

ABSTRACT
Herpes simplex virus (HSV) latency in neurons remains poorly understood, and the heterogeneity of the sensory nervous system complicates mechanistic studies. In this study, we used primary culture of adult trigeminal ganglion (TG) mouse neurons in microfluidic devices and an in vivo model to examine the subtypes of sensory neurons involved in HSV latency. HSV-infected neurofilament heavy-positive (NefH+) neurons were more likely to express latency-associated transcripts (LATs) than infected neurofilament heavy-negative (NefH−) neurons. This differential expression of the LAT promoter correlated with differences in HSV-1 early infection that manifested as differences in the efficiency with which HSV particles reached the cell body following infection at the distal axon. In vivo, we further identified a specific subset of NefH+ neurons which coexpressed calcitonin gene-related peptide α (NefH+ CGRP+ neurons) as the sensory neuron subpopulation with the highest LAT promoter activity following HSV-1 infection. Finally, an early-phase reactivation assay showed HSV-1 reactivating in NefH+ CGRP+ neurons, although other sensory neuron subpopulations were also involved. Together, these results show that sensory neurons expressing neurofilaments exhibit enhanced LAT promoter activity. We hypothesize that the reduced efficiency of HSV-1 invasion at an early phase of infection may promote efficient establishment of latency in NefH+ neurons due to initiation of the antiviral state preceding arrival of the virus at the neuronal cell body. While the outcome of HSV-1 infection of neurons is determined by a broad variety of factors in vivo, neuronal subtypes are likely to play differential roles in modulating the establishment of latent infection.
IMPORTANCE Two pivotal properties of HSV-1 make it a successful pathogen. First, it infects neurons, which are immune privileged. Second, it establishes latency in these neurons. Together, these properties allow HSV to persist for the lifetime of its host. Neurons are diverse and highly organized cells, with specific anatomical, physiological, and molecular characteristics. Previous work has shown that establishment of latency by HSV-1 does not occur equally in all types of neurons. Our results show that the kinetics of HSV infection and the levels of latency-related gene expression differ in certain types of neurons. The neuronal subtype infected by HSV is therefore a critical determinant of the outcome of infection and latency.
KEYWORDS: herpes simplex virus, latency, neurons
INTRODUCTION
Herpes simplex virus 1 and 2 (HSV-1 and HSV-2, respectively) (1) are efficient human pathogens (2) that cause diseases that range from asymptomatic infections and cold sores to blinding stromal keratitis and lethal encephalitis. In developed countries, HSV-1 is the leading cause of infectious blindness and viral encephalitis (1, 3). The HSV life cycle is initiated by infection of the skin, cornea, or mucosal epithelia, where the virus replicates. During primary infection the virus invades sensory free nerve endings (FNEs) that innervate the infected epithelium (4). HSV particles are then retrogradely transported to the neuronal cell body, where the viral DNA is released into the nucleus. HSV DNA persists in the nucleus, and lytic gene expression is repressed through chromatin control (5). The latency-associated transcripts (LATs) and several microRNAs (miRNAs) remain highly expressed, serving to further silence lytic gene expression and promote survival of the infected neuron (4, 5).
Latency in neurons is a major evolutionary advantage for HSV, providing a lifelong reservoir from which virus can reactivate and spread to new susceptible individuals (3). In addition, neurons are an immune-privileged and nondividing cell population, facilitating persistence and obviating the need for the virus to pass into progeny cells. HSV latency is, however, a complex and heterogeneous process with many unknowns (6, 7). First, not all latently infected neurons carry the same amount of viral DNA, and the HSV-1 genome copy number within an infected ganglion can range from the single-copy range to more than 1,000 copies per neuron (8). Second, expression of LATs, an accepted marker of HSV latency, is dispensable for the establishment, maintenance, and reactivation of HSV latency in neurons (6, 7, 9). Moreover, expression of LATs changes during the course of infection (10), and yet only 30% of latently infected neurons express LATs at all (11, 12). Taken together, these data show that HSV latency is not homogeneous in the neurons that populate sensory ganglia.
Although the clinical landscape is shifting, HSV-1 and HSV-2 have evolved distinct anatomical tropisms (1). HSV-1 most frequently infects orolabial epithelia and establishes latency in trigeminal ganglia (TG), while HSV-2 usually infects the genital epithelium and establishes latency in dorsal root ganglia (DRG) (4). TG and DRG are both sensory ganglia, and neurons therein are specialized in conveying various signals to the central nervous system. These include signals for pain, temperature, pressure, and proprioception (13). Sensory neurons within the peripheral nervous system (PNS) have been classified according to their myelination and speed of nerve impulse conduction (13), neurotrophic dependency (14), differential expression of neuropeptides (15), type of sensations conveyed (16), and, more recently, large-scale single-cell RNA sequencing (17). This diversity of sensory neurons seems to be a likely major contributor to the heterogeneity of HSV latency. Indeed, HSV-1 LATs are preferentially expressed in neuropeptide-rich TG neurons in mice (18). This observation was subsequently refined, pointing to a sensory neuron population recognized by an antibody termed A5 (19). It was proposed that A5-positive neurons are less permissive for HSV-1 productive infection than other subpopulations, linking the presence of LATs with the absence of viral productive infection (20–22). However, the A5 mouse monoclonal antibody is an IgM which recognizes poorly defined carbohydrates. This antibody is therefore a limited reagent for further mechanistic investigations.
In this study, we explored how adult sensory neuron heterogeneity may affect acute infection, latency-associated transcription, and establishment of HSV-1 latency. Our approach was to classify sensory neurons by their expression of neuronal intermediate filaments, known as neurofilaments (23, 24). Neurofilaments are the major cytoskeletal component of myelinated neurons. In the PNS, neurofilaments are heteropolymers composed of four subunits, neurofilament heavy, medium, and light (NefH, NefM, and NefL, respectively) and peripherin. The known function of neurofilaments is to confer a threshold thickness to axons, which contributes to the process of myelination (23, 24). In this manner, neurons expressing neurofilaments have bigger cell bodies, a higher axon caliber, and a higher speed of conduction. On the other hand, neurofilament-negative neurons have small cell bodies and thin axons, and Schwann cells wrap these axons without myelination, forming Remak bundles (24, 25). In this study, we used microfluidic chambers in conjunction with murine TG neurons and recombinant viruses, as well as in vivo latency and reactivation models. With these systems, we have garnered evidence for a new hypothesis regarding the relationship between neurofilaments, HSV infection, and latency. In NefH-positive (NefH+) neurons, HSV-1 particles are slow to infect the neuronal cell bodies, but many neurons of this type exhibit high LAT promoter activity during latency. In contrast, HSV-1 rapidly infects the cell bodies of NefH-negative (NefH−) neurons, and relatively few neurons of this type show LAT promoter activity during HSV-1 latency. Our approach has therefore elucidated how the heterogeneity of the PNS correlates with the range of outcomes following infection with HSV.
RESULTS
Establishment and characterization of microfluidic neuronal cultures.
An important question in HSV-1 pathogenesis is whether the intrinsic neuronal heterogeneity within peripheral nervous system tissues influences the establishment of viral latency. To begin to address this, we developed a protocol with which adult mouse TG neurons could recapitulate their compartmentalization within microfluidic devices. In this system, neurites sprout into a compartment distal from the cell bodies, eventually forming a dense, polarized, axonal network (26). Growth of the majority of neuronal subtypes of the TG was promoted by providing a combination of neurotrophic factors (nerve growth factor [NGF], brain-derived neurotrophic factor [BDNF], glial cell-derived neurotrophic factor [GDNF], and soluble GDNF family receptor α1 [GFRα1]). This allowed all subtypes of neurons to contribute to the axonal compartment and created a robust trophic environment, a critical factor in the establishment of HSV-1 latency (27). Adult sensory neurons are uniformly positive for the microtubule β3-tubulin (β3-Tub) but differ in their expression of neurofilaments and are thus categorized as neurofilament heavy protein positive (NefH+) or negative (NefH−) (17). Enumeration of the populations of sensory neurons present in the microfluidics devices was possible upon comparison of NefH+ neurons to total (β3-Tub-positive [β3-Tub+]) neurons in the cell body chamber (Fig. 1A). Our culture conditions routinely yielded a ratio of NefH+/NefH− neurons of ∼30:70 after 3 days in culture, which is representative of these subpopulations in human and murine TG (19, 28) Within the distal axon compartments (Fig. 1B), we observed that NefH+ axons also represented ∼30% of the total surface covered by all β3-Tub-positive axons. Thus, we considered our microfluidic cultures to be an accurate representation of the TG neuron population in vivo and a well-suited model of polarized neurons for our studies.
