

ABSTRACT
Hepatitis C virus (HCV) replication and assembly occur at the specialized site of endoplasmic reticulum (ER) membranes and lipid droplets (LDs), respectively. Recently, several host proteins have been shown to be involved in HCV replication and assembly. In the present study, we demonstrated the important relationship among osteopontin (OPN), the ER, and LDs. OPN is a secreted phosphoprotein, and overexpression of OPN in hepatocellular carcinoma (HCC) tissue can lead to invasion and metastasis. OPN expression is also enhanced in HCV-associated HCC. Our recent studies have demonstrated the induction, proteolytic cleavage, and secretion of OPN in response to HCV infection. We also defined the critical role of secreted OPN in human hepatoma cell migration and invasion through binding to receptors integrin αVβ3 and CD44. However, the role of HCV-induced OPN in the HCV life cycle has not been elucidated. In this study, we showed a significant reduction in HCV replication, assembly, and infectivity in HCV-infected cells transfected with small interfering RNA (siRNA) against OPN, αVβ3, and CD44. We also observed the association of endogenous OPN with HCV proteins (NS3, NS5A, NS4A/B, NS5B, and core). Confocal microscopy revealed the colocalization of OPN with HCV NS5A and core in the ER and LDs, indicating a possible role for OPN in HCV replication and assembly. Interestingly, the secreted OPN activated HCV replication, infectivity, and assembly through binding to αVβ3 and CD44. Collectively, these observations provide evidence that HCV-induced OPN is critical for HCV replication and assembly.
IMPORTANCE Recently, our studies uncovered the critical role of HCV-induced endogenous and secreted OPN in migration and invasion of hepatocytes. However, the role of OPN in the HCV life cycle has not been elucidated. In this study, we investigated the importance of OPN in HCV replication and assembly. We demonstrated that endogenous OPN associates with HCV NS3, NS5A, NS5B, and core proteins, which are in close proximity to the ER and LDs. Moreover, we showed that the interactions of secreted OPN with cell surface receptors αVβ3 and CD44 are critical for HCV replication and assembly. These observations provide evidence that HCV-induced endogenous and secreted OPN play pivotal roles in HCV replication and assembly in HCV-infected cells. Taken together, our findings clearly demonstrate that targeting OPN may provide opportunities for therapeutic intervention of HCV pathogenesis.
KEYWORDS: CD44, hepatitis C virus, lipid droplet, osteopontin, integrin receptors
INTRODUCTION
Hepatitis C virus (HCV) is a causative agent of chronic hepatitis which may eventually lead to hepatocellular carcinoma (HCC) (1). The HCV genome is a single plus-stranded RNA approximately 9.6 kb in length which encodes a precursor polyprotein that is cleaved by viral proteases and host cell signal peptidases into mature structural proteins (core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (2). HCV NS3, NS4A, NS4B, NS5A, and NS5B assemble and form a ribonucleoprotein complex (RNP) to facilitate HCV replication (2). HCV replication occurs in the membranous web which is a specialized membranous compartment derived from altered endoplasmic reticulum (ER) membranes (3).
NS3 protein contains N-terminal protease and C-terminal helicase activities, NS4A stimulates NS3 helicase and protease activities, and NS4B induces the membranous web in the ER (3, 4). NS5B is an RNA-dependent RNA polymerase responsible for the amplification of positive-strand HCV RNA (5). NS5A is required to mediate protein-protein interactions essential for HCV replication complex formation (6). NS5A binds to viral RNA as well as to numerous host proteins and colocalizes with HCV core in the close proximity to lipid droplets (LDs), the site of HCV virion assembly (7).
LDs are lipid storage organelles composed of mostly neutral lipids surrounded by a phospholipid monolayer. Recently, it has been demonstrated that LDs are also dynamic, motile organelles that interact with other intracellular membranes such as the ER and probably mitochondria (8). LDs are known to play an essential role in the assembly of HCV virus particles by interaction with HCV core and NS5A (7, 9). NS5A has been proposed to transport the newly synthesized HCV RNA from replication sites to LDs for encapsidation, thus fulfilling the critical function of NS5A in the HCV assembly process (7). The HCV core is mainly located on the surface of LDs and is believed to recruit HCV NS5A and HCV RNAs to LD-associated membrane, resulting in the production of infectious HCV particles (7, 9).
Recent studies demonstrate that several host proteins, such as TIP47, DGAT-1, TBC1D20, Rab1, Rab18, AP2M1, and phospholipase A2, interact with the HCV core/NS5A and LDs and facilitate HCV assembly (10–15). In addition, several host factors, such as annexin A2, early endosomal proteins (Rab5, EEA1, and phosphatidylinositol 4 [PI4] kinase III alpha), ER proteins (RAB1B and TBC1D20), the Golgi complex-associated protein RAB7L1, and vesicle-associated proteins A and B (VAP-A and -B), have been shown to be associated with the sites of HCV RNA replication (16–19). Previous studies have shown that colocalization of core to lipid droplets is tightly controlled by DGAT1 (11). Furthermore, it has been reported that DGAT1 serves as a cellular link between HCV core and NS5A proteins, directing both onto the surface of same subset of LDs (20). LD-associated host protein TIP47 has been shown to interact with NS5A, which is required for efficient HCV RNA replication (10). Recently, we have demonstrated that HCV-induced OPN, a host protein, plays a critical role in the epithelial-to-mesenchymal transition (EMT), cell migration, and invasion of human hepatocytes (21, 22).
OPN is a glycosylated phosphoprotein observed in larger quantities in response to inflammation, liver injury, tumorigenesis, and angiogenesis (23, 24), and it plays a critical role in many physiological and pathophysiological processes (24–27). Previously, it has been shown that OPN promotes a Th1-type immune response by promoting interleukin 12 (IL-12) production through binding to integrin αVβ3 and suppression of IL-10 production by interacting with CD44v, a spliced variant of CD44 on macrophages (25). OPN is mainly linked to tumor metastasis in a variety of cancers, including HCC (23, 26, 27). Previous studies reported that OPN is also detected in the plasma of patients with HCV-related HCC and patients with advanced fibrosis, along with other cancers (27, 28).
Several viruses, such as HCV, polyomavirus, hepatitis B virus (HBV), influenza virus, human T-cell lymphotropic virus type 1 (HTLV-1), dengue virus, and HIV, have been shown to induce OPN (21, 22, 28–35). Our recent studies have demonstrated that HCV induces OPN through Ca2+ signaling, elevation of reactive oxygen species (ROS), and activation of cellular kinases such as p38 mitogen-activated protein kinase (MAPK), Jun N-terminal protein kinase (JNK), PI3K, and MEK1/2 (21). Furthermore, we have demonstrated that secreted OPN binds to αVβ3 and CD44 and induced phosphorylation/activation of Akt, Src, FAK, glycogen synthase kinase 3β (GSK-3β), and β-catenin in HCV-infected cells (21, 22). However, the role of OPN in the HCV life cycle has not been elucidated.
In this study, we determined the potential role of OPN in HCV replication as well as assembly. Our studies showed that secreted OPN binds to cell surface receptors αVβ3 and CD44 and enhance HCV replication and expression of HCV structural and nonstructural proteins which are involved in HCV replication and assembly. Furthermore, we observed the colocalization of endogenous OPN with HCV proteins in LDs and the ER, indicating a possible contribution of OPN in HCV replication and assembly. Collectively, these observations indicate a novel role for OPN in regulating HCV replication and assembly in HCV-infected cells.
RESULTS
OPN activates HCV RNA replication.
HCV RNA is synthesized by replication complexes (RCs) contained within the membranous web that is derived from altered ER membranes (3). RCs consist of the components essential for RNA replication, such as HCV NS proteins, HCV RNA, and host factors (36, 37). To determine if OPN activates HCV RNA replication, HCV-infected Huh7.5 cells were transfected with control small interfering RNA (siRNA) (sicontrol) and siRNA specific for OPN (siOPN). Total cellular RNA was extracted and subjected to quantitative reverse transcription-PCR (RT-PCR). The results showed a significant increase in HCV RNA copy number (∼5.7 × 106) in HCV-infected cells (Fig. 1A). In contrast, we observed reduced HCV RNA copy number (∼5.8 ×102) in HCV-infected cells transfected with siOPN compared to sicontrol (Fig. 1A).
FIG 1.

OPN, CD44, and αVβ3 activate HCV replication. (A) Huh7.5 cells were incubated with HCV (MOI of 1). At day 4 postinfection, cells were transfected with sicontrol, siOPN, siCD44, and siβ3 as described in Materials and Methods. At 72 h post-siRNA transfection, total cellular RNA was extracted and HCV RNA copy number was analyzed by quantitative RT-PCR using HCV gene-specific primers. (B) K2040 (stably expressing HCV subgenomic replicon) cells were transfected with sicontrol and siOPN. At 72 h posttransfection, total cellular RNA was extracted and the HCV RNA copy number was quantified by RT-PCR. The values represent the means ± SDs from three independent experiments performed in triplicate. *, P < 0.05 compared to mock-infected cells (Huh7); **, P < 0.01 compared to HCV-infected Huh7.5 cells transfected with sicontrol. (C and D) Equal amounts of cellular lysates from the siRNA-transfected cells used for panels A and B were immunoblotted using anti-OPN, anti-CD44, anti-β3, anti-NS5A, anti-NS5B, anti-NS3, and anti-core antibodies. Actin and tubulin were used as protein loading controls.
