

ABSTRACT
Human immunodeficiency virus type 1 (HIV-1) can infect nondividing cells via passing through the nuclear pore complex. The nuclear membrane-imbedded protein SUN2 was recently reported to be involved in the nuclear import of HIV-1. Whether SUN1, which shares many functional similarities with SUN2, is involved in this process remained to be explored. Here we report that overexpression of SUN1 specifically inhibited infection by HIV-1 but not that by simian immunodeficiency virus (SIV) or murine leukemia virus (MLV). Overexpression of SUN1 did not affect reverse transcription but led to reduced accumulation of the 2-long-terminal-repeat (2-LTR) circular DNA and integrated viral DNA, suggesting a block in the process of nuclear import. HIV-1 CA was mapped as a determinant for viral sensitivity to SUN1. Treatment of SUN1-expressing cells with cyclosporine (CsA) significantly reduced the sensitivity of the virus to SUN1, and an HIV-1 mutant containing CA-G89A, which does not interact with cyclophilin A (CypA), was resistant to SUN1 overexpression. Downregulation of endogenous SUN1 inhibited the nuclear entry of the wild-type virus but not that of the G89A mutant. These results indicate that SUN1 participates in the HIV-1 nuclear entry process in a manner dependent on the interaction of CA with CypA.
IMPORTANCE HIV-1 infects both dividing and nondividing cells. The viral preintegration complex (PIC) can enter the nucleus through the nuclear pore complex. It has been well known that the viral protein CA plays an important role in determining the pathways by which the PIC enters the nucleus. In addition, the interaction between CA and the cellular protein CypA has been reported to be important in the selection of nuclear entry pathways, though the underlying mechanisms are not very clear. Here we show that both SUN1 overexpression and downregulation inhibited HIV-1 nuclear entry. CA played an important role in determining the sensitivity of the virus to SUN1: the regulatory activity of SUN1 toward HIV-1 relied on the interaction between CA and CypA. These results help to explain how SUN1 is involved in the HIV-1 nuclear entry process.
KEYWORDS: HIV-1, SUN1, capsid, cyclophilin A, nuclear entry
INTRODUCTION
The life cycle of human immunodeficiency virus type 1 (HIV-1) starts from binding to the receptor CD4 and the coreceptor C-C chemokine receptor type 5 (CCR5) or C-X-C chemokine receptor type 4 (CXCR4), followed by membrane fusion and viral core release into the cytoplasm (1–3). The single-stranded genomic RNA is reverse transcribed into double-stranded DNA, which enters the nucleus in the form of a preintegration complex (PIC) (4). The viral capsid (CA) protein plays an important role in determining the pathways utilized by the PIC for nuclear import (5). For example, the wild-type virus can infect nondividing cells because its PIC enters the nucleus through the nuclear pore, while CA mutants with a superstable capsid structure (CA-E45A) or an unstable capsid structure (CA-N57A and CA-N54A/N57A) prefer dividing cells (6). The canonical nuclear import pathway is also dependent on the interaction between CA and cyclophilin A (CypA) (7, 8). The CA-G89V mutant is CypA binding deficient and uses an unidentified pathway for nuclear import (9, 10).
SUN1 and SUN2 are inner nuclear membrane proteins that form the inner part of the linker of nucleoskeleton and cytoskeleton (LINC) complex through interactions with the KASH domain of nesprins in the outer nuclear membrane (11–14). Nesprins themselves interact with cytoskeleton components, such as actins and microtubules, mediated by dynein and kinesin. SUN1 and SUN2 share some functional similarities in both LINC complex formation and cellular activities. For example, interactions between SUN1 and SUN2 isoforms and nesprin isoforms result in the repertoire formation of LINC complexes (11). Both SUN1 and SUN2 play roles in nuclear morphology, chromosome positioning, and DNA damage responses (15–17). Nonetheless, SUN1 has some unique features compared to those of SUN2. While SUN2 is uniformly distributed on the inner nuclear membrane, a large amount of SUN1 is concentrated at the nuclear pore complex (NPC) (18). The nucleoplasmic domain of SUN1 has been reported to bind to lamin A and Nup153 (19), a component of NPC at the nuclear side. Both the nucleoplasmic domain and the perinuclear space (PNS) domain of SUN1 are important for its association with the NPC (18). Phosphorylated SUN1 also participates in mRNA nuclear export, as one of the components of the NXF-1 pathway (20, 21).
It has been reported that the overexpression of SUN2 blocks HIV-1 infection. This activity is exerted mainly via the nucleoplasmic domain of SUN2 (22). Downregulation of endogenous SUN2 did not affect HIV-1 infection efficiency in HeLa cells but impaired the infection efficiency in primary CD4+ T cells, possibly due to the reduced activation and proliferation of these cells upon SUN2 downregulation (22–24).
Given that SUN1 and SUN2 share many functional similarities, we investigated whether SUN1 regulates HIV-1 infection. During the preparation of this paper, Schaller et al. reported that SUN1 overexpression blocks HIV-1 infection in a CA-dependent manner (25). Our results reported here confirm their results. In addition, we provide evidence suggesting that SUN1 regulates HIV-1 infection by a mechanism dependent on the nuclear import process mediated by CypA-CA interaction.
RESULTS
Overexpression of SUN1 inhibits HIV-1 infection.
To explore whether SUN1 overexpression inhibits HIV-1 infection, HEK293A cells were transfected with increasing amounts of a plasmid expressing Flag-tagged SUN1 together with plasmids expressing CD4 and CCR5. The cells were then challenged with the HIV-1 vector NL4-3luc pseudotyped with the HIV-1 envelope of subtype AE, and luciferase activity was measured as an indicator of infection. With increasing expression levels of SUN1-Flag, luciferase activity decreased (Fig. 1A), indicating that SUN1-Flag inhibited NL4-3luc infection in a dose-dependent manner. To exclude the possibility that the Flag tag caused the antiviral activity of SUN1-Flag, SUN1 without a tag was constructed. The untagged SUN1 protein displayed similar inhibitory activity against HIV-1 infection (Fig. 1B).
FIG 1.

SUN1 overexpression inhibits HIV-1 infection. (A to C) Plasmids expressing Flag-tagged or untagged SUN1 were transfected into HEK293A cells together with plasmids expressing CD4 and CCR5 or CXCR4. A plasmid expressing renilla luciferase was included to serve as a control for transfection efficiency and sample handling. At 48 h posttransfection, cells were infected with NL4-3luc vectors pseudotyped with the indicated HIV-1 envelopes. At 48 h postinfection, luciferase activities were measured. The firefly luciferase activity was normalized to the renilla luciferase activity. Control cells or HEK293A cells stably expressing SUN1-Flag were infected with VSV_G-pseudotyped NL4-3luc. At 48 h postinfection, luciferase activities were measured. Data presented are means and SD for three independent experiments. Protein levels in the cell lysates were analyzed by Western blotting. **, P < 0.005; ***, P < 0.001. (D) MT4 cells were infected with the replication-competent NL4-3 virus. At 2 and 4 days postinfection, the viral protein levels in the cell lysates were measured by Western blotting. EV, MT4 cells transduced with an empty lentivector; SUN1, MT4 cells transduced with a lentivector expressing SUN1-Flag.
