

ABSTRACT
Virus-specific CD8 T cell response seems to play a significant role in the outcome of hepatitis delta virus (HDV) infection. However, the HDV-specific T cell epitope repertoire and mechanisms of CD8 T cell failure in HDV infection have been poorly characterized. We therefore aimed to characterize HDV-specific CD8 T cell epitopes and the impacts of viral mutations on immune escape. In this study, we predicted peptide epitopes binding the most frequent human leukocyte antigen (HLA) types and assessed their HLA binding capacities. These epitopes were characterized in HDV-infected patients by intracellular gamma interferon (IFN-γ) staining. Sequence analysis of large hepatitis delta antigen (L-HDAg) and HLA typing were performed in 104 patients. The impacts of substitutions within epitopes on the CD8 T cell response were evaluated experimentally and by in silico studies. We identified two HLA-B*27-restricted CD8 T cell epitopes within L-HDAg. These novel epitopes are located in a relatively conserved region of L-HDAg. However, we detected molecular footprints within the epitopes in HLA-B*27-positive patients with chronic HDV infections. The variant peptides were not cross-recognized in HLA-B*27-positive patients with resolved HDV infections, indicating that the substitutions represent viral escape mutations. Molecular modeling of HLA-B*27 complexes with the L-HDAg epitope and its potential viral escape mutations indicated that the structural and electrostatic properties of the bound peptides differ considerably at the T cell receptor interface, which provides a possible molecular explanation for the escape mechanism. This viral escape from the HLA-B*27-restricted CD8 T cell response correlates with a chronic outcome of hepatitis D infection. T cell failure resulting from immune escape may contribute to the high chronicity rate in HDV infection.
IMPORTANCE Hepatitis delta virus (HDV) causes severe chronic hepatitis, which affects 20 million people worldwide. Only a small number of patients are able to clear the virus, possibly mediated by a virus-specific T cell response. Here, we performed a systematic screen to define CD8 epitopes and investigated the role of CD8 T cells in the outcome of hepatitis delta and how they fail to eliminate HDV. Overall the number of epitopes identified was very low compared to other hepatotropic viruses. We identified, two HLA-B*27-restricted epitopes in patients with resolved infections. In HLA-B*27-positive patients with chronic HDV infections, however, we detected escape mutations within these identified epitopes that could lead to viral evasion of immune responses. These findings support evidence showing that HLA-B*27 is important for virus-specific CD8 T cell responses, similar to other viral infections. These results have implications for the clinical prognosis of HDV infection and for vaccine development.
KEYWORDS: cytotoxic T lymphocyte, epitope mapping, immune escape, immune selection, large hepatitis delta antigen
INTRODUCTION
It is estimated that 15 to 20 million patients worldwide carry hepatitis delta virus (HDV), a virus that causes the most severe form of viral hepatitis, including fulminant hepatitis, and a high rate of cirrhosis (1, 2). The HDV virion is composed of a circular single-stranded RNA consisting of around 1,700 bases and depends on hepatitis B virus (HBV) for progeny virus production and spread. The antigenomic open reading frame (ORF) of HDV encodes the only viral protein, hepatitis delta antigen (HDAg), in two forms, small and large hepatitis delta antigen (S- and L-HDAg). L-HDAg contains the protein sequence of S-HDAg but has an additional 19 amino acids (aa) at the C terminus (3, 4).
Two courses of HDV infection are known (5). HDV and HBV coinfection of patients shows a course similar to that of acute HBV monoinfection, and the majority of patients clear both HBV and HDV. HDV superinfection in chronic HBV infection, in contrast, results in a high rate of HDV persistence. Antibodies recognizing both HDV proteins are detected at low titers during acute infection and reach high levels during chronic infection, but they are not able to neutralize the virus (6). The efficacy of current therapies against HDV, e.g., with PEGylated interferon, is very limited (7).
Liver damage after superinfection of HBV carriers with HDV results in transaminase elevation. This observation, as well as the fact that HDV itself is not cytopathic (8), may indicate that cytotoxic T lymphocytes (CTL) are involved in the destruction of hepatocytes. T cell responses against HDV antigens seem to be weak, but very little is known about T cell responses controlling HDV infection, and T cell epitopes are poorly characterized. Initial studies in animal models have shown that a CD8 T cell response against S- and L-HDAgs can be generated. In mice, we demonstrated that immunization with a plasmid DNA vaccine expressing HDAg primed a functional CD4 and CD8 T cell immune response against both forms of delta antigens (9). These T cells prevented the development of L-HDAg-expressing tumors in 80 to 100% of immunized mice (9). Moreover, a DNA prime and adenoviral vector boost vaccination with L-HDAg protected woodchucks from HDV in the setting of simultaneous infection with woodchuck hepatitis virus (WHV) and HDV (10). Nisini et al. demonstrated a polyspecific but weak CD4 T cell response to HDAg in patients that was related to the resolution of HDV-induced disease activity (11). CD8 T cell responses to two human leukocyte antigen (HLA)-A*02:01-restricted HDAg epitopes have been described in patients with resolved HDV infection that were absent in patients with an actively replicating chronic infection (12).
In recent years, much has been learned about how human viruses establish chronic and long-lasting infections by escaping the T cell immune response. For the clinically most relevant chronic viral infections, i.e., with HIV, HBV, and hepatitis C virus (HCV), it has been demonstrated that, despite differences in the natural course of infection in humans, failure of T cell responses is a typical phenomenon (13–15). Viral immune escape variants may contribute to the failure of T cell responses and allow viral persistence and high-level replication. CD8 T cell responses are restricted by host HLA class I molecules, with enormous allelic variation between individuals and across different populations. Since the repertoire of the CD8 T cell response is dictated by the genetic HLA background of the individual host, diverse peptide epitopes derived from viral proteins maybe presented.
CD8 T cells have been shown to elicit significant selection pressure in chronic viral infection. In HBV (14), HCV (16), and HIV (17, 18) infections, selection of viral escape variants has been reported. For HDV, a previous study suggested evidence for positive selection within the predicted HDV epitopes restricted by HLA-A*02:01 (19); however, no in vitro experiments have been carried out confirming that immune escape functionally impairs the virus-specific T cell response. The aims of this study, therefore, were (i) to characterize the variability of the only HDV protein, L-HDAg, in a large cohort of patients; (ii) to identify HDAg-specific CD8 T cell epitopes for frequent HLA alleles by in silico and in vitro analyses; and (iii) to evaluate whether immune escape of HDV from CD8 T cell responses by mutation of relevant CD8 epitopes contributes to the persistence of HDV after superinfection of HBV carriers.
RESULTS
HDV epitope prediction and MHC binding capabilities of predicted epitopes in vitro.
Two hundred HDV genotype 1 sequences of L-HDAg, retrieved from GenBank, were submitted for epitope prediction using two different online tools for the prediction of major histocompatibility complex (MHC) class I binding epitopes (implemented in the Immune Epitope Database [IEDB] and the SYFPEITHI database). This prediction of L-HDAg was done for frequent MHC class I alleles in the European population (20), as well as HLA-B*27, which, despite its low frequency, has been shown to play a protective role against a number of serious human viral infections, such as HCV and HIV (21–23). Two predicted HLA-B*27-restricted epitopes, i.e., L-HDAg99–108 (RRDHRRRKAL) and L-HDAg103–112 (RRRKALENKK), showed the best percentile scores (Table 1). Although for most of the common HLA alleles no peptides reached average prediction scores that were comparable to those of the peptide epitopes known from other viruses, we selected 15 peptides with the best predicted binding scores to HLA-A*01, -A*02, -A*03, and -A*24 and HLA-B*07 and -B*27 to determine their binding affinities experimentally.
TABLE 1.
Best predicted epitopes on L-HDAg for the selected HLA allelesa
| HLA | Position (aa) | Sequence | Length | Score |
|
|---|---|---|---|---|---|
| IEDB | SYFPEITHI | ||||
| A*01 | 75–83 | RTDQMEVDS | 9 | 3.2 | 17 |
| 94–102 | FTDKERQHD | 9 | 1.3 | 17 | |
| A*02 | 43–51 | KLEEDNPWL | 9 | 1.3 | 23 |
| 143–152 | RVAGPPVGGV | 10 | 4.2 | 23 | |
| 198–206 | ILFPSDPPF | 9 | 0.8 | 16 | |
| A*03 | 53–61 | NIKGILGKK | 9 | 2.7 | 22 |
| 89–97 | PLRGGFTDK | 9 | 2.1 | 28 | |
| A*24 | 49–57 | PWLGNIKGI | 9 | 3.9 | 13 |
| 192–200 | RGFPWDILF | 9 | 1.1 | 13 | |
| 198–206 | ILFPSDPPF | 9 | 2.4 | 11 | |
| B*07 | 68–76 | APPAKRART | 9 | 1.9 | 21 |
| 71–79 | AKRARTDQM | 9 | 3.1 | 12 | |
| 168–176 | VPSMQGVPE | 9 | 3.0 | 16 | |
| B*27 | 99–108 | RRDHRRRKAL | 10 | 0.35 | 24 |
| 103–112 | RRRKALENKK | 10 | 0.20 | 29 | |
HLA binding was assessed by UV-induced peptide exchange and compared to that of a known high-affinity ligand of the respective HLA class I molecules. None of the synthesized peptides predicted to bind to HLA-A*01, -A*02, -A*03, -A*24, or -B*07 bound with an affinity of >25% compared to the respective positive control (Fig. 1). Only the two peptide ligands for HLA-B*27, aa 99 to 108 (RRDHRRRKAL) and 103 to 112 (RRRKALENKK), showed high binding affinities of 120 and 111.7%, respectively, relative to that of KRWIILGLNK, a well-known HLA-B*27-restricted HIV epitope, which was used as positive control.
FIG 1.