FIG 1.
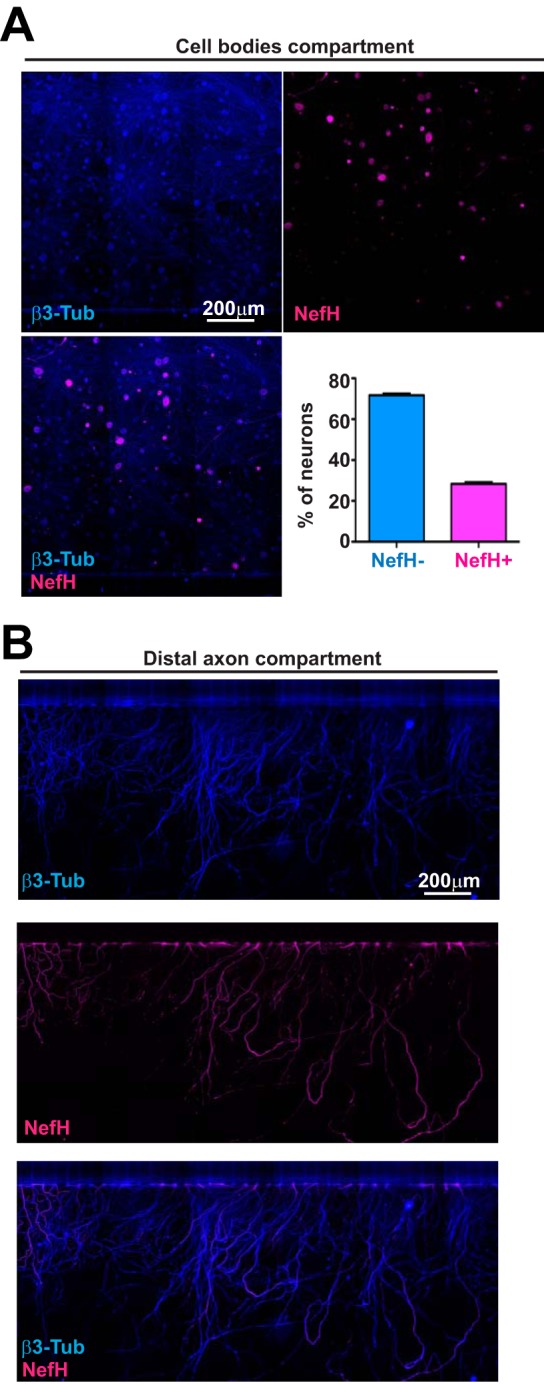
Trigeminal ganglion neuronal subpopulations are identifiable by neurofilament heavy expression. (A) Representative images from the cell bodies compartment of microfluidic devices containing explanted, polarized, adult mouse TG neurons. Neurons polarizing over 3 days in culture were fixed within the devices and immunostained for β3-tubulin (β3-Tub) (blue; upper left) to demarcate all neurons and for neurofilament heavy (NefH; magenta; upper right) to identify NefH+ neurons. The merged image is shown at the lower left. The graph represents the percentage of NefH− versus NefH+ neurons counted from tiles obtained from the central channel in eight replicate chambers. (B) Representative images of TG axons in the distal axon compartment of the microfluidic device depicted in panel A. (Top) β3-Tub (blue); (middle) NefH (magenta); (bottom) merged image.
LAT expression is higher in NefH+ than NefH− neurons.
HSV-1 latency in TG neurons requires infection at the distal axon and silencing of the viral genome within the neuronal nucleus, after which point LAT expression becomes detectable. To determine which neuronal subpopulations are expressing LAT, we utilized KOS/62, an HSV-1 strain in which β-galactosidase (β-Gal) expression is driven by the LAT promoter (10, 29). It should be noted that the region immediately downstream of the LAT promoter is replaced with the β-Gal gene, so that despite accurately reporting LAT promoter activity, KOS/62 is at least partly a LAT-negative virus. Following infection, β-Gal expression in the cell bodies was assessed by immunofluorescence (IF) microscopy. By 5 days postinfection (dpi), β-Gal-positive (β-Gal+) neurons were readily seen (Fig. 2, micrograph). Surprisingly, although NefH+ neurons were the minority population in these cultures, the majority of the β-Gal+ neurons were NefH+ (>80%; Fig. 2, graph). This indicated that NefH+ neurons favor robust LAT promoter activity.
FIG 2.
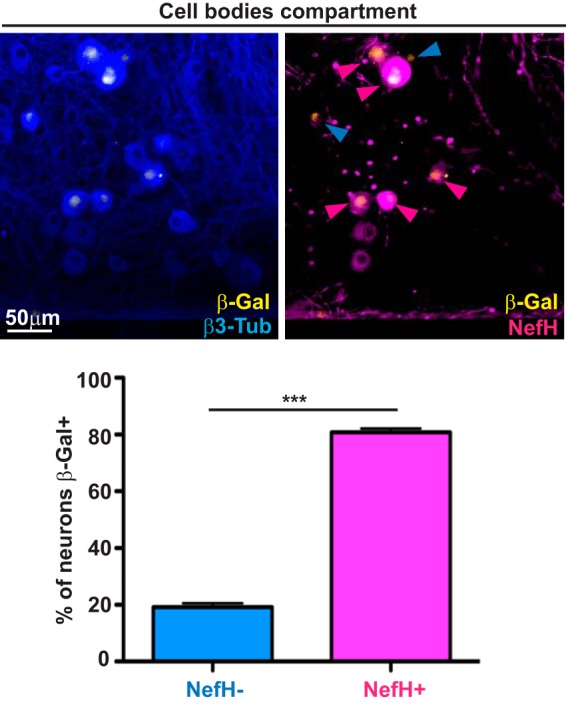
LAT expression is primarily observed in NefH+ neurons following axonal HSV-1 infection. TG neurons cultured in microfluidic devices for 3 days were infected at the distal axon compartment with KOS/62 (MOI = 10). At 5 dpi, the cultures were fixed and immunostained for expression of the LAT promoter-driven β-galactosidase (β-Gal; yellow), β3-Tub (blue), and NefH (magenta). In the right micrograph, the blue arrowheads point to β-Gal+ NefH− neurons and magenta arrowheads point to β-Gal+ NefH+ neurons. The graph represents the percentage of NefH− and NefH+ neurons positive for β-Gal (LAT) expression. The threshold for β-Gal positivity was set using mock-infected chambers. Data are for 8 chambers and are from 3 independent experiments. ***, P < 0.001.
NefH+ neurons have low HSV-1 early-infection efficiency.
We hypothesized that the initial phases of HSV-1 infection may differ during invasion of NefH+ and NefH− sensory neurons. To test this, temperature-shift infections were performed to synchronize infection, such that early-infection efficiency from the distal axon compartment could be compared between neuronal subtypes. HSV-1 KOS first arrived at cell bodies 20 min after infection (Fig. 3, micrographs, left), in agreement with the documented fast axonal transport of HSV-1 to the cell body (using an average speed of 2 μm/s and a maximal distance of 2,000 μm) (30–33). Surprisingly, ∼85% of infected neurons were NefH− at 20 min postadsorption, while only ∼15% of infected neurons were NefH+ (Fig. 3, graph). This percentage of infected NefH+ neurons represented half of the expected frequency based on their proportional presence in the culture (Fig. 1). At 150 min postadsorption, ∼70% of infected neurons were NefH−, while ∼30% were NefH+ (Fig. 3, micrographs, right, and graph). The proportions of neurons infected at this later time point therefore matched the expected 70:30 NefH−/NefH+ ratio (Fig. 1). To confirm this result we repeated these infections using a green fluorescent protein (GFP)-mOrange-tagged HSV-1 strain, strain 17, which showed similar trends (data not shown). Altogether, these results suggest that the early stages of HSV-1 infection are less efficient in NefH+ neurons.
FIG 3.