HIV-1 entry is mediated by binding of the envelope (Env) proteins to the cell surface receptor CD4 and the coreceptor CCR5 or CXCR4 (1). Based on the coreceptor utilized, HIV-1 strains are classified into viruses with X4 tropism and those with R5 tropism. To test whether the antiviral activity of SUN1 is tropism dependent, NL4-3luc vectors pseudotyped with R5-tropic envelopes (AE, BC, and Ad8) and an X4-tropic envelope (Dol) were produced in 293T cells. Overexpression of SUN1 blocked infections by both X4- and R5-tropic viral vectors (Fig. 1C). An NL4-3luc vector pseudotyped with the vesicular stomatitis virus glycoprotein (VSV_G) was also tested on HEK293A cells stably expressing SUN1-Flag. Compared to that in the control cells, the luciferase activity was significantly reduced in the SUN1-expresssing cells (Fig. 1C). These results demonstrated that SUN1 inhibition of HIV-1 infection was independent of the virus entry route.
To probe whether SUN1 inhibits the infection of replication-competent HIV-1, MT4 cells stably expressing SUN1-Flag were challenged with NL4-3 virus. Long-term overexpression of SUN1 in these cells was toxic. We thus analyzed the levels of the viral Gag proteins in the cells at 2 and 4 days postinfection to monitor viral propagation. SUN1 overexpression decreased the viral protein levels (Fig. 1D), indicating that SUN1 inhibited viral replication.
Overexpression of SUN1 inhibits infection by HIV-1 and HIV-2 but not that by SIV or MLV.
We next tested whether SUN1 overexpression blocks infection by other retroviruses. HEK293A cells stably expressing SUN1-Flag were infected with VSV_G-pseudotyped HIV-2, simian immunodeficiency virus (SIV), and murine leukemia virus (MLV) vectors expressing firefly luciferase at serial dilutions. SUN1 specifically inhibited the infection of NL4-3luc and HIV-2-luc but not that of SIVtan-luc or MLV-luc (Fig. 2).
FIG 2.

SUN1 overexpression specifically inhibits infections by HIV-1 and HIV-2 but not those by SIV or MLV. HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were infected with the indicated VSV_G-pseudotyped, luciferase-expressing viral vectors. At 48 h postinfection, luciferase activity was measured. Data presented are means ± SD for three independent experiments.
The N-terminal domain of SUN1 is required for its inhibition of HIV-1 infection.
SUN1 is a type II transmembrane protein composed of an N-terminal nucleoplasmic domain (NTD), a transmembrane domain (TM), and a C-terminal domain (CTD) that is located in the PNS (18). The C-terminal domain is further divided into the membrane-proximal coiled-coil domain and the distal SUN domain (Fig. 3A). To map the domains of SUN1 required for its activity, we generated a series of SUN1 truncation mutants (Fig. 3A) and tested their ability to inhibit NL4-3luc infection. Removal of NTD (ΔN) significantly impaired SUN1's activity, while the NTD alone displayed little inhibitory activity (Fig. 3B), indicating that the NTD is necessary but not sufficient for the activity of SUN1. The NTD-TM mutant (N+TM) also displayed little inhibitory activity (Fig. 3B). However, the N+TM mutant was expressed at a relatively low level compared to that of the wild-type protein or the other mutants (Fig. 3C). Thus, it is hard to draw a conclusion about whether the N+TM mutant is active or not against NL4-3luc. Deletion of the SUN domain (ΔSUN) had little effect on the activity of SUN1 (Fig. 3B). These results indicate that the N-terminal domain of SUN1 is required for its function and that the SUN domain is dispensable.
FIG 3.

The N-terminal domain of SUN1 is required for its inhibition of HIV-1 infection. (A) Schematic representation of SUN1 and its truncation mutants. (B and C) Plasmids expressing Flag-tagged SUN1 or truncation mutants were transiently transfected into HEK293A cells together with plasmids expressing CD4 and CCR5. A plasmid expressing renilla luciferase was included to serve as a control. At 48 h posttransfection, cells were infected with AE-enveloped NL4-3luc. At 48 h postinfection, cells were lysed. (B) Luciferase activities were measured. Firefly luciferase activity was normalized to renilla luciferase activity. Data presented are means and SD for three independent experiments. (C) Expression of the SUN1 mutants was analyzed by Western blotting. The positions of the truncation mutants are indicated with asterisks. EV, empty vector.
SUN1 overexpression inhibits accumulation of 2-LTR and integrated viral DNAs.
To determine at which step HIV-1 infection was blocked by SUN1, HEK293A cells stably expressing SUN1-Flag were infected with VSV_G-pseudotyped NL4-3luc, and the levels of the viral DNA products were measured by quantitative PCR (qPCR) analysis as adapted from previous reports (26, 27). Consistent with the above results, SUN1 overexpression inhibited viral infection (Fig. 4A). Further analysis revealed that comparable levels of the late reverse transcription (RT) product were detected in the control cells and SUN1-expressing cells (Fig. 4B), suggesting that RT was not affected. However, overexpression of SUN1 significantly reduced the level of the 2-long-terminal-repeat (2-LTR) DNA (Fig. 4C), a marker for nuclear entry of the viral cDNA (28–31), as well as that of the integrated viral DNA (Fig. 4D). These results suggest that SUN1 blocked the nuclear import of HIV-1 without affecting reverse transcription.
FIG 4.

Overexpression of SUN1 inhibits accumulation of 2-LTR DNA and integrated viral DNA of HIV-1. HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were infected with VSV_G-pseudotyped NL4-3luc. (A) At 48 h postinfection, the cells were lysed and analyzed for luciferase activity. (B to D) Hirt-extracted DNA or total DNA was obtained at the indicated time points for detection of the late RT product (B), 2-LTR DNA (C), and integrated viral DNA (D) by qPCR. The relative levels of DNA in the control cells were set to 100. Data presented are means and SD for three independent experiments. hpi, hours postinfection; n.s., not significant (P > 0.05).
HIV-1 CA is a determinant for viral sensitivity to SUN1.
The foregoing results showing that overexpression of SUN1 specifically inhibited HIV-1, but not SIV or MLV, prompted us to investigate the viral determinant contributing to the viral sensitivity to SUN1. We generated viral vectors that were chimeras of NL4-3luc and SIVtan (Fig. 5A). Replacement of MA of NL4-3luc with the counterpart from SIVtan impaired the infectivity of the resulting vector, HSIVMA (Fig. 5B). However, the sensitivity of HSIVMA to SUN1 was comparable to that of NL4-3luc (Fig. 5B). Replacement of CA of NL4-3luc with CA from SIV also impaired the infectivity of the resulting vector, HSIVCA (Fig. 5B). In contrast to that of HSIVMA, the sensitivity of HSIVCA was significantly reduced (Fig. 5B). These results indicate that CA is an important determinant for the sensitivity of NL4-3luc to SUN1.
FIG 5.

