

ABSTRACT
Hepatitis B virus (HBV) core protein consists of an N-terminal assembly domain and a C-terminal domain (CTD) with seven conserved serines or threonines that are dynamically phosphorylated/dephosphorylated during the viral replication cycle. Sulfamoylbenzamide derivatives are small molecular core protein allosteric modulators (CpAMs) that bind to the heteroaryldihydropyrimidine (HAP) pocket between the core protein dimer-dimer interfaces. CpAM binding alters the kinetics and pathway of capsid assembly and can result in the formation of morphologically “normal” capsids devoid of viral pregenomic RNA (pgRNA) and DNA polymerase. In order to investigate the mechanism underlying CpAM inhibition of pgRNA encapsidation, we developed an immunoblotting assay that can resolve core protein based on its phosphorylation status and demonstrated, for the first time, that core protein is hyperphosphorylated in free dimers and empty capsids from both mock-treated and CpAM-treated cells but is hypophosphorylated in pgRNA- and DNA-containing nucleocapsids. Interestingly, inhibition of pgRNA encapsidation by a heat shock protein 90 (HSP90) inhibitor prevented core protein dephosphorylation. Moreover, core proteins with point mutations at the wall of the HAP pocket, V124A and V124W, assembled empty capsids and nucleocapsids with altered phosphorylation status. The results thus suggest that core protein dephosphorylation occurs in the assembly of pgRNA and that interference with the interaction between core protein subunits at dimer-dimer interfaces during nucleocapsid assembly alters not only capsid structure, but also core protein dephosphorylation. Hence, inhibition of pgRNA encapsidation by CpAMs might be due to disruption of core protein dephosphorylation during nucleocapsid assembly.
IMPORTANCE Dynamic phosphorylation of HBV core protein regulates multiple steps of viral replication. However, the regulatory function was mainly investigated by phosphomimetic mutagenesis, which disrupts the natural dynamics of core protein phosphorylation/dephosphorylation. Development of an immunoblotting assay capable of resolving hyper- and hypophosphorylated core proteins allowed us to track the phosphorylation status of core proteins existing as free dimers and the variety of intracellular capsids and to investigate the role of core protein phosphorylation/dephosphorylation in viral replication. Here, we found that disruption of core protein interaction at dimer-dimer interfaces during nucleocapsid assembly (by CpAMs or mutagenesis) inhibited core protein dephosphorylation and pgRNA packaging. Our work has thus revealed a novel function of core protein dephosphorylation in HBV replication and the mechanism by which CpAMs, a class of compounds that are currently in clinical trials for treatment of chronic hepatitis B, induce the assembly of empty capsids.
KEYWORDS: hepatitis B virus, antiviral agents, capsid assembly, capsid assembly modulators, protein phosphorylation, viral replication
INTRODUCTION
Hepatitis B virus (HBV) infection affects approximately one-third of the world population, and more than 240 million people are chronically infected by the virus, for which the currently available antiviral therapies fail to provide a cure for the vast majority of patients (1, 2). HBV core protein is a small polypeptide of 183 amino acid residues that exists in hepatocytes as free dimers, as well as structural components of empty capsids and nucleocapsids containing viral RNA progenome or DNA replication intermediates (3). Specifically, core protein dimers first package a pregenomic RNA (pgRNA)-viral DNA polymerase complex into the nucleocapsid, where DNA polymerase converts the pgRNA first into the single-stranded (minus polarity) DNA and then into a partially double-stranded relaxed circular DNA (rcDNA) (4). As illustrated in Fig. 1A, HBV core protein contains two structural domains connected by a 9-amino-acid-residue hinge region. The N-terminal 140 amino acids (aa) form the capsid assembly domain, which is sufficient to assemble into empty capsids. The C-terminal 34 amino acid residues specify the C-terminal domain (CTD), containing multiple arginine-rich motifs and seven conserved serines or threonines that can be dynamically phosphorylated and dephosphorylated during the viral life cycle (5–7). The CTD has been demonstrated to play multiple roles in HBV replication, and execution of the different functions in viral replication is regulated by its dynamic phosphorylation at multiple sites (3, 8–11). Specifically, it has been shown with HBV and duck hepatitis B virus (DHBV) that, whereas the core proteins in the intracellular nucleocapsids are phosphorylated at multiple sites within the CTD, the core proteins in intracellular mature nucleocapsids and secreted virions are hypophosphorylated (12–15). Recently, it was further demonstrated that, while the CTD is unphosphorylated in complete HBV virions, it is phosphorylated in empty virions. However, although nucleocapsid maturation and secretion of complete virions are associated with core protein CTD dephosphorylation, it appears that neither phosphorylation nor dephosphorylation of the CTD is required for virion secretion (16). In addition, it was shown that core protein phosphorylation regulates capsid stability/uncoating and delivery of viral rcDNA into the nucleus for covalently closed circular DNA (cccDNA) synthesis (17–20).
FIG 1.

HBV capsid assembly modulators alter the phosphorylation status of HBV core protein. (A) Schematic representation of the domain structure of HBV core protein. The three major (red) and four minor (orange) phosphor acceptor sites are indicated. (B and C) AML12HBV10 (B) or HepAD38 (C) cells cultured in the absence of Tet were mock treated or treated with 2 μM Bay 41-4109, 5 μM ENAN-34017, or 0.1 μM ETV for 2 and 6 days, respectively. HepG2 cells were transfected with plasmid pCI-HBc-WT (WT), pCI-HBc-3A (3A), or pCI-HBc-7A (7A) and harvested at 72 h posttransfection. Intracellular core proteins were analyzed by a Western blot assay with antibody HBc-170A. β-Actin served as a loading control. (D) AML12HBV10 and HepAD38 cells were mock treated or treated with 2 μM Bay 41-4109 or 5 μM ENAN-34017 for 2 or 8 days, respectively. Intracellular HBV capsids were pelleted by ultracentrifugation and mock treated or treated with protein phosphatase 1 for 16 h at 30°C. HBV core proteins were detected by a Western blot assay with HBc-170A antibody. (E) Wild-type (WT) and mutant core proteins (Cp) with three major or all seven phosphoacceptor sites replaced with A or E were synthesized in RRL, with or without CIAP treatment, and analyzed by a Western blot assay with antibody HBc-170A. The nonspecific band labeled with an asterisk served as a loading control. Hyper-P and Hypo-P, hyper- and hypophosphorylated core protein, respectively.
Due to its unique structure and essential role in viral replication, disruption of nucleocapsid assembly with small molecular core protein allosteric modulators (CpAMs) represents a new frontier in the development of novel antiviral agents against chronic HBV infection (3). Binding of the CpAMs to a hydrophobic pocket, designated the heteroaryldihydropyrimidine (HAP) pocket, at the dimer-dimer interface near the C termini of core protein subunits induces large-scale allosteric conformational changes in core protein subunits and alters the capsid assembly kinetics and pathways (21, 22). While HAPs, such as Bay 41-4109 and GLS4, misdirect capsid assembly to form noncapsid polymers of core proteins (21, 23), all the other chemotypes of CpAMs, including sulfamoylbenzamides (SBAs), benzamines (BAs) and phenylpropenamides (PPAs), induce the formation of empty capsids with distinct quaternary and/or tertiary structure changes and thus preclude viral DNA replication (24–26). However, it is not yet known how the capsid assembly modulators inhibit pgRNA encapsidation and favor empty-capsid assembly. In an effort to understand the mechanism underlying the SBA inhibition of pgRNA encapsidation, we developed a Western blot assay that can resolve HBV core proteins into hyper- and hypophosphorylated species and obtained evidence supporting the notion that core protein dephosphorylation occurs in pgRNA encapsidation and that alteration of the interaction between core protein dimers during capsid assembly changes not only the capsid structure, but also core protein dephosphorylation. Hence, inhibition of pgRNA encapsidation by CpAMs may be due to their disruption of proper dephosphorylation of core protein during nucleocapsid assembly.