Previously, HCV subgenomic replicons (K2040) have been shown to be an ideal system to study HCV replication (38). This system does not allow virus assembly and release. To further confirm the role of OPN in HCV replication, total cellular RNA from Huh7 as well as K2040 cells transfected with siOPN and sicontrol were analyzed by quantitative RT-PCR. The results show significant decrease in HCV RNA replication in K2040 cells transfected with siOPN compared to sicontrol (Fig. 1B). It is well established that HCV NS proteins such as NS3, NS4A, NS4B, NS5A, and NS5B play important role in HCV replication (2). To demonstrate the effect of OPN on HCV NS protein expression, cellular lysates from Fig. 1A were subjected to Western blot analysis using anti-OPN, anti-HCV NS3, anti-HCV NS5A, and anti-HCV NS5B antibodies. The results showed significant reduction in OPN expression in HCV-infected cells transfected with siOPN compared to sicontrol (Fig. 1C, lane 4). We also observed significant reduction in the expression of HCV NS3, NS5A, and NS5B in HCV-infected cells transfected with siOPN compared to sicontrol (Fig. 1C, lanes 3 and 4). In addition, we also observed reduced expression of HCV structural protein and core in HCV-infected cells transfected with siOPN compared to sicontrol (Fig. 1C, lane 3 and 4). However, we did not observe any significant change in the above-mentioned proteins in HCV-infected cells compared to HCV-infected cells transfected with sicontrol (Fig. 1C, lanes 2 and 3). Similarly, cellular lysates from K2040 cells (Fig. 1B) were analyzed using anti-OPN and anti-NS5A antibodies. The results show significant reduction in OPN expression in K2040 cells transfected with siOPN compared to sicontrol (Fig. 1D, lanes 3 and 4). We also observed decreased expression of HCV NS5A protein in K2040 cells transfected with siOPN compared to sicontrol (Fig. 1D, lanes 3 and 4). Taken together, these results suggest that the activation of OPN in HCV-infected cells plays a critical role in HCV NS protein expression and replication, indicating a possible role for OPN in HCV assembly.
Role of cell surface receptors CD44 and αVβ3 in HCV RNA replication.
Previously, we have shown increased expression of endogenous OPN at different time points of HCV postinfection (days 1 to 8) and also demonstrated that OPN was proteolytically cleaved and eventually secreted out in the cell culture supernatant at later time points of HCV infection (21, 22). In addition, we and others have also shown that secreted OPN binds to integrin αVβ3 and CD44 and induces epithelial to mesenchymal transition, cell migration, and invasion (21–24). To determine if CD44 and αVβ3 play an important role in HCV replication, HCV-infected Huh7.5 cells were transfected with sicontrol and siRNAs specific for CD44 (siCD44) and αVβ3 (siβ3). The results showed significant reduction of CD44 and αVβ3 expression in HCV-infected cells transfected with siCD44 and siβ3 compared to sicontrol (Fig. 1C, lanes 5 and 6). Total cellular RNAs from these cells were extracted and HCV replication was analyzed by quantitative RT-PCR. The results show a significantly higher HCV RNA copy number (∼5.7 × 106) in HCV-infected cells than in mock-infected cells; the HCV RNA copy number was reduced in HCV-infected cells transfected with siCD44 (∼1.2 × 103) and siβ3 (∼2.0 ×103) but not with sicontrol (Fig. 1A).
To demonstrate the role of CD44 and αVβ3 in OPN-mediated HCV protein expression, cellular lysates from the above-described cells were analyzed by Western blotting using anti-HCV NS3, anti-HCV NS5A, anti-HCV NS5B, and anti-HCV core antibodies. The results show decreased expression of NS3, NS5A, NS5B, and core in HCV-infected cells transfected with siCD44 and siβ3 compared to sicontrol (Fig. 1C, lanes 5 and 6). Collectively, these results suggest that CD44 and αVβ3 play a critical role in HCV replication.
Exogenous rOPN stimulates HCV replication and HCV protein expression through binding with cells surface receptors CD44 and αVβ3.
Since we have observed that CD44 and αVβ3 knockdown reduced HCV copy number and HCV protein expression, we sought to determine whether exogenously added recombinant OPN (rOPN) plays any role in HCV replication through binding with αVβ3 and CD44. Total cellular RNA was extracted from mock- and HCV-infected cells transfected with sicontrol, siCD44, or siβ3 and treated or not with rOPN. HCV replication was analyzed by quantitative RT-PCR. The results show increased HCV RNA copy number in the presence of rOPN which was significantly reduced in siCD44- and siβ3-transfected cells (Fig. 2A). In contrast, we did not observe any significant increase in HCV copy number in CD44 and αVβ3 siRNA-transfected cells treated with rOPN (Fig. 2A). These results suggest that binding of OPN to receptors CD44 and αVβ3 is required to stimulate HCV replication.
FIG 2.

Exogenous recombinant OPN (rOPN) stimulates HCV replication, infectivity, and assembly. (A) HCV-infected Huh7.5 cells (day 4) were transfected with sicontrol, siCD44, and siβ3. At 24 h post-siRNA transfection, cells were incubated with rOPN (50 nM) for another 48 h. Total RNA was extracted and HCV copy number was analyzed using quantitative RT-PCR. Data represent means ± SDs from three independent experiments performed in duplicate. *, P < 0.05 compared to mock-infected Huh7.5 cells; **, P < 0.01 compared to HCV-infected Huh7.5 cells transfected with sicontrol; ***, P < 0.001 compared to HCV-infected Huh7.5 cells transfected with sicontrol. (B) Equal amounts of cellular lysates from panel A were immunoblotted with the indicated antibodies. (C) The cell culture supernatants collected at various conditions described for panel A were incubated with naive Huh7.5 cells for 6 h, and then cells were washed and replaced with fresh medium. At day 3 postinfection, cellular lysates were immunoblotted with the indicated antibodies. (D) The cells from panel A were suspended in DMEM with 10% fetal calf serum (FCS), lysed by freeze-thaw cycles (4 times) on dry ice and a 37οC water bath, and centrifuged at 4,000 rpm for 5 min. The supernatants were incubated with naive Huh7.5 cells for 6 h and replaced with fresh media. At day 3 postinfection, equal amounts of cellular lysates were immunoblotted with the indicated antibodies. Tubulin was used as a protein loading control. (E) HCV-infected Huh7.5 cells (day 6) were incubated with anti-OPN (1:100) and control isotype goat IgG antibodies. At 24 h posttreatment, total cellular RNA was extracted and HCV copy number was analyzed by quantitative RT-PCR. The values are the means ± SDs from three independent experiments performed in duplicate. *, P < 0.05 compared to mock-infected cells (Huh7.5); **, P < 0.01 compared to HCV-infected Huh7.5 cells neutralized using control IgG antibody. (F) Equal amounts of cellular lysates from panel E were subjected to Western blot analysis using anti-NS5A, anti-NS3, and anti-core antibodies. (G) HCV-infected Huh7.5 cells (day 6) were incubated with FAK inhibitor (inh) PF573228 (2 μM). At 12 h posttreatment, total cellular RNA was extracted and HCV copy number was analyzed by quantitative RT-PCR. The values are means ± SDs from three independent experiments performed in duplicate. *, P < 0.05 compared to mock-infected cells (Huh7.5); **, P < 0.01 compared to HCV-infected Huh7.5 cells treated with an equal amount of DMSO. (H) The cellular lysates from panel G were immunoblotted with the indicated antibodies. Actin and tubulin were used as protein loading controls.
To demonstrate the effect of rOPN through the CD44 and αVβ3 receptors on HCV protein expression, the cellular lysates from Fig. 2A were immunoblotted using the desired antibodies. The results show increased expression of HCV NS3 and core proteins in the presence of rOPN (Fig. 2B, lane 4), which were significantly reduced in CD44 and αVβ3 knockdown cells treated or not with rOPN (Fig. 2B, lanes 5 to 8). We also observed significant knockdown of CD44 and αVβ3 (Fig. 2B, lanes 5 to 8). In contrast, we did not observe any significant change in OPN expression in the presence of rOPN but in the absence of CD44 and αVβ3 (Fig. 2B).
To demonstrate if rOPN plays any role in HCV infectivity through the binding with the CD44 and αVβ3 receptors, the cell culture supernatants collected as described in the legend to Fig. 2A were incubated with naive Huh7.5 cells. The cellular lysates from these cells were immunoblotted with the desired antibodies. We observed increased HCV NS3 and core expression in rOPN-treated cells and significantly reduced expression in CD44 and αVβ3 knockdown cells treated or not with rOPN (Fig. 2C, lanes 5 to 8).
To determine if rOPN has a role in HCV assembly through binding with the CD44 and αVβ3 receptors, the cells from Fig. 2A were lysed using freeze-thaw cycles as described in Materials and Methods. The supernatants were collected and incubated with naive Huh7.5 cells, and cellular lysates were analyzed by Western blotting (Fig. 2C). The findings regarding HCV infectivity (Fig. 2C) and assembly (Fig. 2D) were similar, and collectively, they suggest that rOPN stimulates HCV replication, infectivity, and assembly through binding with the CD44 and αVβ3 receptors.
To neutralize the effect of secreted OPN in HCV RNA replication and HCV protein expression through binding to CD44 and αVβ3, HCV-infected cells were incubated with anti-OPN and isotype control antibodies. Total cellular RNA was extracted, and HCV replication was analyzed using quantitative RT-PCR. The results show decreased HCV RNA copy number in HCV-infected cells incubated with anti-OPN antibody compared to control antibody (Fig. 2E). The cellular lysates from Fig. 2E were subjected to Western blot analysis, and the results show significant reduction in the expression of HCV NS3, NS5A, and core in HCV-infected cells incubated with anti-OPN antibody compared to control IgG antibody (Fig. 2F).