HIV-1 CA is a determinant for viral sensitivity to SUN1. (A and C) Schematic representations of wild-type and chimeric viral vectors. (B and D) HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were infected with VSV_G-pseudotyped viral vectors expressing firefly luciferase. At 48 h postinfection, luciferase activity was measured. Data presented are means and SD for three independent experiments. Fold inhibition of vector expression by SUN1 (FI) was calculated as the luciferase activity in the control cells divided by that in the SUN1-expressing cells and is indicated by numbers above the bars in panels B and D. ns, not significant.
We next constructed HIV-1–MLV chimeric vectors (Fig. 5C) by using previously described strategies (32). Replacement of MA of NL4-3luc with MLV MA-p12 impaired the infectivity of the resulting vector, HMLVMA-p12, but did not affect its sensitivity to SUN1 (Fig. 5D). In contrast, replacing CA of HMLVMA-p12 with MLV CA rendered the resulting vector, HMLVMA-p12-CA, completely resistant to SUN1 (Fig. 5D).
Collectively, these results indicate that CA is a determinant for the sensitivity of NL4-3luc to SUN1 overexpression. The remaining sensitivity of HSIVCA nonetheless indicates that CA is not the only determinant.
SUN1 inhibition of HIV-1 infection is dependent on the nuclear import process.
HIV-1 can infect nondividing cells because its PIC can enter the nucleus through the nuclear pore, while MLV can infect only dividing cells. CA has been reported to be a key viral factor in determining the pathways by which the viral PIC enters the nucleus (32). The above results prompted us to speculate that SUN1 inhibition of HIV-1 infection is dependent on the nuclear import process and thus is cell cycle dependent. To test this idea, we employed HIV-1 mutants which have been reported to fail to infect nondividing cells due to an unstable (N57A and T54A/N57A) or superstable (E45A) CA protein (6, 8). VSV_G-pseudotyped NL4-3luc vectors containing these mutations were produced in 293T cells and quantitated based on CA (p24) content. HEK293A cells were treated with aphidicolin A and then challenged with the same amounts of the viral vectors based on CA content. Compared to the wild-type strain NL4-3luc, the mutants were more sensitive to aphidicolin A (Fig. 6A). These mutants were then analyzed for sensitivity to SUN1. Compared to wild-type strain NL4-3luc, the mutants were much less sensitive to SUN1 (Fig. 6B). These results support the notion that SUN1 inhibition of HIV-1 infection is dependent on the nuclear import process.
FIG 6.
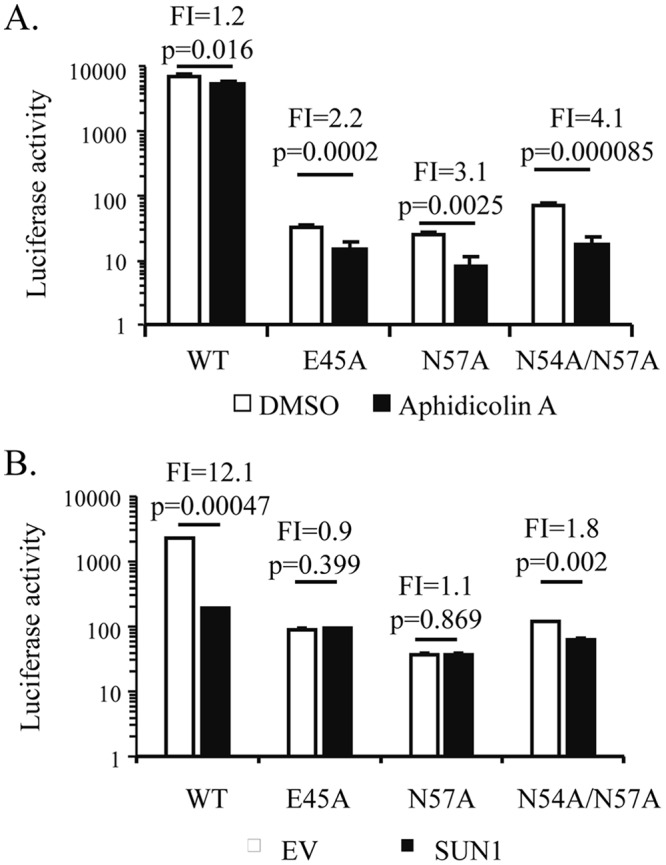
SUN1 inhibition of HIV-1 infection is dependent on the nuclear import process. (A) Dimethyl sulfoxide (DMSO) or aphidicolin A was added to the culture medium of HEK293A cells 2 h before infection. The cells were infected with VSV_G-pseudotyped wild-type (WT) NL4-3luc or its mutants containing the indicated CA mutations. At 48 h postinfection, luciferase activity was measured. (B) HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were infected with the indicated VSV_G-pseudotyped viral vectors. At 48 h postinfection, luciferase activity was measured. Fold inhibition (FI) was calculated as the luciferase activity in the control cells divided by that in the SUN1-expressing cells and is indicated above the bars. Data presented are means and SD for three independent experiments.
SUN1 inhibition of HIV-1 and HIV-2 infections is dependent on interactions between CA and cyclophilin A.
The canonical nuclear import pathway of HIV-1 depends on the interaction between CA and cellular CypA (7, 8). It has been reported that infection by HIV-1, but not that by SIV, is CypA dependent in HeLa cells (33), and we demonstrated that SUN1 inhibited HIV-1 but not SIV (Fig. 2). These results prompted us to hypothesize that SUN1 inhibition of HIV-1 infection might involve CypA-CA interactions. To test this idea, the following two NL4-3luc vectors carrying CA mutants were constructed (Fig. 7A): the CA-G89V mutant, which has been reported to be CypA binding deficient (34, 35), and a chimeric mutant in which the CypA binding loop (CBL) in NL4-3luc CA was replaced with its counterpart from SIVtan. We first confirmed the CypA dependency of the mutant vectors in HEK293A cells. Cells were mock treated or treated with cyclosporine (CsA) to block CA-CypA interactions. CsA treatment significantly reduced the infection efficiency of wild-type NL4-3luc (Fig. 7B). In contrast, the CA-G89V mutant and the CBL chimera were little affected (Fig. 7B). The mutants were then tested for sensitivity to SUN1 in HEK293A cells. Compared to that of wild-type NL4-3luc, the sensitivities of the mutants to SUN1 were significantly reduced (Fig. 7C), suggesting that the diminished CA-CypA interactions rendered the virus less sensitive to SUN1 inhibition. To further substantiate this notion, we analyzed the effect of CsA on SUN1 inhibition of NL4-3luc infection. Control cells and HEK293A cells stably expressing SUN1-Flag were mock treated or treated with increasing concentrations of CsA and then challenged with NL4-3luc. The fold inhibition of NL4-3luc infection by SUN1, calculated as the luciferase activity in the control cells divided by that in the SUN1-expressing cells, decreased with increasing concentrations of CsA (Fig. 7D). These results further support the notion that SUN1 inhibition of HIV-1 infection is dependent on CA-CypA interaction.
FIG 7.