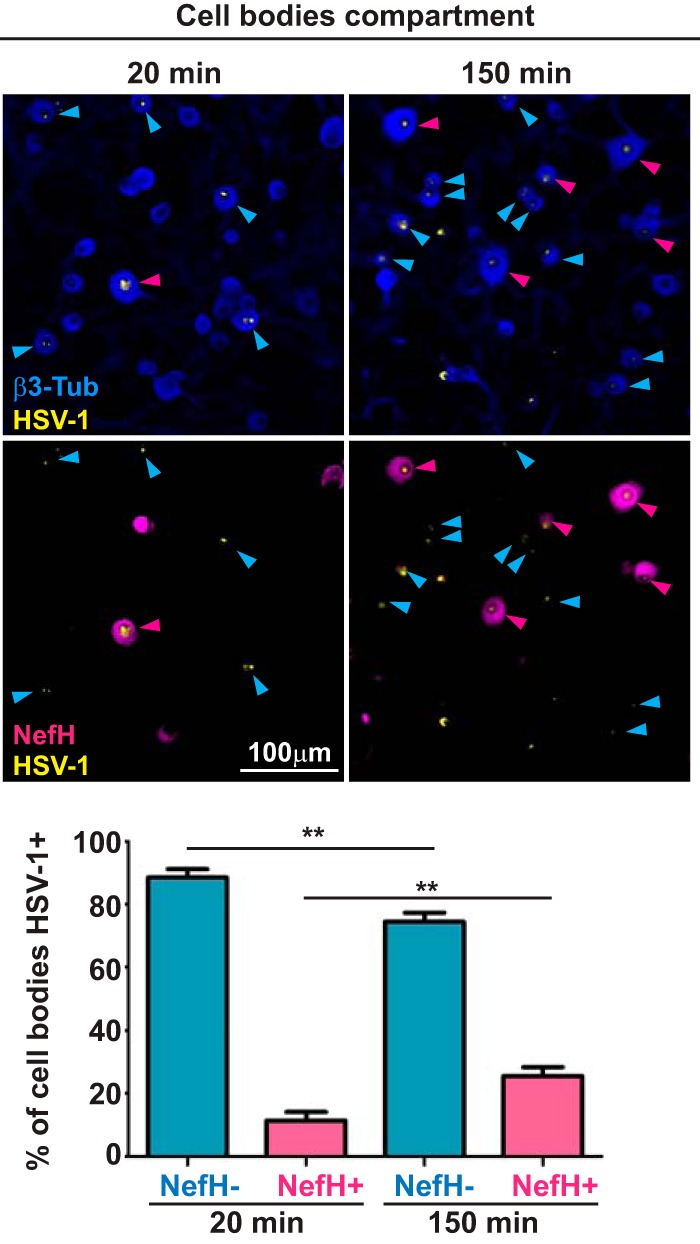
HSV-1 early-infection efficiency is lower in NefH+ neurons. TG neurons cultured in microfluidic devices for 3 days were infected synchronously at the distal axon compartment with KOS (MOI = 200). Cultures were fixed and processed at two time points: 20 min (left) and 150 min (right) postadsorption. Immunostaining for β3-Tub (blue) and NefH (magenta) revealed total neurons and NefH+ neurons, respectively. Immunostaining for HSV-1 is in yellow. Blue arrowheads point to NefH−-infected neurons. Magenta arrowheads point to NefH+-infected neurons. The graph represents the percentage of NefH− and NefH+ cell bodies positive for KOS at the early and late time points. The threshold for KOS detection was set using mock-infected chambers. Data are for 7 chambers for 20 min postadsorption and 7 chambers for 150 min postadsorption from 2 independent experiments. **, P < 0.01.
The A5 antibody identifies the NefH+ and CGRP+ neuronal subtypes.
We next sought to test whether LAT promoter activity was higher in NefH+ neurons than in NefH− neurons in vivo. First, however, we needed to account for the anatomy of the PNS, in that most NefH− neurons end as FNEs, while the majority of NefH+ neurons do not (34). Only a specific subset of NefH+ neurons end as FNEs in the skin and cornea, with those being the lightly myelinated NefH+ calcitonin gene-related peptide α (CGRPα; here referred to as CGRP)-positive (CGRP+) TG (Aδ) neurons that function in mechano/nociception (35, 36). As Aδ neurons express the neuropeptide CGRP (17), we used a transgenic mouse line in which GFP is expressed in its CGRP+ neurons (37, 38). The mice used in this mouse model, here termed CGRP-GFP mice, were bred as heterozygous for the GFP allele and show no detectable phenotype (37). This allowed us to place sensory TG neurons into four categories: NefH+ CGRP+, NefH+ CGRP-negative (CGRP−), NefH− CGRP+, and NefH− CGRP− neurons (Fig. 4A).
FIG 4.
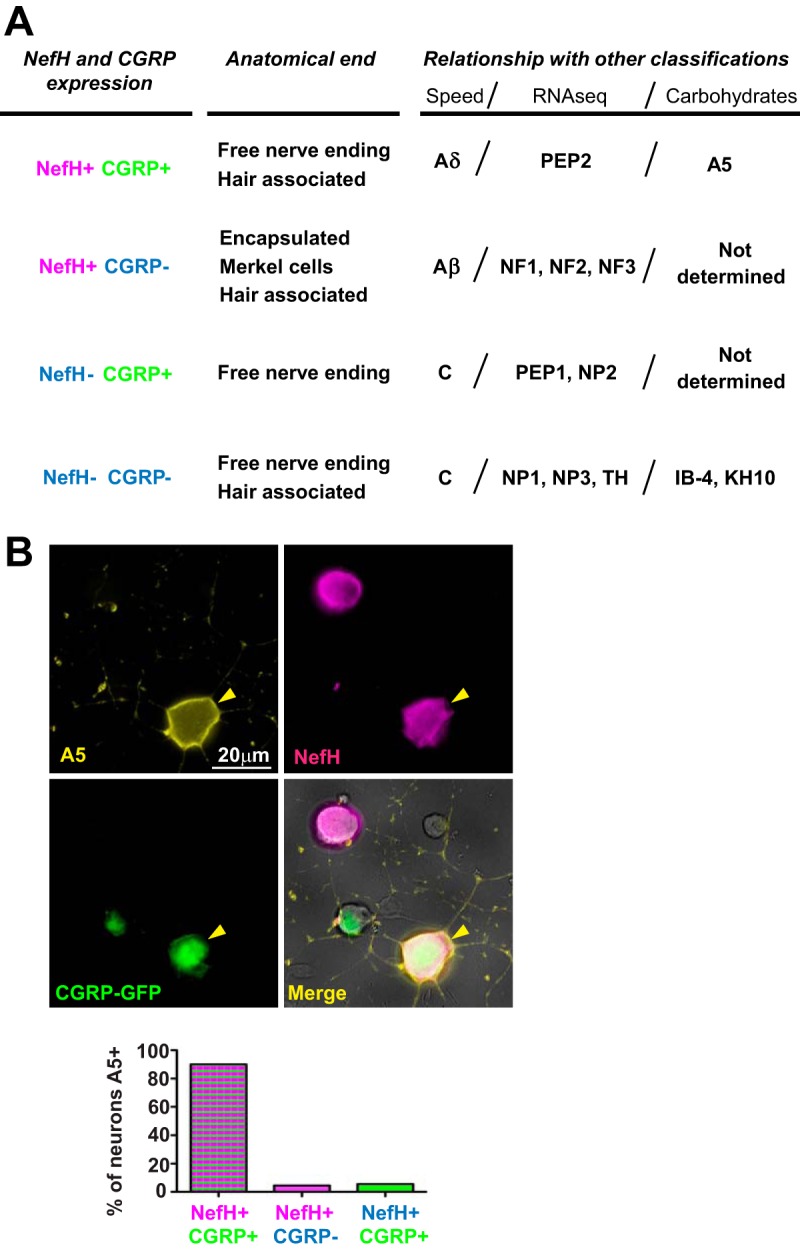
A5+ TG neurons are mainly NefH+ CGRP+ neurons. (A) TG neuron classification based on NefH and CGRP expression. The four defined subpopulations are compared to other existing classifications: anatomical ending, conduction speed, large-scale single-cell RNA-seq, or carbohydrate expression. The information presented is based on previously published data (18–20, 36, 37). (B) Adult CGRP-GFP mouse TG neurons were explanted onto coverslips and immunostained for the A5 cell surface carbohydrate (yellow) and NefH (magenta). CGRP-GFP neurons were green due to GFP expression from the transgenic mouse. Yellow arrowheads point to an A5+ neuron. Neurons were identified by their morphology under phase contrast. The graph represents the percentage of neurons in each subpopulation that were positive for Α5. Data are for >200 neurons from 2 independent experiments.
Curiously, NefH+ CGRP+ (Aδ) TG neurons are intermediate to large, similar in size to those neurons which are recognized by the A5 antibody, which is reactive to a cell surface carbohydrate (19). Previously published studies showed that the highest LAT promoter activity during latency was in A5-positive (A5+) neurons (19). We therefore needed to determine whether NefH+ CGRP+ and A5+ neurons represent the same neuronal subpopulation. We characterized these subpopulations using IF microscopy of cultures of adult TG neurons from CGRP-GFP mice (Fig. 4B, micrographs). A5 immunoreactivity coincided with that of NefH and CGRP, and when quantitated, more than 90% of the A5+ neurons were also NefH+ and CGRP+ (Fig. 4B, graph). This demonstrates that these markers identify the same neuronal subtype, allowing 2 alternate approaches to identify this important subpopulation.