Recently, we have shown that rOPN induces EMT via activating various signaling molecules, such as focal adhesion kinase (FAK), p-Akt, and p-Src (22). To investigate if rOPN binding with the CD44 and αVβ3 receptors stimulates HCV RNA replication through FAK activation, total cellular RNA was extracted from HCV-infected cells treated or not with FAK inhibitors and HCV replication was analyzed using quantitative RT-PCR. We observed reduced copy number in the presence of FAK inhibitor compared to that in untreated HCV-infected cells (Fig. 2G). Cellular lysates from Fig. 2G were immunoblotted with the desired antibodies, and the results show decreased HCV NS3, NS5A, and core expression in FAK inhibitor-treated cells compared to that in HCV-infected cells (Fig. 2H). We did not observe any cytotoxic effect of FAK inhibitors on HCV-infected cells (data not shown). Together, these results suggest that exogenous OPN plays a pivotal role in the activation of HCV replication.
Knockdown of OPN expression reduces HCV infectivity.
Having shown that OPN knockdown significantly reduced the HCV RNA copy number (Fig. 1A), we aimed to demonstrate the role of OPN in virus release and infectivity. Toward this end, we performed a bona fide infectivity assay from the supernatants collected from HCV-infected, OPN-silenced Huh7.5 cells at different time points. Although there was no detected knockdown at 12 h, we observed OPN knockdown at 24 h which was sustained until 48 h and 72 h (Fig. 3A). The focus-forming unit (FFU) assay was performed from the supernatants collected at different time points (24 h, 48 h, and 72 h) and showed downregulation of OPN protein and substantial reduction in the infectivity titers of HCV in OPN knockdown condition compared to HCV control (Fig. 3B and C). This was further strengthened by a quantitative real-time RT-PCR that also showed marked reduction in the HCV RNA copy numbers in the siOPN compared to sicontrol set as well as at different time points for siOPN-transfected cells (Fig. 3D). These results altogether strengthened our conclusions that OPN plays a vital role in HCV replication and assembly as revealed by the real-time RT-PCR and FFU assay.
FIG 3.
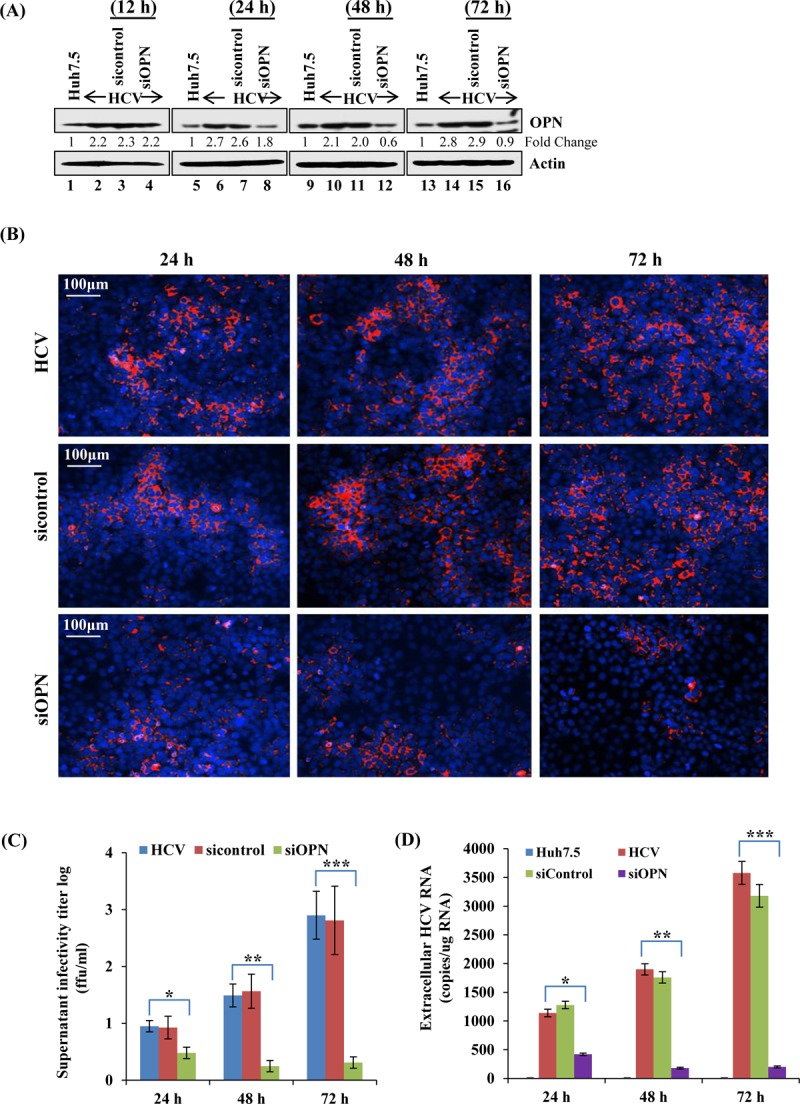
OPN knockdown reduces HCV assembly/release and infectivity. (A) Uninfected and HCV-infected Huh7.5 cells (day 4) were transfected with sicontrol and siOPN, and the knockdown efficiency was analyzed by calculating the fold change in downregulation of OPN protein at the indicated time points by Western blot assay. (B) The supernatants from panel A collected at 24 h, 48 h, and 72 h were used to infect the naive Huh7.5 cells, followed by immunofluorescence staining for the HCV-NS5A protein to calculate the FFU. (C) The graph shows the log viral infectivity titers, obtained from the supernatants collected at the indicated time points upon OPN knockdown. (D) Quantitative real-time RT-PCR was performed from the supernatants (500 μl) used to infect naive Huh7.5 cells in panel B, to obtain the HCV RNA copy numbers in siOPN compared to sicontrol cells. RT-PCR data represent means ± SDs from experiments performed in duplicate. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to HCV-infected Huh7.5 cells).
Overexpression of OPN rescues the activation of HCV replication and HCV protein expression.
Since we have observed that OPN knockdown reduced HCV RNA replication and HCV protein expression, we sought to determine if OPN overexpression rescues the knockdown effect of siOPN. HCV-infected cells were transfected with sicontrol and siOPN followed by overexpression of vector control and Flag-tagged OPN expression plasmids (OPN-fl). The cells transfected with siOPN plus OPN-fl were incubated with and without dimethyl sulfoxide (DMSO) and brefeldin A (BFA) for 8 h. The cellular RNA was extracted and HCV replication was analyzed by quantitative RT-PCR. We observed an increased HCV RNA copy number in cells transfected with sicontrol plus OPN-fl. Interestingly, we observed a reduced HCV copy number in OPN knockdown cells which was rescued in the presence of OPN overexpression (siOPN plus OPN-fl) compared to control vector (Fig. 4A). In addition, we also observed a decreased HCV copy number in the presence of BFA (siOPN plus OPN-fl plus BFA) compared to DMSO (control vehicle) (siOPN plus OPN-fl plus DMSO). BFA was used as a control because it is known to inhibit intracellular transport in the endocytic pathway (Fig. 4A). The used concentration of BFA did not show any significant cytotoxic effect (data not shown) as described earlier (39).
FIG 4.

OPN overexpression rescues HCV RNA replication, HCV infectivity, and assembly. (A) HCV-infected Huh7.5 cells (day 4) were transfected with sicontrol, sicontrol plus Flag-tagged OPN (OPN-fl), siOPN, and siOPN plus OPN-fl. At 48 h posttransfection, cells transfected with siOPN plus OPN-fl were incubated with DMSO and BFA (40 ng/ml) for 8 h. The cells were washed and replaced with fresh medium for 16 additional hours to complete 72 h of transfection. Total RNA was extracted and HCV copy number was analyzed using quantitative RT-PCR. Data represent means ± SDs from three independent experiments performed in duplicate. *, P < 0.05 compared to mock-infected Huh7.5 cells, as well as cells transfected with siOPN plus OPN-fl plus DMSO versus siOPN plus OPN-fl plus BFA. **, P < 0.01 compared to HCV-infected Huh7.5 cells transfected with control vector and sicontrol, as well as cells transfected with siOPN versus siOPN plus OPN-fl; ***, P < 0.001 compared to HCV-infected Huh7.5 cells transfected with sicontrol. (B) Equal amounts of cellular lysates from panel A were immunoblotted using anti-Flag, anti-OPN, anti-NS5A, anti-NS3, and anti-tubulin antibodies. (C) The cell culture supernatants collected for panel A were incubated with naive Huh7.5 cells as described for Fig. 2. At day 3 postinfection, the cellular lysates were immunoblotted with the indicated antibodies. (D) The harvested cells from panel A were resuspended in DMEM with 10% FCS and lysed by freeze-thaw cycling as described for Fig. 2. The collected supernatants were incubated with naive Huh7.5 cells for 6 h. At day 3 postinfection, equal amounts of cellular lysates were immunoblotted with the indicated antibodies. Tubulin was used as a protein loading control.
