

ABSTRACT
Human cytomegalovirus (HCMV) protein pUL38 has been shown to prevent premature cell death by antagonizing cellular stress responses; however, the underlying mechanism remains unknown. In this study, we identified the host protein ubiquitin-specific protease 24 (USP24) as an interaction partner of pUL38. Mutagenesis analysis of pUL38 revealed that amino acids TFV at positions 227 to 230 were critical for its interaction with USP24. Mutant pUL38 TFV/AAA protein did not bind to USP24 and failed to prevent cell death induced by pUL38-deficient HCMV infection. Knockdown of USP24 suppressed the cell death during pUL38-deficient HCMV infection, suggesting that pUL38 achieved its function by antagonizing the function of USP24. We investigated the cellular pathways regulated by USP24 that might be involved in the cell death phenotype by testing several small-molecule compounds known to have a protective effect during stress-induced cell death. The iron chelators ciclopirox olamine and Tiron specifically protected cells from pUL38-deficient HCMV infection-induced cell death, thus identifying deregulated iron homeostasis as a potential mechanism. Protein levels of nuclear receptor coactivator 4 (NCOA4) and lysosomal ferritin degradation, a process called ferritinophagy, were also regulated by pUL38 and USP24 during HCMV infection. Knockdown of USP24 decreased NCOA4 protein stability and ferritin heavy chain degradation in lysosomes. Blockage of ferritinophagy by genetic inhibition of NCOA4 or Atg5/Atg7 prevented pUL38-deficient HCMV infection-induced cell death. Overall, these results support the hypothesis that pUL38 binds to USP24 to reduce ferritinophagy, which may then protect cells from lysosome dysfunction-induced cell death.
IMPORTANCE Premature cell death is considered a first line of defense against various pathogens. Human cytomegalovirus (HCMV) is a slow-replicating virus that encodes several cell death inhibitors, such as pUL36 and pUL37x1, which allow it to overcome both extrinsic and intrinsic mitochondrion-mediated apoptosis. We previously identified HCMV protein pUL38 as another virus-encoded cell death inhibitor. In this study, we demonstrated that pUL38 achieved its activity by interacting with and antagonizing the function of the host protein ubiquitin-specific protease 24 (USP24). pUL38 blocked USP24-mediated ferritin degradation in lysosomes, which could otherwise be detrimental to the lysosome and initiate cell death. These novel findings suggest that iron metabolism is finely tuned during HCMV infection to avoid cellular toxicity. The results also provide a solid basis for further investigations of the role of USP24 in regulating iron metabolism during infection and other diseases.
KEYWORDS: NCOA4, USP24, ferritinophagy, human cytomegalovirus, programmed cell death
INTRODUCTION
Apoptosis is a typical programmed cell death (PCD) which plays an important role in the development and maintenance of the steady state in tissues (1). Apoptosis also occurs as an antiviral response, by eliminating infected cells to prevent viral spread and persistent infection (2). Human cytomegalovirus (HCMV) is a slow-replicating betaherpesvirus that requires 48 to 72 h to begin producing progeny at peak levels in fibroblasts. It is therefore essential for HCMV to block premature cell death to allow it to complete its replication cycle. Indeed, HCMV has evolved multiple mechanisms to accomplish this. Some well-studied examples are as follows. pUL37x1, also known as viral mitochondrial inhibitor of apoptosis (vMIA), suppresses apoptosis by binding and inhibiting the proapoptotic factor Bax (3–8). The UL36-encoded viral inhibitor of caspase-8-induced apoptosis (vICA) inhibits Fas-mediated apoptosis by binding to the prodomain of caspase-8 and preventing its activation (9). HCMV also expresses a long noncoding RNA, β2.7 RNA, which binds to complex I of the respiratory transport chain to maintain its stability and sustain ATP production and cell viability (10). Mouse cytomegalovirus (MCMV) encodes multiple functionally conserved proteins, including the UL36 homolog M36 (11–13), the UL37x1 counterpart m38.5 (6, 14, 15), a viral inhibitor of Bak oligomerization by open reading frame m41.1 (16), and a viral inhibitor of RIP activation (vIRA) by M45 (17–19), to block apoptosis or necroptosis (20).
The HCMV UL38 open reading frame is located within the intron region of UL37 but is transcribed on its own to produce a protein of 331 amino acids (21). Terhune et al. reported that a mutant virus lacking the pUL38-coding sequence induced host cell apoptosis (22). Cells infected by the pUL38-deficient virus underwent a series of morphological changes, including cell shrinkage, membrane blebbing, vesicle release, and chromatin condensation and fragmentation (22). Further studies found that pUL38 was able to prevent endoplasmic reticulum (ER) stress-induced cell death by modulating the unfolded-protein response (23, 24). pUL38 was reported to interact with tuberous sclerosis protein 2 (TSC2) and activate mTORC1 (23), but we showed that this was not responsible for the cell death-inhibiting activity of pUL38 (25). The molecular mechanism underlying the antiapoptotic function of pUL38 thus requires further investigation.
In the current study, we identified ubiquitin-specific protease 24 (USP24) as a novel interaction partner of pUL38. Mutant pUL38 lacking its USP24 binding capacity failed to prevent cell death induced by pUL38-deficient HCMV infection. Inhibition of USP24 expression by RNA interference (RNAi) rescued cell viability during pUL38-deficient HCMV infection, demonstrating that pUL38 achieved its function by inhibiting the function of USP24. We also showed that pUL38 regulated ferritin degradation in the lysosome during HCMV infection by antagonizing USP24, which in turn inhibited autophagic ferritin degradation, thus maintaining lysosome integrity and cellular viability.
RESULTS
pUL38 interacts with USP24.
To further dissect the molecular mechanism of the antiapoptotic function of pUL38, we performed affinity purification in HEK293T cells to isolate pUL38 interaction partners (Table 1). A high-molecular-mass band of over 170 kDa that specifically copurified with pUL38 and pUL381–239 was repeatedly observed in multiple purification attempts (Fig. 1A). This band was identified as USP24 by mass spectrometry. A previous study also identified USP24 as a possible pUL38 interaction protein by affinity purification and mass spectrometry analysis in HCMV-infected fibroblasts (23), but it was not validated by additional experiments. We confirmed the interaction by coimmunoprecipitation (co-IP) and immunoblotting analysis. Flag-tagged pUL38 and pUL381–239 effectively pulled down USP24, detected by a USP24-specific antibody, while Flag-tagged green fluorescent protein (GFP) failed to do so (Fig. 1B, left panels). Reciprocal co-IP showed that pUL38 and pUL381–239 were specifically pulled down by a USP24 antibody (Fig. 1B, right panels). We also analyzed the interaction between pUL38 and USP24 in HCMV-infected human fibroblasts. pUL38 was copurified with USP24 using a polyclonal USP24 antibody but not using control rabbit IgG (Fig. 1C, top panels), and USP24 was copurified with pUL38 using a mouse monoclonal pUL38 antibody (Fig. 1C, bottom panels). These results demonstrated that pUL38 interacted with USP24, both in isolation and in the context of HCMV infection.