SUN1 inhibition of HIV-1 and HIV-2 infections is dependent on CA-CypA interaction. (A) Sequences of the cyclophilin A binding loop (CBL) of the CA proteins of HIV-1 and SIVtan. (B) HEK293A cells were treated with CsA or DMSO and infected with the same amounts of the indicated VSV_G-pseudotyped virion particles. At 48 h postinfection, luciferase activity was measured. CBL-chimera, the CBL of NL4-3luc was replaced with the counterpart of SIVtan. Fold inhibition (FI) was calculated as the luciferase activity in the DMSO-treated cells divided by that in the CsA-treated cells and is indicated above the bars. (C) HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were infected with the same amounts of the indicated VSV_G-pseudotyped virion particles. Luciferase activity was measured at 48 h postinfection. Fold inhibition (FI) was calculated as the luciferase activity in the control cells divided by that in the SUN1-expressing cells and is indicated above the bars. (D) HEK293A cells transduced with an empty lentivector or a lentivector expressing SUN1-Flag were treated with increasing concentrations of CsA and infected with VSV_G-pseudotyped NL4-3luc. At 48 h postinfection, luciferase activity was measured. Fold inhibition was calculated as the luciferase activity in the control cells divided by that in the SUN1-expressing cells. (E) HEK293A cells were treated with DMSO or CsA at the indicated concentrations and then infected with the indicated VSV_G-pseudotyped viruses. At 48 h postinfection, luciferase activity was measured. (F) Sequences of the CBL of HIV-1 and HIV-2. (G) HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing SUN1-Flag were challenged with the indicated VSV_G-pseudotyped virion particles. Luciferase activity was measured at 48 h postinfection. Fold inhibition (FI) was calculated as described for panel C and is indicated above the bars. (H) HEK293A cells transduced with an empty lentivector (EV) or a lentivector expressing TRIMCyp-Flag were infected with the indicated VSV_G-pseudotyped virion particles and treated with CsA or DMSO. At 48 h postinfection, luciferase activity was measured. Fold inhibition (FI) was calculated as the luciferase activity in the control cells divided by that in the TRIMCyp-expressing cells and is indicated above the bars. Data presented are means and SD for three independent experiments.
SUN1 overexpression inhibited infection not only by HIV-1 but also by HIV-2 (Fig. 2). It has been reported that HIV-2 is not sensitive to CsA, although HIV-2 can be inhibited by TRIMCyp (24, 36, 37). It was not clear whether SUN1 overexpression also inhibits HIV-2 in a manner dependent on the interaction between CA and CypA. We first analyzed the sensitivity of HIV-2 infection to CsA. In contrast to NL4-3luc infection, HIV-2-luc infection was little affected by CsA treatment, even at a relatively high concentration (Fig. 7E), which is consistent with the previously reported results. Comparison of the CA sequences of HIV-1 and HIV-2 revealed that the G89 and P90 residues of HIV-1 CA, which are important for CA binding to CypA, are conserved in HIV-2 (Fig. 7F). We thus constructed HIV-2-luc mutants containing CA-G87A and CA-P88A and analyzed their sensitivity to SUN1. HIV-2-luc containing the CA-G92A mutation was also constructed to serve as a control. Infections by HIV-2 and the G92A mutant were significantly inhibited by SUN1 (Fig. 7G). In contrast, the G87A and P88A mutants were totally resistant to SUN1 (Fig. 7G). In an attempt to correlate the sensitivities of the viruses to SUN1 and their interactions with CypA, we tested the sensitivity of these mutants to TRIMCyp. The infections by HIV-2 and the G92A mutant were significantly inhibited by TRIMCyp (Fig. 7H). In contrast, the infections by the G87A and P89A mutants were slightly enhanced rather than reduced in TRIMCyp-expressing cells (Fig. 7H). Furthermore, TRIMCyp inhibition of HIV-2 and the G92A mutant was abolished by CsA treatment, suggesting that TRIMCyp inhibition occurred through the CypA-CA interaction (Fig. 7H). These results imply that the sensitivity of HIV-2 to SUN1 is dependent on the interaction between HIV-2 CA and the CypA domain of TRIMCyp and that SUN1 overexpression inhibits HIV-2 infection in a manner dependent on the interaction between CA and CypA.
Downregulation of endogenous SUN1 impairs nuclear entry of NL4-3luc but not that of the G89V mutant.
To evaluate the role of endogenous SUN1 in HIV-1 infection, SUN1 was downregulated by use of two small interfering RNAs (siRNAs) targeting different sites in HEK293A cells (Fig. 8A). The cells were infected with VSV_G-pseudotyped NL4-3luc and the G89V mutant. The infection levels, as indicated by luciferase activity, were either little affected or slightly increased for both viruses (Fig. 8B). We further measured the levels of the late RT product, the 2-LTR DNA, the integrated viral DNA, and the viral mRNAs to evaluate the effects of SUN1 downregulation on viral nuclear import and related events. For NL4-3luc, downregulation of SUN1 did not affect the levels of the late RT product (Fig. 8C) or the 2-LTR DNA (Fig. 8D). In contrast, the levels of the integrated viral DNA were significantly reduced (Fig. 8E). In line with the above results showing that SUN1 downregulation did not affect luciferase activity, the levels of the mRNAs for Gag, Vif, and Nef-luc were barely affected by SUN1 downregulation (Fig. 8F to H). For the G89V mutant, SUN1 downregulation did not significantly affect the levels of the late RT product (Fig. 8I), the 2-LTR DNA (Fig. 8J), or the integrated viral DNA (Fig. 8K). Collectively, these results indicate that endogenous SUN1 is involved in the nuclear entry process of HIV-1 and may affect multiple aspects of viral infection (see below for further discussion).
FIG 8.

Downregulation of endogenous SUN1 impairs nuclear entry of NL4-3luc but not that of the G89V mutant. HEK293A cells were transfected with a control siRNA (siCtrl) or siRNAs targeting different sites of SUN1. (A) Downregulation of endogenous SUN1 expression was confirmed by Western blotting. (B) Cells were infected with VSV_G-pseudotyped NL4-3luc or NL4-3luc-G89V. At 48 h postinfection, luciferase activity was measured. Fold inhibition was calculated as the luciferase activity in the control cells divided by that in the cells transfected with the SUN1-targeting siRNA and is indicated above the bars. (C to H) Cells were infected with VSV_G-pseudotyped NL4-3luc. At the indicated time points, Hirt-extracted DNA and total DNA were used for detection of the late RT product (C), 2-LTR DNA (D), and integrated viral DNA (E). The relative levels of DNA in the control cells were set to 100. At 48 h postinfection, the cells were harvested and analyzed for mRNA levels for Gag (F), Vif (G), and Nef-luc (H). Fold inhibition was calculated as the relative integrated viral DNA level in the control cells divided by that in the cells transfected with the SUN1-targeting siRNA and is indicated above the bars in panel E. (I to K) Cells were infected with VSV_G-pseudotyped NL4-3luc-G89V. At the indicated time points, Hirt-extracted DNA and total DNA were used for detection of the late RT product (I), 2-LTR DNA (J), and integrated viral DNA (K). The relative levels of DNA in the control cells were set to 100. Data presented are means and SD for three independent experiments. hpi, hours postinfection; ns, not significant (P > 0.05); *, P < 0.05; ***, P < 0.001.