To determine the expression level of HCV proteins involved in HCV replication, cellular lysates from Fig. 4A were immunoblotted using the desired antibodies. Similar results were obtained (Fig. 4B). The reduced HCV protein expression in OPN knockdown cells was rescued in the presence of OPN overexpression (Fig. 4B, compare lane 5 to lane 6). In addition, we observed reduced HCV NS3, NS5A, and core expression in the presence of BFA (Fig. 4B, compare lane 7 to lane 8). The detection of OPN using anti-Flag and anti-OPN antibodies showed the overexpression and rescued levels of OPN (Fig. 4B). In contrast, we did not observe any significant change in OPN expression in the presence of BFA (Fig. 4B, compare lane 8 to lane 7).
To demonstrate if OPN knockdown or overexpression has an effect on HCV infectivity, cell culture supernatant collected from Fig. 4A were incubated with naive Huh7.5 cells and the cellular lysates were immunoblotted using anti-NS3, anti-NS5A, anti-core, and anti-tubulin antibodies. We observed reduced HCV protein expression in OPN knockdown cells which was rescued upon OPN-fl overexpression (Fig. 4C, compare lane 5 to lane 6). Moreover, the rescued effect of OPN was impaired in the presence of BFA (Fig. 4C, compare lane 7 to lane 8).
To further demonstrate if OPN knockdown or overexpression affects HCV assembly, the cells from Fig. 4A were lysed using freeze-thaw cycles as described in Materials and Methods. The supernatants were collected and incubated with naive Huh7.5 cells, and the cellular lysates were subjected to Western blot analysis using the desired antibodies. The findings regarding HCV infectivity (Fig. 4C) and assembly (Fig. 4D) were similar, and collectively, they suggest that OPN specifically regulates HCV replication, protein expression, infectivity, and assembly.
Colocalization of endogenous OPN with HCV proteins.
It is well established that HCV NS proteins assemble to form ribonucleoprotein complex (RNP) to facilitate HCV RNA replication (2). Since we observed that knockdown of OPN reduced HCV replication and overexpression of OPN rescued HCV replication (Fig. 1 and 4), we hypothesized that OPN may interact with HCV NS proteins and regulate HCV replication. To determine if OPN associates with HCV NS proteins and facilitate HCV RNA replication, we performed colocalization studies. Mock- and HCV-infected cells at day 7 postinfection were immunostained with anti-OPN, anti-NS5A, anti-NS5B, and anti-NS3 antibodies. We observed colocalization of OPN with these HCV NS proteins in HCV-infected cells (Fig. 5A to F), suggesting a possible role for OPN in activation of the HCV replication process. In addition, we also observed that OPN associates with HCV core protein, indicating a possible role for OPN in HCV assembly (Fig. 5G and H).
FIG 5.

Colocalization of OPN with HCV proteins. (A, C, E, and G) Mock- and HCV-infected cells (day 7) were fixed and permeabilized as described in Materials and Methods. The cells were immunostained using anti-OPN, anti-NS5A, anti-NS5B, anti-NS3, and anti-core antibodies for 1 h at room temperature, followed by incubation with secondary antibodies for OPN (anti-goat Alexa Fluor 546), NS5A and NS5B (anti-rabbit Alexa Fluor 488), and NS3 and core (anti-mouse Alexa Fluor 488). DAPI was used as a nuclear stain and is shown in blue. Boxed areas were enlarged and white arrows represent the colocalization of OPN with HCV proteins (NS5A, NS5B, NS3, and core) in yellow. Magnification: ×60. (B, D, F, and H) A minimum of 10 cells were analyzed in 3 different optical regions to calculate the averages of the colocalization (yellow spots) using Pearson's correlation coefficient. Error bars indicate SDs. *, P < 0.05.
Interaction of OPN with HCV proteins.
To demonstrate the interaction of OPN with HCV proteins, cellular lysates from mock- and HCV-infected cells were immunoprecipitated using anti-OPN antibody, followed by Western blotting using anti-NS5A, anti-NS5B, anti-NS3, and anti-core antibodies. The results show that OPN was able to pull down these HCV proteins (Fig. 6A). Similarly, we performed reverse coimmunoprecipitation (co-IP) using representative HCV NS protein, anti-NS3, and one HCV anti-core antibody and conducted Western blot analysis using anti-OPN antibody. The results show that HCV proteins were able to pull down OPN in HCV-infected cells, in contrast to control IgG (Fig. 6A, bottom blots). Similarly, we observed the interaction of OPN with HCV NS proteins in HCV subgenomic replicon-expressing cells, K2040 cells (Fig. 6B). In contrast, we did not observe OPN interactions with core in K2040 cells, which do not express HCV core (Fig. 6B). Collectively, these results suggest the interaction of OPN with HCV NS proteins, indicating a functional role for OPN in HCV replication; however, OPN′s interactions with HCV core protein indicate a possible role for OPN in HCV assembly.
FIG 6.
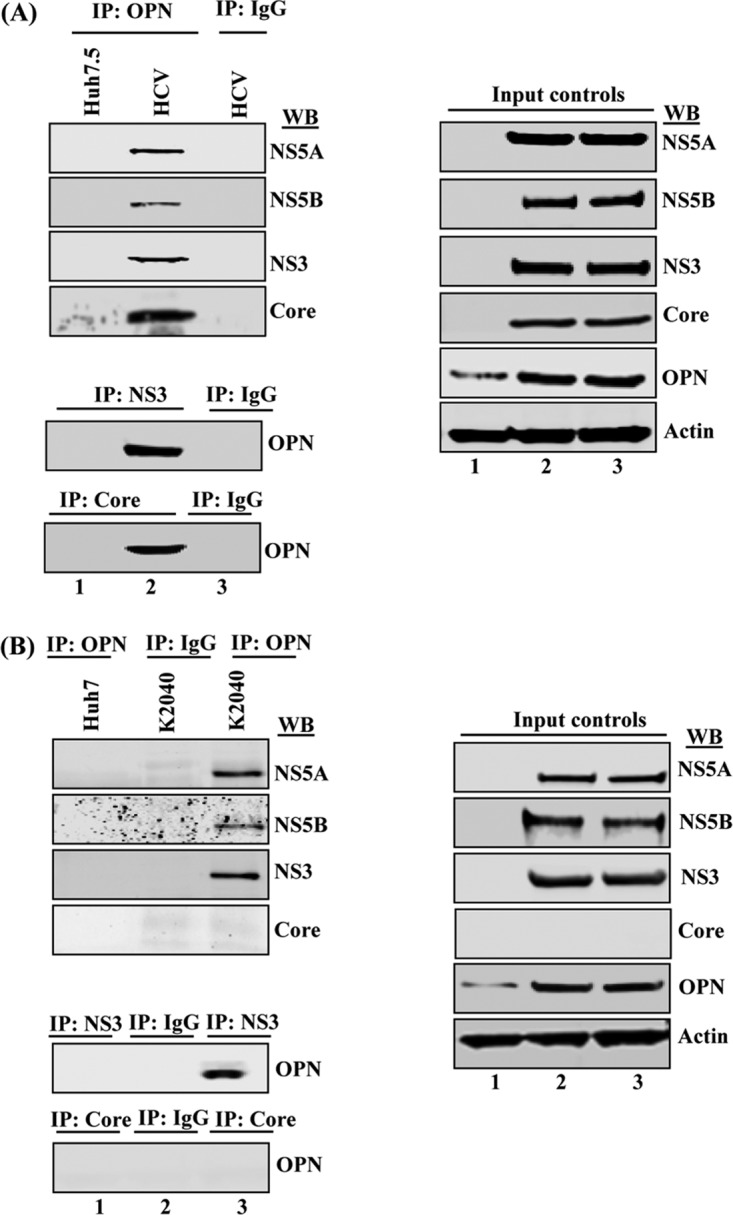
OPN interacts with HCV proteins. (A) Equal amounts of cellular lysates from mock- and HCV-infected Huh7.5 cells were immunoprecipitated with anti-OPN (1:100 dilution) and immunoblotted with anti-NS5A, anti-NS5B, anti-NS3, and anti-core antibodies (lanes 1 and 2). Isotype anti-goat IgG was used as a control (lane 3). (B) Cellular lysates from Huh7 and K2040 cells were immunoprecipitated using anti-OPN and Western blotted (WB) with the above-named HCV antibodies (lanes 1 and 3), including isotype control goat IgG (lane 2). Cellular lysates from HCV-infected cells and K2040 cells were utilized to perform reverse co-IP using anti-NS3 and anti-core antibodies and immunoblotted with anti-OPN (A and B). Right panels show the input controls.
OPN colocalizes with NS5A and NS4A/B in the ER.
Recent studies suggest that HCV induces double membrane vesicles (DMVs), in which viral NS proteins and RNA are protected from host defense processes and participate in HCV RNA replication (40). Studies also suggest that only combined expression of all HCV NS proteins can induce DMVs, in which NS5A plays critical role apart from NS4B (4, 40). To determine if OPN associates with NS5A and NS4 in the ER and regulates HCV replication, mock- and HCV-infected cells were immunostained with anti-OPN, anti-PDI (ER marker), anti-HCV NS5A, and anti-NS4 antibodies. The results show colocalization of OPN with NS5A and NS4A/B in the ER in HCV-infected cells, in contrast to mock-infected cells (Fig. 7A to D, white dots indicated by white arrows). In contrast, we did not observe the colocalization of OPN with EEA1 (endosome marker), used as a negative control (Fig. 7E, absence of yellow dots). However, colocalization of OPN with NS5A is shown in magenta in Fig. 7E and quantified in Fig. 7F. Taken together, these results suggest that HCV-induced OPN associates with NS5A and NS4A/B in the ER and may stimulate HCV RNA replication.
FIG 7.