TABLE 1.
pUL38 protein partners identified by mass spectrometry
| Accession no. | Description | Size (kDa) | Peptide count | Cover (%) |
|---|---|---|---|---|
| Q9UPU5 | Ubiquitin carboxyl-terminal hydrolase 24 | 294.3 | 31 | 12.02 |
| Q5JSZ5 | Proline-rich coiled-coil protein 2B | 243.0 | 4 | 2.11 |
| Q6Y7W6 | GRB10-interacting GYF protein 2 | 150.0 | 3 | 2.54 |
| B4DE59 | cDNA FLJ60424, highly similar to junction plakoglobin | 62.6 | 3 | 4.97 |
| F5GWP8 | Keratin, type I cytoskeletal 17 | 66.3 | 3 | 4.74 |
| B4DW52 | Highly similar to actin, cytoplasmic 1 | 38.6 | 3 | 12.68 |
| P60709 | Actin, cytoplasmic 1 | 41.7 | 3 | 11.73 |
| Q1KLZ0 | HCG15971, isoform CRA_a | 41.7 | 3 | 11.73 |
| Q14207 | Nuclear protein of the ATM locus | 154.3 | 2 | 0.49 |
| B4DVW1 | Ataxia-telangiectasia locus (NPAT) | 52.6 | 2 | 1.45 |
| P35579 | Myosin 9 | 226.6 | 1 | 0.51 |
| A8MT79 | Putative zinc-alpha-2-glycoprotein-like 1 | 229.8 | 1 | 4.90 |
| P25311 | Zinc-alpha-2-glycoprotein | 34.3 | 1 | 3.36 |
| P68871 | Hemoglobin subunit beta | 16.0 | 1 | 8.84 |
| Q6ZR08 | Dynein heavy chain 12, axonemal | 356.9 | 1 | 0.26 |
| P46379 | Large proline-rich protein BAG6 | 119.4 | 1 | 1.15 |
| Q9Y312 | Protein AAR2 homolog | 43.5 | 1 | 2.34 |
| P50454 | Serpin H1 | 46.4 | 1 | 2.87 |
| O15382 | Branched-chain amino-acid aminotransferase, mitochondrial | 44.3 | 1 | 1.79 |
| P54753 | Ephrin type B receptor 3 | 110.3 | 1 | 0.70 |
FIG 1.
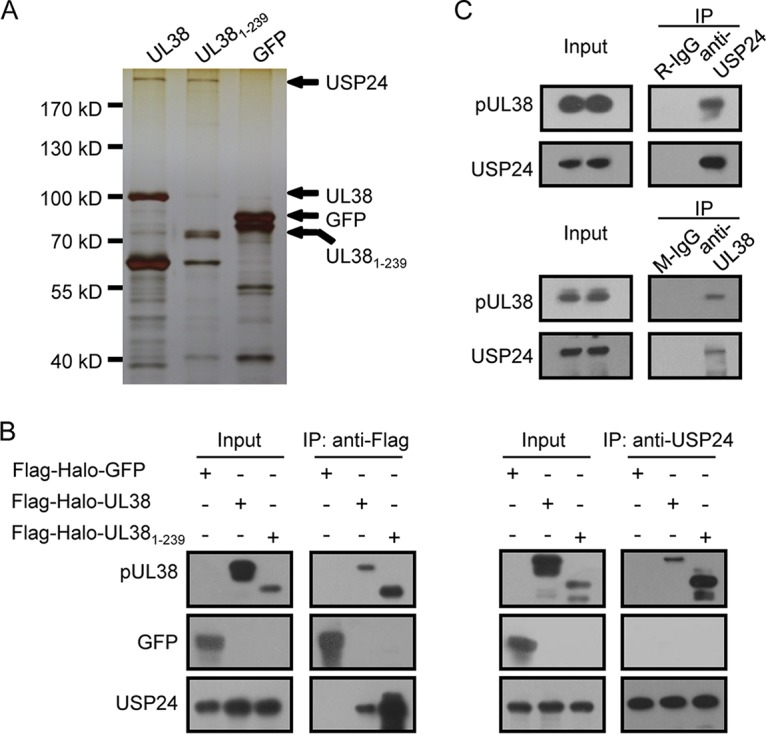
USP24 is an interaction partner of pUL38. (A) Protein complexes associated with each bait protein were isolated by affinity purification and separated by gel electrophoresis. The specific bands were distinguished by silver staining. Bait proteins Flag-Halo-UL38 (UL38), Flag-Halo-UL381–239 (UL381–239), and Flag-Halo-GFP (GFP) and key captured proteins are indicated. The Halo tag was originally used for tandem affinity purification, but one-step purification with the Flag tag was efficient enough for mass spectrometry analysis. (B) pUL38 interacts with USP24 in HEK293T cells. HEK293T cells were transfected with Flag-Halo-GFP-, Flag-Halo-UL38-, or Flag-Halo-UL381–239-expressing plasmid. Cell lysates were collected 48 h later and subjected to immunoprecipitation with anti-Flag beads (left panels) or with anti-USP24 antibody (right panels). Cell lysates and eluted proteins from the immunoprecipitation were analyzed by immunoblotting with the indicated antibodies. (C) pUL38 interacts with USP24 during HCMV infection. MRC5 cells were infected with HCMV at an MOI of 3. Cell lysates were prepared at 48 hpi, and immunoprecipitation was performed using a mouse monoclonal antibody against pUL38 (anti-pUL38) or a rabbit polyclonal antibody against USP24 (anti-USP24). Mouse IgG (M-IgG) or rabbit IgG (R-IgG) was used as a control. Cell lysates and eluted proteins from the immunoprecipitation were analyzed by immunoblotting with the indicated antibodies.
Interaction between pUL38 and USP24 is essential for blocking cell death.
To determine if the physical interaction between pUL38 and USP24 contributed to its antiapoptotic activity, we mapped the key residues of pUL38 required for its interaction with USP24. We previously showed that while pUL381–239 was fully functional, further deletion of another 14 amino acids from 226 to 239 rendered it incapable of blocking cell death (25). We reasoned that the amino acids in this region might mediate the pUL38-USP24 interaction and thus contribute to its cell death-inhibiting activity. We generated three pUL38 mutants, TFV228-230AAA, DR231-232AA, and DS233-234AA, by sequentially replacing each two or three amino acids with alanines. Co-IP and immunoblotting showed that DR/AA and DS/AA mutant pUL38 interacted with USP24 similarly to wild-type pUL38, but the TFV/AAA mutant protein failed to interact with USP24 (Fig. 2A and B). Importantly, DR/AA and DS/AA mutants retained the ability to prevent pUL38-deficient-HCMV-induced cell death in human fibroblasts, but the TFV/AAA mutant protein, which was unable to bind to USP24, failed to prevent cell death (Fig. 2C). Immunoblotting analysis showed that expression of the cell death marker cleaved poly(ADP-ribose) polymerase (PARP) was reduced in pUL38- but not pUL38 TFV-expressing cells during pUL38-deficient HCMV infection (Fig. 2D), indicating that the interaction between pUL38 and USP24 was indispensable for the antiapoptotic function of pUL38.
FIG 2.

The TFV motif in pUL38 is required for interaction with USP24 and cell death inhibition activity. (A and B) HEK293T cells were transfected with plasmids expressing Flag-tagged GFP, pUL38, or pUL38 mutant proteins as indicated. Cell lysates were prepared 48 h later, and immunoprecipitation was performed with anti-Flag beads (A) or anti-USP24 and protein A beads (B). Cell lysates and eluted proteins were then analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were transduced with lentivirus expressing the indicated proteins. Cell samples were prepared at 72 or 96 hpi with wild-type (wt) or pUL38-deficient HCMV. Cell viability was assessed by CellTiter-Glo luminescent cell viability assay. The data shown represent the mean ± SD from three independent experiments. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. (D) MRC5 cells were transduced with lentivirus expressing proteins as indicated. Cell lysates were prepared at 72 hpi with wild-type or pUL38-deficient HCMV at an MOI of 3. Samples were assayed by immunoblotting with the indicated antibodies.
USP24 downregulation prevents pUL38-deficient HCMV infection-induced cell death.