DISCUSSION
Here we show that overexpression of SUN1 blocked infections by HIV-1 (Fig. 1) and HIV-2 (Fig. 2). In contrast, infections by SIV and MLV were affected little (Fig. 2), excluding the possibility that decreased HIV-1 infection resulted from a general cytotoxicity of SUN1 overexpression. SUN1 overexpression reduced the levels of 2-LTR DNA and integrated viral DNA without significantly affecting the levels of the late RT product (Fig. 4), suggesting that SUN1 blocked the nuclear entry of the viral DNA. We further showed that HIV-1 CA is a determinant for viral sensitivity to SUN1 (Fig. 5). During the course of our work, Schaller et al. (25) reported that SUN1 overexpression inhibits the infection of HIV-1 by reducing the accumulation of the 2-LTR DNA in a CA-dependent manner. Our findings reported here are consistent with theirs.
Schaller et al. reported that SUN1 knockout did not affect infection by NL4-3-GFP (25). They evaluated infection by measuring the percentage of green fluorescent protein (GFP)-positive cells, which is the overall result of many steps. SUN1 has been implicated to function in processes such as chromatin attachment to the nuclear membrane (38) and mRNA export (20, 21). Thus, the effect of SUN1 knockout on HIV-1 nuclear entry was not specifically evaluated. In addition, in the knockout cells, the function of SUN1 could be compensated. To investigate whether endogenous SUN1 is involved in the process of HIV-1 nuclear entry, we used siRNAs to downregulate SUN1 expression and analyzed the levels of the late RT product, 2-LTR DNA, integrated viral DNA, viral mRNAs, and luciferase activity. Consistent with the results of Schaller et al., SUN1 downregulation did not seem to affect luciferase activity (Fig. 8B). SUN1 downregulation did not affect the levels of the late RT product (Fig. 8C) or the 2-LTR DNA (Fig. 8D). However, the level of the integrated viral DNA was significantly reduced (Fig. 8E). Since the nuclear viral DNA includes both integrated DNA and 2-LTR DNA, these results indicate that SUN1 downregulation impaired nuclear entry. It has been reported that loss of SUN1 results in the detachment of chromatin from the nuclear envelope (38–40). Association with the nuclear envelope is important for bringing the euchromatin close to the nuclear pore complex, which would facilitate integration of the incoming viral DNA (41). The seemingly unchanged 2-LTR DNA level may thus be a consequence of the increased level of linear viral DNA that failed to integrate into the host genome. It seems puzzling that SUN1 downregulation reduced the nuclear entry of the viral DNA but did not affect the viral mRNA levels (Fig. 8F to H) or the luciferase activity (Fig. 8B). One possible explanation is that in cells with downregulated SUN1, the viral DNA is integrated into transcriptionally active sites and thus active transcription compensates for the reduced levels of the integrated DNA. We propose that endogenous SUN1 is involved in both the nuclear entry and integration processes of HIV-1 and that downregulation of SUN1 affected these two processes and related events, such as formation of the circular DNA and transcription of the integrated DNA.
The mechanism by which SUN1 overexpression blocks HIV-1 infection is not fully understood. Nonetheless, our results provide some clues. Since MLV infects only dividing cells, while HIV-1 infects both nondividing and dividing cells (32), one can speculate that the different activities of SUN1 against HIV-1 and MLV might result, at least partially, from the different viral sensitivities to the cell cycle. NL4-3luc vectors with CA mutants (N57A, E45A, and N54A/N57A) which prefer to infect dividing cells were resistant to SUN1 overexpression (Fig. 6), supporting the notion that SUN1 inhibition of HIV-1 infection is dependent on the cell cycle and the process of nuclear import. Considering the association of SUN1 with the nuclear pore complex, it seems plausible that SUN1 overexpression and downregulation both affect the nuclear pore complex and thus impair the nuclear import process.
CA was shown to be a determinant for viral sensitivity to SUN1 (Fig. 5 to 7), although the remaining sensitivity of the chimeric vector HSIVCA to SUN1 overexpression indicates that CA is not the only determinant. Two possible mechanisms can be speculated: (i) CA is directly targeted by SUN1 during the nuclear entry process of the virus, and (ii) CA-mediated nuclear import of the PIC but not CA itself is targeted by SUN1. Schaller et al. reported that in vitro-assembled HIV-1 CA-NC nanotubes interacted with SUN1, possibly reflecting a direct interaction between SUN1 and CA (25). However, SUN1 also interacted with the CA-NC nanotubes from an HIV-1 strain that was insensitive to SUN1, suggesting that the interaction between CA and SUN1 may not be the only determinant for the sensitivity of the virus to SUN1. Consistent with their results, we observed that the N-terminal domain of SUN1 bound to bacterially expressed CA-NC from both the SUN1-sensitive virus HIV-1 and the SUN1-insensitive virus SIV (X. Luo and G. Gao, unpublished results). CA might determine the pathway by which the virus enters the nucleus, and thus its sensitivity to SUN1.
The nuclear import pathways of HIV-1 depend on the interaction between CA and cellular CypA, while infection by SIV is independent of CypA (8, 33). This might account for the different effects of SUN1 overexpression on HIV-1 and SIV infections. Blocking the CA-CypA interaction by use of CsA significantly reduced the activity of SUN1, and the CBL chimera and the CA-G89V mutant of HIV-1, which failed to bind CypA, were resistant to SUN1 (Fig. 7). SUN1 downregulation reduced the level of the integrated DNA of NL4-3luc but not that of the G89V mutant. In addition, the sensitivities of HIV-2 and its mutants with mutations of the conserved residues in the CBL to SUN1 correlated well with their sensitivities to overexpression and TRIMCyp (Fig. 7G and H). These results support the notion that the CA-CypA interaction is important for the function of SUN1 in nuclear entry. We propose that SUN1 acts downstream of the CA-CypA-mediated nuclear import pathways. Disruption of the CA-CypA interaction might result in a change in the selection of the nuclear import pathways.
In summary, we show here that overexpression of SUN1 inhibits the nuclear entry of HIV-1 DNA in a manner dependent on the interaction between viral CA and cellular CypA. Downregulation of endogenous SUN1 gave similar phenotypes. We propose that SUN1 is involved in the nuclear entry of HIV-1 DNA and that overexpression and downregulation of SUN1 affect the nuclear pore complex and thus the nuclear entry of the viral DNA. These findings will help us to better understand the roles of SUN1 in HIV-1 infection.
MATERIALS AND METHODS
Plasmids.
pCDH-SUN1-Flag expresses SUN1 with a Flag tag at the C terminus. To generate pCDH-SUN1-Flag, the coding sequence for SUN1 was PCR amplified from pCDNA3.1-SUN1 (a generous gift from Zhaocai Zhou of the Shanghai Institute of Biochemistry and Cell Biology) and cloned into the lentivector pCDH-CMV-MCS-EF1-puro (CD550B-1; SBI). A lentivector expressing untagged SUN1 was constructed by engineering a stop codon immediately before the Flag coding sequence in pCDH-SUN1-Flag. To express CD4, CCR5, and CXCR4, the coding sequences were PCR amplified from a human cDNA library and cloned into pcDNA4 (Invitrogen). To generate SUN1 truncation mutants, the coding sequences were PCR amplified by use of downstream primers containing the Flag tag coding sequence and cloned into pCDH-CMV-MCS-EF1-puro.