Colocalization of OPN with NS5A and NS4 in the ER. (A, C, and E) Mock-infected (Huh7.5) and HCV-infected cells (day 7) were permeabilized and immunostained using anti-NS5A, anti-NS4, anti-PDI (ER marker), anti-EEA1 (endosome marker), and anti-OPN antibodies for 1 h at room temperature. The cells were incubated with secondary antibodies for NS5A (anti-rabbit Alexa Fluor 633), PDI and EEA1 (anti-rabbit Alexa Fluor 488), OPN (anti-goat Alexa Fluor 546 for panels A and E and Alexa Fluor 633 for panel C), and NS4 (anti-mouse Alexa Fluor 546). DAPI was used as a nuclear stain and is shown in gray. Boxed areas were enlarged and white arrows show the colocalization of OPN with NS5A or NS4 in the ER (white dots) (A and C). Magenta color represents the colocalization of OPN with NS5A in HCV-infected cells and shown by white arrows (E). Magnification: ×60. (B, D, and F) The mean fluorescent intensities were analyzed for NS5A-ER-OPN, OPN-ER-NS4A/B, and NS5A-OPN (white/magenta spots) for three different fields, with a minimum of 10 cells each, with the metamorph pixel intensity calculator. Error bars represent SDs. **, P < 0.01. (G) Panel showing the possible color combination where Y, W, M, and S represent yellow, white, magenta, and cyan, respectively.
HCV induces association of OPN with LDs.
LDs are believed to be the site of HCV assembly where HCV core and NS5A proteins colocalize (7, 9). Interestingly, the role of OPN in HCV assembly has not been explored. Our results show a reduction in HCV assembly in HCV-infected cells transfected with siOPN (Fig. 4D). To demonstrate if OPN associates with LDs and facilitates HCV assembly, mock- and HCV-infected cells were immunostained with antibodies against OPN, HCV NS5A, and HCV core and LD-specific staining dye BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene). We observed increased LD formation in HCV-infected cells compared to that in mock-infected cells (Fig. 8A and C), which supports our recent finding related to HCV-induced LD formation (41). The results show substantial colocalization of OPN with LDs and NS5A in HCV-infected cells compared to mock-infected cells (Fig. 8A and B, white puncta shown by red arrows). Similarly, we also observed colocalization of OPN with LDs and core (Fig. 8C and D, white puncta shown by red arrows). The graphs in Fig. 8B and D show mean fluorescent intensity of colocalization of OPN-LD-NS5A/core. The colocalization of OPN with LD and core suggests a possible role for OPN in HCV assembly, whereas colocalization of OPN with LD and NS5A suggests a possible role for OPN in the recruitment of NS5A to LDs, the site of HCV assembly. Collectively, these observations suggest that HCV-induced OPN might be critical to facilitate HCV assembly.
FIG 8.

Colocalization of OPN with LDs, NS5A, and core. (A and C) Mock- and HCV-infected cells (day 7) were permeabilized and immunostained using BODIPY dye (LD marker) as well as anti-OPN, anti-NS5A, and anti-core antibodies for 1 h at room temperature. The cells were washed and incubated with secondary antibodies for NS5A (anti-rabbit Alexa Fluor 633), core (anti-mouse Alexa Fluor 633), and OPN (anti-goat Alexa Fluor 546). BODIPY dye was visualized at 488 nm. DAPI was used as a nuclear stain and is shown in gray. Red arrows indicate the colocalization of OPN with LDs and NS5A/core proteins in white. Magnification: ×60. (B and D) The mean fluorescent intensities were analyzed for LD-OPN-NS5A/core (white spots) for three different fields with a minimum of 10 cells each with the metamorph pixel intensity calculator. Error bars represent SDs. * or **, P < 0.05 or P < 0.01, respectively.
Association of OPN with LD in close proximity to ER during HCV infection.
The ER membrane or DMVs have been revealed to be the site of HCV replication (3, 40), whereas LDs are thought to participate in HCV assembly (7, 9). To decipher if HCV-induced OPN bridges LDs and ER and facilitates HCV assembly, mock- and HCV-infected cells were immunostained using anti-OPN, anti-PDI, and anti-core antibodies plus BODIPY dye. We observed substantial colocalization of OPN with LDs in close proximity to the ER in HCV-infected cells compared to mock-infected cells (Fig. 9A and B, white puncta shown by red arrows). The HCV infection was shown by HCV core (Fig. 9A, rightmost column, magenta). The graph in Fig. 9B shows mean fluorescent intensity of colocalization of OPN-LD-ER.
FIG 9.

Colocalization of OPN with LDs and ER. (A) Mock- and HCV-infected cells (day 7) were immunostained using BODIPY dye, anti-OPN, anti-PDI, and anti-core antibodies for 1 h at room temperature, followed by incubation with secondary antibodies for OPN (anti-goat Alexa Fluor 546), PDI (anti-rabbit Alexa Fluor 405), and core (anti-mouse Alexa Fluor 633). BODIPY dye was visualized at 488 nm. Red arrows indicate the colocalization of OPN with LDs and ER in white. HCV core represents HCV infection in cells and is shown in magenta (right panel). (B) The mean fluorescent intensities were analyzed for the colocalization of OPN-LD-ER (white spots) for three different fields with a minimum of 10 cells each with the metamorph pixel intensity calculator. Error bars represent SDs. *, P < 0.05. (C) Colocalization of OPN with NS5A and core. Mock- and HCV-infected cells (day 7) were immunostained with anti-OPN, anti-NS5A, and anti-core antibodies for 1 h at room temperature, followed by incubation with secondary antibodies for OPN (anti-goat Alexa Fluor 488), NS5A (anti-rabbit Alexa Fluor 633), and core (anti-mouse Alexa Fluor 546). Boxed areas were enlarged, and red arrows indicate the colocalization of OPN with NS5A and core in white. Magnification: ×60. (D) The mean fluorescent intensities were analyzed for the colocalization of OPN-core-NS5A (white spots) for three different fields with a minimum of 10 cells each with the metamorph pixel intensity calculator. Error bars represent SDs. *, P < 0.05.
Colocalization of OPN with HCV NS5A and core.
Since HCV core and NS5A have been shown to be involved in HCV assembly (7, 9, 40), we sought to determine if OPN associates with HCV NS5A and core to facilitate virus assembly. Mock- and HCV-infected cells were immunostained using anti-OPN, anti-core, and anti-NS5A antibodies. In contrast to the case with mock-infected cells, confocal imaging revealed substantial colocalization of OPN, NS5A, and core in HCV-infected cells (Fig. 9C, bottom image in “Enlarge” column). The graph in Fig. 9D shows mean fluorescent intensity of colocalization of OPN-core-NS5A. Collectively, these results suggest that OPN may bridge LDs and ER and associate with HCV NS5A and core to facilitate the HCV assembly.
DISCUSSION
HCV replication and assembly occur at the interface of LDs and ER membranes (7). However, the exact details of these processes are still obscure. In this study, we defined the important relationship among OPN, HCV proteins, the ER, and LDs. Our comprehensive studies demonstrate the role of OPN in HCV replication as well as assembly processes. HCV RNA replicates in a specialized membranous compartment or DMVs derived from altered ER membranes (3, 7, 40), and HCV virion assembly occurs at the surface of LDs (7, 42). Studies show that HCV NS2, NS3, NS4A/B, NS5A, and NS5B are involved in HCV replication (2). HCV core and NS5A have been shown to associate with LDs for virus assembly (7, 9). Moreover, HCV NS5A is believed to transport the HCV RNA genome from RCs to LD surfaces, where HCV core initiates HCV RNA encapsidation (4, 7). Previously, several host factors, such as annexin A2, early endosomal proteins (Rab5, EEA1, and PI4-kinase III alpha), and ER- and Golgi complex-associated proteins (RAB1B, TBC1D20, and RAB7L1), have been shown to be associated at the sites of HCV RNA replication (16–19). In addition, TIP47, DGAT-1, TBC1D20, Rab1, Rab18, AP2M1, and phospholipase A2 are known to play important roles in the HCV assembly process (10–15). These studies support a model in which viral and host proteins may promote the interaction between the ER and LDs.
Previously, OPN has been studied extensively in the context of tumor metastasis, and it has been detected in the plasma of patients with HCV-associated HCC (26, 27). OPN exists in the form of the extracellular matrix as well as secreted multifunctional cytokine (23–27). Recently, we have shown induction and proteolytic cleavage of full-length OPN (∼75 kDa) into various cleaved products (∼55, ∼42, and ∼36 kDa) in HCV-infected cells (21). We and others have also demonstrated that secreted OPN binds to αVβ3, a widely studied member of the family of αVβ integrins, and the cell surface adhesion molecule CD44, to initiate cellular signals that enable EMT and tumor progression (21–23, 26, 27). In vivo studies have shown that liver from HCV-infected patients exhibits EMT gene expression, which may expose or alter gap junction proteins important for HCV entry or cell-to-cell transmission of HCV (43). Interestingly, our recent studies demonstrated a potential role for OPN in HCV-induced EMT (21, 22). However, the role of OPN in the HCV life cycle has not been explored.
In the present study, we demonstrated that the activation of OPN regulates HCV replication and assembly, which is consistent with previous observations reports of OPN regulation of HIV-1 replication (34). Studies have shown that OPN ligands induce HCV replication, whereas an inhibitor of OPN (OPN aptamers) represses HCV replication (28). A striking observation made in our studies is the regulation of HCV replication by endogenous OPN as well as secreted forms of OPN through αVβ3 integrin- and CD44-mediated signaling pathways. OPN is also known to be involved in various pathophysiological events and has been studied as a secreted protein (23–27). In this study, we have observed a significant reduction in HCV protein expression as well as HCV RNA replication in HCV-infected cells expressing siOPN, siβ3, and siCD44 which was further rescued by endogenous OPN overexpression.