RESULTS
CpAM treatment alters the phosphorylation status of intracellular HBV core protein.
Although we demonstrated previously that the CTD of HBV core protein did not play a role in CpAM-induced assembly of structurally altered empty capsids, its role in CpAM-induced suppression of pgRNA encapsidation remains to be determined (27). Considering the critical role of the core protein CTD and its phosphorylation in pgRNA encapsidation (9–11), it is conceivable that binding of CpAMs at the HAP pocket may disrupt the dynamic phosphorylation/dephosphorylation of core protein subunits and consequently interfere with pgRNA packaging. Obviously, investigation of this hypothesis requires accurate determination of the core protein phosphorylation status in the different forms of intracellular capsids. Unfortunately, unlike DHBV core protein, which can be resolved into 2 to 4 distinct bands based on their CTD phosphorylation status (12–14), regular SDS-PAGE cannot resolve phosphorylated and dephosphorylated HBV core proteins. However, as shown in Fig. 1B and C, Western blot assay using a NuPAGE 12% Bis-Tris protein gel (Invitrogen) with morpholineethanesulfonic acid (MES) SDS running buffer and probing with a rabbit polyclonal antibody against a peptide (aa 170 to 183) of HBV core protein (HBc-170A) revealed doublet HBV core protein bands with molecular masses of approximately 22 and 21 kDa. As anticipated, the core protein bands were detected in the lysates of AML12HBV10 or HepAD38 cells cultured only in the absence of tetracycline (Tet) to induce HBV pgRNA transcription and HBV DNA replication. Interestingly, while treatment with the HBV polymerase inhibitor entecavir (ETV) did not alter the doublet bands, treatment with either the SBA derivative ENAN-34017 or the HAP derivative Bay 41-4109 dramatically reduced the faster-migrating, but the not slower-migrating, core protein species. As controls for differentially phosphorylated (or dephosphorylated) core proteins, wild-type and mutant core proteins with alanine substitutions for three major (3A) or all seven (7A) phosphoacceptor amino acid residues in the CTD were expressed in HepG2 cells. As shown in Fig. 1B and C, lanes 7 to 9, those three core proteins ran with distinct electrophoresis mobilities under these experimental conditions. Accordingly, we speculated that the doublet bands revealed by the anti-HBV core antibody in AML12HBV10 and HepAD38 cells might represent hyper- and hypophosphorylated core proteins. In support of this notion, we further demonstrated that treatment of HBV capsids prepared from mock-treated and ENAN-34017- or Bay 41-4109-treated AML12HBV10 or HepAD38 cells with protein phosphatase 1 (PP1) converted the slower-migrating core protein species into the faster-migrating species (Fig. 1D). Moreover, as shown in Fig. 1E, HBV core protein synthesized in rabbit reticulocyte lysate (RRL) was hyperphosphorylated (28), and treatment with calf intestinal alkaline phosphatase (CIAP) increased its electrophoresis mobility (Fig. 1E, compare lanes 2 and 3). Similar to the results presented in Fig. 1B and C, replacement of three major (3A) or all seven (7A) phosphoacceptor sites with alanines to mimic the partially or completely dephosphorylated core protein incrementally increased the electrophoresis mobility. On the other hand, replacement of three major or all seven phosphorylation sites with glutamic acids to mimic phosphorylated core proteins (3E or 7E) provided electrophoresis mobility similar to that of the wild-type core protein. Taken together, those results indicate that we had established Western blot assay conditions that were able to resolve HBV core protein based on its phosphorylation status.
Characterization of core protein phosphorylation status with three antibodies against HBV core proteins.
To further investigate the properties of differentially phosphorylated core protein species and to determine the extent of their phosphorylation, Western blot assays were performed with three antibodies that recognize epitopes in different regions of the core protein, with full-length core proteins expressed in Escherichia coli as a control for nonphosphorylated core protein. As shown in Fig. 2A, antibody HBc-170A recognized only full-length core protein, but not core proteins with deletions of 6 or more amino acid residues from the C terminus. As anticipated, the Dako antibody, which recognizes an epitope in the assembly domain, could detect full-length and also the C-terminally truncated core proteins (Fig. 2A, middle). Antibody 10E11 recognizes the N-terminal 2 to 8 amino acid residues of the core protein. As shown in Fig. 2B to D, all three antibodies can detect both the hyper- and hypophosphorylated core proteins from AML12HBV10 cell lysates. Similar to antibody HBc-170A, antibody 10E11 can also demonstrate that CpAM treatment reduced the amounts of hypophosphorylated core protein (Fig. 2D). Moreover, compared to core protein expressed in E. coli, the hypophosphorylated core protein species in AML12HBV10 lysates migrated with slightly lower mobility. The results thus suggest that the observed hypophosphorylated core protein in replicating HBV cells may not be completely dephosphorylated.
FIG 2.

Characterization of core protein phosphorylation status with three antibodies against HBV core proteins. (A) AML12 cells were cotransfected with pTet-off and vector plasmid (pTRE2) or a plasmid expressing either wild-type full-length core protein (pTRE2-HBc183) or the indicated C-terminally truncated core proteins. The cells were harvested at 48 h posttransfection. (Top and middle) Core protein expression was detected by Western blotting assay with antibody HBc170A (top) or anti-core antibody from Dako (middle). (Bottom) β-Actin served as a loading control. (B) Cell lysates from AML12HBV10 cells cultured in the presence or absence of Tet for 2 days and HepG2 cells transfected with plasmid pCI-HBc-WT, pCI-HBc-3A, or pCI-HBc-7A and harvested at 72 h posttransfection, as well as HepG2 cell lysate containing 10 ng HBV core protein expressed in E. coli, were resolved by SDS-PAGE. HBV core protein was detected with antibody HBc170A or Dako antibody. β-Actin served as a loading control. (C) Cell lysates from AML12HBV10 cells cultured in the presence or absence of Tet for 2 days and cell lysates of AML12HBV10 cells (cultured in the presence of Tet) containing 10 ng HBV core protein expressed in E. coli were detected by a Western blot assay with antibody 10E11. β-Actin served as a loading control. (D) AML12HBV10 cells were mock treated or treated with 2 μM Bay 41-4109, 5 μM ENAN-34017, or 0.1 μM ETV for 2 days. Intracellular core proteins were analyzed by a Western blot assay with antibody 10E11. β-Actin served as a loading control. Non-P, nonphosphorylated core protein.
Characterization of the phosphorylation status of intracellular capsids in CpAM-treated cells.