Corneal innervation is heterogeneous.
One of the most widely used in vivo models of HSV-1 infection is the corneal infection model. In this system, the corneal epithelium is scarified before HSV-1 infection, allowing virus direct access to the nerves innervating the cornea. The innervation of the corneal epithelium has been well studied, and based on this literature, we have synthesized a model of corneal epithelium innervation (Fig. 5A) (39–41). This model predicts that the majority of innervating FNEs are NefH− CGRP−, terminate at the surface of the cornea, and are greatly branched. The NefH− CGRP+ neuronal termini lie in the middle-outer layer of the epithelium. The NefH+ CGRP+ FNE population innervates the innermost layer of the corneal epithelium. To address this model directly, we stained the corneal epithelium of CGRP-GFP mice for β3-tubulin (Fig. 5B). In agreement with the model, CGRP− FNEs were the most abundant and the most branched FNEs. Despite our inability to stain directly for NefH in these tissues, we observed two different types of CGRP+ FNEs. One type had a straight morphology (CGRP + long; Fig. 5B), and according to the literature, these FNEs are most likely NefH− CGRP+ FNEs (39–41). The second type had a short unbranched morphology (CGRP+ short; Fig. 5B), and these FNEs are most likely NefH+ CGRP+ FNEs (39–41). This result suggests that the model proposed is an accurate representation of corneal innervation in mice. Together, these results present evidence of the heterogeneity of corneal epithelium innervation, which likely has a critical impact on HSV-1 infection and the establishment of latency following corneal infection in humans and animal model systems.
FIG 5.
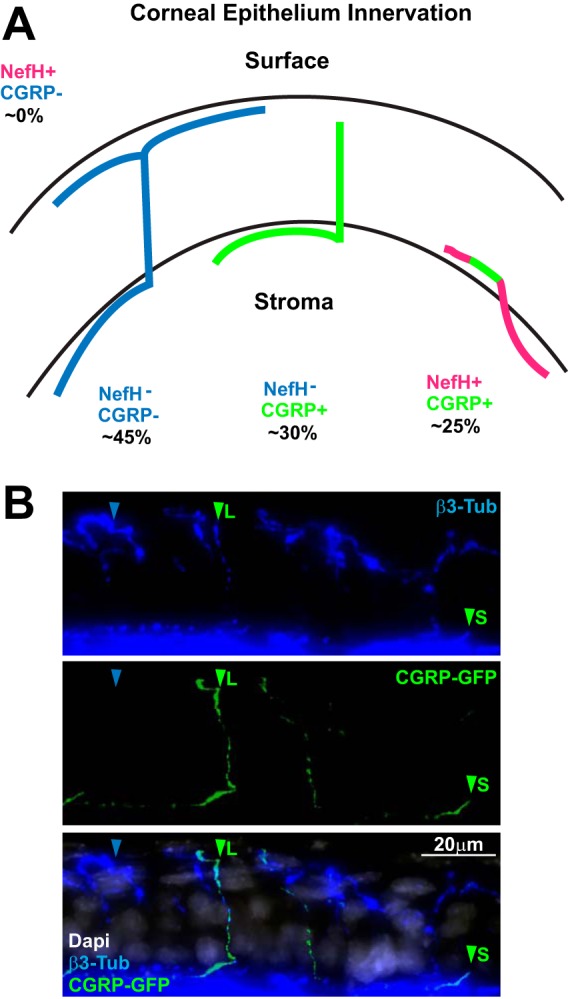
The innervation of mouse corneal epithelium is heterogeneous. (A) Overview of mouse corneal epithelial innervation based on previously published data (39–41). (B) A representative z-stack projection of corneal epithelium from adult CGRP-GFP mice. Tissue sections were immunostained for β3-Tub (blue), and CGRP+ neurons were green from GFP expression in the transgenic mouse. The merged image is shown at the bottom (DAPI [4′,6-diamidino-2-phenylindole] is in white). Blue arrowheads point to CGRP− FNEs. Green arrowheads point to CGRP+ FNEs. L, long; S, short.
LAT promoter activity is highest in Aδ (NefH+ CGRP+) neurons.
Given this model of corneal epithelium innervation, we wished to assess which neurons supported the highest LAT promoter activity. We infected CGRP-GFP mice with KOS/62 and analyzed LAT expression by β-Gal IF in the TG tissue at ≥21 dpi. By IF, the majority of neurons with LAT promoter activity were NefH+ (Fig. 6A), in agreement with in vitro results (Fig. 2). Moreover, the NefH+ CGRP+ neurons comprised nearly 60% of β-Gal-expressing neurons in the TG (Fig. 6A and B). We also found a notable amount of NefH+ CGRP− neurons with LAT promoter activity (∼25%; Fig. 6B). This was very unexpected, since these neurons do not innervate the corneal epithelium (Fig. 5A). The presence of latent HSV in these neurons likely reflects infection of the conjunctiva, interneuronal HSV-1 spread, or possibly, round-trip infection (42). Together, these results suggest that NefH+ neurons are more likely to harbor genomes with high LAT promoter activity in vivo, especially in the Aδ (NefH+ CGRP+) neurons that innervate the innermost layer of the corneal epithelium.
FIG 6.
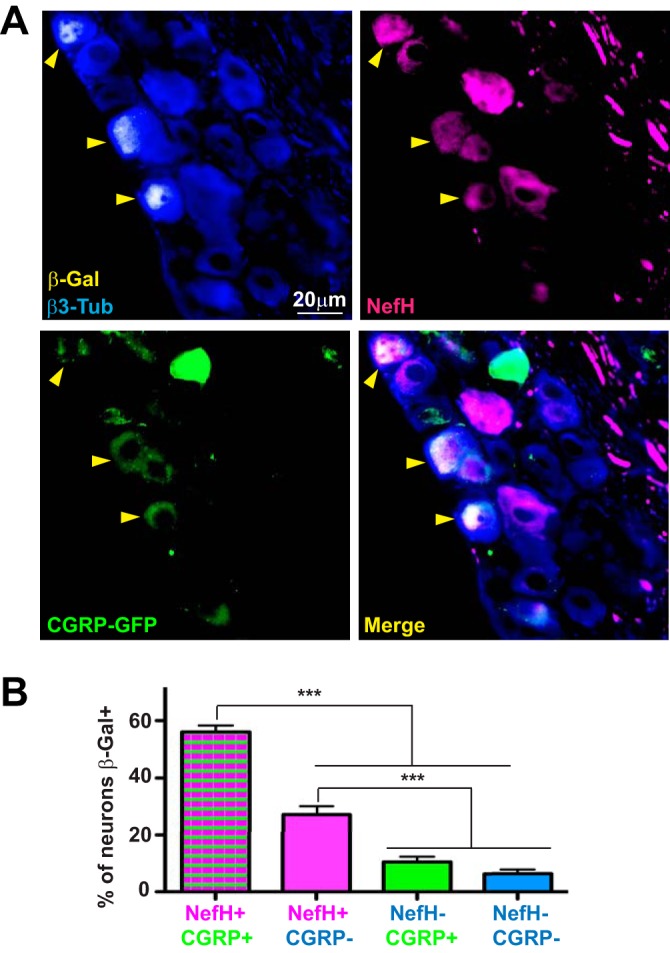
HSV-1 LAT expression is predominantly observed in NefH+ CGRP+ sensory neurons following corneal infection. (A) Representative image of TGs from adult CGRP-GFP mice corneally infected with KOS/62 (1 × 106 PFU/eye) for >21 days. Tissue sections were immunostained for β-Gal (yellow), β3-Tub (blue), and NefH (magenta). CGRP+ neurons were green owing to GFP expression from the transgenic mouse. Yellow arrowheads point to β-Gal+ neurons in which LAT promoter activity is evident. The merged image is shown at the bottom right. (B) The graph represents the percentage of each subpopulation of neurons positive for β-Gal. The threshold for β-Gal detection was set using mock-infected tissue. No fewer than 6 random sections per TG were analyzed. Data are for 12 TGs from 2 independent experiments. ***, P < 0.001. Bar, 20 μm.
HSV molecular reactivation occurs from both NefH+ and NefH− neurons.