Our recent studies suggested that secreted OPN binding with receptors αVβ3 and CD44 in HCV-infected cells or incubation of uninfected cells with exogenous rOPN activates several signaling molecules, such as FAK, Src, and Akt, to induce EMT and cell migration (21, 22). In this study, we observed increased HCV replication, protein expression, infectivity, and assembly in the presence of rOPN. Also, we found reduced expression of NS5A in the presence of FAK inhibitor and neutralization of secreted OPN, implying that OPN binding to αVβ3 and CD44 could follow the signaling pathways to stimulate HCV RNA replication similar to those it does in EMT (21, 22). This area of investigation needs more in-depth study, which we are carrying out. We have further performed viral infectivity assays to show that HCV infectious titer as well as RNA copy number drastically decreases in the absence of OPN compared to that in wild-type cells (Fig. 3). Collectively, our observations indicate a critical role for both endogenous and secreted OPN in HCV replication through binding with αVβ3 and CD44.
It has been suggested that the ER-derived membranous web or DMVs is the specialized site formed mainly by HCV NS4B, whereas HCV NS proteins NS3 to NS5B assemble to facilitate HCV replication (2, 40). HCV NS5A is one of the nonstructural proteins which are involved in HCV replication (6). Here we report for the first time the association of OPN with HCV NS5A, NS5B, NS3, and core. Moreover, we observed OPN′s association with NS5A and NS4 in the ER, implying that OPN might interact with HCV NS proteins to facilitate ribonucleoprotein (RNP) complex formation, which is required for productive HCV replication, as shown for several other host proteins previously (16–19).
Lipid droplets have been shown to participate in the assembly of several pathogens, including HCV (9). HCV core is known to coat the surface of LDs, a critical step for the recruitment of viral RNA replication complexes to the vicinity of LDs, and this process is essential for the encapsidation of viral RNA into progeny virions (7, 9). HCV NS5A has been proposed to function as a vehicle to transport RNA from the ER, an HCV replication site, to LDs and plays a vital role in RNA replication and viral assembly (4, 7). A striking observation in our study is that OPN associates with LDs and the ER, and also associates with NS5A and core together, which strongly indicates a role for OPN in HCV replication and assembly (Fig. 9). Previous studies suggest that HCV exploits an extensive network of host factors for productive infection and propagation and links the host factors in the assembly of various viruses, for example, HIV (44). It may be possible that HCV-induced OPN along with HCV NS5A and core acts as a bridge between the ER and LDs to facilitate virus assembly, which needs further investigation. These observations are consistent with earlier studies in which PLA2G4C has also been shown to bridge the steps of RNA replication and HCV assembly by translocation of replication complexes to LDs (15). TIP47, a host protein, functions as a bridging protein in HIV and HCV (10, 44) and also has been shown to be required for the dengue virus capsid protein to associate with LDs and promote viral assembly (45). A recent report also supports our finding: the authors have shown that phosphatidylserine-specific phospholipase A1, a host factor, is involved in HCV assembly (46).
In summary, we have investigated the role of OPN in HCV replication and assembly. We have also identified a dual and unique role for endogenous OPN as well as secreted OPN in HCV replication and assembly (Fig. 10). Endogenous OPN associates with HCV proteins, whereas secreted OPN binds to αVβ3 and CD44 in an autocrine/paracrine manner and stimulates HCV replication and assembly. In addition, we have demonstrated potential involvement of endogenous OPN in HCV replication and assembly through the association of OPN with LDs along with viral proteins NS5A, NS4A/B, and core in the ER (Fig. 10). HCV protein(s) and OPN association represents a unique target for development of antivirals because it is more difficult for HCV to develop escape mutations against therapeutics that target host factors. These findings of HCV host dependencies may lead to novel antiviral strategies and help to facilitate further insights into viral pathogenesis and potential therapeutic targets.
FIG 10.
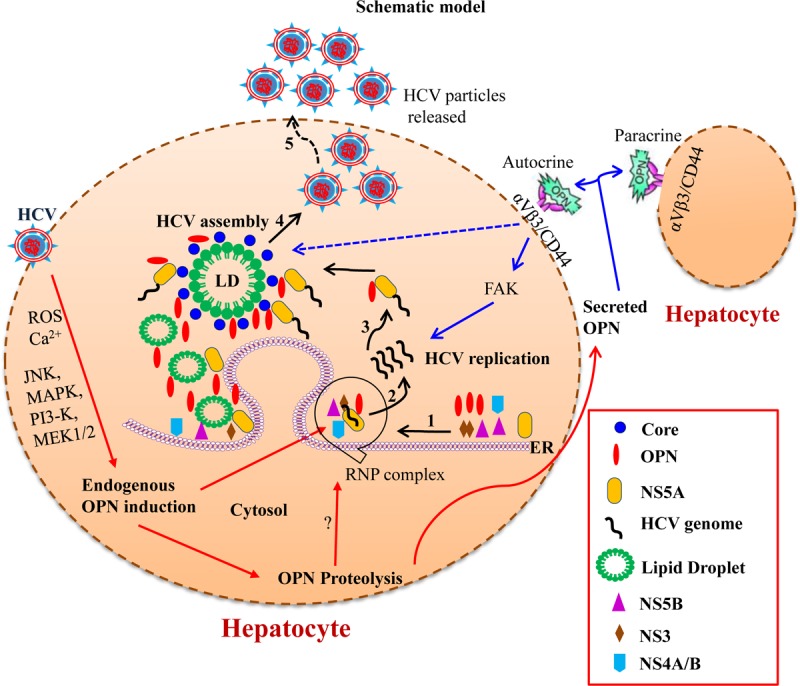
Model illustrating the role of OPN in HCV replication and assembly. Recently, we have shown the induction of OPN via Ca2+ signaling in the ER, induction of ROS in the mitochondria, activation of kinases (MAPK, PI3K, and MEK1/2), and transcription factors AP-1 and SP-1 in HCV-infected cells (21). The precursor form of the OPN is cleaved via activation of calpain proteases, and the cleaved forms are secreted out of the cells (21, 22). In this study, we show that activated endogenous OPN interacts with HCV nonstructural proteins (NS3, NS4A/B, NS5A, and NS5B) in the ER to facilitate HCV replication (steps 1 and 2). In addition, OPN also interacts with HCV NS5A, core, and LDs to regulate HCV assembly process (steps 3 and 4). Interestingly, the secreted forms of OPN bind to cell surface receptors αVβ3 and CD44 to regulate HCV replication and assembly through FAK-mediated increased expression of HCV NS and core proteins.
MATERIALS AND METHODS
Plasmids, antibodies, and reagents.
Wild-type (J6/JFH-1) and replication-defective (JFH-1/GND) HCV genotype 2a cDNAs were obtained from Charles Rice (Rockefeller University, NY). Full-length Flag-OPN DNA (pDest490-OPN-a) was purchased from Addgene, Cambridge, MA.
All antibodies were purchased and used according to the manufacturers' protocols: anti-HCV NS3 and anti-HCV NS5A (Virogen, Watertown, MA), anti-HCV NS5B (Alexis Biochemicals, Plymouth Meeting, PA), antiactin and anti-β-tubulin (Sigma, St. Louis, MO), anti-HCV core (Thermo Scientific Inc., Rockford, IL), anti-HCV NS4 (Abcam, Cambridge, MA), anti-human OPN (R & D Systems, Inc., Minneapolis, MN), anti-integrin αVβ3, anti-PDI (ER marker), anti-EEA1 (endosome marker; Cell Signaling Technology, Danvers, MA), and anti-CD44 (anti-HCAM) (Santa Cruz Biotechnology, Dallas, TX). The control siRNA and siRNAs against OPN, CD44, and integrin αVβ3 were purchased from Santa Cruz Biotechnology. Human recombinant OPN (rOPN) and lipid droplet staining dye BODIPY (4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) were purchased from R & D Systems, Inc., Minneapolis, MN, and Invitrogen, CA, respectively. FAK inhibitor (PF573228) was purchased from Sigma Chemicals. Anti-HCV NS5A (rabbit polyclonal) antibody used for immunostaining was a generous gift from Craig Cameron, Pennsylvania State University, PA.
Cell lines.
The human hepatoma cell line Huh7 and subline Huh7.5 were obtained from A. Siddiqui, The University of California, San Diego (UCSD), CA, and C. Rice, Rockefeller University, NY, respectively. These cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 U of penicillin/ml, and 100 μg of streptomycin sulfate/ml. K2040 cells stably expressing HCV subgenomic replicon (a gift from Michael Gale, University of Washington, Seattle, WA) were also cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 500 μg of G418 per ml. K2040 is the Huh7 cell line which contains an adaptive mutation of lysine at position 2040 in the HCV subgenomic replicon. The cells were incubated at 37°C in a 5% CO2 incubator.
HCV cell culture infection system and titration of infectious HCV.
J6/JFH-1 RNA was in vitro transcribed and electroporated into Huh7.5 cells as described previously (47). The cell-free virus was propagated in Huh7.5 cell culture as described previously (48). The expression of HCV proteins in HCV-infected cells was analyzed using Western blotting. HCV cell culture supernatants were collected at appropriate time points and were used to infect naive Huh7.5 cells at a multiplicity of infection (MOI) of 1 for 5 to 6 h at 37°C and 5% CO2 (47, 48). The viral titer in culture supernatant was expressed as FFU/ml, which was observed by the average number of HCVNS5A-positive foci detected at the highest dilutions as defined previously (48). The cell culture supernatant collected from Huh7.5 cells expressing JFH-1/GND (replication-defective virus) was used as a negative control.