While the results presented above indicated that CpAM treatment reduced the amount of hypophosphorylated core proteins, it did not reveal the core protein phosphorylation status in the different HBV capsids in cells. Accordingly, HBV capsids from the cytoplasmic lysates of mock-treated and ENAN-34017-treated cells were separated by sucrose gradient centrifugation, and the fractions positive for core protein were subjected to analysis of capsid electrophoresis mobility, HBV DNA content, and core protein phosphorylation by particle gel and Western blot assays (Fig. 3). As shown in Fig. 3A, capsids from mock-treated AML12HBV10 cells sedimented faster than those from ENAN-34017-treated cells and peaked at fractions 8 or 9 and 10 or 11, respectively. However, the viral DNA-containing capsids in both mock- and ENAN-34017-treated cells sedimented at similar velocities and peaked at fraction 7. As expected, the total amount of DNA-containing capsids was significantly reduced in ENAN-34017-treated cells. To determine the core protein phosphorylation status in empty and DNA-containing capsids, the viral capsids from fractions 5 to 14 were further concentrated by ultracentrifugation and subjected to particle gel and Western blot analyses. First, electrophoresis in a 1.5% native agarose gel resolved HBV capsids into two distinct bands (Fig. 3B, middle). The slower-migrating band appeared between fractions 5 and 13 and peaked at fraction 9, while the faster-migrating band appeared between fractions 9 and 14 and peaked at fraction 10. HBV DNA was detected in fractions 5 to 9 and peaked at fractions 6 and 7 (Fig. 3B, bottom). These results demonstrate that, while there are two populations of empty capsids with higher and lower electrophoresis mobilities, the DNA-containing capsids have similar (or slightly higher) electrophoresis mobility with slower-migrating empty capsids. Second, Western blot assays revealed two distinct populations of core proteins in the mock-treated cells (Fig. 3B, top). The hyperphosphorylated (slower-migrating) core protein was detected in fractions 7 to 13 and peaked at fraction 9, whereas the hypophosphorylated (faster-migrating) core protein was detected in fractions 5 to 8 and peaked at fraction 6. Based on the cosedimentation profiles, we speculated that the core proteins in both the slower- and faster-migrating empty capsids were hyperphosphorylated but that they were hypophosphorylated in DNA-containing capsids. Third, a particle gel assay revealed that capsids in ENAN-34017-treated cells comigrated with the fast-migrating empty capsids in mock-treated cells and peaked at fraction 10. HBV DNA could be detected in fractions 5 to 10 and peaked at fraction 7. However, similar to what was seen in mock-treated cells, a Western blot assay detected two distinct species of core proteins in ENAN-34017-treated cells. While the hyperphosphorylated core proteins cosedimented with empty capsids, hypophosphorylated core proteins cosedimented with the residual DNA-containing capsids. Hence, our results demonstrated that empty capsids and DNA-containing capsids had distinct biophysical and biochemical properties, and irrespective of ENAN-34017 treatment, core proteins were predominantly hyperphosphorylated in empty capsids but hypophosphorylated in DNA-containing capsids.
FIG 3.

HBV core protein is hyperphosphorylated in empty capsids but hypophosphorylated in DNA-containing capsids. (A) AML12HBV10 cells were cultured in Tet-free medium and left untreated or treated with 5 μM ENAN-34017 for 2 days. Intracellular capsids were sedimented on a sucrose gradient (15 to 30%) with a Beckman SW28 rotor. Eighteen equal-volume (1.5-ml) fractions were collected from the bottom of the tube, and. 50 μl of each fraction was applied to a nylon membrane. The membrane was probed with an antibody against HBc (Dako) to detect HBc and then hybridized with a minus-strand-specific full-length HBV riboprobe to detect HBV DNA. Relative amounts of HBV core protein and DNA were quantified with a Li-Cor Odyssey system and a Typhoon phosphorimager system, respectively. The dot blot images for each of the assays were derived from two consecutive rows of a 96-well dot blot manifold. (B) HBV capsids in the remaining samples of fractions 5 to 14 (highlighted by the red box in panel A) were pelleted by ultracentrifugation and dissolved in TNE buffer. One half of the sample from each fraction was used for Western blot analysis of core protein with antibody HBc-170A and the other half for analyses of capsids and their associated HBV DNA in 1.5% native agarose gel assays. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively.
Core protein in DNA-containing capsids is hypophosphorylated.
To further confirm the observation that HBV core proteins are hypophosphorylated in DNA-containing capsids but hyperphosphorylated in empty capsids in human hepatocyte-derived cells, we analyzed core protein phosphorylation profiles in capsids prepared from HepG2 cells transfected with a plasmid supporting HBV replication (pHBV-1.3) and a plasmid that expresses core protein alone (pCMV-HBcAg) and thus assembles only empty capsids.
Similar to the capsids from AML12HBV10 cells, capsids from HepG2 cells transfected with the two different plasmids sedimented at similar velocities in sucrose gradient centrifugations and peaked at fraction 8 or 9 (Fig. 4A). As anticipated, HBV DNA was detected only in pHBV-1.3-transfected cells, and the DNA-containing capsids sedimented faster than the major population of capsids and peaked at fraction 6 (Fig. 4A and B). To determine the core protein phosphorylation status, HBV capsids in fractions 4 to 13 of pHBV-1.3-transfected cells (Fig. 4B) and pCMV-HBcAg-transfected cells (Fig. 4C) were further concentrated by ultracentrifugation and subjected to particle gel and Western blot analyses. As shown in Fig. 4B and C, the particle gel assay resolved capsids from the transfected cells into two or three distinct bands. Due to their existence in cells transfected with both plasmids, the slower- and faster-migrating capsids that peaked at fractions 8 and 9 should be empty capsids. However, capsids (red arrow) that migrated between the faster-migrating (blue arrows) and the slower-migrating (green arrows) capsids appeared only in cells transfected with the wild-type HBV genome (pHBV-1.3) and cosedimented with core DNA and peaked at fractions 5 and 6. Those capsids are thus most likely DNA-containing capsids. Western blot assays revealed that the core proteins in capsids from pCMV-HBcAg-transfected cells were exclusively hyperphosphorylated, whereas both hyper- and hypophosphorylated core proteins could be detected in capsids from pHBV1.3-transfected cells. The results thus indicate that in both human and mouse hepatocytes, the core proteins associated with both slower- and faster-migrating empty capsids are predominantly hyperphosphorylated, whereas the hypophosphorylated core proteins predominantly cosedimented with DNA-containing capsids. This observation is consistent with the findings from genetic studies that viral DNA synthesis is associated with core protein dephosphorylation (12–15).
FIG 4.

Core proteins in DNA-containing nucleocapsids are hypophosphorylated. HepG2 cells were transfected with plasmid pHBV-1.3 or pCMV-HBcAg and harvested at 72 h posttransfection. Intracellular capsids were sedimented on a sucrose gradient (15 to 30%) with a Beckman SW28 rotor. (A) HBV core protein/capsids and core DNA in each fraction were detected by a dot blot assay. The dot blot images for each of the assays were derived from two consecutive rows of a 96-well dot blot manifold. (B and C) The indicated fractions of HBV capsids in the remaining samples were pelleted by ultracentrifugation and dissolved in TNE buffer. One half of the sample from each fraction was used for Western blot detection of core protein with antibody HBc-170A. The other half of the sample was used for analyses of capsids and HBV DNA in a 1.5% native agarose gel assay. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively. The capsids migrating between the slow and fast capsids are indicated by the red arrow. (C, top, far-right lane) For Western blot detection of HBV core protein from pCMV-HBcAg-transfected cells, pHBV-1.3-transfected cell lysate was used as a control for HBV core protein phosphorylation status.
Core protein of pgRNA-containing capsids is hypophosphorylated.
To rigorously determine the phosphorylation status of core proteins in pgRNA-containing capsids, we first established an AML12-derived stable cell line, designated AML12HBVpolY63F, that Tet-inducibly expresses pgRNA encoding Y63F mutant DNA polymerase that is deficient in priming negative-strand DNA synthesis and thus accumulates only pgRNA-containing capsids. Comparative analyses of capsids derived from AML12HBV10 and AML12HBVpolY63F cells by sucrose gradient centrifugation indicated that, while the total capsids from both cell lines peaked at fraction 6, the DNA-containing capsids from AML12HBV10 cells and pgRNA-containing capsids from AML12HBVpolY63F cells sedimented at similar velocities and peaked at fraction 4 (Fig. 5A). Western blot and particle gel assays further revealed that, while the slower- and faster-migrating capsids cosedimented with the hyperphosphorylated core proteins, the capsids (red arrows) that migrate between the faster-migrating (blue arrows) and the slower-migrating (green arrows) capsids appeared to cosediment with viral DNA or pgRNA and hypophosphorylated core proteins (Fig. 5B and C). Moreover, although the mature DNA-containing capsids sedimented faster than the single-stranded-DNA-containing capsids (Fig. 5B, bottom), the core protein phosphorylation status among those capsids could not be rigorously distinguished, due to the existence of large amounts of empty capsids. These results suggest that, similar to DNA-containing capsids, core proteins of pgRNA-containing capsids are also hypophosphorylated.