LAT expression peaks during HSV-1 latency, but several studies have shown that HSV latency can occur without LAT expression, and it has even been proposed that LAT expression occurs in only 30% of latently infected neurons (11, 12). Thus, the higher frequency of detectable LAT promoter expression in NefH+ CGRP+ TG neurons may reflect either a preferential establishment of HSV-1 latency in these neurons or the fact that NefH+ CGRP+ neurons simply promote higher expression of LAT. To test whether NefH+ CGRP+ TG neurons are a bona fide HSV-1 reservoir, we infected CGRP-GFP mice with HSV-1 KOS, and at 21 dpi we harvested and dissociated TG neurons in the absence of trophic support. These dissociated neurons were cultured with trichostatin A and were monitored for 24 h to assess phase I (early) molecular reactivation, as judged by VP16 expression (43). In this scenario, the cultures maintain neuronal identity without the complication of secondary infection. IF revealed that VP16 was expressed in two different subpopulations: NefH− CGRP− neurons, the most abundant and most arborized neurons in the corneal epithelium, and also in NefH+ CGRP+ neurons (Fig. 7). These results show that upon corneal epithelium infection, HSV-1 is capable of establishing latency in, and at least molecularly reactivating from, different subpopulations of TG neurons, especially NefH− CGRP− and NefH+ CGRP+ neurons.
FIG 7.
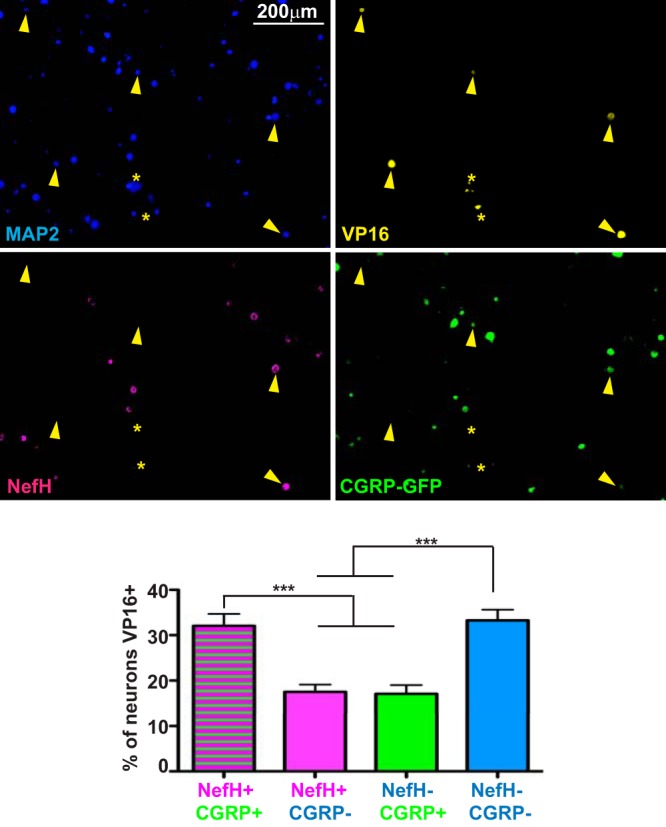
HSV-1 is capable of establishing latency in multiple neuronal populations. Representative images of dissociated TG neurons from adult CGRP-GFP mice infected corneally with KOS/62 (1 × 106 PFU/eye) for >21 days. Neurons were seeded without trophic support and in the presence of trichostatin A to ensure full reactivation from all latently infected neurons. Twenty-four hours later, neurons were immunostained for MAP2 to label all neurons (blue), VP16 (yellow), and NefH (magenta). CGRP+ neurons were identified by GFP emission (green). Yellow arrowheads point to VP16+ neurons. Yellow asterisks mark nonspecific staining or infected, syncytial neurons that were excluded from quantitation. The graph represents the percentage of neurons of each subpopulation that were VP16 positive. The threshold for VP16 detection was set using a culture of mock-infected neurons. Data are for 12 TG from 2 independent experiments. ***, P < 0.001.
DISCUSSION
Sensory neurons are a heterogeneous population (17), and this study has elucidated how this heterogeneity affects the activity of the LAT promoter. The Margolis group has examined this previously (10, 18, 19), and our results concur with and extend their observations. Our results in vivo show that NefH+ CGRP+ TG neurons are more prone to express LAT during latency. This is consistent with the hypothesis that these are the most frequent latently infected neuronal subpopulation. NefH+ CGRP+ TG neurons largely overlap the group previously identified as A5+ TG neurons (19), with >90% of A5+ neurons being NefH+ CGRP+ neurons. These neurons are the least permissive subpopulation in TG, express high levels of LATs (22), and likely represent a crucial category of neurons for HSV latency. The mechanism behind this pattern of HSV-1 infection in these neurons, however, remains obscure. The current study provokes the hypothesis that early-infection efficiency in TG neurons impacts the balance of lytic and latent infection directly or indirectly in concert with the generation of the antiviral state. This study also elucidates the innervation of the corneal epithelium and how the anatomic distribution of specific subpopulations likely plays a critical role in determining the outcomes of HSV infection during human corneal infection and especially when using murine corneal models.
Our experiments with microfluidic chambers showed that NefH+ neurons are more likely to exhibit high LAT promoter activity, as judged by the LAT promoter-regulated expression of β-Gal in strain KOS/62 (10, 29). While β-Gal is only a surrogate readout for LAT expression, there are some similarities, in that both the LAT intron and β-Gal are stable in sensory neurons (44–46). Given that β-Gal has been estimated to have a half-life of 24 to 48 h in neurons (44), it seems reasonable to attribute the differential presence of β-Gal at latency time points to differential LAT promoter activity. Another caveat of our study (and previous studies [19, 22]) is that KOS/62 is a LAT-deficient HSV-1 strain. LAT is used by HSV-1 to decrease viral protein production, facilitate HSV-1 entry in latency, induce neuronal survival, and induce an efficient reactivation (reviewed in references 7 and 8). It seems reasonable, therefore, to hypothesize that the absence of LAT in KOS/62 would mostly impact neurons with higher LAT promoter expression. Assuming this to be so, the survival and establishment of latency in NefH+ neurons would therefore be compromised by KOS/62, strengthening our results that actually showed a higher number of NefH+ neurons expressing the LAT promoter.
Microfluidic chamber experiments also showed that HSV-1 early-infection efficiency is lower in NefH+ neurons. There are a number of possible explanations for these data. First, NefH+ and NefH− have different carbohydrate signatures (19), and it is possible that HSV-1 binding and adsorption are slower in NefH+. It seems unlikely that this is due to differential Nectin-1 (HSV receptor) expression, since transcriptome sequencing (RNA-seq) data show that Nectin-1 levels are comparable between NefH+ and NefH− neurons (17). Since we used cold adsorption synchronization of HSV infection at 4°C, we also considered the caveat that a lower temperature might differentially affect uptake into NefH+/NefH− neurons. RNA-seq, however, also suggests that cold receptors are highly expressed in NefH− neurons (17), suggesting that this population would be the most affected by low-temperature exposure. Infection and transport of the HSV capsid and tegument, however, appear to be normal in these neurons.
Another hypothesis to explain the low infection efficiency in NefH+ neurons is that the expression levels of neurofilaments affect early events in neuronal infection. Microtubules and actin filaments are obligate components of the axonal cytoskeleton, but neurofilaments are dispensable (47); their only known function is to confer thickness to the axon. Neurofilaments are the most abundant component of the cytoskeleton of myelinated neurons (23, 24), organizing the internal structure of the axons and cell bodies, thereby affecting their physiological properties. It is plausible that the presence of neurofilaments could significantly alter the transport of HSV and the efficiency of establishment of latency. In squid axons (30), HSV-1 particles are transported retrogradely with a uniform high speed, but squid axons are homogeneous and do not express neurofilaments. There is greater heterogeneity in mammalian sensory neurons, and correspondingly, there is greater variation observed in the retrograde transport of HSV. That said, fast retrograde axonal transport still appears to be the dominant pathway to the cell bodies (31–33). In our study, we observed that HSV-1 took less time to reach the cell bodies of NefH− neurons than those of NefH+ neurons. If the efficiency of uptake into these cells were equal (discussed above), this would implicate the slow transport of HSV-1 in NefH+ neurons. In fact, neurofilaments are actually the paradigm of slow axonal transport using dyneins and kinesins in a stop-and-go manner (48, 49). Indeed, transgenic mice overexpressing human or mouse NefH show reduced total axonal retrograde transport rates (50, 51). HSV-1 may move slowly in NefH+ axons because it competes with neurofilaments for the same axonal transport machinery or because it is physically delayed in NefH+ axons by neurofilaments that fill spaces that are necessary for HSV-1 transport. The fact that some NefH+ cell bodies are infected at 20 min postadsorption supports the neurofilament-delayed retrograde transport of HSV-1 hypothesis. Trigeminal sensory neurons differentially express neurofilaments, with moderate expression in Aδ neurons (lightly myelinated neurons) and heavy expression in Aβ neurons (highly myelinated neurons) (13), which could potentially result in differentially delayed retrograde transport of HSV.