The lentivector pNL4-3luc (obtained from Nathaniel Landau through the NIH AIDS Research and Reference Reagent Program) and the retrovector pMLV-luc have been described previously (42, 43). Lentivectors pHIV-2-GFP and pSIVtan-GFP were kindly provided by P. D. Bieniasz (44). The coding sequence for green fluorescent protein (GFP) was replaced with that for firefly luciferase to generate pHIV-2-luc and pSIVtan-luc.
The HIV-1 CA mutants, including CA-G89V, CA-N54A/N57A, CA-N57A, and CA-E45A, were each generated by overlap PCR and then cloned into pNL4-3luc. For construction of the HSIVCA chimera, the CA coding sequence in pNL4-3luc was replaced with the counterpart from SIVtan. To generate the HSIVMA chimera, the MA coding sequence in pNL4-3luc was replaced with that from SIVtan. For construction of the HMIVMA-p12 chimera, the MA coding sequence in NL4-3luc was replaced with that for MA-p12 from MLV. For construction of HMIVMA-p12-CA, the coding sequence for MA-CA204 in NL4-3luc was replaced with that for MLV MA-p12-CA.
Cell culture and viruses.
293T and HEK293A cells were maintained in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco). To generate cells stably overexpressing SUN1-Flag, the empty vector pCDH-CMV-MCS-EF1-puro (pCDH for short) and pCDH-SUN1-Flag were packaged into VSV_G-pseudotyped lentivectors in 293T cells and used to transduce HEK293A or MT4 cells. Puromycin-resistant HEK293A and MT4 cells were pooled for future experiments.
NL4-3luc vectors pseudotyped with HIV-1 envelopes of subtypes AE, BC, Ad8, and Dol were produced by transfection of 293T cells with pNL4-3luc (2 μg per 60-mm dish) and the envelope-expressing plasmid (1 μg per 60-mm dish) by use of Lipotransfectin 2010 (Invitrogen) following the manufacturer's instructions. VSV_G-pseudotyped NL4-3luc vectors were produced by cotransfection of plasmids pNL4-3luc (2 μg per 60-mm dish) and pVSV_G (1 μg per 60-mm dish) into 293T cells. For the CA mutant NL4-3luc vectors, the virion particles were quantified by use of an HIV-Ag/Ab enzyme-linked immunosorbent assay (ELISA) kit following the manufacturer's instructions (Wantai Beijing, Beijing, China). For comparison of the infectivities of the viral vectors, the same amounts of virions were used to infect cells. The firefly luciferase activity in the recipient cells was measured by use of a luciferase assay system (Promega). The amounts of the virions varied slightly in different experiments. For the convenience of statistical analysis, the sensitivity of the luminometer was adjusted such that the relative light units in the control cells infected with wild-type NL4-3luc were almost the same in different independent experiments.
To infect HEK293A cells with HIV-1 Env-pseudotyped NL4-3luc vectors, the cells were transfected with plasmids expressing CD4 and CCR5 or CXCR4 together with an empty vector or the SUN1-expressing plasmid. The renilla luciferase-expressing plasmid pCMV-hRL (Promega) was included to serve as a control for transfection efficiency and sample handling. At 48 h posttransfection, the cells were infected with the NL4-3luc vectors. Cells were lysed, and luciferase activities were measured by use of a dual-luciferase reporter assay system (Promega). The firefly luciferase activity was normalized to the renilla luciferase activity.
The proviral plasmid pNL4-3 was obtained from the NIH AIDS Research and Reference Reagent Program. Replication-competent HIV-1 strain NL4-3 was produced by transfecting pNL4-3 into 293T cells. Culture supernatant was harvested at 48 h posttransfection, and the virion particles were quantified using an HIV-Ag/Ab ELISA kit following the manufacturer's instructions (Wantai Beijing, China). The amount of virions containing 1 ng p24 was used to infect MT4 cells.
To disrupt the CypA-CA interaction, CsA was added upon viral infection, to a final concentration of 5 μM, until the measurement of luciferase activity. To induce cell cycle arrest, aphidicolin A was added 2 h before the viral infection, to a final concentration of 2 μg/ml, and removed right upon the viral supernatant being replaced with fresh medium.
To downregulate the expression of endogenous SUN1, 2 × 106 HEK293A cells were seeded into 60-mm culture dishes the day before transfection and transfected with siRNAs at a final concentration of 0.1 μM by use of Lipofectamine 2000 (Invitrogen). The target sequences of the siRNAs were as follows: siCtrl, 5′-UUCUCCGAACGUGUCACGUTT-3′; siSUN1-1, 5′-GCACAAACAAAUCAGCUUU-3′; and siSUN1-2, 5′-GGUAACUGCUGGGCAUUUA-3′.
Hirt DNA extraction, RNA extraction, and real-time PCR.
Cells were infected with VSV_G-pseudotyped NL4-3luc virus. The virus stocks were pretreated with DNase I at 37°C for 30 min to prevent possible contamination from the plasmids used for transfection. Cells in 60-mm culture dishes were lysed in 800 μl Hirt lysis buffer (0.6% SDS, 100 mM Tris-HCl, pH 7.5, 10 mM EDTA) at room temperature for 10 min, followed by addition of 200 μl 5 M NaCl. The mixtures were incubated at 4°C overnight and centrifuged at 10,000 rpm for 1 h. DNA was extracted with phenol-chloroform twice and with chloroform once and then precipitated with ethanol. The DNA pellet was dissolved in 100 μl double-distilled water (ddH2O).
The late RT product and 2-LTR DNA in the Hirt-extracted DNA were quantified by qPCR, using mitochondrial DNA as an internal control. The PCR conditions were as follows: 40 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 20 s. Sequences of the PCR primers were as follows: primers for the late RT product, U5-gag forward (5′ untranslated region [5′UTR]; positions 557 to 575) (5′-TGTGTGCCCGTCTGTTGTG-3′) and U5-gag reverse (5′UTR; positions 684 to 699) (5′-GAGTCCTGCGTCGAGA-3′); primers for 2-LTR DNA, U5-U3 forward (3′UTR; positions 11240 to 11262) (5′-CCCTCAGACCCTTTTAGTCAGTG-3′) and U5-U3 reverse (5′UTR; positions 77 to 98) (5′-TGGTGTGTAGTTCTGCCAATCA-3′); and primers for mitochondrial DNA, Mito-F (5′-ACCCACTCCCTCTTAGCCAATATT-3′) and Mito-R (5′-GTAGGGCTAGGCCCACCG-3′).