Having demonstrated the role of the pUL38-USP24 interaction in preventing cell death, we determined whether pUL38 acted by antagonizing the activity of USP24 using two USP24-specific short hairpin RNAs (shRNAs) expressed in human fibroblasts. Both shRNAs effectively downregulated USP24 protein expression, with shUSP24-1 being more efficient (Fig. 3D). Knockdown of USP24 by RNAi had no apparent effect on the morphology of wild-type-HCMV-infected human fibroblasts (Fig. 3A and B, left panels) but changed the morphology of pUL38-deficient-HCMV-infected MRC5 cells (Fig. 3A and B, right panels). More spindle-shaped adherent fibroblasts were observed in cells expressing USP24 shRNA (shUSP24-1 or shUSP24-2) than in control shRNA (shc)-expressing cells. These cells expressed higher levels of GFP driven by an expression cassette built into the HCMV genome and were likely to be more viable than the rounded cells expressing control shRNA (Fig. 3A and B, right panels). The phenotype was more obvious in shUSP24-1-expressing cells than in shUSP24-2-expressing cells, likely because the former downregulated USP24 more efficiently (Fig. 3D). These observations were confirmed by cell viability assays (Fig. 3C). Immunoblotting analysis showed that expression of the cell death marker cleaved PARP was reduced in USP24 shRNA-expressing cells during pUL38-deficient HCMV infection (Fig. 3D). These results suggest that pUL38 prevented cell death by antagonizing a function of USP24 during HCMV infection.
FIG 3.

HCMV pUL38 prevents cell death by inhibiting the function of USP24. (A and B) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or USP24-specific shRNA shUSP24-1 (A) or shUSP24-2 (B). Cells were then infected with wild type (wt) or pUL38-deficient HCMV (UL38 mut) at an MOI of 3. Images were taken at 72 hpi. (C) MRC5 cells were transduced and infected as for panels A and B, and cell viability was assessed at 72 or 96 hpi by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (D) MRC5 cells were transduced and infected as for panels A and B. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies.
Inhibition of USP24 makes cells less sensitive to ER stress-induced cell death.
We previously showed that pUL38 blocked ER stress-induced cell death when expressed alone in human fibroblasts (25). We therefore examined the role of USP24 in this process. As expected, MRC5 cells expressing pUL38 were resistant to cell death caused by the ER-stress inducers tunicamycin (TM) and thapsigargin (TG) (Fig. 4A). However, the TFV mutant that could not bind USP24 failed to prevent cell death. The viability of these cells was thus similar to that of the control vector cells (Fig. 4A), and PARP cleavage was not suppressed (Fig. 4B). We also determined whether USP24 knockdown was sufficient to prevent ER stress-induced cell death. As shown in Fig. 4C and D, the cell viability was higher in USP24-specific shRNA (shUSP24-1)-expressing cells than in control shRNA (shc)-expressing cells after TM and TG treatment, and PARP cleavage was reduced in shUSP24-1-expressing cells under the same experimental conditions. These results indicated that the pUL38-USP24 interaction was required for pUL38 to prevent ER stress-induced cell death in isolation, and suppression of USP24 was sufficient to reduce ER stress-induced cell death.
FIG 4.

pUL38 prevents ER stress-induced cell death by inhibiting the function of USP24. (A) MRC5 cells were transduced with lentivirus expressing the indicated proteins. Cell viability was assessed at 24 h after treatment with thapsigargin (TG) (2 μM) or tunicamycin (TM) (2 μg/ml) by CellTiter-Glo luminescent cell viability assay. The viability of dimethyl sulfoxide (DMSO)-treated cells was set as 100%, and the viabilities of TG- and TM-treated samples were normalized to that of DMSO-treated cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (B) MRC5 cells were transduced with lentivirus expressing the indicated proteins. Cell samples were prepared at 18 or 24 h after treatment with TG (2 μM) and analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or USP24-specific shRNA (shUSP24-1). Cell viability was assessed at 24 h after treatment with TG (2 μM) or TM (2 μg/ml) by CellTiter-Glo luminescent cell viability assay. The viability of DMSO-treated cells was set as 100%, and the viabilities of TG- and TM-treated samples were normalized to that of DMSO-treated cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (D) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or USP24-specific shRNA (shUSP24-1). Cell samples were prepared at 24 h after treatment with TG (2 μM) or TM (2 μg/ml) and analyzed by immunoblotting with the indicated antibodies.
Iron chelators prevent cell death induced by infection with pUL38-deficient HCMV.
We determined the events or pathways downstream of USP24 that initiated cell death during pUL38-deficient HCMV infection by screening small molecules known to inhibit ER stress-induced cell death by neutralizing reactive oxygen species (ROS). These molecules included Tiron, which showed a protective effect based on cell morphology and immunoblotting analysis of PARP cleavage (Fig. 5A). Tiron increased the number of spindle-shaped adherent cells (Fig. 5A) and reduced PARP cleavage (Fig. 5B), both indicating increased cell survival. Notably, however, two other commonly used ROS scavengers, Trolox and N-acetyl-l-cysteine (NAC), failed to prevent the cell-death phenotype (Fig. 5A and B), suggesting that Tiron protected against cell death via mechanisms other than neutralizing ROS. Tiron is known to act as an iron chelator, and we therefore determined if another iron chelator, ciclopirox olamine (CPX), could also protect the cells from pUL38-deficient-HCMV-induced cell death. As shown in Fig. 5C, CPX treatment increased cell viability similarly to Tiron treatment in our system, and both compounds reduced PARP cleavage (Fig. 5D). Previous studies indicated that HCMV replication was inhibited by iron chelators such as desferrioxamine (26, 27). However, the virus titer was unaffected in wild-type-HCMV-infected cells and was slightly upregulated in pUL38-deficient-HCMV-infected cells at 5 days postinfection following treatment with CPX and Tiron, potentially related to increased cell viability (Fig. 5E). These results indicated that iron was involved in pUL38-deficient-HCMV-induced cell death.
FIG 5.

CPX and Tiron rescue the viability of cells infected with pUL38-deficient virus. (A) MRC5 cells were infected with wild-type (wt) or pUL38-deficient (UL38 mut) HCMV at an MOI of 3. Tiron (200 μM), Trolox (25 μg/ml), NAC (5 mM), or DMSO (control) was added to cells infected with pUL38-deficient virus at the time of infection. Images were taken at 72 hpi. (B) Cells were infected and treated as for panel A. Cell lysates were collected and analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were treated with Tiron (200 μM), CPX (0.5 μM), or DMSO and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed at the indicated time points by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (D) MRC5 cells were mock infected or infected with-wild type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3 and then treated as for panel C. Cell lysates were collected at 72 hpi, and cleaved PARP and FTH1 were analyzed by immunoblotting. Antibodies to viral proteins, as indicated, and the cellular protein tubulin were used as infection and loading controls, respectively. (E) Growth analysis of wild-type (wt) or pUL38-deficient (mut) HCMV infection of MRC5 cells at an MOI of 3 with DMSO, CPX (0.5 μM), or Tiron (200 μM) treatment. Cell-free viruses in the supernatants were collected at the indicated days postinfection (dpi), and the virus titers were determined by 50% tissue culture infective dose (TCID50) assay.
pUL38 and USP24 regulate ferritin degradation in lysosomes during HCMV infection.