In human HSV infections, skin biopsy specimens show that HSV-1 and HSV-2 infect cells within the epidermis, largely sparing the dermis (52–54). The epidermis is innervated by specific neurons ending as FNEs, while the dermis is innervated by specific neurons associated with Merkel cells, encapsulated mechanoreceptors, or hair follicles (34). These facts suggest that sensory neurons are differently exposed to and infected by HSV, making neuroanatomy a key parameter during HSV infection. For our study of HSV-1 LAT promoter activity during latency, we have used the corneal scarification model in mice. First, we placed sensory neurons into four different groups in a Punnett square fashion, based on the presence or absence of NefH and CGRP (Fig. 4), and then we studied their anatomical location in the corneal epithelium (Fig. 5). Surprisingly, NefH+ CGRP+ neurons were the most likely to activate the LAT promoter and to yield reactivatable virus, a finding which is somewhat at odds with their location deep within the corneal epithelium. It is possible that corneal scarification artificially allows HSV to directly access these FNEs. This also highlights a key difference between certain corneal infection models, whether or not scarification is performed. In addition, such models also use different virus strains; HSV-1 McKrae is the favored strain for nonscarification, and almost all other HSV-1 strains require scarification for robust infection (55, 56). Moreover, these models likely allow viral access to distinct subpopulations of neurons. Scarification may therefore promote infection of a different set of neurons relative to human corneal infections. It was also surprising to see NefH+ CGRP− neurons with prominent LAT promoter activity, since these are mostly associated with hair or encapsulated mechanoreceptor structures in the dermis (34). These neurons are unlikely targets for direct infection by HSV-1 in corneas since they do not form FNEs therein. As shown above, however, this subtype does get infected, most likely through round-trip infection of the periocular skin or through the conjunctiva. The NefH− CGRP− neurons are the most abundant, branched, and anatomically exposed neurons in the corneal epithelium (39–41) and epidermis (15, 57). Their abundance, position, and shape would predict that they are highly infected by HSV in the cornea. Surprisingly, these cells have the lowest numbers of LAT-positive cells in vivo. This may be because scarification damages these nerves and renders them less viable. Alternatively, these NefH− nerves may not support latent infection due to their fast infection. As discussed in detail below, HSV could arrive at the cell body prior to establishment of the antiviral state, promoting lytic rather than latent infection. The overall finding of high-frequency LAT promoter activity in NefH+ neurons in vivo is consistent with data from our microfluidic chambers. This is an important consistency, since the chambers normalize for anatomic variances and support the notion that it is an intrinsic property of NefH+ neurons that promotes LAT promoter activity and latency rather than the localization of the FNEs.
While LAT promoter activity is favored in neurons expressing NefH, HSV-1 is nonetheless capable of establishing latency in NefH+ and NefH− neurons, as evidenced by the early-phase reactivation assay. More importantly, the data suggest that the LAT promoter is mainly expressed in a specific subpopulation of sensory neurons (NefH+ CGRP+ neurons) during latency. The unique anatomy of these neurons and their slower early infection may give them sufficient time to mount an antiviral response, as timing is a critical determinant of the outcome of infection by neurotropic viruses. The low early-infection efficiency of HSV-1 in NefH+ neurons may allow more time for development of an antiviral response from the infected axon and for signals to be transmitted to its own cell body prior to arrival of the capsid. Similarly, low early-infection efficiency would give the cell bodies of NefH+ neurons time to respond to the beta interferon secreted from surrounding infected neuronal and nonneuronal cells (58, 59). The low expression of HSV genes in these neurons may promote rapid expression of LAT and repression of gene expression through chromatin association, antiviral activity, and miRNAs. In NefH− neurons, HSV-1 infects rapidly from FNEs to the cell body and would promote higher viral protein production. This may favor latency with low LAT promoter activity once HSV gene expression has been repressed, most likely by chromatin control, miRNAs, and intrinsic/innate immunity (58, 60). These models remain to be formally proven but provide a framework for ongoing experiments.
MATERIALS AND METHODS
Viruses and animals.
The wild-type HSV-1 strain KOS and its derivative, KOS/62 (obtained from Todd Margolis, Washington University School of Medicine, St. Louis, MO), were used throughout this study. KOS/62 contains a β-galactosidase gene cassette inserted into both copies of the LAT locus, resulting in a LAT deletion virus with β-galactosidase expression from the LAT promoter (10, 29). All viruses were propagated and plaqued on Vero cells using standard practices, as previously described (61).
C57BL/6J mice were purchased from The Jackson Laboratory and bred in-house. Calca tm1.1 (EGFP/HBEGF) Mjz mice with a transmembrane GFP sequence inserted in the calcitonin locus were kindly provided by Mark Zylka (University of North Carolina, Neuroscience Center). Calca tm1.1 (EGFP/HBEGF) Mjz mice were used as heterozygotes with respect to the GFP-containing locus by crossing them with C57BL/6J mice. These mice, as heterozygotes, show no detectable phenotype (37, 38). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (62). The protocol was approved by the Dartmouth IACUC (permit number leib.da.1#2m13a). No surgery was performed, and all efforts were made to minimize suffering.
Microfluidics culture of adult mouse TG neurons.
TG neurons from adult mice were isolated and cultured largely as previously reported (26). Microfluidic devices (Standard Neuron Device 150 [SND150]; Xona Microfluidics) were attached to 24- by 60-mm coverslips. Devices were incubated with 10% fetal bovine serum-supplemented Dulbecco modified Eagle medium (HyClone) overnight and then were coated with poly-d-lysine (BD Biosciences) at 500 μg/ml in Hanks balanced salt solution lacking calcium and magnesium (HBSS; HyClone) overnight. The devices were then washed three times with HBSS and coated with natural mouse laminin (Invitrogen) at a concentration of 18 μg/ml in HBSS overnight. TG neurons were isolated as described previously, with a few modifications (26). Mice 5 to 10 weeks old were euthanized using approved methods and transcardially perfused with phosphate-buffered saline (PBS; HyClone). TG were harvested and enzymatically digested in a solution consisting of 40 U/ml of papain (Worthington) in HBSS with 2.75 mM l-cysteine (Sigma) and 8% NaHCO3 diluted 1:1,000 (Sigma) for 20 min at 37°C on a rotator. This was followed by a similar incubation in a solution of 5 mg/ml of collagenase type II (Invitrogen) and 5.5 mg/ml of neutral protease (Worthington) dissolved in HBSS. TG were then triturated in Neurobasal-A (NB-A) working medium (Neurobasal-A [Invitrogen], 2% B27 [StemCell], 1% penicillin-streptomycin [Pen-Strep; HyClone]). The resulting homogenate was spun over a two-layer density gradient made with Percoll and NB-A working medium, as described above. Neurons were pelleted by a 10-min centrifugation at 1,300 × g, the upper layers were discarded, and the pellet was washed three times in NB-A working medium. Neurons were resuspended in minimal volumes of NB-A complete medium including the antimitotic 5-fluoro-2-deoxyuridine (FUDR; Sigma) for a minimum of 3 days prior to use. NB-A complete medium consisted of Neurobasal-A, 2% B27, 1% GlutaMAX (Invitrogen), 1% Pen-Strep, 25 ng/ml of brain-derived neurotrophic factor (BDNF; Peprotech), 50 ng/ml of nerve growth factor (NGF; Invitrogen), 50 ng/ml of glial cell-derived neurotrophic factor (GDNF; R&D Systems), and 100 ng/ml of GDNF family receptor α1 Fc (GFRα1 Fc; R&D Systems). Two TG were used per microfluidic device.
HSV-1 infections. (i) In vitro.
Infections in microfluidic devices were performed based on an average of 1,000 neurons growing from the cell body compartment to the distal axon compartment (26). Infection of neurons in culture for 3 days was performed at a multiplicity of infection (MOI) of 10 at the distal axon in a volume of 200 μl with KOS/62. The chambers were incubated for 1 h to allow viral adsorption to axons, and then the medium was replaced at the distal axon compartment. Secondary infection at the cell body compartment was prevented by using human neutralizing antibodies (1:50; catalog number 14-402E; Lonza, BioWhittaker). Synchronized infections at the distal axon compartment were performed by prechilling neurons cultivated in the microfluidic devices on ice for 20 min. Distal axon compartments were then infected for 20 min at 4°C with HSV-1 KOS (MOI = 200) to allow viral adsorption; finally, the distal axon compartment medium was replaced with 37°C medium and infections were continued at 37°C for 20 min or 150 min.