FIG 5.

Core proteins in pgRNA-containing nucleocapsids are hypophosphorylated. AML12HBV10 and AML12HBVpolY63F cells were cultured in Tet-free medium for 2 days. Intracellular capsids were sedimented on a sucrose gradient (15 to 50%) with a Beckman SW28 rotor at 24,000 rpm for 16 h. Sixteen equal-volume (2-ml) fractions were collected from the bottoms of the tubes, and 50 μl of each fraction was applied to a nylon membrane. The membrane was probed with an antibody against HBc (Dako; B0586) to detect HBV core protein and then hybridized with a minus-strand-specific full-length HBV riboprobe to detect HBV DNA or with a plus-strand-specific full-length HBV riboprobe to detect HBV RNA. Relative amounts of HBV core protein, DNA, or RNA were quantified with a Li-Cor Odyssey system and a Typhoon phosphorimager system, respectively. The dot blot images for each of the assays were derived from two consecutive rows of a 96-well dot blot manifold. (B and C) HBV capsids in the remaining samples of fractions 2 to 10 were pelleted by ultracentrifugation and dissolved in TNE buffer. One-tenth of the sample from each fraction was used for Western blot analysis of core protein with antibody HBc-170A or for analyses of capsids and their associated HBV DNA or pgRNA in a 1.5% native agarose gel assay. HBV DNA was extracted from one-third of the sample from each fraction derived from AML12HBV10 cells and analyzed by Southern blotting hybridization assay. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively. The capsids migrating between the slow and fast capsids are indicated by red arrows.
Core-protein-free dimers are hyperphosphorylated.
It has been speculated that HBV core protein is co- or posttranslationally phosphorylated and that its proper phosphorylation is required for capsid assembly and pgRNA encapsidation (13, 14, 28). However, the phosphorylation status of HBV core-protein-free dimers in HBV-infected cells has not been determined. The fact that the vast majority of intracellular core protein exists in assembled capsids makes this a technically difficult question to be answered. Recently, it was found that the tyrosine residue at position 132 of core protein stabilizes the interaction between HBV core protein dimers, and this interaction is essential for the assembly of capsids (29). When this tyrosine is replaced with an alanine, the mutant (Y132A) core protein becomes deficient in capsid assembly and exists as free dimers (22). Indeed, as demonstrated in Fig. 6A, sucrose gradient centrifugation analyses showed that, while the core protein in the cytoplasmic lysates of HepG2 cells transfected with pHBV-1.3 sedimented as assembled capsids, the core protein in the lysates of HepG2 cells transfected with pHBV1.3/coreY132A was detected in the top fractions of the sucrose gradient. In addition, particle gel assays failed to detect assembled capsids in pHBV-1.3/coreY132A-transfected HepG2 cells (Fig. 6B). Hence, it is conceivable that the phosphorylation status of Y132A mutant core protein in transfected cells should reflect the phosphorylation status of wild-type HBV core-protein-free dimers. We therefore compared the phosphorylation profiles of core proteins in cells transfected with pHBV-1.3 and pHBV-1.3/coreY132A, with those of cells transfected with plasmids expressing wild-type core protein, HBc3A or HBc7A as controls, by a Western blot assay (Fig. 6C). In agreement with the results presented above, in addition to a dominant hyperphosphorylated core protein band, a minor hypophosphorylated core protein band could also be detected in cells transfected with pHBV-1.3. However, only the hyperphosphorylated core protein species were detected in cells transfected with the plasmid pHBV-1.3/coreY132A or pCI-HBc. As anticipated, core proteins from cells transfected with plasmid pCI-HBc-3A or pCI-HBc-7A migrated faster than the hyperphosphorylated wild-type core protein. The results thus suggest that the core-protein-free dimers in HBV-infected hepatocytes are most likely hyperphosphorylated.
FIG 6.

Sucrose gradient centrifugation analysis of Y132A mutant core protein. (A) HepG2 cells were transfected with plasmid pHBV-1.3 or pHBV-1.3/coreY132A and harvested at 72 h posttransfection. Intracellular capsids were sedimented on a sucrose gradient (15 to 30%) with a Beckman SW28 rotor. HBV core protein/capsids in each fraction were detected by a dot blot assay and quantified with a Li-Cor Odyssey system. The sucrose density of each fraction is also plotted. The dot blot images for each of the assays were derived from two consecutive rows of a 96-well dot blot manifold. (B) HBV capsids in the cytoplasmic lysates of HepG2 cells mock transfected or transfected with plasmid pHBV-1.3 or pHBV-1.3/coreY132A were detected by 1.5% native agarose gel particle assay. (C) HepG2 cells were mock transfected or transfected with the indicated plasmids and harvested at 72 h posttransfection. Intracellular HBV core protein was analyzed by a Western blot assay with antibody HBc-170A.
Inhibition of pgRNA encapsidation by an HSP90 inhibitor prevented core protein dephosphorylation.
Because core-protein-free dimers, the building blocks of pgRNA-containing capsids, are hyperphosphorylated but pgRNA-containing capsids contain hypophosphorylated core proteins, it is reasonable to hypothesize that dephosphorylation of core protein occurs during pgRNA packaging. Because the heat shock protein 90 (HSP90) chaperone complex is required for viral polymerase to bind the ε RNA element, an essential step for packaging of the viral polymerase-pgRNA complex into nucleocapsids and priming of negative-strand DNA synthesis (30), inhibition of HSP90 ATPase with geldanamycin derivatives can efficiently prevent pgRNA encapsidation (31). Taking advantage of this observation, we examined the effects of the HSP90 inhibitor 17-dimethylamino geldanamycin (17-DMAG) and ENAN-34017, alone or in combination, on pgRNA encapsidation and core protein dephosphorylation in AML12HBV10 cells. As expected, both 17-DMAG and ENAN-34107 did not alter the amounts of total viral RNA but reduced the amounts of encapsidated pgRNA (Fig. 7A and B). Also, consistent with previous reports (27, 31) and the results presented in Fig. 1, while 17-DMAG treatment did not alter the migration of HBV capsids in the native agarose gel electrophoresis, ENAN-34017 treatment induced the formation of faster-migrating capsids (Fig. 7C). However, compared with mock-treated controls, both 17-DMAG and ENAN-34107 treatments reduced the amounts of core-associated HBV DNA (Fig. 7C, bottom) and hypophosphorylated HBV core protein (Fig. 7D). The latter observation is consistent with the notion that core proteins in empty capsids are hyperphosphorylated and strongly suggests that dephosphorylation of core protein occurs during the encapsidation of pgRNA.
FIG 7.