Iron is an essential element for life. Key enzymes in the respiratory chain and DNA metabolism require iron for their function. However, excess iron can be detrimental to life through initiation of ROS-dependent or -independent cell death (28, 29). The autophagic degradation of ferritin, a major intracellular iron storage protein, which is also called ferritinophagy, turns out to be a key mechanism maintaining iron homeostasis (30, 31). Upon iron depletion or starvation, nuclear receptor coactivator 4 (NCOA4) binds to ferritin heavy chain (FTH1) and targets it to lysosomes for degradation, thus releasing the stored iron. In addition to supplying cells with sufficient amounts of iron, uncontrolled ferritin degradation and increased amounts of labile free iron could also sensitize cells to cell death (32, 33). Based on this knowledge, we determined whether autophagic ferritin degradation was modulated by pUL38 and USP24, which could potentially explain the involvement of iron in pUL38-deficient-HCMV-induced cell death. NCOA4 protein levels were higher in pUL38-deficient-HCMV-infected cells than in the wild-type-HCMV-infected cells at 72 hpi (Fig. 6A, lanes 8 and 9), and the faster-migrating FTH1 band, corresponding to a lysosomal FTH1 degradation product (31), was increased in pUL38-deficient-HCMV-infected cells compared with wild-type-HCMV-infected cells (Fig. 6A, lanes 8 and 9), demonstrating decreased inhibition of lysosomal ferritin degradation in the absence of pUL38. Complementary expression of pUL38, but not the pUL38 TFV mutant, blocked ferritin degradation during pUL38-deficient HCMV infection in MRC5 cells, indicating that the interaction between pUL38 and USP24 was indispensable for pUL38 to regulate lysosomal ferritin degradation (Fig. 6B). As expected, FTH1 colocalized with the lysosome marker LAMP1 in pUL38-deficient- but not in wild-type-HCMV-infected cells (Fig. 6C and D). Knockdown of USP24 decreased NCOA4 protein levels and lysosomal FTH1 degradation at 72 hpi (Fig. 6A, lanes 9 and 12), suggesting that USP24 played a crucial role in directing FTH1 for lysosomal degradation. As expected, knockdown of USP24 reduced the colocalization between FTH1 and LAMP1 in pUL38-deficient-HCMV-infected cells (Fig. 6C and D), reflecting the decreased ferritinophagy. USP24 is a deubiquitinase and stabilizes substrates by removing ubiquitin. We therefore measured NCOA4 stability by cycloheximide (CHX) chase assay and showed that the stability of NCOA4 protein decreased after USP24 knockdown (Fig. 6E and F). Overall, these observations suggest that pUL38 and USP24 regulated NCOA4 stability and ferritin degradation in lysosomes during HCMV infection.
FIG 6.

pUL38 antagonizes the function of USP24 and prevents ferritin degradation in lysosomes. (A) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or USP24-specific shRNA (shUSP24-1) and then mock infected or infected with wild type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3. Cell lysates were collected at the indicated time points after virus infection and analyzed by immunoblotting with the indicated antibodies. (B) MRC5 cells were transduced with empty vector (vector), or with pUL38- or pUL38 TFV/AAA-expressing lentivirus and then mock infected or infected with wild type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were transduced with mCherry-FTH1- and shc/shUSP24-1-expressing lentiviruses and then infected with wild-type (wt) or pUL38-deficient (mut) HCMV at an MOI of 0.3. Cells were fixed at 96 hpi and stained with the indicated antibodies. Images were taken using an Olympus FV1200 laser scanning microscope. (D) Colocalization of mCherrry-FTH1 and LAMP1 was assessed using ImageJ. Data shown represent the mean ± SD for nine cells. ***, P < 0.001; ns, not significant. (E) RNAi knockdown of USP24 reduces NCOA4 protein stability. MRC5 cells were transduced with shc/shUSP24-1-expressing lentivirus and then treated with 100 μg/ml cycloheximide (CHX). Cell lysates were collected at the indicated time points and analyzed by immunoblotting. (F) Quantification of NCOA4 protein levels in two independent CHX chase experiments. Data shown represent the mean ± SD from two independent experiments.
Inhibition of autophagic ferritin degradation prevents pUL38-deficient HCMV infection-induced cell death.
To clarify the relationship between USP24-mediated ferritinophagy and pUL38-deficient-HCMV-induced cell death, we determined whether the cell death phenotype could be prevented by blocking ferritinophagy via genetic inhibition of NCOA4, Atg5/Atg7, or FTH1 itself. Elimination of NCOA4 expression by RNAi knockdown significantly blocked cell death (Fig. 7A), PARP cleavage, and ferritin degradation in lysosomes in MRC5 cells infected with pUL38-deficient HCMV (Fig. 7B). Atg5 and Atg7 are critical for the formation of the autophagosome (34). Knockdown of Atg5 or Atg7 successfully blocked pUL38-deficient HCMV infection-induced cell death, possibly because Atg5 or Atg7 depletion diminished lysosomal activation and subsequent ferritinophagy (Fig. 7C to E). Interestingly, knockdown of FTH1 itself also blocked pUL38-deficient-HCMV-induced cell death (Fig. 7F and G), suggesting that lysosomal degradation of FTH1 was a pivotal process for pUL38-deficient-HCMV-induced cell death. These results, together with the evidence that pharmacologic treatment with CPX and Tiron successfully blocked pUL38-deficient HCMV infection-induced cell death (Fig. 5C and D), suggested that autophagic ferritin degradation and the release of free iron into the lysosome was essential for pUL38-deficient HCMV infection-induced cell death. pUL38 prevented these events by antagonizing the activity of USP24.
FIG 7.

Inhibition of ferritinophagy prevents pUL38-deficient HCMV infection-induced cell death. (A) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or NCOA4-specific shRNA (shNCOA4-1 or shNCOA4-2) and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed at the indicated time points by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. **, P < 0.01; ***, P < 0.001. (B) Control and NCOA4 knockdown MRC5 cells were mock infected or infected with wild-type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or Atg5- or Atg7-specific shRNA (shAtg5/shAtg7) and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed at the indicated time points by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (D) Control and Atg5 or Atg7 knockdown MRC5 cells were mock infected or infected with wild-type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies. (E) Knockdown efficiencies of Atg5 and Atg7 were quantified by qRT-PCR. (F) MRC5 cells were transduced with lentivirus expressing control shRNA (shc) or FTH1-specific shRNA (shFTH1-1 or shFTH1-2) and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed at the indicated time points by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. **, P < 0.01; ***, P < 0.001. (G) Control and FTH1 knockdown MRC5 cells were mock infected or infected with wild-type (wt) or pUL38-deficient (mut) HCMV at an MOI of 3. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies.
pUL38 inhibits LMP-mediated cell death.
Intralysosomal iron produced by ferritin degradation in lysosomes induces lysosomal membrane permeabilization (LMP) and cathepsin leakage (35). The release of lysosomal cathepsin into the cytosol induces caspase activation and apoptosis (36–38). As expected, cathepsin L leakage was increased in cells infected with pUL38-deficient HCMV compared with wild-type HCMV (Fig. 8A). Furthermore, cathepsin L leakage was decreased by treatment with iron chelators in cells infected with pUL38-deficient HCMV (Fig. 8B). In addition, combination treatment with the cathepsin inhibitors pepstatin A and E64d increased the viability (Fig. 8C) and reduced PARP cleavage (Fig. 8D) of cells infected with pUL38-deficient HCMV. These findings suggest that pUL38 inhibited LMP-mediated cell death by regulating ferritin degradation in lysosomes.
FIG 8.
