

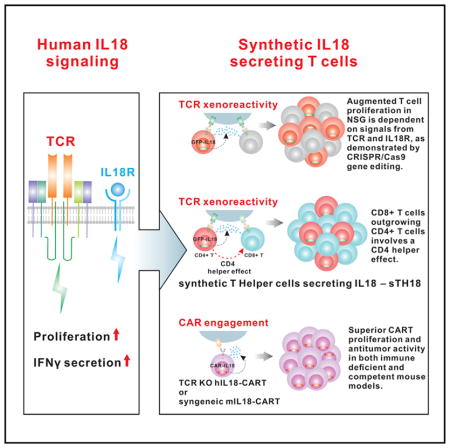
SUMMARY
The effects of transgenically encoded human and mouse IL-18 on T cell proliferation and its application in boosting chimeric antigen receptor (CAR) T cells are presented. Robust enhancement of proliferation of IL-18-secreting human T cells occurred in a xenograft model, and this was dependent on TCR and IL-18R signaling. IL-18 augmented IFN-γ secretion and proliferation of T cells activated by the endogenous TCR. TCR-deficient, human IL-18-expressing CD19 CAR T cells exhibited enhanced proliferation and antitumor activity in the xenograft model. Antigen-propelled activation of cytokine helper ensemble (APACHE) CAR T cells displayed inducible expression of IL-18 and enhanced antitumor immunity. In an intact mouse tumor model, CD19-IL-18 CAR T cells induced deeper B cell aplasia, significantly enhanced CAR T cell proliferation, and effectively augmented antitumor effects in mice with B16F10 melanoma. These findings point to a strategy to develop universal CAR T cells for patients with solid tumors.
In Brief
Hu et al. create IL-18-secreting chimeric antigen receptor T (IL-18-CAR T) cells to significantly boost CAR T cell proliferation and antitumor activity.
INTRODUCTION
Following the initial reports of clinical success using chimeric antigen receptor (CAR) T cell therapy to treat B cell leukemia and lymphoma (Kochenderfer et al., 2010; Porter et al., 2011), there have been intense efforts to improve the design and clinical translation of CAR T cells. In addition to T cell receptor (TCR) engagement and costimulatory signaling, cytokines also play a fundamental role in modulating T cell function. Therefore, an appealing strategy is to engineer cytokine production to promote CAR functions, such as interkeulin-12 (IL-12) (Chmielewski et al., 2011; Pegram et al., 2012; Zhang et al., 2015), IL-15 (Hurton et al., 2016), and interferon-β (IFN-β) (Zhao et al., 2015).
We hypothesized that synthetic IL-18 expression might promote CAR T cell functions. IL-18 was initially characterized as an inducer of interferon-γ (IFN-γ) expression in T cells (Nakamura et al., 1989, 1993) and has been shown to activate lymphocytes and monocytes without eliciting severe dose-limiting toxicity in clinical trials (Robertson et al., 2006). We previously reported that human recombinant (r)IL-18 could significantly enhance engraftment of human CD8+ T cells in a xenograft model (Carroll et al., 2008). In the current study, we discovered potent IL-18-mediated effects on human and mouse T cell proliferation and that synthetic IL-18 secretion by CAR T cells significantly enhanced CAR T cell expansion and antitumor activity.
RESULTS
IL-18 Enhances CAR T Cell Proliferation In Vitro and In Vivo
To engineer IL-18-expressing CAR T cells, we introduced a human IL-18 transgene into anti-mesothelin (SS1) CAR (SS1-IL-18) and anti-CD19 CAR constructs (CD19-IL-18) (Figure 1A). The resulting SS1-IL-18 construct led to efficient SS1 CAR expression (Figure S1A) and secretion of bioactive human IL-18 (Figures S1B and S1C) and modestly enhanced the lytic activity of CAR T cells (Figure S1D). To evaluate IL-18-CAR T proliferation in vitro, we restimulated CAR T cells with artificial antigen-presenting cells (aAPCs)—K562-expressing mesothelin (K562-Meso) or CD19 (K562-CD19). Robust IL-18 secretion was observed (Figure S1E), and IL-18-CAR T cells displayed enhanced secretion of IFN-γ and several other cytokines (Figure S1F). Interestingly, SS1-IL-18 CAR T cells displayed significantly increased proliferation relative to SS1 CAR T cells (Figure 1B). However, CD19-IL-18 CAR T cells expanded similarly as the CD19 CAR T cell control (Figure 1B), which is perhaps related to our previous finding that the SS1 CAR has constitutive ligand-independent signaling, whereas the CD19 CAR does not have constitutive signaling (Frigault et al., 2015).