(ii) In vivo.
Mice were anesthetized intraperitoneally with ketamine (90 mg/kg of body weight) and xylazine (10 mg/kg). The corneas were bilaterally scarified with a 25-gauge syringe needle, and virus was inoculated by adding 1 × 106 PFU per eye in a 3-μl volume. To reduce the pain caused at the cornea, the mice were then injected with buprenorphine (0.41 mg/kg).
Early-phase reactivation.
The corneas of adult CGRP GFP+ or CGRP GFP-negative mice were infected, using corneal scarification, with HSV-1 KOS (1 × 106 PFU/eye). After 21 days, latently infected TGs were dissected and dissociated as described above. The resulting adult TG neurons were seeded onto 12-mm coverslips that had been precoated with poly-d-lysine and laminin, in the absence of any trophic support and in the presence of trichostatin A (Sigma-Aldrich) diluted to 600 nM in dimethyl sulfoxide for 24 h.
Immunofluorescence microscopy. (i) Antibodies.
The primary antibodies used were anti-β3-tubulin (1:500; clone TuJ-1 MAB1195; R&D), anti-neurofilament heavy (1:1,000; catalog number 40-1278; Proteus Bioscience), anti-β-galactosidase (1:100; catalog number 100-4136; Rockland), anti-HSV-1 (1:500; Dako), anti-A5 (1:5; catalog number FE-A5; Developmental Study Hybridoma Bank), anti-MAP2 (1:200; catalog number ab32454; Abcam), and anti-VP16 (1:500; clone 1-21; catalog number sc-7545; Santa Cruz). The secondary antibodies, all of which were from Invitrogen, were Alexa Fluor 350-conjugated goat anti-mouse immunoglobulin (catalog number A-11045), Alexa Fluor 350-conjugated goat anti-rabbit immunoglobulin (catalog number A-21068), Alexa Fluor 555-conjugated goat anti-mouse immunoglobulin (catalog number A-32727), Alexa Fluor 555-conjugated goat anti-rabbit immunoglobulin (catalog number A-32732), and Alexa Fluor 647-conjugated goat anti-chicken IgY (catalog number A-21449). For detection of β-Gal in TG sections, an amplification step was performed using goat anti-rabbit biotin conjugate (1:500; catalog number 31820) and Alexa Fluor 555-conjugated streptavidin (catalog number S-32355), both from Thermo Fisher.
(ii) Neuron cultures.
Cultures in microfluidic devices were fixed using 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) for 10 min. Devices remained attached during the entire process of staining and microscope analysis. Chambers were washed three times with 0.1 M PB and incubated with 1% bovine serum albumin (BSA) and 1% Triton X-100 in 0.1 M PB for 1 h and then overnight with primary antibodies diluted in 1% BSA and 1% Triton X-100 in 0.1 M PB. On the next day, the chambers were washed three times with 1% Triton X-100 in 0.1 M PB for 10 min each time, and then the chambers were incubated with secondary antibodies diluted in 1% BSA and 1% Triton X-100 in 0.1 M PB for 2 h. Finally, the chambers were washed with 1% Triton X-100 in 0.1 M PB for 10 min and in 0.1 M PB for an additional 10 min. After the final wash, the chambers were immediately imaged using an automated AxioVision Observer Z1 (Zeiss) microscope. Image tiles comprising the entire central channels of the microfluidic devices were acquired, and images were analyzed using ZEN2012 or NIH Fiji software. Phase I reactivation studies were performed similarly, except that poly-d-lysine- and laminin-precoated 12-mm coverslips were used. After the final washes, the coverslips were mounted in FluorSave reagent (Calbiochem). Random tile images were acquired for analysis. A5/NefH staining was performed similarly, but the A5 antibody was used without Triton X-100 permeabilization, after which the NefH antibody was incubated for an additional 1 h in the presence of Triton X-100.
(iii) Immunohistochemistry.
Mice were euthanized at 21 dpi by approved methods and transcardially perfused with PBS. TG were harvested and fixed ex vivo with 4% formaldehyde (PFA; Fisher Scientific) in 0.1 M PB for 3 h. They were washed 3 times in 0.1 M PB and incubated in 15% sucrose overnight at 4°C and in 30% sucrose overnight at 4°C. Cryoprotected ganglia were embedded in Tissue-Tek OCT compound (Sakura). The tissue was sectioned, using a Leica CM1860 cryostat, into 15-μm transverse plane sections, which were mounted directly onto charged glass slides (Thermo Scientific) and allowed to dry for 2 h.TG sections were carefully rehydrated with 0.1 M PBS (3 times) and incubated with 1% BSA and 1% Triton X-100 in 0.1 M PB for 1 h, and immunostaining was performed as described above. Tissue sections were mounted in FluorSave reagent, a coverslip was applied, and images were acquired and analyzed.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism software. For two-group conditions, data were analyzed using an unpaired t test. For multiple-group conditions, two-way analysis of variance followed by a Bonferroni posttest was performed.
ACKNOWLEDGMENTS
We thank Todd Margolis and Mark Zylka for the generous provision of reagents. We thank Todd Margolis and Gregory Smith for helpful discussions. We are grateful to the Dartmouth College Center for Comparative Medicine and Research for excellent animal husbandry. Brian North and Jesse Mehrbach provided superb technical support.
This work was made possible by funding from the Hitchcock Foundation of the Dartmouth Hitchcock Medical Center (to J.R.C.) and from NEI R01 EY09083 and NIAID P01 AI098681 (to D.A.L.). Support and input from the BioMT COBRE (P20-GM113132) are also acknowledged.
REFERENCES
- 1.Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed). 2013. Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, Nahmias AJ, Berman SM, Markowitz LE. 2006. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296:964–973. doi: 10.1001/jama.296.8.964. [DOI] [PubMed] [Google Scholar]
- 3.Whitley RJ, Roizman B. 2001. Herpes simplex virus infections. Lancet 357:1513–1518. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham AL, Diefenbach RJ, Miranda-Saksena M, Bosnjak L, Kim M, Jones C, Douglas MW. 2006. The cycle of human herpes simplex virus infection: virus transport and immune control. J Infect Dis 194(Suppl):S11–S18. doi: 10.1086/505359. [DOI] [PubMed] [Google Scholar]
- 5.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 6.Nicoll MP, Proença JT, Efstathiou S. 2012. The molecular basis of herpes simplex virus latency. FEMS Microbiol Rev 36:684–705. doi: 10.1111/j.1574-6976.2011.00320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 8.Sawtell NM. 1997. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J Virol 71:5423–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks K, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Margolis TP, Sedarati F, Dobson AT, Feldman LT, Stevens JG. 1992. Pathways of viral gene expression during acute neuronal infection with HSV-1. Virology 189:150–160. doi: 10.1016/0042-6822(92)90690-Q. [DOI] [PubMed] [Google Scholar]
- 11.Mehta A, Maggioncalda J, Bagasra O, Thikkavarapu S, Saikumari P, Valyi-Nagy T, Fraser NW, Block TM. 1995. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal ganglia of latently infected mice. Virology 206:633–640. doi: 10.1016/S0042-6822(95)80080-8. [DOI] [PubMed] [Google Scholar]
- 12.Chen X-P, Mata M, Kelley M, Glorioso JC, Fink DJ. 2002. The relationship of herpes simplex virus latency associated transcript expression to genome copy number: a quantitative study using laser capture microdissection. J Neurovirol 8:204–210. doi: 10.1080/13550280290049642. [DOI] [PubMed] [Google Scholar]
- 13.Purves D, Augustine GJ, Fitzpatrick D, Hall WC, LaMantia A-S, White LE (ed). 2012. Neuroscience, 5th ed Sinauer Associates, Sunderland MA. [Google Scholar]
- 14.Marmigère F, Ernfors P. 2007. Specification and connectivity of neuronal subtypes in the sensory lineage. Nat Rev Neurosci 8:114–127. doi: 10.1038/nrn2057. [DOI] [PubMed] [Google Scholar]
- 15.Zylka MJ, Rice FL, Anderson DJ. 2005. Topographically distinct epidermal nociceptive circuits revealed by axonal tracers targeted to Mrgprd. Neuron 45:17–25. doi: 10.1016/j.neuron.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 16.Belmonte C, Viana F. 2008. Molecular and cellular limits to somatosensory specificity. Mol Pain 4:14. doi: 10.1186/1744-8069-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. 2015. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 18.Margolis TP, Dawson CR, LaVail JH. 1992. Herpes simplex viral infection of the mouse trigeminal ganglion. Immunohistochemical analysis of cell populations. Invest Ophthalmol Vis Sci 33:259–267. [PubMed] [Google Scholar]