MHC binding assay of the highest-ranking predicted L-HDAg epitopes. Binding to the indicated HLA molecules was assessed by UV-induced ligand exchange. A total of 15 different predicted epitopes were tested for binding HLA-A*01:01 (n = 2), HLA-A*02:01 (n = 3), HLA-A*03:01 (n = 2), HLA-A*24:02 (n = 3), HLA-B*07:02 (n = 3), and HLA-B*27:05 (n = 2). L-HDAg198–206 was tested with HLA-A*2:01 and -A*24:02. Binding of predicted peptide epitopes is shown as percent binding of the indicated epitopes with high binding affinities for the respective HLA molecules. Means and standard deviations (SD) are shown. Negative controls (Neg. Cont.) included exchange of a nonbinder and UV illumination in the absence of any peptide.
Detection of HDV-specific CD8 T cells in patients with resolved HDV infections.
To determine if the predicted peptide epitopes would be recognized in HDV infection, in a first set of experiments, we analyzed peripheral blood mononuclear cells (PBMCs) of one HLA-B*27-positive and three HLA-B*27-negative patients who had resolved HDV infections. Using an overlapping peptide library spanning the whole L-HDAg (Table 2) and divided into 8 peptide pools (A to H), we detected a T cell response only in the HLA-B*27-positive patient A (Fig. 2). The CD8+ T cell response, shown by intracellular cytokine staining (ICS), was induced by peptides of pool D (0.53% gamma interferon-positive [IFN-γ+] CD8+ T cells compared to 0.04% relative to negative-control peptides). Restimulation of the cells with the single peptides of pool D showed that the response was present only after stimulation with peptide D14. This 16-mer peptide (L-HDAg98–113 [ERRDHRRRKALENKKK]) includes the sequences of two HLA-B*27:05 peptide ligands, L-HDAg99–108 (RRDHRRRKAL) and L-HDAg103–112 (RRRKALENKK), both of which we had predicted and proven to bind to this allele (Table 1 and Fig. 1). Four HLA-B*27-negative patients with resolved HDV infections and five HLA-B*27-positive patients with chronic HDV infections (HDV RNA positive), as well as two HLA-B*27-positive individuals without HBV, HCV, or HDV infection (anti-HDV and HDV RNA negative) used as controls, showed no response to the 16-mer or to the single peptide epitopes (Fig. 3).
TABLE 2.
Library of HDV peptidesa
| Pool | Peptide name | Position (aa) | Sequence |
|---|---|---|---|
| A | A1 | 1–16 | MSRSESRKNRGGREEL |
| A | A2 | 2–17 | SRSESRKNRGGREELL |
| A | A3 | 10–23 | RGGREELLEQWVAGRK |
| A | A4 | 18–33 | EQWVAGRKKLEELERD |
| B | B5 | 26–41 | KLEELERDLRKTKKKL |
| B | B6 | 34–49 | LRKTKKKLKKIEDENP |
| B | B7 | 42–57 | KKIEDENPWLGNIKGI |
| B | B8 | 50–65 | WLGNIKGILGKKDKDG |
| C | C9 | 58–73 | LGKKDKDGEGAPPAKR |
| C | C10 | 66–81 | EGAPPAKRARTDQMEV |
| C | C10a | 66–81 | EGAPPAKRARTDRMEV |
| C | C11 | 74–89 | ARTDQMEVDSGPGKRP |
| D | D11a | 74–89 | ARTDRMEVDSGPGKRP |
| D | D12 | 82–97 | DSGPGKRPLRGGFTDK |
| D | D13 | 90–105 | LRGGFTDKERRDHRRR |
| D | D14 | 98–113 | ERRDHRRRKALENKKK |
| E | E15 | 106–121 | KALVNKKKQLSAGGKN |
| E | E16 | 114–129 | QLSAGGKNLSKEEEEE |
| E | E17 | 122–137 | LSKEEEEELRRLTEED |
| E | E18 | 130–145 | LRRLTEEDERRERRVA |
| F | F19 | 138–153 | ERRERRVAGPPVGGVN |
| F | F20 | 146–161 | GPPVGGVNPLEGGSRG |
| F | F21 | 154–169 | PLEGGSRGAPGGGFVP |
| F | F22 | 162–177 | APGGGFVPNLQGVPES |
| G | G23 | 170–185 | NLQGVPESPFSRTGEG |
| G | G23a | 170–185 | NLQGVPESPFARTGEG |
| G | G24 | 178–193 | PFSRTGEGLDIRGNQG |
| G | G24a | 178–193 | PFARTGEGLDIRGNQG |
| H | H25 | 186–201 | LDIRGNQGFPQDTLFP |
| H | H26 | 194–209 | FPQDTLFPADPPLSPQ |
| H | H27 | 199–214 | LFPADPPLSPQSCRPQ |
Overlapping 16-mer peptides spanning the whole HDAg open reading frame of 214 amino acids that were used for T cell stimulation.
FIG 2.

Peptide recognition by T cells from patients with resolved HDV infections. PBMCs were stimulated with the indicated peptides after in vitro expansion and surface staining for T cell markers. ICS was performed and analyzed by flow cytometry. (A) CD8 and IFN-γ staining after stimulation with pools A to H of overlapping 16-mer peptides spanning the whole L-HDAg and the indicated single peptides from pool D. Peptide D14 was L-HDAg98–113 (ERRDHRRRKALENKKK). (B and C) Representative dot plots of CD8 T cell responses of patient B (B) and patient C (C) to the 16-mer peptide D14, as well as the two indicated 10-mer candidate epitopes L-HDAg99–108 (RRDHRRRKAL) and L-HDAg103–112 (RRRKALENKK) within the 16-mer peptide. Negative controls were treated exactly the same as the other samples from the respective patients but were not stimulated with peptide prior to intracellular IFN-γ staining.
FIG 3.

HLA-B*27-restricted epitope recognition by HLA-B*27-negative patients with resolved infections, HLA-B*27-positive patients with chronic HDV infections, and HLA-B*27- positive healthy individuals. (A) PBMCs from four HLA-B*27-negative patients with resolved HDV infections (rHDV) were stimulated with the pool of peptides, including the 16-mer peptide (D14) spanning both identified HLA-B*27-restricted epitopes, and evaluated for IFN-γ production by CD8 T cells. (B) PBMCs from five HLA-B*27-positive patients with chronic HDV infections (cHDV) were expanded with a 16-mer peptide spanning both HLA-B*27-restricted epitopes, as well as single 10-mer epitopes (cHDV-1 to -3) or only 10-mers (cHDV-4 and -5). IFN-γ production was evaluated by surface and intracellular staining after stimulation. (C) The same experiment as in panel B was performed on PBMC samples from 2 HLA-B*27-positive individuals without HBV, HCV, or HDV infection (Healthy-1 and -2). Negative controls (Neg. Ctrl.) were treated exactly like the other samples from the respective patients but were not stimulated with peptide prior to intracellular IFN-γ staining. Positive controls (Pos. Ctrl.) were stimulated with phorbol myristate acetate (PMA)/ionomycin prior to intracellular IFN-γ staining.
To confirm the CD8 T cell response observed in patient A, we were able to gain access to two additional HLA-B*27-positive individuals (patients B and C) with resolved HDV infections (Fig. 2B and C), while PBMCs from patient A were no longer available. Stimulation of PBMCs from both patients B and C with the 16-mer peptide (aa 98 to 113 [ERRDHRRRKALENKKK]) resulted in an IFN-γ response similar to what had been the case with patient A. Interestingly, patient B recognized the HLA-B*27:05 ligand L-HDAg99–108 (RRDHRRRKAL) (Fig. 2B) and patient C recognized L-HDAg103–112 (RRRKALENKK) (Fig. 2C), indicating a differential CD8 T cell response in HLA-B*27-positive individuals. Our HLA-B*27+ patients were recruited from Germany, Italy, and Spain, where the HLA-B*27 subtypes (alleles) B*27:05 and B*27:02 are prevalent. Since these two HLA-B*27 subtypes may influence the targeting of virus-specific CD8+ T cell epitopes (24), we analyzed the presentation of the epitope L-HDAg103–112 by HLA-B*27:05 and -B*27:02. Of note, the epitope was presented by both HLA-B*27 subtypes (Fig. 4).
FIG 4.

The epitope HLA-B*27 L-HDAg103–112 is restricted by HLA-B*27 subtypes HLA-B*27:05 (HLA-B*27 prototype) and HLA-B*27:02 (minor Mediterranean HLA-B*27 subtype). PBMCs were expanded in the presence of peptide RRRKALENKK for 10 days. Then, intracellular IFN-γ staining was performed after direct stimulation with peptide (left) or stimulation with EBV-immortalized B cell lines (BCL) that were positive for HLA-B*27:05 or HLA-B*27:02 or negative for HLA-B*27, and the cells were loaded with peptide overnight and then washed extensively (bottom) or not loaded with peptide (top). Cells were also stimulated with PMA/ionomycin as positive controls.
According to prediction and binding assays, in our hands, the two previously described HLA-A*02-restricted peptides (L-HDAg26–34 and L-HDAg43–51) (12) had low prediction scores and no capacities for binding to the HLA-A*02 molecule in vitro. Nevertheless, we intended to confirm the two epitopes in 4 HLA-A*02-positive patients with resolved HDV infections. We observed no specific T cell response in PBMCs of all 4 patients after 10 days of culture (Fig. 5).
FIG 5.

Analysis of T cell responses of HLA-A*02-positive patients to previously described HLA-A*02-restricted HDV epitopes. PBMCs from 4 HLA-A*02-positive patients with resolved HDV infections were stimulated with previously described HLA-A*02-restricted HDV epitopes, L-HDAg43–51 (KLEDENPWL) and L-HDAg26–34 (KLEDLERDL), in a 10-day culture. T cell surface marker staining, as well as ICS, were performed and analyzed by flow cytometry. The percentage of IFN-γ+/CD8+ T cells is indicated in each diagram. Patient number 4 was not stimulated with L-HDAg26–34 (KLEDLERDL). Negative controls were treated exactly like the other samples from the respective patients but were not stimulated with peptide prior to intracellular IFN-γ staining. Positive controls were stimulated with PMA/ionomycin prior to intracellular IFN-γ staining.
Molecular footprints for viral immune escape in HLA-B*27 epitopes L-HDAg99–108 and L-HDAg103–112.