To detect the integrated viral DNA, total DNA was extracted by use of a DNeasy Blood & Tissue kit (Qiagen). For cells from one 60-mm culture dish, the total DNA was dissolved in 100 μl AE buffer. Integrated viral DNA was detected by nested PCR. The first round of PCR was performed with primers int-1F and int-1R, which bind to the Alu and HIV-1 gag DNA sequences, to amplify the Alu-HIV DNA. The PCR products were used as templates for the second round of qPCR, using primers int-2F and int-2R. The PCR conditions for the first round were as follows: 15 cycles of 95°C for 10 s, 63°C for 10 s, and 72°C for 2 min 50 s. The PCR conditions for the second round were as follows: 40 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 20 s. The sequences of the primers were as follows: int-1F (Alu), 5′-GCCTCCCAAAGTGCTGGGATTACAG-3′; int-1R (gag; positions 1486 to 1505), 5′-GTTCCTGCTATGTCACTTCC-3′; int-2F (5′UTR; positions 518 to 539), 5′-TTAAGCCTCAATAAAGCTTGCC-3′; and int-2R (5′UTR; positions 628 to 647), 5′-GTTCGGGCGCCACTGCTAGA-3′.
All qPCRs were done with 2× SuperReal Premix Plus with SYBR green I (lot Q6220; Tiangen). The amplification specificity was confirmed by melting point analysis and sequencing of the PCR products.
To measure the viral mRNA levels, HEK293A cells infected with VSV_G-pseudotyped NL4-3luc were harvested and RNA was extracted by use of TRIzol (Ambion) reagent. One microgram of RNA was digested with DNase I at 37°C for 30 min and used as the template for reverse transcription-qPCR detection. The PCR primer sequences were as follows: for unspliced gag mRNA, Gag-FP (5′-CCTATAGTGCAGAACATCC-3′) and Gag-RP (5′-CAAAACTCTTGCCTTATGGC-3′); for singly spliced vif mRNA, Vif-FP (5′-GGCGACTGGGACAGC-3′) and Vif-RP (5′-CACACAATCATCACCTGCC-3′); for multiply spliced nef-luc mRNA, Nef-luc-FP (5′-ACAGTCAGACTCATCAAGCTTCT-3′) and Nef-luc-RP (5′-CGGGTCCCCTCGGGATT-3′); and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, GAPDH-FP (5′-TCGGAGTCAACGGATTTG-3′) and GAPDH-RP (5′-GCATCGCCCCACTTGATT-3′).
Western blotting.
HEK293A cells were lysed in passive lysis buffer (Promega) for at least 15 min on ice (for one 35-mm culture dish, 200 μl passive lysis buffer was used). The lysates were boiled in loading buffer for 15 min and clarified by centrifugation at 12,000 rpm for 10 min. Supernatants were loaded into SDS-PAGE gels, electrophoresed at a constant voltage of 100 V, and transferred to a polyvinylidene difluoride (PVDF) membrane at a constant current of 200 mA for 2 h. The membrane was blocked with 5% skim milk in Tris-buffered saline (TBS) for 1 h. Primary antibodies were diluted in 2.5% skim milk in TBS plus 0.1% Tween 20 (TBST) and incubated with the membrane for 2 h at room temperature or overnight at 4°C. Dilutions for the primary antibodies were as follows: anti-p24, 1:10,000; anti-SUN1, 1:2,000; anti-Flag, 1:10,000; and anti-actin, 1:10,000. A horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibody (Promega) and an HRP-conjugated anti-rabbit IgG secondary antibody (ZSGB-bio) were diluted 1:10,000 and incubated with the membrane for 1 h at room temperature. The membrane was incubated with ECL buffer and then exposed to X-ray films.
Antibodies and reagents.
A human SUN1-specific rabbit monoclonal antibody (Abcam), Flag-specific antibody M2 (Sigma-Aldrich), and a β-actin-specific antibody (ZSGB-bio) were commercially available. An HIV-1 p24-specific mouse monoclonal antibody (P5F1) was a generous gift from Yong-Tang Zheng, Kunming, China. CsA (ab120114) and aphidicolin A (ab142400) were commercially obtained from Abcam.
Statistical analysis.
Statistical analyses were performed in Microsoft Excel, using the t test. Unless otherwise indicated, the data presented are means ± standard deviations (SD) for three independent experiments.
ACKNOWLEDGMENTS
We thank Zhaocai Zhou of the Shanghai Institute of Biochemistry and Cell Biology for providing the SUN1-expressing plasmid and Paul Bieniasz for providing GFP-based lentivectors pHIV-2 and pSIVtan.
This work was supported by grants to G.G. from the National Health and Family Planning Commission of China (grant 2017ZX10201101-001-005) and the National Natural Science Foundation of China (grant 81530066).
REFERENCES
- 1.Wilen CB, Tilton JC, Doms RW. 2012. HIV: cell binding and entry. Cold Spring Harb Perspect Med 2:a006866. doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 3.Yap MW, Nisole S, Stoye JP. 2005. A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol 15:73–78. doi: 10.1016/j.cub.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 4.Hu WS, Hughes SH. 2012. HIV-1 reverse transcription. Cold Spring Harb Perspect Med 2:a006882. doi: 10.1101/cshperspect.a006882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamashita M, Perez O, Hope TJ, Emerman M. 2007. Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS Pathog 3:1502–1510. doi: 10.1371/journal.ppat.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. 2002. Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc Natl Acad Sci U S A 99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ. 2011. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7:e1002439. doi: 10.1371/journal.ppat.1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sokolskaja E, Sayah DM, Luban J. 2004. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol 78:12800–12808. doi: 10.1128/JVI.78.23.12800-12808.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med 9:1138–1143. doi: 10.1038/nm910. [DOI] [PubMed] [Google Scholar]
- 11.Jahed Z, Soheilypour M, Peyro M, Mofrad MR. 2016. The LINC and NPC relationship—it's complicated! J Cell Sci 129:3219–3229. doi: 10.1242/jcs.184184. [DOI] [PubMed] [Google Scholar]
- 12.Padmakumar VC, Libotte T, Lu W, Zaim H, Abraham S, Noegel AA, Gotzmann J, Foisner R, Karakesisoglou I. 2005. The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 118:3419–3430. doi: 10.1242/jcs.02471. [DOI] [PubMed] [Google Scholar]
- 13.Razafsky D, Hodzic D. 2009. Bringing KASH under the SUN: the many faces of nucleo-cytoskeletal connections. J Cell Biol 186:461–472. doi: 10.1083/jcb.200906068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Shi Z, Jiao S, Chen C, Wang H, Liu G, Wang Q, Zhao Y, Greene MI, Zhou Z. 2012. Structural insights into SUN-KASH complexes across the nuclear envelope. Cell Res 22:1440–1452. doi: 10.1038/cr.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YL, Burke B. 2017. LINC complexes and nuclear positioning. Semin Cell Dev Biol 2017:S1084–9521(17)30455-X. doi: 10.1016/j.semcdb.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 16.Lei K, Zhang X, Ding X, Guo X, Chen M, Zhu B, Xu T, Zhuang Y, Xu R, Han M. 2009. SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc Natl Acad Sci U S A 106:10207–10212. doi: 10.1073/pnas.0812037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lei K, Zhu X, Xu R, Shao C, Xu T, Zhuang Y, Han M. 2012. Inner nuclear envelope proteins SUN1 and SUN2 play a prominent role in the DNA damage response. Curr Biol 22:1609–1615. doi: 10.1016/j.cub.2012.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Q, Pante N, Misteli T, Elsagga M, Crisp M, Hodzic D, Burke B, Roux KJ. 2007. Functional association of Sun1 with nuclear pore complexes. J Cell Biol 178:785–798. doi: 10.1083/jcb.200704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen ZJ, Wang WP, Chen YC, Wang JY, Lin WH, Tai LA, Liou GG, Yang CS, Chi YH. 2014. Dysregulated interactions between lamin A and SUN1 induce abnormalities in the nuclear envelope and endoplasmic reticulum in progeric laminopathies. J Cell Sci 127:1792–1804. doi: 10.1242/jcs.139683. [DOI] [PubMed] [Google Scholar]