HCV-infected cells at day 4 postinfection were transfected with siRNA for 72 h; however, day 5 postinfection cells were incubated with rOPN for 48 h to maintain day 7 postinfection at the time of cell's harvesting.
The focus-forming unit assay was performed, from the supernatants collected from HCV-infected OPN-silenced Huh7.5 cells at different time points (24 h, 48 h, and 72 h postinfection), by infecting naive Huh7.5 cells in 8-well chambered glass slides (Thermo Scientific, IL). The cells were incubated with supernatant for 5 to 6 h at 37°C, followed by replacements with fresh DMEM. The HCV infection was determined 3 days postinfection by immunofluorescence staining for HCV-NS5A protein and the viral titer expressed as the focus-forming units per milliliter of supernatant.
Immunoprecipitation and Western blotting.
For immunoprecipitation, mock- and HCV-infected cells were lysed in cell lysis buffer (142.5 mM KCl, 5 mM MgCl2, 10 mM HEPES [pH 7.2], 1 mM EGTA, 1% NP-40, protease inhibitor cocktail [10 μl/ml; Thermo Scientific, IL], 1 mM ATP, and 1 mM dithiothreitol [DTT]) for 30 min on ice. Cellular lysates were immunoprecipitated using anti-OPN (10 μg/ml) overnight at 4°C. The immune complexes were incubated with protein G-Sepharose (GE Healthcare, Piscataway, NJ) for 1 h at 4°C, washed 3 or 4 times using the above-described lysis buffer containing 0.6% NP-40, and boiled in SDS sample buffer for 5 min.
For Western blot analysis, mock-infected (Huh7.5) and HCV-infected cells were harvested and cellular lysates were prepared by incubation in radioimmunoprecipitation (RIPA) lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM sodium orthovanadate, 1 mM sodium formate, 10 μl/ml of protease inhibitor cocktail) for 30 min on ice. Equal amounts of protein from cellular lysates were subjected to SDS-PAGE. Gels were electroblotted onto nitrocellulose membranes (Thermo Scientific, IL) in transfer buffer (25 mM Tris, 192 mM glycine, and 20% methanol). The nitrocellulose membranes were incubated for 1 h in blocking buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween 20, 5% dry milk), probed with primary antibody for 1 h at room temperature, and washed twice for 4 to 5 min with blocking buffer without milk, followed by incubation with secondary antibody for 1 h at room temperature. After an additional washing step with blocking buffer, immunoblots were visualized by the Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE). The fold change of protein band intensity was analyzed by Odyssey infrared imaging system software.
For HCV assembly experiments, trypsinized cells were washed with phosphate-buffered saline (PBS) by centrifugation at 1,500 rpm and cell pellets were resuspended in complete DMEM. The cells were lysed by freeze-thaw cycling (4 times) on dry ice and a 37°C water bath as described previously (49). Furthermore, cells were centrifuged at 4,000 rpm, and collected supernatants were incubated with naive cells for 6 h and replaced with fresh medium for further incubation. The cellular lysates were Western blotted to examine the HCV assembly processes.
Quantitative real-time RT-PCR.
Total RNA was extracted from mock- and HCV-infected cells using TRIzol (Invitrogen, CA). HCV RNA was quantified by real-time RT-PCR using an ABI PRISM 7500 sequence detector (Applied Biosystems, CA). Amplifications were conducted in triplicate using HCV-specific primers and 6-carboxyfluorescein (FAM)- and tetrachloro-6-carboxyfluorescein (TAMRA)-labeled probes (Applied Biosystems). The sequences for the primers and probes were designed using Primer Express software (Applied Biosystems). HCV sense primer (5′-CGGGAGAGCCATAGTGG-3′), antisense primer (5′-AGTACCACAAGGCCTTTCG-3′), and TaqMan probe (5′-FAM-CTGCGGAACCGGTGAGTACAC-TAMRA-3′) were used. Amplification reactions were performed in a 25-μl mix using RT-PCR core reagent kit and template RNA. Reactions were performed in a 96-well spectrofluorometric thermal cycler under the following conditions: 2 min at 50°C, 30 min at 60°C, 10 min at 95°C, and 44 cycles of 20 s at 95°C and 1 min at 62°C. Fluorescence was monitored during every PCR cycle at the annealing step. At the termination of each PCR run, the data were analyzed using the automated system and amplification plots were generated. To determine the HCV RNA copy number, standards ranging from 101 to 108 copies/μg were used for comparison.
RNA interference.
Mock- and HCV-infected cells were transfected with control siRNA (sicontrol), siOPN, siCD44, and siβ3 according to the manufacturer's protocols (Santa Cruz Biotechnology). Each siRNA consisted of a pool of three to five target-specific 19- to 25-nucleotide (nt) siRNAs designed to knock down expression of the targeted gene. For siRNA transfections, two solutions were prepared. For solution A, 60 pmol of siRNA duplex was mixed with 100 μl of siRNA transfection medium. For solution B, 6 μl of transfection reagent was added to 100 μl of siRNA transfection medium. Solutions A and B were incubated at room temperature for 20 min and then were combined and incubated again for 20 min at room temperature. The combined solutions were then added to the cells and incubated for 5 to 7 h at 37°C and 5% CO2, and then transfection solution was replaced with 3 ml of complete DMEM with 10% FBS.
Laser scanning confocal microscopy.
Mock- and HCV-infected cells on coverslips were washed with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, permeabilized for 5 min using 0.2% Triton X-100, and blocked for 45 min with 5% bovine serum albumin (BSA) in PBS. Next, the cells were incubated with primary antibody against the specific protein for 1 h at room temperature or overnight at 4°C, followed by incubation with Alexa Fluor-labeled secondary antibody (Molecular Probes) for 1 h. For LD immunostaining, the above-described blocked cells were incubated with BODIPY (20 μM) for 30 min at room temperature. After a washing with PBS, cells were mounted with antifade reagent DAPI (4,6-diamidino-2 phenylindole) (Invitrogen, CA) and examined under a laser scanning confocal microscope (Olympus FV10i).
Pearson's correlation coefficients were calculated for quantitative colocalization measurement using ImageJ software. Values can range from 1 to −1, with 1 indicating a complete positive correlation, −1 a negative correlation, and 0 no correlation.
The mean fluorescent intensities were analyzed for three different fields with a minimum of 10 cells each with the metamorph pixel intensity calculator. The mean colocalization pixel intensities were measured with respect to the uninfected control in a graph, and a paired t test was used to obtain the P values.
Statistical analysis.
Error bars show the standard deviations of the means of data from three individual trials. Two-tailed unpaired t tests were used to compare experimental settings to those of the respective controls. In all tests, a P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Charles Rice (Rockefeller University, NY) for the generous gift of HCV genotype 2a J6/JFH-1 infectious cDNA and the Huh7.5 cell line. We thank Craig Cameron (Pennsylvania State University, PA) for the generous gift of HCV NS5A antibody.
This work was supported in part by the National Institutes of Health (grant DK106244) and by the Rosalind Franklin University of Medicine and Science-H. M. Bligh Cancer Research Fund.