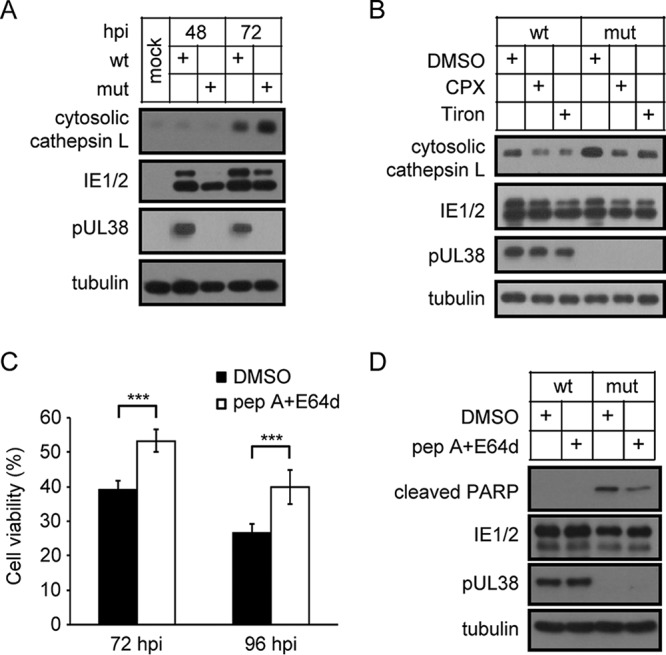
pUL38 inhibits cathepsin leakage-mediated cell death. (A) MRC5 cells were infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell samples were collected 72 h later. Cytosolic proteins were separated and analyzed by immunoblotting with the indicated antibodies. (B) MRC5 cells were treated with Tiron (200 μM), CPX (0.5 μM), or DMSO and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell samples were collected 72 h later. Cytosolic proteins were separated and analyzed by immunoblotting with the indicated antibodies. (C) MRC5 cells were treated with pepstatin A (pep A) (25 μM) plus E64d (10 μM) or with DMSO and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed at the indicated time points by CellTiter-Glo luminescent cell viability assay. The viability of cells infected with wild-type HCMV infected cells was set as 100%, and the viability of cells infected with pUL38-deficient HCMV was normalized to that of wild-type-HCMV-infected cells. Data shown represent the mean ± SD from three independent experiments. ***, P < 0.001. (D) MRC5 cells were treated with pepstatin A (pep A) (25 μM) plus E64d (10 μM) or with DMSO and then infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell lysates were collected at 72 hpi and analyzed by immunoblotting with the indicated antibodies.
DISCUSSION
Programmed cell death plays a pivotal role in regulating HCMV replication (5, 22, 39), and a panel of viral proteins, including pUL36, pUL37x1, pUL38, and some noncoding RNAs, are utilized to overcome PCD (3–10, 22, 25, 40–43). Although pUL38 has been shown to prevent cell death by targeting the unfolded-protein response pathway (24), the detailed molecular mechanism remains elusive.
In the current study, we identified the host protein USP24 as a novel interaction partner of pUL38 by immunoprecipitation-mass spectrometry analysis (Fig. 1). This interaction is likely to be an important factor enabling pUL38 to block cell death, given that a mutant pUL38 protein incapable of binding USP24 failed to block cell death (Fig. 2 and 4A and B). pUL38 fulfills its cell death inhibition activity by antagonizing USP24, and RNAi knockdown of USP24 rescued cell viability during pUL38-deficient HCMV infection or ER stress (Fig. 3 and 4C and D).
Mechanistically, pUL38 restrained the positive effect of USP24 on ferritinophagy, whereby ferritin is degraded in lysosomes and releases free iron. Our results were consistent with the hypothesis that pUL38 suppresses ferritinophagy by modulating the activity of USP24, which in turn reduces the release of free iron that could cause cell death during HCMV infection. First, the iron chelators CPX and Tiron specifically protected cells from pUL38-deficient HCMV infection-induced cell death (Fig. 5), indicating that iron plays a role in PCD under these conditions. Second, more FTH1 was colocalized with lysosomes (Fig. 6C), and the lysosomal degradation of FTH1 was enhanced in pUL38-deficient-HCMV-infected cells (Fig. 6A), indicating increased ferritinophagy. Third, knockdown of USP24 reduced the lysosomal degradation of FTH1 (Fig. 6A) and prevented cell death during pUL38-deficent HCMV infection (Fig. 3), suggesting that pUL38 blocks cell death by inhibiting USP24-mediated ferritinophagy. Fourth, a mutant pUL38 protein incapable of binding to USP24 was unable to suppress ferritinophagy or cell death (Fig. 2) (Fig. 6B). Finally, inhibition of ferritinophagy by knockdown of NCOA4, Atg5/Atg7, or FTH1 itself prevented pUL38-deficient HCMV infection-induced cell death (Fig. 7). Overall, these results support our hypothesis that pUL38 antagonizes USP24 and prevents premature cell death during HCMV infection by suppressing ferritinophagy (Fig. 9).
FIG 9.

Model of pUL38- and USP24-mediated regulation of ferritin degradation in lysosomes and cell death. pUL38 binds to USP24 in cells infected with wild-type HCMV to inhibit its function in stabilizing NCOA4. NCOA4 is stabilized by USP24 in cells infected with pUL38-deficient HCMV. Increased NCOA4 binds to ferritin and promotes ferritin degradation in lysosomes, resulting in iron accumulation in the lysosomes and lysosomal membrane permeabilization. Release of cathepsin into the cytosol induces caspase activation and cell death.
USP24 is a deubiquitinase (44), and it was identified as a potential interaction partner of pUL38 (23) in the current study. Single-nucleotide polymorphisms in the USP24 gene have been reported to be associated with Parkinson's disease (45, 46). Furthermore, several studies have demonstrated that USP24 regulated cell survival in various contexts through modulating the protein stability of some of its substrates, such as DDB2 (47), Mcl-1 (48), and p53 (49, 50). However, to the best of our knowledge, the role of USP24 in modulating ferritinophagy and iron metabolism has not previously been reported.
Iron is essential for various cellular biochemistry processes, forming complexes such as hemoglobin and myoglobin. Replication of and chronic infection with many viruses, including HCMV, also rely on iron, and HCMV replication was inhibited by treatment with an iron chelator (26, 27). The HCMV viral protein US2 was reported to increase cellular iron uptake by targeting the transferrin receptor inhibitor HFE for proteasome-mediated degradation (51). When iron absorption is enhanced during infection, excess iron must be controlled to avoid iron overload-induced cell death. Ferritin is a globular protein complex consisting of 24 subunits, including FTH1 and ferritin light chain (FTL), with important roles in iron storage and homeostasis (52). Ferritin has been reported to be degraded in the lysosome under iron-depleted conditions, which is crucial for iron extraction from ferritin and utilization by cells (53). However, uncontrolled lysosomal release of iron induces LMP and cathepsin leakage-mediated cell death during oxidative stress (32, 35). Lysosomal cell death is executed mainly as a result of the cytosolic leakage of lysosomal cathepsin proteases, leading to necrosis or apoptosis, depending on the degree of leakage and the specific cellular context (36, 54–57). In the current study, cytosolic leakage of lysosomal cathepsin was increased in pUL38-deficient-HCMV-infected cells and decreased by treatment with iron chelators (Fig. 8A and B). In addition, inhibition of cathepsin activity by pepstatin A and E64d successfully rescued the viability of cells infected with pUL38-deficient HCMV (Fig. 8C and D), thus supporting our hypothesis that pUL38 antagonizes USP24 and suppresses ferritinophagy to block cathepsin leakage-induced premature cell death during HCMV infection.
NCOA4 was recently identified as an adaptor for ferritin degradation via ferritinophagy, a target-specific autophagic turnover of ferritin (31). Here, we showed that the host protein USP24 also regulated ferritinophagy, at least during HCMV infection. The HCMV protein pUL38 binds to and suppresses USP24, which in turn inhibits ferritinophagy and the premature death of infected cells. Further studies are needed to determine whether USP24 modulates ferritinophagy and iron metabolism under other physiological and pathological conditions and during other infections and to establish the precise mechanisms whereby USP24 modulates ferritinophagy.
MATERIALS AND METHODS
Plasmids and reagents.
The following lentiviral overexpression vectors were derived from pLKO.DCMV.tetO (58). All of the following plasmids carried a 3×Flag tag at their N termini. pLKO-3×Flag-UL38 expressed the pUL38 protein, and pLKO-3×Flag-GFP expressed the green fluorescent protein. Amino acids 228 to 230, 231 to 232, and 233 to 234 of pUL38 were mutated to alanines by overlapping PCR and cloned into the pLKO.DCMV.tetO vector, producing pLKO-3×Flag-UL38 TFV/AAA, pLKO-3×Flag-UL38 DR/AA, and pLKO-3×Flag-UL38 DS/AA. Primers for introducing the mutations are listed in Table 2. pLKO-3×Flag-Halo-UL38, pLKO-3×Flag-Halo-UL381–239, and pLKO-3×Flag-Halo-GFP all carried a 3×Flag tag and a Halo tag at their N termini. All plasmids were verified by restriction enzyme analysis and DNA sequencing.