Several studies have demonstrated that viral mutations leading to sequence variation within T cell epitopes are selected as a result of immune pressure. These mutations can result in failure of T cell recognition and may be responsible for viral persistence. We therefore compared the coding sequences for L-HDAg by direct sequencing in a cohort of 104 patients with chronic HDV infection who were HLA typed. Sequence alignment and subsequent phylogenetic analysis indicated that all the isolates were HDV genotype 1 (data not shown). Within the 214-aa L-HDAg, we observed high variability, e.g., at amino acid positions 9, 121, and 191, whereas aa 10, 99, and 115 were conserved in all the isolates (data not shown). These analyses indicated that the candidate HLA-B*27-restricted epitopes (aa 99 to 108 and 103 to 112) are located in a relatively conserved region of L-HDAg.
Comparing HLA-B*27-positive and -negative patients, we found a higher rate of substitutions within the verified HLA-B*27 epitopes L-HDAg99–108 and L-HDAg103–112 in HDV isolates from the six HLA-B*27-positive individuals (Fig. 6). Sequence analysis indicated that two amino acid substitutions, R105K and K106M, were significantly enriched in isolates from HLA-B*27-positive patients (P = 0.002). Two additional residues within this region, L-HDAg aa 100 and 112, showed some variations in both HLA-B*27-positive and -negative patients that were present not only in genotype 1, but also in all 8 genotypes of HDV following comprehensive analysis of 621 available isolates from GenBank (Fig. 7). We also analyzed amino acid sequences upstream of L-HDAg99–108 and downstream of L-HDAg103–112 in isolates from HLA-B*27-positive versus HLA-B*27-negative patients with chronic HDV infections. These analyses indicated no enrichment of mutations in the flanking regions of these two epitopes, as described previously (25). The L-HDAg sequence analysis was also extended to another 13 predicted epitopes restricted by HLA-A*01, -A*02, -A*03, -A*24, or -B*07 (Table 1). Importantly, for none of these candidate epitopes was a higher frequency of viral mutations observed in isolates from patients positive for the respective HLA type compared to patients negative for the alleles. These results are consistent with those of prediction and binding assays.
FIG 6.
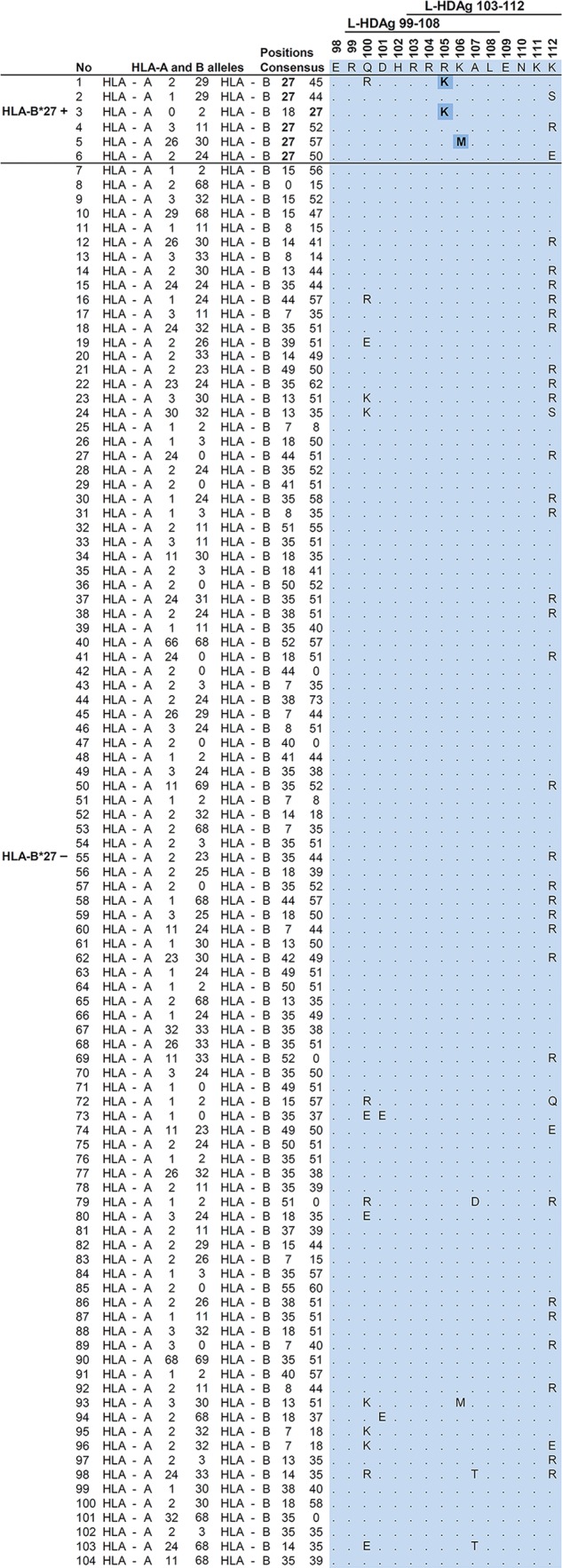
Viral sequence variations within the HLA-B*27-restricted epitopes. Amino acid sequences spanning the two overlapping HLA-B*27-restricted epitopes, L-HDAg99–108 (RRDHRRRKAL) and L-HDAg103–112 (RRRKALENKK), were subtracted from HDV isolate sequence analyses. The amino acid sequence alignments were sorted by HLA-B*27 haplotype. The first six lines show the sequence data from individual HLA-B*27-positive patients. The rest of the list shows the frequencies of the amino acid variations found in 98 HLA-B*27-negative patients. Apart from variability at aa 100 and 112 present in both HLA-B*27-positive and -negative patients, variations within the two epitopes were observed in 6 out of 98 HLA-B*27-negative patients versus 3 out of 6 HLA-B*27-positive patients. Fisher's exact test was used to make contingency tables and to calculate the P values for each amino acid–HLA-B*27 correlation. P values smaller than 0.05 were considered statistically significant. Substitutions at residues 105 and 106, reflecting immune escape mutations inside these epitopes, are shaded.
FIG 7.

Amino acid substitutions at aa 100 of L-HDAg in HDV genotypes 1 to 8. The most frequent viral protein sequence variations in the region binding to HLA-B*27 within HDV genotype 1 and other HDV genotypes (2 to 8) are listed.
Notably, two of the three HLA-B*27-positve patients (Fig. 6, no. 2 and 6) with wild-type epitopes had resolved the HDV infection in follow-up after more than 3 years of persistent infection.
To identify additional minor variants, next-generation sequencing was performed in HDV isolates from the HLA-B*27-positive patients and the six randomly selected HLA-B*27-negative patients. The analyses of 43,724 validated sequences demonstrated that the substitutions identified by direct sequencing were present in 100% of the haplotype 4/5 HLA-B*27-positive patients but in 0/6 HLA-B*27-negative patients. Additional low-frequency substitutions (0.3 to 0.8% of the sequences) in the L-HDAg epitope coding region were observed only in HLA-B*27-positive cases (Table 3). Haplotypes at position 112 showed a polymorphism, K/R, that was observed in both HLA-B*27-positive and -negative cases (Fig. 6). In two HLA-B*27-negative cases, a mixture of 112K and R was observed in minor proportions (Table 3). Thus, distinct molecular footprints were identified in the L-HDAg coding sequences of HDV isolates in HLA-B*27-positive patients.
TABLE 3.
Deep-sequencing analysis of 11 samples of patients (5 HLA-B*27 positive and 6 HLA-B*27 negative) using UDPS
| HLA status | Patient | 99–112 sequencea | Epitope polymorphism(s), proportion(s) (%) | No. of haplotypes | No. of reads |
|---|---|---|---|---|---|
| HLA-B*27 positive | H09 | RQDHRRRKALENKS | K111R, 0.8; K112S, 100 | 14 | 3,428 |
| H20 | RQDHRRRKALENKR | 26 | 3,346 | ||
| S17 | RQDHRRKKALENKK | R105K, 100; K111R, 0.8 | 29 | 4,244 | |
| H36 | RQDHRRRKALENKE | Q100R, 0.8; K112E, 100 | 17 | 3,959 | |
| It06 | RQDHRRRMALENKK | K106M, 100; R105G, 0.3 | 15 | 2,341 | |
| HLA-B*27 negative | H26 | RQDHRRRKALENKR | 9 | 3,447 | |
| H40 | RQDHRRRKALENKR | 41 | 2,422 | ||
| It04 | RQDHRRRKALENKR | 22 | 4,457 | ||
| It20 | RQDHRRRKALENKK | K112R, 18.7 | 46 | 2,487 | |
| S10 | RQDHRRRKALENKK | K112R, 2 | 12 | 3,397 | |
| E18 | RQDHRRRKALENKK | 19 | 3,277 |
The master sequence corresponding to the epitope region (HDAg codons 99 to 112), with amino acid positions affected by polymorphisms in boldface and substitutions in relation to the canonical epitope sequence observed in the master sequence underlined.
Impacts of footprints within HLA-B*27 epitopes L-HDAg99–108 and L-HDAg103–112 on the CD8 T cell response.
To investigate whether the amino acid variations detected in our patient cohort had an impact on the T cell response, we tested the variant peptides for cross-recognition in the HLA-B*27+ patients with resolved HDV infections (Fig. 8). Further characterization of L-HDAg103–112 using PBMCs from patient C indicated equal recognition of the 10-mer epitope and the corresponding 9-mer (L-HDAg104–112 [RRKALENKK]) (Fig. 8A). Variation at position 112 (K112R), observed in both HLA-B*27-positive and -negative patient groups, did not influence CD8 T cell responses. Therefore, we investigated the impacts of R105K and K106M substitutions in both 10-mer and 9-mer epitopes with lysine (K) at position 112. While R105K substitutions affected the corresponding T cell response in none of the epitopes, the K106M substitution completely impaired the specific T cell response to 10-mer and 9-mer epitopes. Equivalent results were obtained when an IFN-γ enzyme-linked immunosorbent spot (ELISpot) assay instead of intracellular cytokine staining was performed as a readout (Fig. 9). This indicated that for patients recognizing the epitope L-HDAg103–112, only the variation K106M is able to allow HDV to escape virus-specific immunity.
FIG 8.