REFERENCES
- 1.Di Bisceglie AM. 1997. Hepatitis C and hepatocellular carcinoma. Hepatology 26:34S–38S. doi: 10.1002/hep.510260706. [DOI] [PubMed] [Google Scholar]
- 2.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 3.Egger D, Wolk B, Gosert R, Bianchi L, Blum HE, Moradpour D, Bienz K. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 76:5974–5984. doi: 10.1128/JVI.76.12.5974-5984.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyers NL, Fontaine KA, Kumar GR, Ott M. 2016. Entangled in a membranous web: ER and lipid droplet reorganization during hepatitis C virus infection. Curr Opin Cell Biol 41:117–124. doi: 10.1016/j.ceb.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 6.Penin F, Brass V, Appel N, Ramboarina S, Montserret R, Ficheux D, Blum HE, Bartenschlager R, Moradpour D. 2004. Structure and function of the membrane anchor domain of hepatitis C virus nonstructural protein 5A. J Biol Chem 279:40835–40843. doi: 10.1074/jbc.M404761200. [DOI] [PubMed] [Google Scholar]
- 7.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 8.Beller M, Thiel K, Thul PJ, Jackle H. 2010. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett 584:2176–2182. doi: 10.1016/j.febslet.2010.03.022. [DOI] [PubMed] [Google Scholar]
- 9.Boulant S, Targett-Adams P, McLauchlan J. 2007. Disrupting the association of hepatitis C virus core protein with lipid droplets correlates with a loss in production of infectious virus. J Gen Virol 88:2204–2213. doi: 10.1099/vir.0.82898-0. [DOI] [PubMed] [Google Scholar]
- 10.Vogt DA, Camus G, Herker E, Webster BR, Tsou CL, Greene WC, Yen TS, Ott M. 2013. Lipid droplet-binding protein TIP47 regulates hepatitis C virus RNA replication through interaction with the viral NS5A protein. PLoS Pathog 9:e1003302. doi: 10.1371/journal.ppat.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV Jr, Ott M. 2010. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat Med 16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nevo-Yassaf I, Yaffe Y, Asher M, Ravid O, Eizenberg S, Henis YI, Nahmias Y, Hirschberg K, Sklan EH. 2012. Role for TBC1D20 and Rab1 in hepatitis C virus replication via interaction with lipid droplet-bound nonstructural protein 5A. J Virol 86:6491–6502. doi: 10.1128/JVI.00496-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salloum S, Wang H, Ferguson C, Parton RG, Tai AW. 2013. Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog 9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. 2012. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog 8:e1002845. doi: 10.1371/journal.ppat.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu S, Pei R, Guo M, Han Q, Lai J, Wang Y, Wu C, Zhou Y, Lu M, Chen X. 2012. Cytosolic phospholipase A2 gamma is involved in hepatitis C virus replication and assembly. J Virol 86:13025–13037. doi: 10.1128/JVI.01785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Backes P, Quinkert D, Reiss S, Binder M, Zayas M, Rescher U, Gerke V, Bartenschlager R, Lohmann V. 2010. Role of annexin A2 in the production of infectious hepatitis C virus particles. J Virol 84:5775–5789. doi: 10.1128/JVI.02343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger KL, Cooper JD, Heaton NS, Yoon R, Oakland TE, Jordan TX, Mateu G, Grakoui A, Randall G. 2009. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci U S A 106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. 2007. TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J Biol Chem 282:36354–36361. doi: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- 19.Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol 79:13473–13482. doi: 10.1128/JVI.79.21.13473-13482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camus G, Herker E, Modi AA, Haas JT, Ramage HR, Farese RV Jr, Ott M. 2013. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J Biol Chem 288:9915–9923. doi: 10.1074/jbc.M112.434910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iqbal J, McRae S, Banaudha K, Mai T, Waris G. 2013. Mechanism of hepatitis C virus (HCV)-induced osteopontin and its role in epithelial to mesenchymal transition of hepatocytes. J Biol Chem 288:36994–37009. doi: 10.1074/jbc.M113.492314. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Iqbal J, McRae S, Mai T, Banaudha K, Sarkar-Dutta M, Waris G. 2014. Role of hepatitis C virus induced osteopontin in epithelial to mesenchymal transition, migration and invasion of hepatocytes. PLoS One 9:e87464. doi: 10.1371/journal.pone.0087464. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Rangaswami H, Bulbule A, Kundu GC. 2006. Osteopontin: role in cell signaling and cancer progression. Trends Cell Biol 16:79–87. doi: 10.1016/j.tcb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Chakraborty G, Jain S, Kundu GC. 2008. Osteopontin promotes vascular endothelial growth factor-dependent breast tumor growth and angiogenesis via autocrine and paracrine mechanisms. Cancer Res 68:152–161. doi: 10.1158/0008-5472.CAN-07-2126. [DOI] [PubMed] [Google Scholar]
- 25.Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H. 2000. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287:860–864. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 26.Wai PY, Kuo PC. 2008. Osteopontin: regulation in tumor metastasis. Cancer Metastasis Rev 27:103–118. doi: 10.1007/s10555-007-9104-9. [DOI] [PubMed] [Google Scholar]
- 27.Cao DX, Li ZJ, Jiang XO, Lum YL, Khin E, Lee NP, Wu GH, Luk JM. 2012. Osteopontin as potential biomarker and therapeutic target in gastric and liver cancers. World J Gastroenterol 18:3923–3930. doi: 10.3748/wjg.v18.i30.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi SS, Claridge LC, Jhaveri R, Swiderska-Syn M, Clark P, Suzuki A, Pereira TA, Mi Z, Kuo PC, Guy CD, Pereira FE, Diehl AM, Patel K, Syn WK. 2014. Osteopontin is up-regulated in chronic hepatitis C and is associated with cellular permissiveness for hepatitis C virus replication. Clin Sci (Lond) 126:845–855. doi: 10.1042/CS20130473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whalen KA, Weber GF, Benjamin TL, Schaffhausen BS. 2008. Polyomavirus middle T antigen induces the transcription of osteopontin, a gene important for the migration of transformed cells. J Virol 82:4946–4954. doi: 10.1128/JVI.02650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao L, Li T, Wang Y, Pan Y, Ning H, Hui X, Xie H, Wang J, Han Y, Liu Z, Fan D. 2008. Elevated plasma osteopontin level is predictive of cirrhosis in patients with hepatitis B infection. Int J Clin Pract 62:1056–1062. doi: 10.1111/j.1742-1241.2007.01368.x. [DOI] [PubMed] [Google Scholar]
- 31.Morimoto J, Sato K, Nakayama Y, Kimura C, Kajino K, Matsui Y, Miyazaki T, Uede T. 2011. Osteopontin modulates the generation of memory CD8+ T cells during influenza virus infection. J Immunol 187:5671–5683. doi: 10.4049/jimmunol.1101825. [DOI] [PubMed] [Google Scholar]
- 32.Sarkis S, Belrose G, Peloponese JM Jr, Olindo S, Cesaire R, Mesnard JM, Gross A. 2013. Increased osteopontin expression in HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) patient cells is associated with IL-17 expression. J Clin Virol 58:295–298. doi: 10.1016/j.jcv.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Chagan-Yasutan H, Lacuesta TL, Ndhlovu LC, Oguma S, Leano PS, Telan EF, Kubo T, Morita K, Uede T, Dimaano EM, Hattori T. 2014. Elevated levels of full-length and thrombin-cleaved osteopontin during acute dengue virus infection are associated with coagulation abnormalities. Thromb Res 134:449–454. doi: 10.1016/j.thromres.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown A, Islam T, Adams R, Nerle S, Kamara M, Eger C, Marder K, Cohen B, Schifitto G, McArthur JC, Sacktor N, Pardo CA. 2011. Osteopontin enhances HIV replication and is increased in the brain and cerebrospinal fluid of HIV-infected individuals. J Neurovirol 17:382–392. doi: 10.1007/s13365-011-0035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chagan-Yasutan H, Saitoh H, Ashino Y, Arikawa T, Hirashima M, Li S, Usuzawa M, Oguma S, EF OT, Obi CL, Hattori T. 2009. Persistent elevation of plasma osteopontin levels in HIV patients despite highly active antiretroviral therapy. Tohoku J Exp Med 218:285–292. doi: 10.1620/tjem.218.285. [DOI] [PubMed] [Google Scholar]
- 36.Targett-Adams P, Hope G, Boulant S, McLauchlan J. 2008. Maturation of hepatitis C virus core protein by signal peptide peptidase is required for virus production. J Biol Chem 283:16850–16859. doi: 10.1074/jbc.M802273200. [DOI] [PubMed] [Google Scholar]
- 37.El-Hage N, Luo G. 2003. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral proteins and RNA. J Gen Virol 84:2761–2769. doi: 10.1099/vir.0.19305-0. [DOI] [PubMed] [Google Scholar]
- 38.Sumpter R Jr, Wang C, Foy E, Loo YM, Gale M Jr. 2004. Viral evolution and interferon resistance of hepatitis C virus RNA replication in a cell culture model. J Virol 78:11591–11604. doi: 10.1128/JVI.78.21.11591-11604.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goueslain L, Alsaleh K, Horellou P, Roingeard P, Descamps V, Duverlie G, Ciczora Y, Wychowski C, Dubuisson J, Rouille Y. 2010. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J Virol 84:773–787. doi: 10.1128/JVI.01190-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McRae S, Iqbal J, Sarkar-Dutta M, Lane S, Nagaraj A, Ali N, Waris G. 2016. The hepatitis C virus-induced NLRP3 inflammasome activates the sterol regulatory element-binding protein (SREBP) and regulates lipid metabolism. J Biol Chem 291:3254–3267. doi: 10.1074/jbc.M115.694059. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Bartenschlager R, Penin F, Lohmann V, Andre P. 2011. Assembly of infectious hepatitis C virus particles. Trends Microbiol 19:95–103. doi: 10.1016/j.tim.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Bukong TN, Kodys K, Szabo G. 2013. Human ezrin-moesin-radixin proteins modulate hepatitis C virus infection. Hepatology 58:1569–1579. doi: 10.1002/hep.26500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez-Vergès S, Camus G, Blot G, Beauvoir R, Benarous R, Berlioz-Torrent C. 2006. Tail-interacting protein TIP47 is a connector between Gag and Env and is required for Env incorporation into HIV-1 virions. Proc Natl Acad Sci U S A 103:14947–14952. doi: 10.1073/pnas.0602941103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carvalho FA, Carneiro FA, Martins IC, Assuncao-Miranda I, Faustino AF, Pereira RM, Bozza PT, Castanho MA, Mohana-Borges R, Da Poian AT, Santos NC. 2012. Dengue virus capsid protein binding to hepatic lipid droplets (LD) is potassium ion dependent and is mediated by LD surface proteins. J Virol 86:2096–2108. doi: 10.1128/JVI.06796-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo M, Pei R, Yang Q, Cao H, Wang Y, Wu C, Chen J, Zhou Y, Hu X, Lu M, Chen X. 2015. Phosphatidylserine-specific phospholipase A1 involved in hepatitis C virus assembly through NS2 complex formation. J Virol 89:2367–2377. doi: 10.1128/JVI.02982-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Presser LD, McRae S, Waris G. 2013. Activation of TGF-beta1 promoter by hepatitis C virus-induced AP-1 and Sp1: role of TGF-beta1 in hepatic stellate cell activation and invasion. PLoS One 8:e56367. doi: 10.1371/journal.pone.0056367. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gastaminza P, Kapadia SB, Chisari FV. 2006. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]