TABLE 2.
Primers used to introduce mutations into the pUL38-coding sequence
| Mutant | Primers used to introduce mutationsa |
|---|---|
| TFV/AAA | 5′-CAGCGAGTCGCGATCGGCGGCGGCCATAGGAATTTTCCCTTCGC-3′ |
| 5′-GCCGCCGCCGATCGCGACTCGCTG-3′ | |
| DR/AA | 5′-CAGCGAGTCGGCGGCCACGAAGGTCATAGGAATTTTCCCTTCGC-3′ |
| 5′-ACCTTCGTGGCCGCCGACTCGCTG-3′ | |
| DS/AA | 5′-ATTGGCTCGCAGGGCGGCGCGATCCACGAAGGTCATAGGAATTT-3′ |
| 5′-GTGGATCGCGCCGCCCTGCGAGCCAAT-3′ |
Underlining indicates nucleotide substitutions introduced into the pUL38-coding sequence.
The iron chelators Tiron and ciclopirox olamine (CPX), the ROS scavengers Trolox and N-acetyl-l-cysteine (NAC), the ER stress inducers tunicamycin (TM) and thapsigargin (TG), the cathepsin inhibitors pepstatin A and E64d, and the eukaryote translation inhibitor cycloheximide (CHX) were purchased from Sigma-Aldrich. Primary antibodies used in this study included the following: anti-GFP, anti-alpha-tubulin, and anti-USP24 (Proteintech); anti-cleaved poly(ADP-ribose) polymerase (PARP) and anti-LAMP1 (Cell Signaling Technology); anti-Flag (Abmart); anti-pUL44 (Virusys); anti-NCOA4 (Sigma-Aldrich); anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Hangzhou Xianzhi); anti-ferritin heavy chain (FTH1) (Santa Cruz); anti-cathepsin L (R&D Systems); anti-IE1/2 (a generous gift from Jay Nelson, Oregon Health & Science University); and anti-pUL38 and anti-pp28 (gifts from Thomas Shenk, Princeton University). Alexa Fluor 647 goat anti-rabbit IgG(H+L) secondary antibody was purchased from Invitrogen.
Cell culture, transfection, and transduction.
HEK293T cells (kindly provided by Bin Li, Shanghai Jiao Tong University), primary human foreskin fibroblasts (HFFs), and human embryonic lung fibroblasts (MRC5 cells) were routinely cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS). HFFs and MRC5 cells were obtained from ATCC. 293T cells were transfected with the indicated plasmid together with the packaging plasmids 9.2ΔR and vesicular stomatitis virus G (VSV-G) to produce lentivirus stocks. MRC5 cells were transduced with lentivirus supplemented with 5 μg/ml Polybrene and selected with 2 μg/ml puromycin (Sigma-Aldrich) for approximately 3 days to generate a pool of cells expressing the protein of interest.
Viruses.
Bacterial artificial chromosome (BAC) pAD-GFP carried the green fluorescent protein (GFP)-tagged genome of the HCMV AD169 strain and was used to produce wild-type virus ADwt (59). BAC pADpmUL38 was used to produce pUL38-deficient virus ADpmUL38. pADpmUL38 carried multiple point mutations and was constructed by two-step linear recombination as previously described (24, 59). Briefly, in the first step, the PCR fragment containing a GalK/kanamycin dual-marker cassette was used to replace the UL38 region of pAD-GFP. In the second step, linear recombination, the marker cassette was replaced by the PCR fragment of the UL38 gene that contained the desired mutations, including nonsense mutations at His6 and Met12,and a 2-bp insertion (GC) at Met96 to introduce frameshift mutation (24).
To reconstitute the virus, 2 μg of the indicated BAC DNA and 1 μg of the pp71-overexpressing plasmid were transfected into HFFs as described previously (21). At 24 hours after transfection, the culture medium was replaced with fresh medium. The supernatant was collected when 100% of the cell monolayer was lysed.
shRNA knockdown.
shRNA-expressing lentiviral vectors were constructed based on the pLKO.1 vector. The shRNA targeting sequences used in this study are listed in Table 3. Lentivirus preparation and transduction were the same as described above. Pools of MRC5 cells stably expressing the respective shRNAs were used in the experiments.
TABLE 3.
shRNA targeting sequences
| Name | shRNA targeting sequence |
|---|---|
| shUSP24-1 | GAAACTCAGGGTTGATACT |
| shUSP24-2 | GGAAGCTTCTGCTCTTGATAC |
| shFTH1-1 | GCTATCTTCCAGATTCCTTAA |
| shFTH1-2 | CCTGTCCATGTCTTACTACTT |
| shNCOA4-1 | GGTATTGTAGCTGTCCCTTTC |
| shNCOA4-2 | GCAAACCTGCCAGTGGTTATC |
| shAtg5 | AGATTGAAGGATCAACTATTT |
| shAtg7 | GCTTCCAGAAATGGCATTTAG |
Assays of cell death/apoptosis.
Confluent MRC5 cells were infected at a multiplicity of infection (MOI) of 3 in DMEM containing 10% FBS. At the indicated time points, cells were analyzed for morphological and biochemical changes indicating premature cell death as previously reported (22, 24). These assays included microscopic analysis for cell morphology and immunoblotting analysis for elevated PARP cleavage. For microscopic analysis, MRC5 cells were examined under a phase-contrast microscope at 72 h postinfection (hpi) for their cell morphology. Dying cells were detached from the culture plate and fragmented, while surviving cells remained attached and maintained a fibroblast-like morphology. For induction of elevated PARP cleavage, cell lysates were analyzed by immunoblotting using the antibody specific to the cleaved forms of PARP.
IP and immunoblotting.
Coimmunoprecipitation (co-IP) was done either in MRC5 cells infected with HCMV at an MOI of 3 at 48 hpi or in 293T cells transfected with plasmids of interest. Cells were lysed in lysis buffer, containing 120 mM NaCl, 40 mM HEPES (pH 7.4), 1 mM EDTA, 0.3% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS) (Sigma-Aldrich), protease inhibitor cocktail (PIC) (Roche), and phosphatase inhibitor cocktail (PHIC) (Sigma-Aldrich), with rotation for 1 h at 4°C. After centrifugation at 12,000 rpm for 15 min at 4°C, supernatants were taken and incubated with Flag antibody-conjugated magnetic beads (Sigma-Aldrich) for 1 h at room temperature, washed with wash buffer (120 mM NaCl, 40 mM HEPES, 1 mM EDTA, 0.3% CHAPS) five times, and eluted with 0.2 mg/ml Flag peptide (Sigma-Aldrich). The eluted proteins were analyzed by immunoblotting.
For mass spectrometry analysis, 10 plates of 293T cells were used for immunoprecipitation. The eluted proteins were concentrated with an Amicon Ultra centrifugal filter (Millipore) and separated in a 4 to 12% SDS-containing polyacrylamide gel, and the specific bands were cut and sent to Shanghai PSI Biotechnology Co., Ltd., for mass spectrometry analysis.
Proteins were analyzed by immunoblotting as described previously (24). Briefly, cells were washed with ice-cold phosphate-buffered saline (PBS) once and then lysed in protein sample buffer supplemented with protease inhibitor cocktail (PIC) (Roche) and phosphatase inhibitor cocktail (PHIC) (Sigma-Aldrich). Proteins from equal cell numbers were separated by electrophoresis on an SDS-containing polyacrylamide gel, transferred to a polyvinylidene difluoride (PVDF) membrane (0.45 μm; Millipore), hybridized with primary antibodies, reacted with horseradish peroxidase (HRP)-conjugated secondary antibodies, and visualized with Clarity Western ECL substrate (Bio-Rad).
qRT-PCR.