Functional impacts of amino acid variations in L-HDAg103–112 and L-HDAg99–108 on HDV-specific CD8 T cell response. (A) The 10-mer L-HDAg103–112 (RRRKALENKK) or the 9-mer L-HDAg104–112 (RRKALENKK) peptide was used to stimulate IFN-γ responses of CD8+ T cells from a donor with resolved HDV infection (patient C). The functional impacts of K112R, R105K, and K106M amino acid substitutions on the T cell response were analyzed by flow cytometry using the respective 10-mer (left) and 9-mer (right) peptides for stimulation. (B) Either highly prevalent L-HDAg99–108 RQDHRRRKAL and RRDHRRRKAL variants of HDV or rare Q/R100K and Q/R100E variants were used to stimulate IFN-γ responses of CD8+ T cells from a donor with resolved HDV infection (patient B) (2 left columns). The functional impacts of potential immune escape variants R105K and K106M (indicated in red) on the T cell response were analyzed by flow cytometry (2 right columns). The negative control was treated exactly like the other samples from the respective patients but was not stimulated with peptide prior to intracellular IFN-γ staining.
FIG 9.

Mutation K106M leads to viral escape. PBMCs from patient C 2 and 3 years after HDV clearance (top) were expanded in the presence of peptide L-HDAg104–112 for 10 days and were then analyzed by intracellular IFN-γ staining after stimulation with peptide L-HDAg104–112, as well as the two viral variants K106M (■) and R105K (▲) in increasing concentrations. The ELISpot assay was also performed after stimulation with wild-type or variant peptide in increasing concentrations (bottom). Tests 2 years after HDV clearance were performed singly due to limitations of cell numbers; tests 3 years after HDV clearance were performed in triplicate (the data are presented as means ± standard deviations). For statistical analysis of responses to variant versus consensus, 2-tailed Student t tests were performed. A P value of less than 0.05 was considered significant. *, P < 0.05; ***, P < 0.001.
In addition to the variations R105K and K106M within the L-HDAg99–108 epitope, we also observed variations at position 100 (boldface), the second amino acid in that epitope, throughout all the studied isolates (RRDHRRRKAL, RQDHRRRKAL, REDHRRRKAL, and RKDHRRRKAL) (Fig. 6). To address the impacts of these amino acid substitutions on T cell recognition, we tested the peptide variants in HLA-B*27-positive patient B, with resolved HDV infection, for cross-recognition (Fig. 8B). This assay confirmed that L-HDAg99–108 induced CD8 T cell responses irrespective of aa 100 variations (R[R/Q/E/K]DHRRRKAL) (Fig. 8B, left). However, IFN-γ production was not observed when PBMCs were stimulated with peptide variants either at position 105 (R105K) or at position 106 (K106M) (Fig. 8B, right). This matched our finding that amino acid substitutions R105K and K106M were observed exclusively in HLA-B*27-positive patients with chronic HDV infections, whereas substitutions at position 100 were observed in patients with diverse HLA haplotypes (Fig. 6) and are also prevalent in other HDV genotypes (Fig. 7). Taken together, our data provide strong evidence that two residues, 105 and 106, within the novel HLA-B*27-restricted CD8 T cell epitope L-HDAg99–108 are selected under immune pressure by the CD8 T cell response and that the variants confer an immune escape on HDV.
In silico structural analysis of six representative peptide variants bound to HLA-B*27:05.
To gain a detailed molecular understanding of the HLA-B*27:05 binding mode of the newly identified epitopes and their T cell activation properties, we performed a molecular dynamics (MD)-based analysis of the binding properties of six representative peptides bound to HLA-B*27:05: the epitope L-HDAg99–108 (RRDHRRRKAL), the naturally occurring L-HDAg peptide variants mutated at amino acid position 100 (Q/E/K100), and the immune escape variants R105K and K106M (Table 4 and Fig. 10 to 12).
TABLE 4.
Experimental and calculated MHC binding and TCR activity data
| Peptide | Abbreviation | MHC binding (%)a | T cell activity (%)b | Eint (kcal/mol)c |
|---|---|---|---|---|
| RRDHRRRKAL | WT | 127.2 ± 13.3 | 0.59 | −686.96 ± 15.39 |
| RQDHRRRKAL | R100Q | 61.0 ± 13.8 | 0.57 | −668.75 ± 16.85 |
| REDHRRRKAL | R100E | 62.6 ± 12.4 | 0.52 | −570.54 ± 41.39 |
| RKDHRRRKAL | R100K | 40.2 ± 5.3 | 0.50 | −727.40 ± 3.17 |
| RRDHRRKKALd | R105K | 92.7 ± 3.7 | 0.08 | −678.31 ± 14.93 |
| RRDHRRRMALd | K106 M | 133.0 ± 2.7 | 0.18 | −484.66 ± 24.61 |
Binding (±standard deviation) of MHC binding of two replicates relative to the positive control (KRWIILGLNK).
Compare Fig. 10.
Average (±standard deviation) of the interaction energies (Eint) of two independent MD simulations.
The escape mutations are indicated in bold letters.
FIG 10.

Representative structures of L-HDAg99–108 peptide epitopes carrying natural variants R/Q100 or immune escape variants R105K and K106M bound to HLA-B*27:05. For better visualization, all bound peptide ligands are oriented from the C to the N terminus (left to right). The HLA-B*27:05 molecule is shown in cartoon representation in gray, and important residues are shown in atom-type colored licorice representation (cyan carbon atoms). The peptides are shown in licorice (orange) representation or alternatively as a solvent-accessible surface area colored according to the electrostatic potential (i.e., blue, positively charged; red, negatively charged; white, neutral areas of the surface). Hydrogen bonds are shown in magenta. (A and B) Structures of the bound R100 (A) and Q100 (B) L-HDAg99–108 peptides undergoing alternative binding site interactions; the insets show enlarged views. (C and D) Structures of the bound wild-type L-HDAg99–108 peptide (C) and its variant R105K (D). The residues R105 and R104 are boxed in yellow. (E to H) Structures of the bound wild-type (E and G) and variant K106M (F and H) peptides that lose their interaction with one of the HLA-B*27:05 α-helices. The insets provide enlarged views of the binding site interactions. (G and H) Structural representations of the MHC binding site, with the bound peptide shown as a surface representation.
FIG 11.

Inter- and intramolecular hydrogen bonds of the L-HDAg99–108 epitope (A) and the K106M variant of the L-HDAg99–108 epitope (B) monitored over the simulation time. A vertical black line indicates that at least one hydrogen bond is formed at a specific time frame during the simulation between the functional groups of the two amino acid residues shown on the y axis. The percentage of simulation time during which a hydrogen bond exists is shown on the right. The L-HDAg99–108 epitope and K106M variant amino acid residues are depicted in orange, and MHC binding pocket amino acid residues are shown in cyan.
FIG 12.

Representative structures of L-HDAg99–108 peptide epitopes carrying the natural variants K/E100 bound to HLA-B*27:05. For better visualization, all the bound peptide ligands are oriented from the C to the N terminus (left to right). The MHC molecule is shown in cartoon representation in gray, and important residues are shown in atom-type colored licorice representation (cyan carbon atoms). The peptides are shown in licorice (orange) representation. Hydrogen bonds are shown in magenta. The insets provide enlarged views of the binding site interactions. (A) Structure of the bound K100 peptide variant. (B) Structure of the bound E100 peptide variant.
In Table 4, the experimental binding and T cell activation data for these peptides are provided, together with the calculated interaction energies. The experimental MHC binding data suggest that R100 is the strongest binder among all the peptides varying at position 100 (Table 4, column 3). This is also reflected in the calculated interaction energies for the Q100 and E100 variants (Table 4, column 5), but not for K100. Thus, two further analyses of the theoretical results were performed. First, a visual analysis of the structural binding properties was performed (Fig. 10 and 12), which showed that all the variants (Q/E/K100) fit less well into the binding site than R100. Second, a quantitative analysis of the hydrogen bond network between the peptides and the HLA-B*27:05 binding site (data not shown) was done, which substantiated the visual observations. The area of the HLA-B*27:05 binding site in which residue 100 is located consists of a very deep pocket with two hydrogen bond acceptors/donors, E69 and T48, at its bottom (Fig. 10A, inset). This pocket is optimally filled by the large R residue, which forms three or four stable hydrogen bonds with E69 and T48, while K, containing only one charged group, interacts alternatively with E69 or T48, with its side chain flipping back and forth between the two residues during the simulation, indicating unstable binding. Q and E are simply too small to form stable interactions within the pocket, as their side chains cannot reach the bottom of the binding site. Therefore, our computational analyses can provide an explanation for the experimentally observed strong binding of the R100 wild type and are consistent with the experimental HLA-B*27:05 binding data.
To estimate the influence of the R105K and K106M mutations on the T cell response, we investigated their effects on the shape and electrostatic potential of the solvent-exposed surface region of the peptides, as this is the region to which the T cell receptor (TCR) binds (Fig. 10 and 12). In addition, the same visual analysis and quantitative hydrogen bond analysis as for the R100 variants were performed (Fig. 10 and 11). For the L-HDAg99–108 epitope (Fig. 10C), the TCR interface is dominated by R105 and R104, forming a very characteristic steric and electrostatic fingerprint, which seems to be important for TCR recognition, as for both variants, which do not show any T cell response, large differences were observed in the TCR interaction region (Fig. 10D and H). The R105K mutation leads directly to strong changes in the electrostatic potential of the peptide in the TCR interaction area (Fig. 10D). In the K106M variant, the effects are more complex and are caused indirectly by a different hydrogen bond pattern within the peptide, which leads to considerable changes in the conformation and thus the shape of the peptide's surface (Fig. 10F and H and 11). The reason behind this different behavior of the mutant is analogous to the observations made for the variations at position 100, as discussed above, namely, that the native residue K106 fits optimally into its MHC binding pocket, whereas the M mutant does not. Thus, K106 forms stable hydrogen bonds with the pocket residues D101 and D98 (Fig. 10E), leading to an optimal peptide-MHC hydrogen bond network, which is stable throughout the simulation (Fig. 11A). On the other hand, M106 cannot interact via hydrogen bonding with the negatively charged D101/D98 (Fig. 10F) and thus moves to the edge of the binding pocket during the simulation. This leads to large structural rearrangements within the peptide and a final, alternative peptide conformation stabilized by strong intrapeptide hydrogen bonds between peptide residues R103 and D101 (Fig. 10F and 11B). Due to this alternative conformation, the peptide-TCR interaction surface differs considerably from that of the original K106 epitope (Fig. 10H versus G), providing a potential explanation for the missing T cell response to the K106M variant and consistent with the effect observed for R105K.