Figure 1. IL18-CAR T Cells Have Enhanced Proliferation In Vitro and In Vivo.

(A) The construct designs.
(B) Population doubling and cell volume of SS1 anti-mesothelin or anti-CD19 CAR T cells following the first restimulation with irradiated K562-Meso or K562-CD19 with exogenous IL-2.
(C) NSG mice (n = 5) bearing an AsPC1 pancreatic flank tumor received CD19 or SS1 CAR T cells (2e6), and 3 weeks later, peripheral blood was analyzed by Trucount.
(D) NSG mice (n = 5) bearing a systemic Nalm6 acute lymphoblastic leukemia (ALL) tumor received 1e6 CD19 or CD19-IL-18 CAR T cells. After 18 days, circulating T cells were assessed.
(E) Tumor-free NSG mice (n = 5) were inoculated with 5e6 SS1 or SS1-IL-18 CAR T cells and, 3 weeks later, analyzed for circulating T cells.
(F and G) CAR T cells were analyzed on day 9 of ex vivo expansion. In vivo expansion of SS1-IL-18 and CD19-IL-18 CAR T cells was determined by harvesting spleens from the mice described in (C) and (D), respectively.
(F) Representative fluorescence-activated cell sorting (FACS) plots of CD8+CAR+ cells.
(G) The percentage of CD4+CAR+ or CD8+CAR+ T cells in spleens from (F).
All data with error bars are presented as mean ± SEM. Student’s t test: **p < 0.01, ***p < 0.001.
Encouraged by enhanced functions of IL-18-CAR T cells in vitro, we moved to investigate SS1-IL-18 CAR T cells in a xenograft model of mesothelin-expressing pancreatic tumor AsPC1 (labeled with click beetle green [CBG] and GFP reporter proteins) in NOD scid gamma (NSG) mice. Given that AsPC1 cells do not express CD19, we were surprised, 3 weeks after T cell injection, to find that both CD19-IL-18 and SS1-IL-18 T cells were substantially elevated in peripheral blood 62-fold to 13,990 cells/μL or 74-fold to 38,210 cells/μL relative to CD19 or SS1 CAR T alone (p < 0.01; Figure 1C). We further confirmed the robust T cell expansion when injecting CD19-IL-18 CAR T cells into NSG mice bearing Nalm6 cells, a CD19+ leukemia cell line (Figure 1D), and SS1-IL-18 CAR T cells into tumor-free NSG mice (Figure 1E). Further analysis indicated that expanded CD4+ and CD8+ T cells include both CAR+ and CAR− populations in the spleen (Figures 1F and 1G), which were polyclonal (Figure S1G). Collectively, these results in the xenograft model suggested that IL-18 promotes target antigen-independent CAR T cell proliferation in vivo in tumor-free and tumor-bearing NSG mice.
Effects of Human IL-18 on T Cell Proliferation and Cytokine Secretion
The observations of CD19-IL-18 CAR T cells proliferating in mice bearing CD19− tumors (Figure 1C) and SS1-IL-18 CAR T cells proliferating in tumor-free mice (Figure 1E) suggest that CAR signaling might be dispensable for IL-18-driven expansion. Therefore, we hypothesized that a functional synergy exists between IL-18 signaling and TCR activation driven by xenogeneic effects. To test this hypothesis in vitro, primary human T cells were stimulated using anti-CD3 only or with rIL-18. Exogenous rIL-18 enhanced increases in T cell volume compared with cells stimulated with anti-CD3 only, and the enhancement in blast transformation after addition of IL-18 was similar to that after addition of anti-CD3/28 beads (Figure 2A). Purified T cells stimulated with anti-CD3 only did not proliferate (Figure 2A); however, anti-CD3 in combination with IL-18 efficiently stimulated proliferation. The costimulatory effect of IL-18 on T cell proliferation was confirmed in T cells isolated from two additional normal donors (Figure 2B).
