Scholar]
- 7.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Du B, Chen X, Ma H. 2009. Convergence of dissipation and impedance analysis of quartz crystal microbalance studies. Anal Chem 81:642–648. doi: 10.1021/ac8019762. [DOI] [PubMed] [Google Scholar]
- 9.Li Y-L, Chandrasekaran V, Carter SD, Woodward CL, Christensen DE, Dryden KA, Pornillos O, Yeager M, Ganser-Pornillos BK, Jensen GJ, Sundquist WI. 2016. Primate TRIM5 proteins form hexagonal nets on HIV-1 capsids. Elife 5:e16269. doi: 10.7554/eLife.16269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kong J, Ma M, He S, Qin X. 2016. Mx oligomer: a novel capsid pattern sensor? Future Microbiol 11:1047–1055. doi: 10.2217/fmb-2016-0004. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez FJD, He S, Perilla JR, Jang S, Schulten K, Engelman AN, Scheres SHW, Zhang P. 2017. CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci Adv 3:e1701264. doi: 10.1126/sciadv.1701264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sowd GA, Serrao E, Wang H, Wang W, Fadel HJ, Poeschla EM, Engelman AN. 2016. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc Natl Acad Sci U S A 113:E1054–E1063. doi: 10.1073/pnas.1524213113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dettwiler S, Aringhieri C, Cardinale S, Keller W, Barabino SM. 2004. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J Biol Chem 279:35788–35797. doi: 10.1074/jbc.M403927200. [DOI] [PubMed] [Google Scholar]
- 15.Price AJ, Fletcher AJ, Schaller T, Elliott T, Lee K, KewalRamani VN, Chin JW, Towers GJ, James LC. 2012. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog 8:e1002896. doi: 10.1371/journal.ppat.1002896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruegsegger U, Blank D, Keller W. 1998. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol Cell 1:243–253. doi: 10.1016/S1097-2765(00)80025-8. [DOI] [PubMed] [Google Scholar]
- 17.Ruepp MD, Aringhieri C, Vivarelli S, Cardinale S, Paro S, Schumperli D, Barabino SM. 2009. Mammalian pre-mRNA 3′ end processing factor CF I m 68 functions in mRNA export. Mol Biol Cell 20:5211–5223. doi: 10.1091/mbc.E09-05-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee K, Mulky A, Yuen W, Martin TD, Meyerson NR, Choi L, Yu H, Sawyer SL, KewalRamani VN. 2012. HIV-1 capsid-targeting domain of cleavage and polyadenylation specificity factor 6. J Virol 86:3851–3860. doi: 10.1128/JVI.06607-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN, Yeager M. 2014. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fricke T, Valle-Casuso JC, White TE, Brandariz-Nuñez A, Bosche WJ, Reszka N, Gorelick R, Diaz-Griffero F. 2013. The ability of TNPO3-depleted cells to inhibit HIV-1 infection requires CPSF6. Retrovirology 10:46. doi: 10.1186/1742-4690-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito A, Henning MS, Serrao E, Dubose BN, Teng S, Huang J, Li X, Saito N, Roy SP, Siddiqui MA, Ahn J, Tsuji M, Hatziioannou T, Engelman AN, Yamashita M. 2016. Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo. J Virol 90:6918–6935. doi: 10.1128/JVI.00019-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrose Z, Lee K, Ndjomou J, Xu H, Oztop I, Matous J, Takemura T, Unutmaz D, Engelman A, Hughes SH, KewalRamani VN. 2012. Human immunodeficiency virus type 1 capsid mutation N74D alters cyclophilin A dependence and impairs macrophage infection. J Virol 86:4708–4714. doi: 10.1128/JVI.05887-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jun S, Ke D, Debiec K, Zhao G, Meng X, Ambrose Z, Gibson GA, Watkins SC, Zhang P. 2011. Direct visualization of HIV-1 with correlative live-cell microscopy and cryo-electron tomography. Structure 19:1573–1581. doi: 10.1016/j.str.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonald D, Vodicka MA, Lucero G, Svitkina TM, Borisy GG, Emerman M, Hope TJ. 2002. Visualization of the intracellular behavior of HIV in living cells. J Cell Biol 159:441–452. doi: 10.1083/jcb.200203150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Franks T, Gibson G, Huber K, Rahm N, De Castillia CS, Luban J, Aiken C, Watkins S, Sluis-Cremer N, Ambrose Z. 2013. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology 10:70. doi: 10.1186/1742-4690-10-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Francis AC, Marin M, Shi J, Aiken C, Melikyan GB. 2016. Time-resolved imaging of single HIV-1 uncoating in vitro and in living cells. PLoS Pathog 12:e1005709. doi: 10.1371/journal.ppat.1005709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamede JI, Cianci GC, Anderson MR, Hope TJ. 2017. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc Natl Acad Sci U S A 114:E7169–E7178. doi: 10.1073/pnas.1706245114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albanese A, Arosio D, Terreni M, Cereseto A. 2008. HIV-1 pre-integration complexes selectively target decondensed chromatin in the nuclear periphery. PLoS One 3:e2413. doi: 10.1371/journal.pone.0002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burdick RC, Hu WS, Pathak VK. 2013. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc Natl Acad Sci U S A 110:E4780–4789. doi: 10.1073/pnas.1315996110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng K, Muranyi W, Glass B, Laketa V, Yant SR, Tsai L, Cihlar T, Muller B, Krausslich HG. 2014. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. Elife 3:e04114. doi: 10.7554/eLife.04114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Iaco A, Santoni F, Vannier A, Guipponi M, Antonarakis S, Luban J. 2013. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 10:20. doi: 10.1186/1742-4690-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ambrose Z, Aiken C. 2014. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology 454-455:371–379. doi: 10.1016/j.virol.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bajar BT, Wang ES, Lam AJ, Kim BB, Jacobs CL, Howe ES, Davidson MW, Lin MZ, Chu J. 2016. Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci Rep 6:20889. doi: 10.1038/srep20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X, Liu H, Xiao H, Conway JA, Hunter E, Kappes JC. 1997. Functional RT and IN incorporated into HIV-1 particles independently of the Gag/Pol precursor protein. EMBO J 16:5113–5122. doi: 10.1093/emboj/16.16.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol 69:2729–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP.. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 38.Hori T, Takeuchi H, Saito H, Sakuma R, Inagaki Y, Yamaoka S. 2013. A carboxy-terminally truncated human CPSF6 lacking residues encoded by exon 6 inhibits HIV-1 cDNA synthesis and promotes capsid disassembly. J Virol 87:7726–7736. doi: 10.1128/JVI.00124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hué S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc Natl Acad Sci U S A 103:5514–5519. doi: 10.1073/pnas.0509996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, Aiken C, Charlton LM, Gronenborn AM, Zhang P. 2011. Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog 7:e1002009. doi: 10.1371/journal.ppat.1002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forshey BM, von Schwedler U, Sundquist WI, Aiken C. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76:5667–5677. doi: 10.1128/JVI.76.11.5667-5677.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hulme AE, Perez O, Hope TJ. 2011. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc Natl Acad Sci U S A 108:9975–9980. doi: 10.1073/pnas.1014522108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 45.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.