Quantitative real-time reverse transcription-PCRs (qRT-PCRs) were performed in an ABI7900HT instrument (Applied Biosystems) using SYBR Premix Ex Taq (TaKaRa). Each sample was analyzed in duplicate with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. Total RNA was extracted using the TRIzol reagent (Invitrogen). cDNA was reverse transcribed with oligo(dT) primer and random 6-mers using the Primerscript RT reagent kit (TaKaRa). cDNA was then quantified using SYBR Premix Ex Taq (TaKaRa) with specific primer pairs for the gene of interest or the GAPDH gene. The sequences of the qRT-PCR primers used in this study are listed in Table 4.
TABLE 4.
Primers used for qRT-PCR
| Name | Primer sequences |
|---|---|
| GAPDH | 5′-CTGTTGCTGTAGCCAAATTCGT-3′ |
| 5′-ACCCACTCCTCCACCTTTGAC-3′ | |
| Atg5 | 5′-AAAGATGTGCTTCGAGATGTGT-3′ |
| 5′-CACTTTGTCAGTTACCAACGTCA-3′ | |
| Atg7 | 5′-ATGATCCCTGTAACTTAGCCCA-3′ |
| 5′-CACGGAAGCAAACAACTTCAAC-3′ |
Cell viability assay.
MRC5 cells were seeded at a density of 8,000 cells per well in 96-well plates and allowed to attach overnight. Cells were infected with wild-type or pUL38-deficient HCMV at an MOI of 3. Cell viability was assessed by using the CellTiter-Glo luminescent cell viability assay (Promega). Cells were lysed by adding 100 μl of reagent per well in 96-well plates at indicated time points. Luminescence was recorded with a Thermo Scientific Varioskan Flash multimode reader. The cell viability of wild-type-HCMV-infected cells was set as 100%. The cell viability of pUL38-deficient-HCMV-infected samples was normalized to that of wild-type-HCMV-infected cells. The results are presented as the mean ± standard deviation (SD) (n = 3). Statistical analyses were performed with the paired Student t test. The threshold of significance was set at a P value of <0.05.
Immunofluorescence microscopy.
Cells grown on glass coverslips were fixed in 2% paraformaldehyde (in PBS) for 20 min at room temperature and permeabilized with 0.1% Triton X-100 (in PBS) for 15 min at room temperature. The cells were blocked with 5% FBS (in PBS) for 20 min before subsequent incubation with primary and fluorescence-labeled secondary antibodies. To visualize the nuclei, cells were counterstained with DAPI (4,6-diamidino-2-phenylindole) (Beyotime) for 10 min. Finally, labeled cells were mounted on slides with Prolong Gold antifade reagent (Invitrogen-Molecular Probes) overnight. Images were captured using an Olympus FV1200 confocal laser scanning microscope at a magnification of ×60.
Detection of lysosomal permeability.
For cytosolic extraction, the final concentration of digitonin was 25 μg/ml. Briefly, cell samples were collected and extracted with digitonin lysis buffer (25 μg/ml digitonin [300410; Calbiochem] in lysis buffer containing 1 mM Pefabloc [76307-100MG; Sigma] and 8 mM dithiothreitol [DTT] at pH 7.5). Cells were gently incubated for 10 min on ice with intermittent upside-down rotation every 2 min. The samples were then spun down (13,000 rpm, 90 s, 4°C), the supernatant was quickly transferred to a new tube, 3× SDS sample buffer was added, and the mixture was boiled at 100°C for 10 min. The samples were analyzed by immunoblotting. Cytosolic cathepsin L was used as a marker for lysosomal membrane permeabilization (LMP) and cathepsin leakage.
ACKNOWLEDGMENTS
We thank all the members of the Herpesvirus and Molecular Virology Research Unit for helpful discussions, Jay Nelson for the IE1/2 antibody, Thomas Shenk for antibodies, and Bin Li for HEK293T cells. We thank the molecular imaging core facility at the Institut Pasteur of Shanghai for the confocal laser scanning microscope.
This work was supported by the National Natural Science Foundation of China (grants 81371826 and 81572002 to Z.Q. and grants 31300148 and 31570169 to B.X.), the Ministry of Science and Technology of China (2016YFA0502101), and the Chinese Academy of Sciences “100 Talents” program to Z.Q. B.X. was supported by the Youth Innovation Promotion Association, CAS.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Gavrieli Y, Sherman Y, Ben-Sasson SA. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cuconati A, White E. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16:2465–2478. doi: 10.1101/gad.1012702. [DOI] [PubMed] [Google Scholar]
- 3.Goldmacher VS. 2002. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie 84:177–185. doi: 10.1016/S0300-9084(02)01367-6. [DOI] [PubMed] [Google Scholar]
- 4.McCormick AL, Smith VL, Chow D, Mocarski ES. 2003. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol 77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reboredo M, Greaves RF, Hahn G. 2004. Human cytomegalovirus proteins encoded by UL37 exon 1 protect infected fibroblasts against virus-induced apoptosis and are required for efficient virus replication. The J Gen Virol 85:3555–3567. doi: 10.1099/vir.0.80379-0. [DOI] [PubMed] [Google Scholar]
- 6.Norris KL, Youle RJ. 2008. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J Virol 82:6232–6243. doi: 10.1128/JVI.02710-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma J, Edlich F, Bermejo GA, Norris KL, Youle RJ, Tjandra N. 2012. Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc Natl Acad Sci U S A 109:20901–20906. doi: 10.1073/pnas.1217094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang A, Hildreth RL, Colberg-Poley AM. 2013. Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J Virol 87:5657–5668. doi: 10.1128/JVI.00145-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormick AL, Roback L, Livingston-Rosanoff D, St Clair C. 2010. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J Virol 84:5108–5123. doi: 10.1128/JVI.01345-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. 2007. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316:1345–1348. doi: 10.1126/science.1142984. [DOI] [PubMed] [Google Scholar]
- 11.Menard C, Wagner M, Ruzsics Z, Holak K, Brune W, Campbell AE, Koszinowski UH. 2003. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J Virol 77:5557–5570. doi: 10.1128/JVI.77.10.5557-5570.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cicin-Sain L, Ruzsics Z, Podlech J, Bubic I, Menard C, Jonjic S, Reddehase MJ, Koszinowski UH. 2008. Dominant-negative FADD rescues the in vivo fitness of a cytomegalovirus lacking an antiapoptotic viral gene. J Virol 82:2056–2064. doi: 10.1128/JVI.01803-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebermann L, Ruzsics Z, Guzman CA, van Rooijen N, Casalegno-Garduno R, Koszinowski U, Cicin-Sain L. 2012. Block of death-receptor apoptosis protects mouse cytomegalovirus from macrophages and is a determinant of virulence in immunodeficient hosts. PLoS Pathog 8:e1003062. doi: 10.1371/journal.ppat.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurak I, Schumacher U, Simic H, Voigt S, Brune W. 2008. Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol 82:4812–4822. doi: 10.1128/JVI.02570-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arnoult D, Skaletskaya A, Estaquier J, Dufour C, Goldmacher VS. 2008. The murine cytomegalovirus cell death suppressor m38.5 binds Bax and blocks Bax-mediated mitochondrial outer membrane permeabilization. Apoptosis 13:1100–1110. doi: 10.1007/s10495-008-0245-2. [DOI] [PubMed] [Google Scholar]
- 16.Cam M, Handke W, Picard-Maureau M, Brune W. 2010. Cytomegaloviruses inhibit Bak- and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ 17:655–665. doi: 10.1038/cdd.2009.147. [DOI] [PubMed] [Google Scholar]