Importantly, both variants (R105K and K106M) still show strong MHC binding (Table 4). For R105K, this is directly reflected in the MHC-peptide interaction energy (Table 4) and is not surprising, as residue 105 is located outside the MHC binding site and thus does not contribute to MHC-peptide binding. For variant K106M, the calculated interaction energy is rather low, which is in contrast to the experimental MHC binding data. This, however, can be explained by the above-discussed conformational rearrangement of the peptide due to the mutation, leading to a very strong stabilization of the internal peptide conformation and thus a very favorable internal energy of the peptide (data not shown), which comes at the cost of weaker MHC interactions. Interestingly, this alternative binding mode of a very rigid peptide conformation interacting moderately with the MHC binding site seems to also lead to very stable bound complexes (the experimental data are shown in Table 4). Thus, in conclusion, our modeling results indicate that the missing T cell response to the R105K and K106M variants of the HLA-B*27:05 epitope L-HDAg99–108 is caused by structural and electrostatic differences at the TCR interface, which are not directly correlated with the peptides' HLA-B*27:05 binding strength.
DISCUSSION
In this study, we identified two overlapping HLA-B*27-restricted CD8 T cell epitopes, L-HDAg99–108 and L-HDAg103–112, in a conserved region of the only HDV antigen. A specific CD8 T cell response to both epitopes was detected in HLA-B*27-positive patients who were able to resolve their HDV infections. Molecular footprinting using a collection of HDV isolates from patients with chronic hepatitis delta provided strong evidence that variations at aa 105 and 106, common to both CD8 T cell epitopes, are selected. Functional T cell analysis confirmed that aa 105 and 106 variants confer immune escape on HDV, and in silico structural modeling of the HLA-B*27 epitope L-HDAg99–108 revealed that both the R105K and the K106M variants dramatically alter the TCR interface, explaining the immune escape.
Antibodies directed against the HDV nucleoprotein have no neutralizing capacity and are not effective in controlling HDV infection (6). Therefore, T cell response plays a major role in eliminating HDV, which occurs in about 10 to 20% of HBV carriers who have been superinfected (26). On the other hand, about 80 to 90% of superinfected patients are not able to eliminate the virus and develop chronic HDV infection. This may be due to primary or secondary failure of the T cell response, which in turn may be caused by T cell exhaustion, by an immune escape of the HDV variants, or simply by an absence of epitopes to certain HLA alleles within HDV proteins.
HDV has a very short genome of 1.7 kb and only one ORF encoding two isoforms of one protein (S-HDAg and L-HDAg). The 214-aa L-HDAg comprises all potential epitopes that may be recognized by T cell responses. In our analysis, the number of peptides predicted to bind any of the major HLA class I molecules was significantly lower than that of proteins from other viruses, such as hepatitis A virus (HAV), HBV, and HCV (27–29). The lack of epitopes may be explained by the fact that the HDV protein could have been derived from a host protein (30) against which T cells are deleted during the selection of T cells in the thymus. It is notable that we did not detect high-affinity HDV epitopes binding to HLA alleles other than HLA-B*27, although we used various approaches. In HLA binding analyses of the predicted peptide epitopes, we observed only low-affinity binding to, e.g., HLA-A*01:01, HLA-A*02, or HLA-B*07:02, alleles with high prevalence in Western countries. The low-affinity binding indicates that these peptides are probably outcompeted by better-binding peptides. A previous study by Huang et al. described two HLA-A*02 epitopes in Asian patients (12), but we could not confirm these findings in 4 HLA-A*02-positive patients with resolved HDV infections in our study. This may be due to differences in the amino acid sequences of genotype 1 isolates prevalent in East Asia. Moreover, these HLA-A*02:01-restricted HDV epitopes are located in a highly variable region of L-HDAg that is not conserved in most European HDV isolates. HDV contains only a few highly conserved regions in its genome. Among these is the region around the two identified HLA-B*27 epitopes, which is conserved throughout all isolates from all genotypes. In fact, our comprehensive analyses of 545 sequences of L-HDAg from all eight HDV genotypes indicated that the region restricted by HLA-B*27 is conserved within all the genotypes, implying a possible cross-genotypic HDV-specific T cell response. This may be a unique feature of HDV and stands in contrast to earlier findings made in studies of HCV-specific T cell responses, where the protective antiviral effect of HLA-B*27 was shown to be restricted to HCV genotype 1 and not to any other prevalent genotypes, such as 3a (31). The low substitution rate in the region may thus be due to viral fitness costs of mutations in the region.
We demonstrated an HLA-B*27-restricted HDV-specific CD8 T cell response in some patients with resolved HDV infections. However, other HDV-infected HLA-B*27-positive patients fail to clear HDV and develop a persistent infection. We therefore hypothesized that viral variants emerged that could not be targeted by the relevant virus-specific CD8 T cells. We addressed this possibility by analyzing a multicentric cohort of patients in which we observed amino acid substitutions in both L-HDAg99–108 and L-HDAg103–112 epitopes abrogating the respective CD8 T cell responses. This sequence variation was absent in HLA-B*27-negative patients. These findings were strengthened by deep sequencing, as we detected minor variations at this location only in the HLA-B*27-positive patients, in addition to the mutations found by conventional sequencing. Further longitudinal studies will be of interest to determine whether such minor species may outgrow the wild-type virus. Classical escape mutations are known as mutations occurring at anchor positions impairing HLA binding capacity and have been detected in other viruses, e.g., HBV and HCV. It is noteworthy that none of the escape mutations described here occur at HLA-B*27 anchor residues. Interestingly, one of the described variants of the L-HDAg103–112 epitope demonstrated a methionine substitution for lysine at residue 106 (the fourth residue of the epitope), which was also reported in an HLA-B*27-restricted HCV epitope, abrogating the T cell response to HCV (31); this may indicate the role of methionine in the T cell interface of HLA-B*27 epitopes, regardless of their origin. Our study is the first report of impairment of the HDV-specific CD8 T cell response due to escape mutation, suggesting that genetic diversity of HDV, at least in part, is driven by immune pressure.
Molecular-modeling studies of the bound complexes of the HDV-specific epitopes of L-HDAg99–108 and its potential immune escape variants of HLA-B*27 provide a potential structural explanation for why variations at the amino acid positions 105 and 106 of the peptide have a strong impact on TCR binding whereas aa 100 variations do not. Analysis of the naturally occurring aa 100 variants showed that these mutations do not affect the TCR binding surface but can significantly change the binding pattern with the HLA-B*27 molecule and thus lead to weaker interactions. This is reasonable, as aa 100 is located in a deep binding pocket of the MHC, four residues away from the two solvent-exposed arginine residues (R104 and R105) crucial for TCR binding. These observations are in accordance with our experimental T cell analyses, in which aa 100 variant peptides induced a T cell response comparable to that of wild-type peptides.
In silico analysis also provided an explanation for the importance of the surface-exposed peptide residues R104 and R105 for TCR binding. This was experimentally confirmed by the lack of T cell response to the R105K and K106M variants, supporting the notion that R104 and R105 are crucial for T cell recognition. Our computational results indicate that the escape mechanism behind the corresponding variants is based on mutation-induced structural (K106M) and electrostatic (R105K) changes in the surface region of the bound peptide, which is essential for T cell recognition. Thus, the structural integrity of the TCR interface seems to be more important than the MHC binding affinity of the variant. This conclusion is supported by the fact that no correlation between the experimental or calculated MHC binding properties of the peptides and their ability to activate T cell responses could be found.
It is important to point out that viral escape may not be the only mechanisms contributing to virus-specific CD8+ T cell failure during persistent infection. Indeed, Huang et al. demonstrated HLA*02-restricted HDV-specific CD8+ T cell responses in two patients with long-term negative HDV RNA (and thus probably recovery from HDV infection) but were not able to demonstrate HDV-specific responses in persistently infected patients, indicating that HDV-specific CD8+ T cells were either not primed in these patients or had undergone T cell exhaustion and deletion (12). In line with these results, we also failed to identify HLA-B*27-restricted HDV-specific CD8+ T cells after antigen-specific expansion in five patients with chronic HDV infections (Fig. 3). Even after performing peptide/MHC multimer bead-based enrichment of HDV-specific CD8+ T cells from five patients with chronic HDV infections, a technique well established in our laboratory for enrichment of low-frequency populations for virus-specific CD8+ T cells (32, 33), we were not able to detect HDV-specific CD8+ T cells in chronically infected patients (Fig. 13). These data support the hypothesis that HDV-specific CD8+ T cells undergo final exhaustion and deletion in patients with chronic HBV-HDV coinfections. In addition, sequestration of HDV-specific CD8+ T cells in the liver may be an alternate explanation for their absence in peripheral blood. In contrast, using this technique, HDV-specific CD8+ T cells were readily detectable in a patient with cleared HDV infection but persistent low-level HBV viremia. These HDV-specific CD8+ T cells were antigen experienced (not naive, as indicated by CCR7 and CD45RA costaining) and displayed high expression of PD1 but varying expression levels of CD127.
FIG 13.

Ex vivo peptide/tetramer bead-based enrichment of L-HDAg104-112-specific CD8+ T cells in patients with resolved versus chronic HDV infections. (A) L-HDAg104-112 peptide/HLA-B*27:05 tetramer bead-based ex vivo enrichment of HDV-specific CD8+ T cells was performed for five patients with chronic HBV/HDV coinfections, as well as one patient with chronic HBV infection and resolved HDV infection (32). Using this extremely sensitive method, we were able to detect HDV-specific CD8+ T cells from a patient with chronic HBV infection and resolved HDV infection. In contrast, we were not able to enrich HDV-specific CD8+ T cells from the five patients with chronic HBV-HDV coinfections. The autologous sequences are also displayed (ND, viral sequencing not done due to low viral load). (B) Ex vivo-enriched HDV-specific CD8+ T cells from the patient with chronic HBV infection and resolved HDV infection were further analyzed for expression of PD1 and CD127. Naive CD8+ T cells were excluded by costaining for CCR7 and CD45RA.