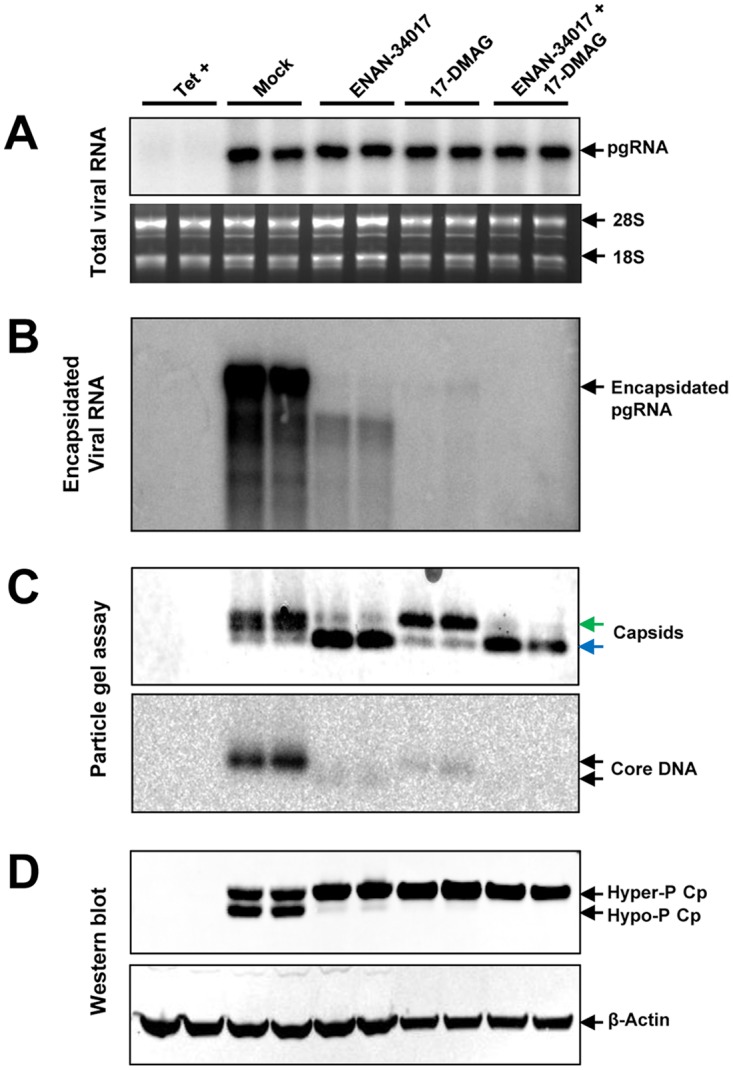
Disruption of pgRNA encapsidation by an HSP90 inhibitor reduced the amount of dephosphorylated core protein. AML12HBV10 cells were mock treated or treated with 5 μM ENAN-34017 or 300 nM 17-DMAG, alone or in combination, in the absence of Tet for 2 days. (A) Total and encapsidated pgRNAs were determined by Northern blotting hybridization assays. 28S and 18S rRNAs served as loading controls. (B) Encapsidated pgRNA was determined by Northern blotting hybridization assay. (C) Cytoplasmic HBV capsids were analyzed by a particle gel assay with 1.8% agarose gel electrophoresis. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively. Capsid-associated HBV DNA was determined by hybridization with a full-length riboprobe specific for minus-strand HBV DNA. (D) The phosphorylation status of core protein was determined by a Western blot assay with antibody HBc-170A. β-Actin served as a loading control.
Investigating the relationship between capsid assembly and core protein phosphorylation.
To further investigate whether interference of core protein dimer-dimer interaction at the HAP pocket during capsid assembly disrupts the dynamic phosphorylation and dephosphorylation of core proteins, we took advantage of well-characterized mutant core proteins with point mutations of critical amino acid residues on the wall of the HAP pocket that mimics CpAM effects on capsid assembly and pgRNA encapsidation (32, 33). Specifically, as reported previously, while the wild-type core protein in pHBV1.3-transfected HepG2 cells supported the assembly of both slow- and fast-migrating capsids, V124A and V124W mutant core proteins supported the assembly of predominantly slow- and fast-migrating capsids, respectively (27) (Fig. 8A). Moreover, HBV DNA replication levels in pHBV-1.3/coreV124A- and pHBV-1.3/coreV124W-transfected cells were approximately 32% and 9% of that in cells transfected with pHBV1.3 (Fig. 8A, particle gel assay). To analyze the core protein phosphorylation status in the capsids assembled from wild-type and mutant core proteins, capsids in the transfected-cell lysates were separated by sucrose gradient ultracentrifugation. As shown in Fig. 8B to D, consistent with the results presented in Fig. 4, the particle gel assay was able to partially resolve capsids from the cells transfected with the wild-type HBV genome (pHBV-1.3) into three distinct bands, i.e., slower- and faster-migrating empty capsids in factions 8 to 14 and DNA-containing capsids that migrate between the two empty-capsid species in fractions 5 to 9 (Fig. 8B). On the other hand, the capsids from pHBV-1.3/coreV124A-transfected cells were resolved into the slower-migrating empty capsids in fractions 8 to 12 and a slightly faster-migrating capsid species in fractions 5 to 8. This result is consistent with the particle gel assay results with total cell lysates (Fig. 8A) showing that V124A core protein did not support the assembly of faster-migrating empty capsids and that HBV DNA replication was arrested at the stage of full-length minus-strand DNA (Fig. 8C, bottom) (33). Also, in agreement with the results shown in Fig. 8A, V124W core protein predominantly supported the assembly of faster-migrating capsids, but DNA-containing capsids could not be detected. Interestingly, Western blot analysis revealed that, whereas DNA-containing capsids from pHBV1.3-transfected cells cosedimented with hypophosphorylated core proteins, DNA-containing capsids from pHBV-1.3/coreV124A-transfected cells cosedimented with both hyper- and hypophosphorylated core proteins. Moreover, the empty capsids from pHBV-1.3/coreV124W-transfected cells were associated with predominantly hyperphosphorylated, but detectable amounts of hypophosphorylated, core protein. These results thus suggest that alteration of HBV capsid assembly via genetic replacement of a key amino acid residue mediating the dimer-dimer interaction alters the phosphorylation status of core protein in both empty and DNA-containing capsids.
FIG 8.

Analyses of effects of core protein mutation on capsid assembly, DNA replication, and core protein phosphorylation. (A) HepG2 cells were transfected with plasmid pHBV-1.3, pHBV-1.3/coreV124A, or pHBV-1.3/coreV124W and harvested at 72 h posttransfection. (Top and middle) HBV capsids and HBV DNA were detected by a 1.5% native agarose gel assay. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively. Encapsidated pgRNA was determined by Northern blotting hybridization. (Bottom) The cytoplasmic HBV DNA replication intermediates were analyzed by Southern blotting hybridization. (B to D) HepG2 cells were transfected with plasmid pHBV-1.3 (B), pHBV-1.3/coreV124A (C), or pHBV-1.3/coreV124W (D) and harvested at 72 h posttransfection. Intracellular capsids were sedimented on a sucrose gradient (15% to 30%). Sucrose density, HBV core protein, and DNA from each of the fractions were determined. Core protein and capsids in fractions 5 (high sucrose density) to 14 (low sucrose density) were analyzed by Western blotting and particle gel assays as described in the legend to Fig. 4. HBV DNA replicative intermediates were analyzed by Southern blotting hybridization. The slow- and fast-migrating capsids are indicated by green and blue arrows, respectively. The capsids migrating between the slow and fast capsids are indicated by red arrows.