- 19.Yang L, Voytek CC, Margolis TP. 2000. Immunohistochemical analysis of primary sensory neurons latently infected with herpes simplex virus type 1. J Virol 74:209–217. doi: 10.1128/JVI.74.1.209-217.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J Virol 81:1872–1878. doi: 10.1128/JVI.02110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imai Y, Apakupakul K, Krause PR, Halford WP, Margolis TP. 2009. Investigation of the mechanism by which herpes simplex virus type 1 LAT sequences modulate preferential establishment of latent infection in mouse trigeminal ganglia. J Virol 83:7873–7882. doi: 10.1128/JVI.00043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertke AS, Swanson SM, Chen J, Kinchington PR, Margolis TP, Imai Y. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuan A, Rao MV, Veeranna Nixon RA. 2012. Neurofilaments at a glance. J Cell Sci 125:3257–3263. doi: 10.1242/jcs.104729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perrot R, Eyer J. 2013. Neurofilaments: properties, functions, and regulation, p 171–236. In Dermietzel R. (ed), The cytoskeleton Neuromethods, vol 79 Humana Press, Totowa, NJ. doi: 10.1007/978-1-62703-266-7_9. [DOI] [Google Scholar]
- 25.Jessen KR, Mirsky R, Lloyd AC. 2015. Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol 7:a020487. doi: 10.1101/cshperspect.a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katzenell S, Cabrera JR, North BJ, Leib DA. 2017. Isolation, purification, and culture of primary sensory neurons. In Mossman K. (ed), Innate antiviral immunity Methods Mol Biol, vol 1656 Humana Press, New York, NY. doi: 10.1007/978-1-4939-7237-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. doi: 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flowerdew SE, Wick D, Himmelein S, Horn AKE, Sinicina I, Strupp M, Brandt T, Theil D, Hüfner K. 2013. Characterization of neuronal populations in the human trigeminal ganglion and their association with latent herpes simplex virus-1 infection. PLoS One 8:e83603. doi: 10.1371/journal.pone.0083603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J Virol 66:2157–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bearer EL, Breakefield XO, Schuback D, Reese TS, LaVail JH. 2000. Retrograde axonal transport of herpes simplex virus: evidence for a single mechanism and a role for tegument. Proc Natl Acad Sci U S A 97:8146–8150. doi: 10.1073/pnas.97.14.8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith G, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J Virol 84:1504–1512. doi: 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abraira VE, Ginty DD. 2013. The sensory neurons of touch. Neuron 79:618–639. doi: 10.1016/j.neuron.2013.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Julius D. 2001. Molecular mechanisms of nociception. Nature 413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 36.Basbaum AI, Bautista DM, Scherrer G, Julius D. 2009. Cellular and molecular mechanisms of pain. Cell 139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCoy ES, Taylor-Blake B, Zylka MJ. 2012. CGRPα-expressing sensory neurons respond to stimuli that evoke sensations of pain and itch. PLoS One 7:e36355. doi: 10.1371/journal.pone.0036355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ. 2013. Peptidergic CGRPα primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belmonte C, Acosta MC, Gallar J. 2004. Neural basis of sensation in intact and injured corneas. Exp Eye Res 78:513–525. doi: 10.1016/j.exer.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 40.De Felipe C, Gonzalez GG, Gallar J, Belmonte C. 1999. Quantification and immunocytochemical characteristics of trigeminal ganglion neurons projecting to the cornea: effect of corneal wounding. Eur J Pain 3:31–39. doi: 10.1016/S1090-3801(99)90186-6. [DOI] [PubMed] [Google Scholar]
- 41.Alamri A, Bron R, Brock JA, Ivanusic JJ. 2015. Transient receptor potential cation channel subfamily V member 1 expressing corneal sensory neurons can be subdivided into at least three subpopulations. Front Neuroanat 9:71. doi: 10.3389/fnana.2015.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Summers BC, Margolis TP, Leib DA. 2001. Herpes simplex virus type 1 corneal infection results in periocular disease by zosteriform spread. J Virol 75:5069–5075. doi: 10.1128/JVI.75.11.5069-5075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8:e1002540. doi: 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith RL, Geller AI, Escudero KW, Wilcox CL. 1995. Long-term expression in sensory neurons in tissue culture from herpes simplex virus type 1 (HSV-1) promoters in an HSV-1-derived vector. J Virol 69:4593–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas DL, Lock M, Zabolotny JM, Mohan BR, Fraser NW. 2002. The 2-kilobase intron of the herpes simplex virus type 1 latency-associated transcript has a half-life of approximately 24 hours in SY5Y and COS-1 cells. J Virol 76:532–540. doi: 10.1128/JVI.76.2.532-540.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brinkman KK, Mishra P, Fraser NW. 2013. The half-life of the HSV-1 1.5-kb LAT intron is similar to the half-life of the 2.0-kb LAT intron. J Neurovirol 19:102–108. doi: 10.1007/s13365-012-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kevenaar JT, Hoogenraad CC. 2015. The axonal cytoskeleton: from organization to function. Front Mol Neurosci 8:44. doi: 10.3389/fnmol.2015.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown A. 2010. Slow axonal transport, p 1–9. In Encyclopedia of neuroscience. Elsevier, New York, NY. doi: 10.1016/B978-008045046-9.02000-3. [DOI] [Google Scholar]
- 49.Brown A, Jung P. 2013. A critical reevaluation of the stationary axonal cytoskeleton hypothesis. Cytoskeleton 70:1–11. doi: 10.1002/cm.21083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collard JF, Côté F, Julien JP. 1995. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 51.Marszalek JR, Williamson TL, Lee MK, Xu Z, Hoffman PN, Becher MW, Crawford TO, Cleveland DW. 1996. Neurofilament subunit NF-H modulates axonal diameter by selectively slowing neurofilament transport. J Cell Biol 135:711–724. doi: 10.1083/jcb.135.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huff JC, Krueger GG, Overall JC, Copeland J, Spruance SL. 1981. The histopathologic evolution of recurrent herpes simplex labialis. J Am Acad Dermatol 5:550–557. doi: 10.1016/S0190-9622(81)70115-4. [DOI] [PubMed] [Google Scholar]
- 53.Cunningham AL, Turner RR, Miller AC, Para MF, Merigan TC. 1985. Evolution of recurrent herpes simplex lesions. An immunohistologic study. J Clin Invest 75:226–233. doi: 10.1172/JCI111678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu J, Koelle DM, Cao J, Vazquez J, Huang ML, Hladik F, Wald A, Corey L. 2007. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J Exp Med 204:595–603. doi: 10.1084/jem.20061792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pasieka TJ, Menachery VD, Rosato PC, Leib DA. 2012. Corneal replication is an interferon response-independent bottleneck for virulence of herpes simplex virus 1 in the absence of virion host shutoff. J Virol 86:7692–7695. doi: 10.1128/JVI.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.BenMohamed L, Osorio N, Khan AA, Srivastava R, Huang L, Krochmal JJ, Garcia JM, Simpson JL, Wechsler SL. 2016. Prior corneal scarification and injection of immune serum are not required before ocular HSV-1 infection for UV-B-induced virus reactivation and recurrent herpetic corneal disease in latently infected mice. Curr Eye Res 41:747–756. doi: 10.3109/02713683.2015.1061024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cabrera JR, Viejo-Borbolla A, Martinez-Martín N, Blanco S, Wandosell F, Alcamí A. 2015. Secreted herpes simplex virus-2 glycoprotein G modifies NGF-TrkA signaling to attract free nerve endings to the site of infection. PLoS Pathog 11:e1004571. doi: 10.1371/journal.ppat.1004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosato PC, Leib DA. 2015. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog 11:e1005028. doi: 10.1371/journal.ppat.1005028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW. 2016. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. mBio 7:e02145-15. doi: 10.1128/mBio.02145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A. 2012. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12:334–345. doi: 10.1016/j.chom.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rader KA, Ackland-Berglund CE, Miller JK, Pepose JS, Leib DA. 1993. In vivo characterization of site-directed mutations in the promoter of the herpes simplex virus type 1 latency-associated transcripts. J Gen Virol 74:1859–1869. doi: 10.1099/0022-1317-74-9-1859. [DOI] [PubMed] [Google Scholar]
- 62.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]