Figure 2. Combined TCR and IL-18 Signaling Promotes CD4+ T Cell Expansion in the Absence of CAR Signaling.

(A–C) Primary T cells from a healthy donor were activated with anti-CD3 beads and varying concentrations (1.56 doubled up to 200 ng/mL) of rIL-18 added on days 0 and 3.
(A) Changes in T cell volume and cell numbers.
(B) IL-18-dependent expansion of T cells from two additional donors stimulated with rIL-18, anti-CD3 beads, and anti-CD3 beads plus rIL-18.
(C) Luminex analysis of secreted IFN-γ and IL-2 with 12 culture conditions indicated. Additional cytokine data are shown in Figure S2A.
(D) Normal donor T cells expanded with anti-CD3/CD28 beads for 10 days were selected by MACS columns to purify CD8+ and CD4+ T cells. Purified CD4+ T cells, CD8+T cells, or bulk T cells were restimulated with anti-CD3 beads and 100 ng/mL rIL-18, added on days 0 and 3, as indicated in the legend.
(E–H) T cells transduced with lentivectors encoding GFP orGFP-IL-18 were selected using MACS columns to purify CD8+ and CD4+ T cells. Tumor-free NSG mice (n = 5) were injected with 2e6 T cells as indicated. The data shown are representative of two independent experiments.
(E) Circulating CD45+ T cells and pictures of spleens from various groups.
(F) Circulating CD45+CD4+ T cells on day 23 following T cell injection and the ratio of CD8+ to CD4+ T cells.
(G) Serum levels of IL-18 and IFN-γ.
(H) Serum from mice on day 23 was analyzed by Luminex to assess the cytokine profile.
All data with error bars are presented as mean ± SEM. Student’s t test: **p < 0.01, ***p < 0.001.
Further characterization of the cytokine profile in IL-18-activated T cells indicated that low concentrations of IL-18 significantly augmented IFN-γ production, whereas IL-2 levels were at least 20-fold lower than that induced by optimal stimulation by anti-CD3/28 beads (Figure 2C). The production of other cytokines was generally comparable between anti-CD3 plus IL-18 and anti-CD3/CD28 bead activation (Figure S2A). Multicolor flow phenotype revealed that IL-18-expanded T cells had a primarily central memory (TCM) phenotype (CCR7+CD45RO+) on day 8 after activation, similar to that obtained with anti-CD3/28 beads (Figure S2B).
Combined TCR and IL-18 Signaling Promotes CD4 T Cell Expansion
To address the specific roles of T cell subsets in these responses, T cells expanded by standard anti-CD3/CD28 beads were sorted by magnetic-activated cell sorting (MACS) to generate CD4+ and CD8+ subpopulations. CD8+ T cells failed to proliferate after restimulation with rIL-18 and anti-CD3 beads, despite a significant increase in size (Figure 2D), and addition of IL-2 (20 U/mL) had no additional effect. In sharp contrast, CD4+ T cells or bulk T cells restimulated with rIL-18 and anti-CD3 beads proliferated efficiently in the absence of CD28 stimulation, at a level comparable with that of combined CD4+ and CD8+ T cells following anti-CD3/CD28 bead restimulation. rIL-18-re-stimulated CD4+ and CD8+ T cells displayed transiently elevated TCM markers but ultimately expressed a primarily effector memory (TEM) phenotype (Figure S2C).
Next, we hypothesized that GFP-IL-18-expressing human T cells would serve as a desirable platform to test the combinatorial effects of IL-18 signaling and TCR xenoreactivity in NSG mice (Figure 2E). GFP-IL-18 CD8+ T cells failed to expand in vivo, whereas GFP-IL-18-expressing CD4+ or bulk T cells exhibited greater than 400-fold expansion between days 15 and 23 after T cell injection (Figure 2E), which was also associated with massively enlarged spleens. The number of expanded CD4+ T cells was comparable whether pure CD4+ T cells or total T cells were injected, suggesting that CD8+ T cells do not have a significant effect on CD4+ T cell expansion (Figure 2F). Interestingly, CD8+ T cells proliferated more than CD4+ T cells in magnitude within bulk T cells (Figure 2F), which is consistent with our previous finding that rIL-18 only enhanced CD8+ T cell engraftment (Carroll et al., 2008).