DISCUSSION
HBV and DHBV core proteins were discovered as phosphoproteins in the late 1980s by using 32P metabolic labeling and phosphatase treatment/immunoblotting assays, respectively. Unlike DHBV core protein, which can be resolved into 2 to 4 distinct species based on their phosphorylation status, the regular immunoblotting assay failed to resolve the differentially phosphorylated HBV core proteins (12–14, 34). In this study, we defined SDS-PAGE conditions that could resolve HBV core proteins into doublet bands with molecular masses of approximately 22 and 21 kDa (Fig. 1B). Conversion of the slower-migrating species into a faster-migrating species by phosphatase treatment convincingly demonstrated that the slower- and faster-migrating bands represent the hyper- and hypophosphorylated core proteins, respectively (Fig. 1D). Moreover, the electrophoresis conditions were also able to separate core proteins expressed in E. coli and in vitro-translated or intracellularly expressed wild-type and mutant core proteins with replacement of the three major or all seven phosphoacceptor sites by alanines or glutamic acids with three antibodies recognizing epitopes located at the N terminus, C terminus and middle of core protein (Fig. 1 and 2). Hence, we have developed a Western blot assay that allows us, for the first time, to conveniently monitor core protein phosphorylation status during viral replication and under antiviral treatment. With this technical advantage, we have made several interesting findings and have investigated their biological implications, as summarized in Fig. 9 and discussed below.
FIG 9.

Schematic representation of dynamic core protein phosphorylation/dephosphorylation during viral replication and the impacts of CpAMs. See the text for a detailed explanation. ssDNA, single-stranded DNA.
First, because the vast majority of core proteins in HBV replicating cells exist in assembled capsids, the phosphorylation status of intracellular core-protein-free dimers, the building blocks of capsids, cannot be unambiguously determined (35). Moreover, the fact that purified capsids from HBV-infected cells contain cellular kinases, such as cyclin-dependent kinase 2 (CDK2), that are able to phosphorylate the S/T-P sites in the HBV and DHBV CTDs under in vitro endogenous kinase assay conditions suggests that core protein phosphorylation can occur after capsid assembly (36, 37). However, to better understand the dynamics and function of core protein phosphorylation during capsid assembly and DNA replication, it is important to know the initial phosphorylation status of core protein dimers. Taking advantage of Y132A mutant core protein, which fails to assemble capsids, we showed that the core-protein-free dimers exist with hyperphosphorylated status (Fig. 6). Considering that the core proteins in all the empty capsids from HBV-replicating cells or cells expressing core protein alone, with or without ENAN-34017 or HSP90 inhibitor treatment, are hyperphosphorylated, significant dephosphorylation of core protein may not occur during the assembly of empty capsids. Moreover, the exclusive existence of hyperphosphorylated core protein in Bay 41-4109-treated cells indicates that the formation of HAP-induced noncapsid core protein polymers also does not require core protein dephosphorylation (Fig. 1). However, it was demonstrated recently that core protein dephosphorylation was required for capsid assembly in RRL in vitro (28). This discrepancy could be due to either the difference in phosphorylated sites in core proteins or factors that regulate the assembly of capsids in the cytoplasm of hepatocytes and RRL.
Second, previous phosphomimetic mutagenesis studies have indicated that phosphorylation of the core protein CTD is required for HBV pgRNA packaging and DNA synthesis (7, 9, 11, 35). For DHBV, CTD phosphorylation is needed for first-strand DNA synthesis and dephosphorylation is required for second-strand DNA synthesis (14, 34, 38, 39). However, a problem with phosphomimetic mutagenesis studies is that the mutations prevent the dynamic phosphorylation/dephosphorylation of core protein during viral replication, and the experimental results derived from such studies may sometimes be misleading. The ability to monitor core protein phosphorylation status without disruption of its natural dynamics, therefore, is much preferred. Using our immunoblotting assay, we consistently demonstrated that the hypophosphorylated core proteins cosediment with viral DNA- or pgRNA-containing capsids (Fig. 3 to 5). These results are consistent with the finding from previous genetic studies that core protein dephosphorylation is required for viral DNA synthesis. However, our findings also imply that at least partial dephosphorylation of core proteins occurs in the assembly of pgRNA-containing capsids or nucleocapsids. Interestingly, side-by-side comparison of pgRNA- and DNA-containing capsids indicated that the two capsid species have similar sedimentation velocities and electrophoresis mobilities and contain core proteins with indistinguishable phosphorylation status (Fig. 5). However, because the current immunoblotting assay cannot reveal the (de)phosphorylation status of core proteins at each of the phosphoacceptor sites in the CTD, the exact phosphorylation profiles of the different capsids and nucleocapsids remain to be determined. Nevertheless, our findings are supported by results showing that inhibition of pgRNA encapsidation by an HSP90 inhibitor significantly reduced the amount of hypophosphorylated core protein (Fig. 6) and are consistent with a recent report suggesting that both hyper- and hypophosphorylation of core protein inhibited pgRNA packaging (28). Moreover, studies are under way to identify the cellular protein phosphatases responsible for the dephosphorylation of core protein (40) and to determine their role in pgRNA encapsidation.
Third, the fact that CpAM treatment inhibits the formation of dephosphorylated capsids implies that disrupting the interaction of core protein dimers interferes with their dephosphorylation. Structural biology and genetic analyses indicate that all the chemotypes of CpAMs reported thus far disrupt HBV capsid assembly by binding to the hydrophobic (HAP) pocket between core protein dimer-dimer interfaces to enhance their interaction (21, 22, 24, 25). As illustrated in Fig. 9, except for HAPs, which induce assembly of noncapsid core protein polymers that are subsequently degraded by proteasomes, all the CpAMs induce the assembly of morphologically “normal” empty capsids. Because the CpAMs enhance the interaction of core protein dimers and consequently accelerate the kinetics of core protein assembly (41), the reduced concentration of cytoplasmic free core protein dimers and/or the accelerated assembly kinetics may disrupt encapsidation of the viral pgRNA-polymerase complex. However, recent mutagenesis studies have shown that alteration of core protein interaction at the dimer-dimer interfaces alters not only capsid assembly kinetics, but also pgRNA packaging (32, 33). By analyzing two mutant core proteins with distinct capsid assembly phenotypes, we showed that modulation of the core protein dimer-dimer interaction changed the core protein phosphorylation profiles. Hence, it is possible that, rather than being mutually exclusive, inhibition of pgRNA encapsidation by CpAMs is due to not only the accelerated capsid assembly kinetics, but also the disrupted core protein dephosphorylation dynamics that resulted from alteration of the interaction between core protein dimer interfaces during capsid assembly.
Finally, the ability to monitor core protein phosphorylation with a convenient immunoblotting assay should allow the investigation of other molecular events in HBV replication that are possibly regulated by dynamic core protein phosphorylation, such as nucleocapsid envelopment and uncoating and delivery of rcDNA into nuclei for cccDNA synthesis (17–20, 34, 42). In addition, studies investigating the role of core protein dephosphorylation in interferon inhibition of HBV pgRNA encapsidation (43, 44) and accelerated decay of DNA-containing capsids (45) are under way.
MATERIALS AND METHODS
Cell cultures and antiviral compounds.
HepAD38 was obtained from Christoph Seeger at Fox Chase Cancer Center, Philadelphia, PA (46). The AML12HBV10 cell line was established in our laboratory as described previously (45). Both HepAD38 and AML12HBV10 are stable cell lines supporting HBV pgRNA transcription and DNA replication in a Tet-inducible manner. The HepG2 cell line was purchased from the ATCC and maintained in Dulbecco's modified Eagle's medium (DMEM)–F-12 medium (1:1) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin. AML12 cells (a gift from Chen Liu at Florida State University, Jacksonville, FL) were maintained in DMEM–F-12 medium (Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. ETV was a gift from William S. Mason at Fox Chase Cancer Center, Philadelphia., PA. ENAN-34017 was synthesized in house (27). Bay41-4109 was a gift from Lai Wei at the Hepatology Institute, People's Hospital, Beijing University, Beijing, China. 17-Dimethylamino geldanamycin was purchased from InvivoGen.