Overall, our results indicate that the number of potential epitopes to the frequent HLA alleles in the 214 aa of the single protein of HDV is very low; only two HLA-B*27-restricted epitopes have been defined so far, whereas for all major HLA alleles present in the European population, no epitope has so far been identified. The low number of epitopes and T cell failure resulting from immune escape may contribute to the high chronicity rate in HDV infection, in addition to the immunoinhibitory environment established by persistent HBV infection that persistent HDV infection is tied to. The hypothesis that HLA-B*27 maybe a protective allele against HDV, as shown in other infections, like HIV and HCV, has to be verified by initiation of a prospective study in a larger cohort with patients who are HLA-B*27 positive or negative. These data also imply that the development of effective antiviral approaches, e.g., immunotherapy, against HDV will be a challenging task.
MATERIALS AND METHODS
Patient cohorts and samples.
Chronically HDV-infected patients (n = 104; anti-HDV+ HDV RNA+/−) were recruited from 8 different medical centers located in Germany (Essen, Freiburg, Hannover, and Munich), Spain, Italy, and Iran. HLA backgrounds, as well as L-HDAg sequences, were determined.
HLA class I typing was performed with a Luminex PCR sequence-specific oligonucleotide (PCR-SSO) probe, using LabType SSO kits (One Lambda, Canoga Park, CA, USA) (20), from genomic DNA extracted from blood samples using a column purification kit (Qiagen, Hilden, Germany). Two-digit HLA typing and, in selected patients, high-resolution typing were done to define four-digit HLA types or subgroups.
HDV phylogenetic analysis and epitope prediction.
Total RNA was extracted from the patient's serum samples and was reverse transcribed into cDNA by reverse transcriptase from Moloney murine leukemia virus (Promega, Madison, WI, USA) at 37°C for 1 h using HDV-specific primer 771R (Table 5). In order to obtain sequences of the L-HDAg-encoding region, cDNA products from the reverse transcription step were amplified by Pfu DNA polymerases (Promega, USA) in a two-step nested PCR. The PCR steps using HDV-specific primers 891-F, 339-R, 912-F, and 1674-R were performed under the following thermal profile: 94°C for 10 min; 35 cycles of 94°C for 30 s, 54°C for 45 s, and 72°C for 90 s; and then an elongation step at 72°C for 7 min. Finally, the PCR products were purified via QIAquick gel extraction (Qiagen) and directly sequenced on an ABI 3730xl DNA analyzer using internal primers 912-F and 1674-R.
TABLE 5.
Primers used in L-HDAg amplification for direct and deep sequencing
| Primer | Sequence | Location (nt) |
|---|---|---|
| 771R | 5′-CGGTCCCCTCGGAATGTTG-3′ | 753–771 |
| 339R | 5′-GCTGAAGGGGTCCTCTGGAGGTG-3′ | 319–341 |
| 1674R | 5′-AGAAAAGAGTAAGAGYACTGAGG-3′ | 1652–1674 |
| 912F | 5′-GAGATGCCATGCCGACCCGAAGAG-3′ | 912–936 |
| 891F | 5′-AGGTCGGACCGCGAGGAGGT-3′ | 891–910 |
| M13 HDV 956a | 5′-GTTGTAAAACGACGGCCAGTTCACTGGGGTCGACAACTCTG-3′ | |
| M13 HDV 1360a | 5′-CACAGGAAACAGCTATGACCGTAGACTCCGGACCTAGGAAGA-3′ | |
| OligoAMIDM13fwb | 5′-CGTATCGCCTCCCTCGCGCCATCAGMIDGTTGTAAAACGACGGCCAGT-3′ | |
| OligoBMIDM13rvb | 5′-CTATGCGCCTTGCCAGCCCGCTCAGMIDCACAGGAAACAGCTATGACC-3′ |
M13 HDV 956 and M13 HDV 1360 primers consist of M13 fagus universal tail at the 5´ ends followed by HDV-specific sequences (underlined).
Specific oligonucleotides for UDPS are indicated in bold followed by multiplex identifiers (MID) and the M13 fagus universal tails.
HDV sequences from this study and NCBI GenBank were aligned using ClustalX2 software (34). A maximum-likelihood phylogenetic tree was constructed under the Tamura-Nei substitution model using MEGA software v6 (35). The reliability of the pairwise comparison and phylogenetic tree analysis was assessed by 1,000-replicate bootstrapping. The genotypes of HDV strains were determined by comparison to full-length L-HDAg reference sequences from GenBank.
Next-generation sequencing was performed in 11 selected samples to identify variations at a detection level of 0.1% and to pin down variations in the region flanked by nucleotide positions 956 to 1,360 by ultradeep pyrosequencing (UDPS) (primer sequences are listed in Table 5). The amplicons had a length of 515 bp and were isolated from 0.9% agarose gels with the QIAquick extraction kit (QIAquick spin handbook; Qiagen, Hilden, Germany) and quantified using Quan-iTPicogreens DNA reagent (Invitrogen). UDPS was performed with the 454/GS-Junior platform (Roche, Branford, CT, USA) using titanium chemistry (GS-Junior titanium sequencing kit). The FASTA file from the GS-Junior was demultiplexed and filtered as recently described (36). The specific region analyzed by UDPS (after discarding primer sequences) covered nucleotides (nt) 977 to 1338 of the HDV genome (362 nt were analyzed). All the sequences were submitted as a new BioProject to GenBank. FASTA reads were filtered and analyzed as recently described (37).
Two hundred L-HDAg sequences of HDV genotype 1 from GenBank were applied for epitope prediction using the IEDB (http://www.iedb.org) and the SYFPEITHI database (http://www.syfpeithi.de) algorithms (38–40) to predict potential binders to the most common alleles in Europeans (41): HLA-A*02, -A*01, -A*03, -A*11, -A*24, -B*35, -B*51, -B*07, -B*18, -B*08, and -B*27.
Analysis of HLA binding capabilities of candidate peptides.
The binding affinities of the predicted epitopes were analyzed with the UV-mediated peptide exchange assay using PeliChange p*HLA-A*01:01, -A*02:01, -A*03:01, -A*24:02, -B*07:02, and -B*27:05 kits (Sanquin, Amsterdam, The Netherlands) as described previously (27, 42). Briefly, peptide exchange reactions were performed by exposure for 30 min of conditional pHLA complexes (0.53 μM) to long-wavelength UV using a 366-nm UV lamp (Camag, Muttenz, Switzerland) in the presence or absence of the indicated peptide (50 μM). Subsequently, the peptide exchange efficiency was analyzed using an HLA class I enzyme-linked immunosorbent assay (ELISA), which detects beta-2 microglobulin of peptide-stabilized HLA class I complexes in a properly diluted exchange reaction mixture. To this end, streptavidin (2 μg/ml) was bound onto polystyrene microtiter wells (Nunc MaxiSorp). After washing and blocking, HLA complex present in exchange reaction mixtures or controls was captured by the streptavidin on the microtiter plate via its biotinylated heavy chain (with incubation for 1 h at 37°C). Nonbound material was removed by washing. Subsequently, horseradish peroxidase (HRP)-conjugated antibody to human beta-2-microglobulin (0.6 μg/ml) was added (with incubation for 1 h at 37°C). After removal of nonbound HRP conjugate by washing, an ABTS [2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt] substrate solution was added to the wells. The reaction was stopped after 8 min (incubation at room temperature) by the addition of a stop solution and read in an ELISA reader at 414 nm. Every peptide was independently exchanged twice. Every exchange mixture was measured in duplicate in the HLA class I ELISA. The absorbances of all the peptides were normalized to the absorbance of a known HLA allele-specific ligand with high affinity for each corresponding allele (representing 100%). Negative controls included an HLA allele-specific nonbinder and UV irradiation of the conditional HLA class I complex in the absence of a rescue peptide.
Polyclonal antigen-specific expansion of T cells.
Patient PBMCs were obtained by standard Ficoll density centrifugation (Biocoll; Biochrom, Germany). Freshly isolated PBMCs (2 × 106) were cultured in 1 ml complete medium (RPMI 1640 medium supplemented with 10% fetal calf serum, 100 U/ml penicillin, 100 g/ml streptomycin) containing anti-CD28 antibody (0.5 μg/ml; BD Biosciences). The cells were stimulated with 31 synthetic overlapping 16-mer peptides (EMC Microcollections, Tübingen, Germany; final concentration, 10 μg/ml per peptide) corresponding to the complete sequence of L-HDAg divided into 8 pools (pools A to H) or individual 10-mer or 11-mer peptides. On day 2, 1 ml complete medium and interleukin-2 (10 U/ml; Roche, Basel, Switzerland) were added. On days 10 and 12, the cells were tested for IFN-γ secretion by intracellular-cytokine staining after restimulation with the peptide pools (day 10) or individual peptides from the positive pool (day 12) as described previously (43). Prior to staining, the cells were cultured for 5 h in the presence of peptides (10 μg/ml) and 5 μg/ml brefeldin A (Sigma-Aldrich, St. Louis, MO, USA). The cells were stained using allophycocyanin (APC)-conjugated anti-CD8 and phycoerythrin (PE)-conjugated anti-CD4 at 4°C for 15 min. After permeabilization, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-IFN-γ monoclonal antibody (MAb) (all reagents were from BD Biosciences) and analyzed by flow cytometry on a FACSCalibur instrument (BD Biosciences). Dead cells were stained using Viaprobe reagent (BD Biosciences) and excluded from analyses. Data files were analyzed with FlowJo 7.6.5 software (Tree Star, Ashland, OR).
Computational modeling of HLA-B*27 binding.