Consistent with the observed T cell proliferation, serum human (h)IL-18 and IFN-γ levels increased in mice engrafted with GFP-IL-18 bulk T cells and GFP-IL-18 CD4+ T cells but not in mice engrafted with GFP-IL-18 CD8+ T cells (Figure 2G). Luminex analysis revealed very high levels of IFN-γ and granulocyte macrophage colony-stimulating factor (GM-CSF) in mice engrafted with IL-18-secreting bulk T cells and IL-18 CD4+T cells (Figure 2H). Levels of IP10, MIP-1A, MIP-1B, and tumor necrosis factor alpha (TNF-α) were also selectively elevated in mice engrafted with IL-18 CD4+ and IL-18 bulk T cells. IL-17 levels were also selectively elevated in mice engrafted with IL-18 CD4+ T cells. In summary, these results suggest that genetically encoded IL-18 acts as a signaling cytokine that interacts synergistically with TCR activation to drive T cell proliferation and Th1-like cytokine secretion, largely dependent on effects through CD4+ helper T cells.
TCR and IL-18R Are Required for IL-18-Mediated T Cell Proliferation In Vivo
Based on the effects of IL-18 in vitro and the dramatic proliferation of GFP-IL-18-expressing T cells in vivo, we hypothesized that both the TCR and IL-18 receptor (IL-18R) are required for IL-18-driven proliferation in vivo. To test this, we engineered TCR-deficient (TCR knockout [KO]) T cells expressing GFP or GFP-IL-18 using CRISPR/Cas9 gene editing. TCR+ GFP-IL-18 T cells expanded dramatically in the blood and spleen compared with TCR+ GFP control T cells, whereas TCR KO GFP-IL-18 T cells did not expand (Figure 3A).
Figure 3. IL-18-Enhanced T Cell Proliferation Requires TCR and IL-18R Signaling.

(A) Tumor-free NSG mice (n = 5) were injected with 2e6 TCR KO or TCR WT GFP-IL-18 T cells or the indicated controls. After 18 days, T cells in the blood (top) or spleens (bottom) were assessed.
(B) Cell-competitive repopulation design. CD19 CAR-IL-18 WT, GFP WT, AmCyan IL-18R KO and DsRed TCR KO T cells were mixed together and injected into tumor-free NSG mice (n = 5).
(C) Purity of TCR KO and IL-18R KO cells after CRISPR/Cas9 gene editing and MACS sorting.
(D) Initial frequency of each cell population shown in (B).
(E–H) The mice were analyzed 3 weeks after T cell injection.
(E) Left: total circulating CD45+ cells, indicating robust expansion. Right: individual populations of T cells.
(F) Expression levels of IL-18Rα on T cells in the blood (left) and spleen (right).
(G) Representative FACS plot of each cell population in the blood.
(H) The ratios of CD19 CAR-IL-18 WT, IL-18R KO AmCyan, and TCR KO DsRed to GFP WT cells in the blood (left) and spleen (right).
All data with error bars are presented as mean ± SEM. Student’s t test: **p < 0.01, ***p < 0.001.
To further investigate the requirement of the IL-18R, we generated IL-18Rα-deficient (IL-18Rα–) T cells using CRISPR/Cas9 gene editing and employed a cell competition assay (Figure 3B) involving CD19 CAR-IL-18 wild-type (WT) cells, GFP WT cells, DsRed TCR KO cells, and AmCyan IL-18R KO T cells. Successful deletion of the TCR and IL-18R was confirmed by flow cytometry, and MACS depletion was performed to remove residual T cells expressing either TCR or IL-18Rα proteins (Figure 3C). Following ex vivo expansion, all four T cell populations were mixed, and flow cytometry analysis confirmed the baseline frequencies of (WT CD19 CAR-IL18):(WT GFP):(TCR KO DsRed):(IL-18R KO AmCyan) cells to be 8.59%:7.45%:6.71%:7.27% within total T cells (Figure 3D). Each NSG mouse received the mixed cell populations consisting of approximately 1e6 cells of each type intravenously. Three weeks later, the number of CD45+ human T cells ranged from 32,800 to 79,600 cells/μL blood (Figure 3E), confirming robust T cell proliferation. Staining for IL-18Rα indicated that some AmCyan-labeled T cells had low levels of residual IL-18R expression (Figure 3F), which likely represents contaminating T cells that did not undergo successful gene editing and were not completely removed by MACS depletion.