- 17.Upton JW, Kaiser WJ, Mocarski ES. 2008. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem 283:16966–16970. doi: 10.1074/jbc.C800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mack C, Sickmann A, Lembo D, Brune W. 2008. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci U S A 105:3094–3099. doi: 10.1073/pnas.0800168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upton JW, Kaiser WJ, Mocarski ES. 2010. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe 7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brune W, Andoniou CE. 2017. Die another day: inhibition of cell death pathways by cytomegalovirus. Viruses 9:E249. doi: 10.3390/v9090249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J Virol 83:3463–3474. doi: 10.1128/JVI.02307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian Z, Xuan B, Gualberto N, Yu D. 2011. The human cytomegalovirus protein pUL38 suppresses endoplasmic reticulum stress-mediated cell death independently of its ability to induce mTORC1 activation. J Virol 85:9103–9113. doi: 10.1128/JVI.00572-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cinatl J Jr, Cinatl J, Rabenau H, Gumbel HO, Kornhuber B, Doerr HW. 1994. In vitro inhibition of human cytomegalovirus replication by desferrioxamine. Antiviral Res 25:73–77. doi: 10.1016/0166-3542(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 27.Gumbel H, Cinatl J Jr, Rabenau H, Vogel JU, Doerr HW, Ohrloff C. 1995. Selective inhibition of replication of human cytomegalovirus by desferrioxamine in vitro and in vivo (case report). Ophthalmologe 92:840–843. [PubMed] [Google Scholar]
- 28.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. 2016. Ferroptosis: process and function. Cell Death Differ 23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dixon SJ, Stockwell BR. 2014. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 30.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, Cantwell J, Luu C, Cornella-Taracido I, Harrington E, Fekkes P, Lei H, Fang Q, Digan ME, Burdick D, Powers AF, Helliwell SB, D'Aquin S, Bastien J, Wang H, Wiederschain D, Kuerth J, Bergman P, Schwalb D, Thomas J, Ugwonali S, Harbinski F, Tallarico J, Wilson CJ, Myer VE, Porter JA, Bussiere DE, Finan PM, Labow MA, Mao X, Hamann LG, Manning BD, Valdez RA, Nicholson T, Schirle M, Knapp MS, Keaney EP, Murphy LO. 2014. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16:1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 32.Kurz T, Gustafsson B, Brunk UT. 2011. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Rad Biol Med 50:1647–1658. doi: 10.1016/j.freeradbiomed.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ III, Kang R, Tang D. 2016. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine B, Klionsky DJ. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 35.Lin Y, Epstein DL, Liton PB. 2010. Intralysosomal iron induces lysosomal membrane permeabilization and cathepsin D-mediated cell death in trabecular meshwork cells exposed to oxidative stress. Invest Ophthalmol Vis Sci 51:6483–6495. doi: 10.1167/iovs.10-5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S, Simon HU. 2008. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 205:685–698. doi: 10.1084/jem.20072152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conus S, Pop C, Snipas SJ, Salvesen GS, Simon HU. 2012. Cathepsin D primes caspase-8 activation by multiple intra-chain proteolysis. J Biol Chem 287:21142–21151. doi: 10.1074/jbc.M111.306399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu ZQ, Jia Y, Chen G. 2014. Possible involvement of cathepsin B/D and caspase-3 in deferoxamine-related neuroprotection of early brain injury after subarachnoid haemorrhage in rats. Neuropathol Appl Neurobiol 40:270–283. doi: 10.1111/nan.12091. [DOI] [PubMed] [Google Scholar]
- 39.Lokensgard JR, Cheeran MC, Gekker G, Hu S, Chao CC, Peterson PK. 1999. Human cytomegalovirus replication and modulation of apoptosis in astrocytes. J Hum Virol 2:91–101. [PubMed] [Google Scholar]
- 40.Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J Virol 69:7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan G, Nogalski MT, Bentz GL, Smith MS, Parmater A, Yurochko AD. 2010. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J Immunol 184:3213–3222. doi: 10.4049/jimmunol.0903025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo X, Huang Y, Qi Y, Liu Z, Ma Y, Shao Y, Jiang S, Sun Z, Ruan Q. 2015. Human cytomegalovirus miR-UL36-5p inhibits apoptosis via downregulation of adenine nucleotide translocator 3 in cultured cells. Arch Virol 160:2483–2490. doi: 10.1007/s00705-015-2498-8. [DOI] [PubMed] [Google Scholar]
- 43.Li HP, Yuan CL, Zho YC. 2015. Human cytomegalovirus inhibits apoptosis involving upregulation of the antiapoptotic protein Bag-1. J Med Virol 87:1953–1959. doi: 10.1002/jmv.24259. [DOI] [PubMed] [Google Scholar]
- 44.Komander D, Clague MJ, Urbe S. 2009. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Bio 10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- 45.Wu YR, Chen CM, Chen YC, Chao CY, Ro LS, Fung HC, Hsiao YC, Hu FJ, Lee-Chen GJ. 2010. Ubiquitin specific proteases USP24 and USP40 and ubiquitin thiolesterase UCHL1 polymorphisms have synergic effect on the risk of Parkinson's disease among Taiwanese. Clin Chim Acta 411:955–958. doi: 10.1016/j.cca.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 46.Li Y, Schrodi S, Rowland C, Tacey K, Catanese J, Grupe A. 2006. Genetic evidence for ubiquitin-specific proteases USP24 and USP40 as candidate genes for late-onset Parkinson disease. Hum Mutat 27:1017–1023. doi: 10.1002/humu.20382. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Lubin A, Chen H, Sun Z, Gong F. 2012. The deubiquitinating protein USP24 interacts with DDB2 and regulates DDB2 stability. Cell Cycle 11:4378–4384. doi: 10.4161/cc.22688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, Courtney SM, Young M, Showalter HD, Sun D, Jakubowiak A, Malek SN, Talpaz M, Donato NJ. 2015. Targeting deubiquitinase activity with a novel small molecule inhibitor as therapy for B-cell malignancies. Blood 125:3588–3597. doi: 10.1182/blood-2014-10-605584. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Nemzow L, Chen H, Lubin A, Rong X, Sun Z, Harris TK, Gong F. 2015. The deubiquitinating enzyme USP24 is a regulator of the UV damage response. Cell Rep 10:140–147. doi: 10.1016/j.celrep.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Gong F. 2016. Involvement of USP24 in the DNA damage response. Mol Cell Oncol 3:e1011888. doi: 10.1080/23723556.2015.1011888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ben-Arieh SV, Zimerman B, Smorodinsky NI, Yaacubovicz M, Schechter C, Bacik I, Gibbs J, Bennink JR, Yewdell JW, Coligan JE, Firat H, Lemonnier F, Ehrlich R. 2001. Human cytomegalovirus protein US2 interferes with the expression of human HFE, a nonclassical class I major histocompatibility complex molecule that regulates iron homeostasis. J Virol 75:10557–10562. doi: 10.1128/JVI.75.21.10557-10562.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison PM, Arosio P. 1996. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- 53.Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. 2011. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol Cell Biol 31:2040–2052. doi: 10.1128/MCB.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aits S, Jaattela M. 2013. Lysosomal cell death at a glance. J Cell Sci 126:1905–1912. doi: 10.1242/jcs.091181. [DOI] [PubMed] [Google Scholar]
- 55.Repnik U, Stoka V, Turk V, Turk B. 2012. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta 1824:22–33. doi: 10.1016/j.bbapap.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 56.Smith B, Randle D, Mezencev R, Thomas L, Hinton C, Odero-Marah V. 2014. Camalexin-induced apoptosis in prostate cancer cells involves alterations of expression and activity of lysosomal protease cathepsin D. Molecules 19:3988–4005. doi: 10.3390/molecules19043988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. 2016. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci 41:274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J Virol 84:3476–3487. doi: 10.1128/JVI.02544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qian Z, Xuan B, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J Virol 82:3452–3465. doi: 10.1128/JVI.01964-07. [DOI] [PMC free article] [PubMed] [Google Scholar]