To investigate the binding properties of selected L-HDAg-derived peptides to HLA-B*27:05 on the structural level, in silico modeling studies were performed. The investigated peptide–HLA-B*27:05 complexes were modeled on the basis of the crystal structure of the peptide–HLA-B*27:05 complex with Protein Data Bank (PDB) accession no. 4G9D (44). Only the α1 and α2 subdomains of the HLA-B*27:05 protein (residues 25 to 206), forming the peptide-binding cleft, were considered during modeling. The peptide sequence was mutated using the IRECS algorithm (45, 46). To allow the HLA-B*27:05 peptide-binding cleft to adapt to the mutated peptide, HLA-B*27:05 side chains were subsequently also remodeled with IRECS. The modeled peptide–HLA-B*27:05 complexes were energy minimized in a neutralized, rectangular box of TIP3P (47) water molecules with a minimum solute distance to the box boundary of 12 Å. The general system setup and the simulations were conducted with AmberTools 14 and the Amber 14 software package (48). The Amber ff99SBILDN force field (49) was chosen. MD simulations were performed using a time step of 1 fs. The SHAKE algorithm (50) was applied to all bonds involving hydrogen atoms. Long-range electrostatic interactions were computed using the particle mesh Ewald (PME) method (using the molecular dynamics program pmemd.cuda_SPFP) (51). A cutoff of 14 Å was used for the computation of nonbonded interactions. Temperature and pressure were controlled by applying the Berendsen thermostat and barostat with default settings (52). The systems were stepwise heated to 300 K while gradually decreasing solute atom restraints, using the NVT (constant number of particles N, constant volume V, and constant temperature T) ensemble. Afterward, NPT (constant number of particles N, constant pressure P, and constant temperature T)-based molecular dynamics simulations were performed for 20 ns for each system.
Trajectory processing and analysis were done with CPPTRAJ (53). MD frames for analysis were extracted every 20 ps from the last 5 ns of the simulations. Representative conformations were obtained by conformational clustering using the average linkage method with a cutoff of 2 Å, and hydrogen bonds and peptide–HLA-B*27:05 interaction energies were calculated by applying the default settings of CPPTRAJ. Electrostatic potentials were calculated using the PDB2PQR server (version 2.1.1) and APBS (version 1.2.1) (54, 55), keeping the default settings. Mapping of electrostatic potentials to the solvent-accessible surface areas of the peptide ligands was performed with VMD (version 1.9) (56). Figures showing peptide–HLA-B*27:05 complexes were also generated with VMD (version 1.9).
Study approval.
Local ethics committees approved this study according to the 1975 Declaration of Helsinki guidelines. The main approaches for analyzing the HDV-specific T cell immune responses in HDV-infected patients were approved by the Ethics Committee of the Faculty of Medicine of Duisburg-Essen University (10-4472). The protocol for isolating and analyzing genomic DNA (genome-wide association study) was also approved by the Ethics Committee of the Medical School of Hannover (OE 9515/no. 3388 and no. 5292M). The Ethics Committee of the Albert-Ludwigs-Universität Freiburg, no. 369-15, approved the protocol for investigation of viral escape from the dominant virus-specific CD8+ T cell response in HBV monoinfection and HBV-hepatitis delta virus coinfection and strategies to restore the antiviral CD8+ T cell response. All the patients who participated in the study were interviewed and informed about the concept of the study and possible side effects before they agreed to participate by giving written informed consent.
Accession number(s).
The newly determined sequences are available under GenBank accession numbers MF175257 to MF175360, BioProject accession number PRJNA434630, and SRA accession number SRP133994.
ACKNOWLEDGMENTS
This work was financially supported by internal funds of the Medical Faculty of the University of Duisburg-Essen/Technical University of Munich and by a Registry Grant from the European Association for the Study of the Liver (EASL). H. Karimzadeh was supported by a grant from the German Academic Exchange Service (DAAD). The Deutsche Forschungsgemeinschaft (DFG) financed parts of the study by support to H. Karimzadeh and B. Raziorrouh (SFB-TR179/TP03), U. Protzer (SFB-TR179/TP14 and TRR36/B13), M. Glaser (SFB 1035/A10), C. Neumann-Haefelin (SFB-TRR-179/TP 2), and D. Hoffmann and J. Timm (TRR60/B1). Instituto de Salud Carlos III grant PI14/01416, along with the European Regional Development Fund (ERDF), partly cofinanced this study by providing support to D. Tabernero.
We thank all the people who volunteered for this research and the staff working in the diagnostic sections, institutes of virology, University Hospital Essen, and TU Munich, Germany, for their close collaboration, recruiting the samples, and technical assistance. We are also grateful to Ulrich Spengler, Svenja Hardtke, and Thomas Schirdewahn for providing information from patients, including those with resolved HDV infection. We appreciate the help and support from the German Center for Infection Research (DZIF) regarding this work.
H. Karimzadeh, M. Roggendorf, M. Rizzetto, and J. Timm assisted with proof of concept and design; H. Karimzadeh, A. D. Kosinska, M. M. Kiraithe, M. Fiedler, V. Oberhardt, E.S. Alizei, J. Y. Mok, M. Nguyen, J. Grabowski, A. Olivero, A. Heinold, M. Homs, and F. Rodríguez-Frías conducted various aspects of the experimental work; B. Budeus, and D. Hoffmann prepared statistical analysis; M. Glaser and I. Antes performed the molecular modeling studies; H. Karimzadeh, A. D. Kosinska, M. Hofmann, C. Neumann-Haefelin, F. Rodríguez-Frías, D. Tabernero, W. J. E. van Esch, A. Smedile, T. Bauer, B. Raziorrouh, H. Keyvani, J. Schulze zur Wiesch, M. Buti, S. M. Alavian, H. Wedemeyer, U. Protzer, and M. Roggendorf analyzed and interpreted the data; H. Karimzadeh, A. D. Kosinska, M. Homs, I. Antes, U. Protzer, and M. Roggendorf wrote the manuscript; C. Neumann-Haefelin, M. Fiedler, M. Buti, T. Bauer, B. Raziorrouh, D. Hoffmann, H. Wedemeyer, M. Rizzetto, M. Fiedler, U. Protzer, and J. Timm provided critical revision of intellectual content; M. Fiedler, H. Wedemeyer, M. Hofmann, and M. Buti facilitated recovered patient recruitment; M. Fiedler, H. Karimzadeh, J. Grabowski, M. Roggendorf, H. Keyvani, J. Schulze zur Wiesch, and S. M. Alavian handled ethics and sample recruitment; and C. Neumann-Haefelin, U. Protzer, I. Antes, and M. Roggendorf supervised the study.
REFERENCES
- 1.Rizzetto M. 2015. Hepatitis D virus: introduction and epidemiology. Cold Spring Harb Perspect Med 5:a021576. doi: 10.1101/cshperspect.a021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gomes-Gouvea MS, Soares MC, Bensabath G, de Carvalho-Mello IM, Brito EM, Souza OS, Queiroz AT, Carrilho FJ, Pinho JR. 2009. Hepatitis B virus and hepatitis delta virus genotypes in outbreaks of fulminant hepatitis (Labrea black fever) in the western Brazilian Amazon region. J Gen Virol 90:2638–2643. doi: 10.1099/vir.0.013615-0. [DOI] [PubMed] [Google Scholar]
- 3.Weiner AJ, Choo QL, Wang KS, Govindarajan S, Redeker AG, Gerin JL, Houghton M. 1988. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J Virol 62:594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang JG, Cullen J, Lemon SM. 1992. Immunoblot analysis demonstrates that the large and small forms of hepatitis delta virus antigen have different C-terminal amino acid sequences. J Gen Virol 73:183–188. doi: 10.1099/0022-1317-73-1-183. [DOI] [PubMed] [Google Scholar]
- 5.Rizzetto M, Smedile A. 2002. Hepatitis D, p 863–875. Lippincott, Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Rizzetto M, Hoyer BH, Purcell RH, Gerin JL. 1984. Hepatitis delta virus infection, p 371–379. Grune & Stratton, Orlando, FL. [Google Scholar]
- 7.Wedemeyer H, Yurdaydin C, Dalekos GN, Erhardt A, Cakaloglu Y, Degertekin H, Gurel S, Zeuzem S, Zachou K, Bozkaya H, Koch A, Bock T, Dienes HP, Manns MP. 2011. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 364:322–331. doi: 10.1056/NEJMoa0912696. [DOI] [PubMed] [Google Scholar]
- 8.Wang D, Pearlberg J, Liu YT, Ganem D. 2001. Deleterious effects of hepatitis delta virus replication on host cell proliferation. J Virol 75:3600–3604. doi: 10.1128/JVI.75.8.3600-3604.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mauch C, Grimm C, Meckel S, Wands JR, Blum HE, Roggendorf M, Geissler M. 2001. Induction of cytotoxic T lymphocyte responses against hepatitis delta virus antigens which protect against tumor formation in mice. Vaccine 20:170–180. doi: 10.1016/S0264-410X(01)00252-3. [DOI] [PubMed] [Google Scholar]
- 10.Fiedler M, Kosinska A, Schumann A, Brovko O, Walker A, Lu M, Johrden L, Mayer A, Wildner O, Roggendorf M. 2013. Prime/boost immunization with DNA and adenoviral vectors protects from hepatitis D virus (HDV) infection after simultaneous infection with HDV and woodchuck hepatitis virus. J Virol 87:7708–7716. doi: 10.1128/JVI.00645-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nisini R, Paroli M, Accapezzato D, Bonino F, Rosina F, Santantonio T, Sallusto F, Amoroso A, Houghton M, Barnaba V. 1997. Human CD4+ T-cell response to hepatitis delta virus: identification of multiple epitopes and characterization of T-helper cytokine profiles. J Virol 71:2241–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YH, Tao MH, Hu CP, Syu WJ, Wu JC. 2004. Identification of novel HLA-A*0201-restricted CD8+ T-cell epitopes on hepatitis delta virus. J Gen Virol 85:3089–3098. doi: 10.1099/vir.0.80183-0. [DOI] [PubMed] [Google Scholar]