- 20.Li P, Noegel AA. 2015. Inner nuclear envelope protein SUN1 plays a prominent role in mammalian mRNA export. Nucleic Acids Res 43:9874–9888. doi: 10.1093/nar/gkv1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li P, Stumpf M, Muller R, Eichinger L, Glockner G, Noegel AA. 2017. The function of the inner nuclear envelope protein SUN1 in mRNA export is regulated by phosphorylation. Sci Rep 7:9157. doi: 10.1038/s41598-017-08837-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donahue DA, Amraoui S, di Nunzio F, Kieffer C, Porrot F, Opp S, Diaz-Griffero F, Casartelli N, Schwartz O. 2016. SUN2 overexpression deforms nuclear shape and inhibits HIV. J Virol 90:4199–4214. doi: 10.1128/JVI.03202-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donahue DA, Porrot F, Couespel N, Schwartz O. 2017. SUN2 silencing impairs CD4 T cell proliferation and alters sensitivity to HIV-1 infection independently of cyclophilin A. J Virol 91:e02303-16. doi: 10.1128/JVI.02303-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lahaye X, Satoh T, Gentili M, Cerboni S, Silvin A, Conrad C, Ahmed-Belkacem A, Rodriguez EC, Guichou JF, Bosquet N, Piel M, Le Grand R, King MC, Pawlotsky JM, Manel N. 2016. Nuclear envelope protein SUN2 promotes cyclophilin-A-dependent steps of HIV replication. Cell Rep 15:879–892. doi: 10.1016/j.celrep.2016.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaller T, Bulli L, Pollpeter D, Betancor G, Kutzner J, Apolonia L, Herold N, Burk R, Malim MH. 2017. Effects of the inner nuclear membrane proteins SUN1/UNC-84A and SUN2/UNC-84B on the early steps of HIV-1 infection. J Virol 91:e00463-17. doi: 10.1128/JVI.00463-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedrich B, Li G, Dziuba N, Ferguson MR. 2010. Quantitative PCR used to assess HIV-1 integration and 2-LTR circle formation in human macrophages, peripheral blood lymphocytes and a CD4+ cell line. Virol J 7:354. doi: 10.1186/1743-422X-7-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Butler SL, Johnson EP, Bushman FD. 2002. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J Virol 76:3739–3747. doi: 10.1128/JVI.76.8.3739-3747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierson TC, Kieffer TL, Ruff CT, Buck C, Gange SJ, Siliciano RF. 2002. Intrinsic stability of episomal circles formed during human immunodeficiency virus type 1 replication. J Virol 76:4138–4144. doi: 10.1128/JVI.76.8.4138-4144.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki Y, Misawa N, Sato C, Ebina H, Masuda T, Yamamoto N, Koyanagi Y. 2003. Quantitative analysis of human immunodeficiency virus type 1 DNA dynamics by real-time PCR: integration efficiency in stimulated and unstimulated peripheral blood mononuclear cells. Virus Genes 27:177–188. doi: 10.1023/A:1025732728195. [DOI] [PubMed] [Google Scholar]
- 31.Zhang R, Mehla R, Chauhan A. 2010. Perturbation of host nuclear membrane component RanBP2 impairs the nuclear import of human immunodeficiency virus-1 preintegration complex (DNA). PLoS One 5:e15620. doi: 10.1371/journal.pone.0015620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashita M, Emerman M. 2004. Capsid is a dominant determinant of retrovirus infectivity in nondividing cells. J Virol 78:5670–5678. doi: 10.1128/JVI.78.11.5670-5678.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG. 1994. Functional association of cyclophilin A with HIV-1 virions. Nature 372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- 34.Braaten D, Franke EK, Luban J. 1996. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol 70:3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franke EK, Yuan HEH, Luban J. 1994. Specific incorporation of cyclophilin-A into HIV-1 virions. Nature 372:359–362. doi: 10.1038/372359a0. [DOI] [PubMed] [Google Scholar]
- 36.Mamede JI, Damond F, Bernardo A, Matheron S, Descamps D, Battini JL, Sitbon M, Courgnaud V. 2017. Cyclophilins and nucleoporins are required for infection mediated by capsids from circulating HIV-2 primary isolates. Sci Rep 7:45214. doi: 10.1038/srep45214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Billich A, Hammerschmid F, Peichl P, Wenger R, Zenke G, Quesniaux V, Rosenwirth B. 1995. Mode of action of SDZ NIM 811, a nonimmunosuppressive cyclosporin A analog with activity against human immunodeficiency virus (HIV) type 1: interference with HIV protein-cyclophilin A interactions. J Virol 69:2451–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumoto A, Sakamoto C, Matsumori H, Katahira J, Yasuda Y, Yoshidome K, Tsujimoto M, Goldberg IG, Matsuura N, Nakao M, Saitoh N, Hieda M. 2016. Loss of the integral nuclear envelope protein SUN1 induces alteration of nucleoli. Nucleus 7:68–83. doi: 10.1080/19491034.2016.1149664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chi YH, Haller K, Peloponese JM Jr, Jeang KT. 2007. Histone acetyltransferase hALP and nuclear membrane protein hsSUN1 function in de-condensation of mitotic chromosomes. J Biol Chem 282:27447–27458. doi: 10.1074/jbc.M703098200. [DOI] [PubMed] [Google Scholar]
- 40.Xiong H, Rivero F, Euteneuer U, Mondal S, Mana-Capelli S, Larochelle D, Vogel A, Gassen B, Noegel AA. 2008. Dictyostelium Sun-1 connects the centrosome to chromatin and ensures genome stability. Traffic 9:708–724. doi: 10.1111/j.1600-0854.2008.00721.x. [DOI] [PubMed] [Google Scholar]
- 41.Lusic M, Siliciano RF. 2017. Nuclear landscape of HIV-1 infection and integration. Nat Rev Microbiol 15:69–82. doi: 10.1038/nrmicro.2016.162. [DOI] [PubMed] [Google Scholar]
- 42.Mu X, Fu Y, Zhu Y, Wang X, Xuan Y, Shang H, Goff SP, Gao G. 2015. HIV-1 exploits the host factor RuvB-like 2 to balance viral protein expression. Cell Host Microbe 18:233–242. doi: 10.1016/j.chom.2015.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, Zhang X, Han Y, Wang X, Gao G. 2016. M2BP inhibits HIV-1 virion production in a vimentin filaments-dependent manner. Sci Rep 6:32736. doi: 10.1038/srep32736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]