Antibodies.
A rabbit polyclonal antibody against HBV core protein was purchased from Dako (B0586). A monoclonal antibody (10E11) recognizing 2 to 8 amino acid residues of HBV core protein was purchased from Abcam (ab8639). A rabbit polyclonal antibody against the C-terminal 14 amino acids (aa 170 to 183) of HBV core protein was generated at GenScript, Piscataway, NJ, USA. Unlike the previous batch of the antibody designated HBc-170, which predominantly recognized dephosphorylated core protein (28, 47), the new batch of antibody used in this study recognized both full-length HBV core proteins with phosphorylated and dephosphorylated CTDs, as shown in Fig. 1 and 2. To distinguish the new batch of antibody, it was designated HBc-170A. Antibody against β-actin was obtained from CST (catalog no. 4967).
Plasmids.
Wild-type HBV replicons pHBV-1.3 and pCMV-HBcAg and pHBV-1.3-derived plasmids expressing mutant core proteins (V124A, V124W, or Y132A), pTRE2 (Clontech), and a derived plasmid expressing full-length and C-terminally truncated HBV core proteins were described previously (27). The plasmids pCI-HBc-WT, pCI-HBc-3A, pCI-HBc-7A, pCI-HBc-3E,a nd pCI-HBc-7E were described previously (28).
Establishment of a stable cell line expressing pgRNA encoding DNA polymerase with a Y63F mutation.
The DNA polymerase Y63F substitution was introduced into pTRE_HBV_DE (48) by an overlapping-PCR strategy. Specifically, DNA fragments were amplified first with primers F1 (5′-AAGTCGAGCTCGGTACCCGGGTCGA-3′) and R63 (5′-GGTACAGTAGAAGAAAAAAGACCAGTAA-3′) and primers F63 (5′-TTACTGGTCTTTTTTCTTCTACTGTACC-3′) and R1 (5′-AAAGGTTGTGGAATTCCACTGCATG-3′). A ligation PCR was performed using these two DNA fragments as templates with the F1 and R1 primers. The resulting DNA fragment was digested with SphI and EcoRI to replace the corresponding fragment of pTRE_HBV_DE, yielding plasmid pTRE_HBV_DEpolY63F. AML12 cells were cotransfected with the plasmids pTRE_HBV_DEpolY63F and pTet-off (Clontech) at a molar ratio of 10:1. The transfected cells were selected with 400 μg/ml G418 in the presence of 1 μg/ml tetracycline. G418-resistant colonies were picked and expanded into cell lines. HBV pgRNA transcription was induced by culturing cells in tetracycline-free medium, and the levels of pgRNA-containing capsids were determined by Northern blotting hybridization and particle gel assays. One of the cell lines with the highest level of pgRNA-containing capsid assembly, designated AML12HBVpolY63F, was chosen for this study.
Analyses of HBV capsids, core DNA, and RNA.
The cytoplasmic HBV capsids and associated viral DNA were analyzed by a native agarose gel electrophoresis-based assay (27). Southern blot analysis of HBV core DNA from transfected HepG2 cells was done as described previously (27). Total cellular RNA was extracted with TRIzol reagent (Invitrogen) following the manufacturer's directions. Encapsidated HBV RNA was extracted as previously described (45). HBV mRNA and encapsidated HBV RNA were analyzed by Northern blot hybridization with an [α-32P]UTP-labeled full-length minus-strand RNA probe.
Western blot assay.
Cells in the wells of a 12-well plate were lysed in 150 μl of 1× LDS loading buffer (Invitrogen), and a total of 25 μl of the cell lysate was resolved in a NuPAGE 12% Bis-Tris protein gel (Invitrogen) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Invitrogen). The membrane was blocked with phosphate-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk and probed with the appropriate primary antibody. The bound antibody was revealed by IRDye secondary antibodies and visualized with a Li-Cor Odyssey system.
In vitro transcription/translation.
The pCI-derived plasmids were used to express the HBV core proteins in a TNT-coupled RRL in vitro translation system (Promega) as described previously (28). One unit of calf intestinal alkaline phosphatase (New England BioLabs) per microliter of the final reaction mixture volume was added at the end of translation, and the reaction mixture was incubated for 16 h at 37°C. HBV core proteins were analyzed by a Western blot assay with antibody HBc-170A.
Ultracentrifugation analysis of HBV capsids.
Cells in a 10-cm plate were lysed in 2 ml of cell lysis buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.5% NP-40) at room temperature for 10 min. The cell lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant was loaded onto a 20% sucrose cushion and centrifuged at 46,000 rpm for 3 h (Beckman; SW55 rotor). The pellet was dissolved in 200 μl TNE buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 1 mM EDTA) containing protease and phosphatase inhibitors (Invitrogen) overnight and then loaded onto a 15% to 30% linear sucrose gradient in TNE buffer and spun at 27,000 rpm for 4 h (Beckman; SW28 rotor). Fractions (1.5 ml/each) were collected from the bottom of the centrifugation tube, and 50 μl of each fraction was applied to dot blot assays to detect viral capsids with anti-HBcAg antibody (Dako; B0586) and core DNA by hybridization. The sucrose concentration of each fraction was measured with a Densito 30PX (Mettler Toledo). The rest of each fraction was mixed with 3 ml TNE buffer and pelleted by centrifugation at 46,000 rpm for 3 h. The pellet was dissolved in 40 μl TNE buffer. Twenty microliters of capsid solution was resolved in a NuPAGE 12% Bis-Tris protein gel (Invitrogen) to detect hyper- or hypophosphorylated HBcAg by probing with anti-HBcAg antibody (HBc-170A); 20 μl of capsid solution was fractionated on a nondenaturing 1.5% agarose gel and transferred to a nitrocellulose membrane. HBV core protein/capsids (HBc) were detected by probing the membrane with anti-HBcAg antibody (Dako; B0586). Core DNA was detected by hybridization.
Protein phosphatase treatment.
AML12HBV10 or HepAD38 cells cultured in 10-cm plates were lysed by the addition of 2 ml of cell lysis buffer at room temperature for 10 min. The cell lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C. The supernatant was loaded onto a 20% sucrose cushion and centrifuged at 46,000 rpm for 3 h (Beckman; SW55 rotor). The pellet was dissolved in 85 μl TNE buffer. One half of the capsid samples were treated with8 units per microliter protein phosphatase 1 (New England BioLabs) for 16 h at 30°C. The other half were mock treated. HBV core proteins were analyzed by a Western blot assay with antibody HBc-170A.
ACKNOWLEDGMENTS
We thank Eain A. Murphy for critical readings of and comments on the manuscript.
The National Institute of Allergy and Infectious Diseases (NIAID), NIH, provided funding to Ju-Tao Guo under grant number R01AI113267. NIAID provided funding to Jianming Hu under grant number R01AI043453. This work was partially supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under award no. W81XWH-17-1-0600.
The opinions, interpretations, conclusions, and recommendations are ours and are not necessarily endorsed by the Department of Defense.