A representative FACS plot revealed that the final frequencies of (WT CD19 CAR-IL18):(WTGFP):(TCR KO DsRed):(IL-18R KO AmCyan) were 22.0%:18.0%:5.82%:0.43% (Figure 3G). Interestingly, the ratio of WT CD19 CAR-IL-18 to WT GFP T cells following in vivo expansion was essentially unchanged (Figure 3H). In contrast, the proportion of IL-18R KO AmCyan T cells decreased significantly in the blood and spleen, and it was even more dramatic for TCR KO DsRed T cells (Figure 3H). These data indicate that both the TCR and IL-18R are necessary for CAR-IL-18-mediated in vivo proliferation of human T cells and that neither alone is sufficient, as tested in the NSG model.
IL-18 Signaling Significantly Enhances CAR T Cell Proliferation and Antitumor Activity
Given that IL-18 signaling augments TCR signaling, we hypothesized that IL-18 would also augment CAR T cell proliferation because the CAR contains the basic TCR signaling domains. To avoid potential antigen non-specific T cell activation and graft versus host disease (GVHD) associated with endogenous TCR reactivity (Labrecque et al., 2001), only TCR-deficient (TCR KO) human T cells (Ren et al., 2017) were used in xenograft experiments. In vitro experiments indicated that TCR KO CD19 CAR and TCR KO SS1 CAR T cells had equivalent proliferation, with or without the IL-18 transgene, after initial activation by anti-CD3/CD28 bead activation and IL-2 (Figure S3A). In these experiments, the TCR was disrupted on days 3–4 of culture so that the endogenous TCR was present during the baseline stimulation by anti-CD3/28 beads, and later in culture, the T cells became CD3-deficient (data not shown). However, after aAPC restimulation with the surrogate antigens CD19 or mesothelin, IL-18 expression promoted a modest increase in proliferation in TCR KO CD19-IL-18 CAR T cells (Figure 4A) and SS1-IL-18 CAR T cells (Figure S3B). Robust IL-18 secretion was observed during initial ex vivo expansion and after aAPC restimulation (Figures S3C and S3D). In addition, TCR KO-IL-18-CAR T cells displayed enhanced secretion of IFN-γ and several other cytokines (Figure S3E). Restimulated TCR KO IL-18-CAR T cells displayed transiently elevated TCM markers but ultimately expressed a primarily TEM phenotype (Figure S3F).
Figure 4. IL-18 Costimulation Significantly Enhances Proliferation and Antitumor Activity of Human TCR-Deficient and Syngeneic Murine CD19 CAR T Cells.

(A) Expansion kinetics and volume of TCR KO CAR T cells following the first (with IL-2) and second (without IL-2) restimulation with irradiated K562-CD19cells.
(B and C) NSG mice were injected intravenously with 1e6 Nalm6 ALL cells expressing CBG-GFP. On day 7 after tumor injection, mice (n = 5) received the indicated TCR KO CAR T cells (1e6). The data shown are representative of two independent experiments.
(B) Live animal imaging of tumor growth.
(C) Tumor growth curves of four groups (left) and a magnified plot of TCR KO CD19-GFP and TCR KOCD19-IL-18 CAR T cohorts (right). Additional data include body weight, survival, number of CD45+CD3 KO T cells, IL-18 plasma concentration, percentage of CD3 KO cells among total CD45+ cells, distribution of CD4+ and CD8+ T cells within CD19 CAR T cells, and memory phenotype in CD4+ and CD8+ CAR T cells.
(D) Study of murine CD19-IL-18 CAR T cells in the B16F10 melanoma tumor model that was derived from the B6 mouse in 1954. Lymphocyte-replete C57BL/6 (CD45.2 strain) mice were inoculated subcutaneously (s.c.) with 2e6 B16F10 tumor cells expressing murine CD19 (B16-mCD19). One week later, mice were randomized and treated with 5e6 murine CD19-GFP (n = 10) or CD19-IL18 (n = 10) CAR T cells (CD45.1 origin) or left untreated (n = 7). The mice were monitored for tumor growth, B cell frequency, survival, body weight, CD45.1 T cells, and murine IL-18 plasma concentration.