- 13.Neumann-Haefelin C, Oniangue-Ndza C, Kuntzen T, Schmidt J, Nitschke K, Sidney J, Caillet-Saguy C, Binder M, Kersting N, Kemper MW, Power KA, Ingber S, Reyor LL, Hills-Evans K, Kim AY, Lauer GM, Lohmann V, Sette A, Henn MR, Bressanelli S, Thimme R, Allen TM. 2011. Human leukocyte antigen B27 selects for rare escape mutations that significantly impair hepatitis C virus replication and require compensatory mutations. Hepatology 54:1157–1166. doi: 10.1002/hep.24541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kefalakes H, Budeus B, Walker A, Jochum C, Hilgard G, Heinold A, Heinemann FM, Gerken G, Hoffmann D, Timm J. 2015. Adaptation of the hepatitis B virus core protein to CD8(+) T-cell selection pressure. Hepatology 62:47–56. doi: 10.1002/hep.27771. [DOI] [PubMed] [Google Scholar]
- 15.Fitzmaurice K, Petrovic D, Ramamurthy N, Simmons R, Merani S, Gaudieri S, Sims S, Dempsey E, Freitas E, Lea S, McKiernan S, Norris S, Long A, Kelleher D, Klenerman P. 2011. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut 60:1563–1571. doi: 10.1136/gut.2010.228403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timm J, Walker CM. 2015. Mutational escape of CD8+ T cell epitopes: implications for prevention and therapy of persistent hepatitis virus infections. Med Microbiol Immunol 204:29–38. doi: 10.1007/s00430-014-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CR, Rizza CR, McMichael AJ. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 18.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 19.Wang SY, Wu JC, Chiang TY, Huang YH, Su CW, Sheen IJ. 2007. Positive selection of hepatitis delta antigen in chronic hepatitis D patients. J Virol 81:4438–4444. doi: 10.1128/JVI.02847-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinemann FM. 2009. HLA genotyping and antibody characterization using the Luminex multiplex technology. Transfus Med Hemother 36:273–278. doi: 10.1159/000228834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzmaurice K, Hurst J, Dring M, Rauch A, McLaren PJ, Gunthard HF, Gardiner C, Klenerman P. 2015. Additive effects of HLA alleles and innate immune genes determine viral outcome in HCV infection. Gut 64:813–819. doi: 10.1136/gutjnl-2013-306287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLaren PJ, Carrington M. 2015. The impact of host genetic variation on infection with HIV-1. Nat Immunol 16:577–583. doi: 10.1038/ni.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neumann-Haefelin C, McKiernan S, Ward S, Viazov S, Spangenberg HC, Killinger T, Baumert TF, Nazarova N, Sheridan I, Pybus O, von Weizsäcker F, Roggendorf M, Kelleher D, Klenerman P, Blum HE, Thimme R. 2006. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 43:563–572. doi: 10.1002/hep.21049. [DOI] [PubMed] [Google Scholar]
- 24.Nitschke K, Barriga A, Schmidt J, Timm J, Viazov S, Kuntzen T, Kim AY, Lauer GM, Allen TM, Gaudieri S, Rauch A, Lange CM, Sarrazin C, Eiermann T, Sidney J, Sette A, Thimme R, Lopez D, Neumann-Haefelin C. 2014. HLA-B*27 subtype specificity determines targeting and viral evolution of a hepatitis C virus-specific CD8+ T cell epitope. J Hepatol 60:22–29. doi: 10.1016/j.jhep.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirvani-Dastgerdi E, Amini-Bavil-Olyaee S, Alavian SM, Trautwein C, Tacke F. 2015. Comprehensive analysis of mutations in the hepatitis delta virus genome based on full-length sequencing in a nationwide cohort study and evolutionary pattern during disease progression. Clin Microbiol Infect 21:510.e11–e23. doi: 10.1016/j.cmi.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Farci P, Niro GA. 2012. Clinical features of hepatitis D. Semin Liver Dis 32:228–236. doi: 10.1055/s-0032-1323628. [DOI] [PubMed] [Google Scholar]
- 27.Schulte I, Hitziger T, Giugliano S, Timm J, Gold H, Heinemann FM, Khudyakov Y, Strasser M, Konig C, Castermans E, Mok JY, van Esch WJ, Bertoletti A, Schumacher TN, Roggendorf M. 2011. Characterization of CD8+ T-cell response in acute and resolved hepatitis A virus infection. J Hepatol 54:201–208. doi: 10.1016/j.jhep.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Brinck-Jensen NS, Vorup-Jensen T, Leutscher PD, Erikstrup C, Petersen E. 2015. Immunogenicity of twenty peptides representing epitopes of the hepatitis B core and surface antigens by IFN-gamma response in chronic and resolved HBV. BMC Immunol 16:65. doi: 10.1186/s12865-015-0127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong DK, Dudley DD, Afdhal NH, Dienstag J, Rice CM, Wang L, Houghton M, Walker BD, Koziel MJ. 1998. Liver-derived CTL in hepatitis C virus infection: breadth and specificity of responses in a cohort of persons with chronic infection. J Immunol 160:1479–1488. [PubMed] [Google Scholar]
- 30.Brazas R, Ganem D. 1996. A cellular homolog of hepatitis delta antigen: implications for viral replication and evolution. Science 274:90–94. doi: 10.1126/science.274.5284.90. [DOI] [PubMed] [Google Scholar]
- 31.Neumann-Haefelin C, Timm J, Schmidt J, Kersting N, Fitzmaurice K, Oniangue-Ndza C, Kemper MN, Humphreys I, McKiernan S, Kelleher D, Lohmann V, Bowness P, Huzly D, Rosen HR, Kim AY, Lauer GM, Allen TM, Barnes E, Roggendorf M, Blum HE, Thimme R. 2010. Protective effect of human leukocyte antigen B27 in hepatitis C virus infection requires the presence of a genotype-specific immunodominant CD8+ T-cell epitope. Hepatology 51:54–62. doi: 10.1002/hep.23275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt J, Neumann-Haefelin C, Altay T, Gostick E, Price DA, Lohmann V, Blum HE, Thimme R. 2011. Immunodominance of HLA-A2-restricted hepatitis C virus-specific CD8+ T cell responses is linked to naive-precursor frequency. J Virol 85:5232–5236. doi: 10.1128/JVI.00093-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nitschke K, Flecken T, Schmidt J, Gostick E, Marget M, Neumann-Haefelin C, Blum HE, Price DA, Thimme R. 2015. Tetramer enrichment reveals the presence of phenotypically diverse hepatitis C virus-specific CD8+ T cells in chronic infection. J Virol 89:25–34. doi: 10.1128/JVI.02242-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 35.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quer J, Gregori J, Rodriguez-Frias F, Buti M, Madejon A, Perez-Del-Pulgar S, Garcia-Cehic D, Casillas R, Blasi M, Homs M, Tabernero D, Alvarez-Tejado M, Munoz JM, Cubero M, Caballero A, del Campo JA, Domingo E, Belmonte I, Nieto L, Lens S, Munoz-de-Rueda P, Sanz-Cameno P, Sauleda S, Bes M, Gomez J, Briones C, Perales C, Sheldon J, Castells L, Viladomiu L, Salmeron J, Ruiz-Extremera A, Quiles-Perez R, Moreno-Otero R, Lopez-Rodriguez R, Allende H, Romero-Gomez M, Guardia J, Esteban R, Garcia-Samaniego J, Forns X, Esteban JI. 2015. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J Clin Microbiol 53:219–226. doi: 10.1128/JCM.02093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homs M, Rodriguez-Frias F, Gregori J, Ruiz A, Reimundo P, Casillas R, Tabernero D, Godoy C, Barakat S, Quer J, Riveiro-Barciela M, Roggendorf M, Esteban R, Buti M. 2016. Evidence of an exponential decay pattern of the hepatitis delta virus evolution rate and fluctuations in quasispecies complexity in long-term studies of chronic delta infection. PLoS One 11:e0158557. doi: 10.1371/journal.pone.0158557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim Y, Ponomarenko J, Zhu Z, Tamang D, Wang P, Greenbaum J, Lundegaard C, Sette A, Lund O, Bourne PE, Nielsen M, Peters B. 2012. Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530. doi: 10.1093/nar/gks438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. 1999. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 40.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. 2008. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res 36:W509–W512. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller CR, Ehninger G, Goldmann SF. 2003. Gene and haplotype frequencies for the loci hLA-A, hLA-B, and hLA-DR based on over 13,000 German blood donors. Hum Immunol 64:137–151. doi: 10.1016/S0198-8859(02)00706-1. [DOI] [PubMed] [Google Scholar]
- 42.Rodenko B, Toebes M, Hadrup SR, van Esch WJ, Molenaar AM, Schumacher TN, Ovaa H. 2006. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc 1:1120–1132. doi: 10.1038/nprot.2006.121. [DOI] [PubMed] [Google Scholar]
- 43.Kern F, Surel IP, Brock C, Freistedt B, Radtke H, Scheffold A, Blasczyk R, Reinke P, Schneider-Mergener J, Radbruch A, Walden P, Volk HD. 1998. T-cell epitope mapping by flow cytometry. Nat Med 4:975–978. doi: 10.1038/nm0898-975. [DOI] [PubMed] [Google Scholar]
- 44.Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38:425–436. doi: 10.1016/j.immuni.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 45.Hartmann C, Antes I, Lengauer T. 2007. IRECS: a new algorithm for the selection of most probable ensembles of side-chain conformations in protein models. Protein Sci 16:1294–1307. doi: 10.1110/ps.062658307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hartmann C, Antes I, Lengauer T. 2009. Docking and scoring with alternative side-chain conformations. Proteins 74:712–726. doi: 10.1002/prot.22189. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 48.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. 2014. AMBER 14. University of California, San Francisco, CA. [Google Scholar]
- 49.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. 2010. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryckaert J-P, Ciccotti G, Berendsen HJC. 1977. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys 23:327–341. doi: 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- 51.Salomon-Ferrer R, Götz AW, Poole D, Grand SL, Walker RC. 2013. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput 9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 52.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. 1984. Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 53.Roe DR, Cheatham TEI. 2013. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 54.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. 2004. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 57.Trolle T, Metushi IG, Greenbaum JA, Kim Y, Sidney J, Lund O, Sette A, Peters B, Nielsen M. 2015. Automated benchmarking of peptide-MHC class I binding predictions. Bioinformatics 31:2174–2181. doi: 10.1093/bioinformatics/btv123. [DOI] [PMC free article] [PubMed] [Google Scholar]