REFERENCES
- 1.Tang L, Zhao Q, Wu S, Cheng J, Chang J, Guo JT. 2017. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin Drug Discov 12:5–15. doi: 10.1080/17460441.2017.1255195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang J, Guo F, Zhao X, Guo JT. 2014. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B 4:248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. 2015. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 121:82–93. doi: 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeger C, Mason WS. 2015. Molecular biology of hepatitis B virus infection. Virology 479-480:672–686. doi: 10.1016/j.virol.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roossinck MJ, Siddiqui A. 1987. In vivo phosphorylation and protein analysis of hepatitis B virus core antigen. J Virol 61:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeh CT, Ou JH. 1991. Phosphorylation of hepatitis B virus precore and core proteins. J Virol 65:2327–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol 69:1025–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venkatakrishnan B, Zlotnick A. 2016. The structural biology of hepatitis B virus: form and function. Annu Rev Virol 3:429–451. doi: 10.1146/annurev-virology-110615-042238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung J, Hwang SG, Chwae YJ, Park S, Shin HJ, Kim K. 2014. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol 88:8754–8767. doi: 10.1128/JVI.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. doi: 10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- 11.Lewellyn EB, Loeb DD. 2011. Serine phosphoacceptor sites within the core protein of hepatitis B virus contribute to genome replication pleiotropically. PLoS One 6:e17202. doi: 10.1371/journal.pone.0017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pugh J, Zweidler A, Summers J. 1989. Characterization of the major duck hepatitis B virus core particle protein. J Virol 63:1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perlman DH, Berg EA, O'Connor PB, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 102:9020–9025. doi: 10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol 81:1641–1649. doi: 10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su PY, Yang CJ, Chu TH, Chang CH, Chiang C, Tang FM, Lee CY, Shih C. 2016. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci Rep 6:38959. doi: 10.1038/srep38959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ning X, Basagoudanavar SH, Liu K, Luckenbaugh L, Wei D, Wang C, Wei B, Zhao Y, Yan T, Delaney W, Hu J. 2017. Capsid phosphorylation state and hepadnavirus virion secretion. J Virol 91:e00092-17. doi: 10.1128/JVI.00092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selzer L, Kant R, Wang JC, Bothner B, Zlotnick A. 2015. Hepatitis B virus core protein phosphorylation sites affect capsid stability and transient exposure of the C-terminal domain. J Biol Chem 290:28584–28593. doi: 10.1074/jbc.M115.678441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. 1999. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol 145:45–55. doi: 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kock J, Kann M, Putz G, Blum HE, Von Weizsacker F. 2003. Central role of a serine phosphorylation site within duck hepatitis B virus core protein for capsid trafficking and genome release. J Biol Chem 278:28123–28129. doi: 10.1074/jbc.M300064200. [DOI] [PubMed] [Google Scholar]
- 20.Barrasa MI, Guo JT, Saputelli J, Mason WS, Seeger C. 2001. Does a cdc2 kinase-like recognition motif on the core protein of hepadnaviruses regulate assembly and disintegration of capsids? J Virol 75:2024–2028. doi: 10.1128/JVI.75.4.2024-2028.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkatakrishnan B, Katen SP, Francis S, Chirapu S, Finn MG, Zlotnick A. 2016. Hepatitis B virus capsids have diverse structural responses to small-molecule ligands bound to the heteroaryldihydropyrimidine pocket. J Virol 90:3994–4004. doi: 10.1128/JVI.03058-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klumpp K, Lam AM, Lukacs C, Vogel R, Ren S, Espiritu C, Baydo R, Atkins K, Abendroth J, Liao G, Efimov A, Hartman G, Flores OA. 2015. High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A 112:15196–15201. doi: 10.1073/pnas.1513803112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stray SJ, Bourne CR, Punna S, Lewis WG, Finn MG, Zlotnick A. 2005. A heteroaryldihydropyrimidine activates and can misdirect hepatitis B virus capsid assembly. Proc Natl Acad Sci U S A 102:8138–8143. doi: 10.1073/pnas.0409732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katen SP, Tan Z, Chirapu SR, Finn MG, Zlotnick A. 2013. Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure. Structure 21:1406–1416. doi: 10.1016/j.str.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JA, Qin N. 2017. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci Rep 7:42374. doi: 10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campagna MR, Liu F, Mao R, Mills C, Cai D, Guo F, Zhao X, Ye H, Cuconati A, Guo H, Chang J, Xu X, Block TM, Guo JT. 2013. Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol 87:6931–6942. doi: 10.1128/JVI.00582-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu S, Zhao Q, Zhang P, Kulp J, Hu L, Hwang N, Zhang J, Block TM, Xu X, Du Y, Chang J, Guo JT. 2017. Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J Virol 91:e00519-17. doi: 10.1128/JVI.00519-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng Voitenleitner SC, Delaney WET, Hu J. 2016. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-terminal domain and regulated by phosphorylation. J Virol 90:5830–5844. doi: 10.1128/JVI.00394-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bourne CR, Katen SP, Fulz MR, Packianathan C, Zlotnick A. 2009. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry 48:1736–1742. doi: 10.1021/bi801814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, Seeger C. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc Natl Acad Sci U S A 93:1060–1064. doi: 10.1073/pnas.93.3.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu J, Flores D, Toft D, Wang X, Nguyen D. 2004. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J Virol 78:13122–13131. doi: 10.1128/JVI.78.23.13122-13131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan Z, Maguire ML, Loeb DD, Zlotnick A. 2013. Genetically altering the thermodynamics and kinetics of hepatitis B virus capsid assembly has profound effects on virus replication in cell culture. J Virol 87:3208–3216. doi: 10.1128/JVI.03014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A. 2015. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol 89:3275–3284. doi: 10.1128/JVI.03545-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlicht HJ, Bartenschlager R, Schaller H. 1989. The duck hepatitis B virus core protein contains a highly phosphorylated C terminus that is essential for replication but not for RNA packaging. J Virol 63:2995–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gazina EV, Fielding JE, Lin B, Anderson DA. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol 74:4721–4728. doi: 10.1128/JVI.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albin C, Robinson WS. 1980. Protein kinase activity in hepatitis B virus. J Virol 34:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. 2012. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 86:12237–12250. doi: 10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu M, Summers J. 1994. Phosphorylation of the duck hepatitis B virus capsid protein associated with conformational changes in the C terminus. J Virol 68:2965–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu M, Summers J. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J Virol 68:4341–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCluskey A, Sim AT, Sakoff JA. 2002. Serine-threonine protein phosphatase inhibitors: development of potential therapeutic strategies. J Med Chem 45:1151–1175. doi: 10.1021/jm010066k. [DOI] [PubMed] [Google Scholar]
- 41.Katen SP, Chirapu SR, Finn MG, Zlotnick A. 2010. Trapping of hepatitis B virus capsid assembly intermediates by phenylpropenamide assembly accelerators. ACS Chem Biol 5:1125–1136. doi: 10.1021/cb100275b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo F, Zhao Q, Sheraz M, Cheng J, Qi Y, Su Q, Cuconati A, Wei L, Du Y, Li W, Chang J, Guo JT. 2017. HBV core protein allosteric modulators differentially alter cccDNA biosynthesis from de novo infection and intracellular amplification pathways. PLoS Pathog 13:e1006658. doi: 10.1371/journal.ppat.1006658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wieland SF, Eustaquio A, Whitten-Bauer C, Boyd B, Chisari FV. 2005. Interferon prevents formation of replication-competent hepatitis B virus RNA-containing nucleocapsids. Proc Natl Acad Sci U S A 102:9913–9917. doi: 10.1073/pnas.0504273102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. 2003. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 77:1885–1893. doi: 10.1128/JVI.77.3.1885-1893.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu C, Guo H, Pan XB, Mao R, Yu W, Xu X, Wei L, Chang J, Block TM, Guo JT. 2010. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J Virol 84:9332–9340. doi: 10.1128/JVI.00918-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. 1997. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 41:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo H, Mao R, Block TM, Guo JT. 2010. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol 84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo H, Jiang D, Zhou T, Cuconati A, Block TM, Guo JT. 2007. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J Virol 81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]