All data with error bars are presented as mean ± SEM. In some cases, the magnitude of the SEM is less that the size of the symbol. Student’s t test: *p < 0.05, **p < 0.01, ***p < 0.001.
After confirming enhanced proliferation of TCR KO IL-18-CAR T cells in vitro, we next tested their antitumor activity in NSG mice bearing Nalm6 leukemia cells expressing CBG-GFP. TCR KO CD19-IL-18 CAR T cells effectively cleared detectable tumor cells by day 10, in contrast to TCR KO CD19-GFP CAR T cells (Figure 4B). Notably, control TCR KO SS1-IL-18 CAR T cells had no effect on tumor progression, indicating that IL-18 was not sufficient for the antitumor effect (Figure 4C). NSG mice treated with TCR KO CD19-IL-18 CAR T cells had a transient decrease in body weight on day 8 that returned to baseline levels and remained stable thereafter (Figure 4C), consistent with a transient cytokine release syndrome. Remarkably, mice injected with TCR KO CD19-IL-18 CAR T cells remained viable for over 185 days after T cell injection, except for one mouse that exhibited tumor relapse on day 101 (Figures 4B and 4C) because of downregulation of CD19 expression (data not shown). Additional analysis revealed no emergence of contaminating TCR+ CD19-IL-18 CAR T cells during treatment, significant proliferation of TCR KO CD19-IL-18 compared with CD19-GFP CAR T cells (350 versus 10.5 cells/μL blood), a high level of IL-18 in the plasma (77,500 pg/mL on day 10), persistence of CD4+ CD19− IL-18 CAR T cells, and a primarily TEM phenotype (Figure 4C). Subsequently, the number of T cells and IL-18 concentrations gradually decreased to below 1 CD45+CD3− T cell/μL and approximately 100 pg/mL in tumor-free surviving mice. These results collectively demonstrate that IL-18 can significantly enhance TCR KO CD19 CAR T cell proliferation and induces superior antitumor activity without TCR-mediated GVHD during therapy and that systemic IL-18 levels wane following elimination of the tumor burden.
Although the NSG mice have been the most widely used pre-clinical model of CAR T cell therapy in the study of human T cell adoptive transfer, it would be meaningful to extend the investigation of IL-18-CAR T cells to an immune-competent model. Therefore, we engineered murine IL-18-expressing anti-mouse CD19 (murine CD19-IL-18) CAR T cells for adoptive transfer to C57BL/6 mice bearing B16F10 melanoma tumors stably expressing mouse CD19 (Figure 4D). Murine CD19-IL-18 CAR T cells have superior engraftment in lymphoreplete mice compared with CD19 CAR T cells. In addition, murine CD19-IL-18 CAR T cells had augmented antitumor efficacy, as measured by tumor volume, and significantly enhanced survival. Furthermore, profound B cell aplasia on day 7 after T cell infusion was observed with CD19-IL-18 CAR T cells. Consistent with this, murine CD19-IL-18 CAR T cells resulted in high levels of IL-18 in the plasma on day 7, which decreased rapidly afterward. Consistent with systemic IL-18 levels, mouse body weight decreased slightly on day 7 and then returned to baseline levels. Except for the death of one mouse of 10 mice on day 9, potentially because of systemic IL-18 toxicity, there were no other clinical symptoms observed during the study.
These results indicate that murine CD19-IL-18 CAR T cells have superior on-target effects, as judged by tumor elimination and B cell aplasia, with acceptable toxicity in an immune-competent B16F10 tumor model. Nevertheless, we believe that a safer approach may be to minimize the potential toxicity associated with systemic IL-18 exposure observed in both xenograft and syngeneic models by designing an inducible system for IL-18 CAR T cells. Therefore, we created a nuclear factor of activated T-cells (NFAT)-based inducible system for coupling IL-18 production with CAR signaling along with simultaneous disruption of the endogenous TCR. We designated this approach antigen-propelled activation of cytokine helper ensemble (APACHE) CAR T cells. APACHE CAR T cells have inducible IL-18 secretion upon an encounter with surrogate antigen and enhanced anti-tumor activity in the Nalm6 tumor model (Figure S4).
DISCUSSION
Except for B cell tumors treated with CD19 CAR T cells, a major limitation of current CAR T cell approaches has been insufficient expansion and persistence of the infused T cells, as exemplified by several trials (Brown et al., 2016; Hege et al., 2017; Kershaw et al., 2006; Lamers et al., 2006; Park et al., 2007; Pule et al., 2008; Tanyi et al., 2017). IL-18 has been shown to regulate both innate and adaptive immune responses through its effects on natural killer cells, monocytes, dendritic cells, T cells, and B cells (Dinarello et al., 2013). The ultimate rationale for our studies is to enhance CAR T cell proliferation and to utilize IL-18 as a pro-inflammatory cytokine to recruit a second wave of immune cells to the tumor microenvironment to mediate antitumor effects toward cancer cells that are resistant to the direct effects of CAR T cells.
A strength of the NSG xenograft model is that it enables investigation of human IL-18-mediated augmentation of CAR T cell proliferation. There are differences between mouse and human immunology; human T cells are more responsive to IL-18 than mouse T cells. This might help explain why the in vitro effects of human IL-18 on T cells are more potent than those of mouse IL-18 (Carroll et al., 2008; Montufar-Solis et al., 2007). Because of the immune deficiency of the NSG model and lack of a representative tumor microenvironment, further investigation is underway to comprehensively evaluate the safety of IL-18-CAR T cells, interaction with tumor infiltrating T and natural killer (NK) cells, and modulation of the tumor microenvironment in mice with intact host immune systems.
IL12, a cytokine related to IL-18, has also been shown to augment the functional effects of CAR T cells (Chmielewski et al., 2014; Pegram et al., 2012). Further studies will be required to determine the relative merits and risks of these two approaches. An inducible form of IL-12 was tested in tumor infiltrating lymphocytes (TIL) cells given to melanoma patients and was found to be too toxic (Zhang et al., 2012). One potential advantage of IL-18 is that it is monomeric, whereas IL-12 is heterodimeric and more susceptible to changes in potency, resulting in lethal toxicity (Cohen, 1995). Many biologic effects of IL-12 and IL-18 are overlapping, so it is unlikely that combining these approaches would be synergistic.
In summary, we demonstrate that CAR T cells can be engineered to secrete IL-18 that supports in vivo engraftment and persistence of CAR T cells. Our observations demonstrate that IL-18 has a previously unappreciated form of signaling that is primarily selective to human CD4+ T cells, and, therefore, these IL-18-expressing CD4 T cells can be categorized as synthetic T helper cells producing IL-18 (sTh18). These findings may have therapeutic implications because the IL-18 CAR T cells can support the proliferation of CD8+ T cells and because increasing evidence points to the importance of adoptively transferred CD4+ T cells in antitumor effects (Hunder et al., 2008; Hung et al., 1998). Finally, our results point to a new approach to develop universal CAR T cells derived from allogeneic T cells that are devoid of the endogenous TCR.
EXPERIMENTAL PROCEDURES
Generation of IL-18-CAR T Cells
All CAR designs are 4-1BB-based second generation. The sequence of human IL-18 (Uniprot: Q14116) and murine IL-18 following a T2A linker were synthesized from Integrated DNA Technologies and Invitrogen. The parental vector pTRPE or MSGV was used to generate the constructs CAR-GFP, CAR-IL-18, GFP, and GFP-IL-18. Human normal donor or murine CD45.1 T cells were ex vivo-expanded by anti-CD3/CD28 beads with exogenous IL-2 and transduced with a lentivector or retroviral vector as reported previously (Milone et al., 2009). The generation of an anti-mouse CAR targeting CD19 using the 1D3 rat anti-mouse hybridoma (Kochenderfer et al., 2009) is described in the Supplemental Experimental Procedures. Expression of the anti-mouse CD19 CAR was similar in mouse T cells expressing the CD19 CAR or the CD19-mIL-18 CAR constructs. B16F10 tumor cells expressing mouse CD19 were generated by retroviral transduction and enriched to purity by cell sorting three times for mouse CD19.
Mouse Experiments
The experiments were conducted under Institutional Animal Care and Use Committee (IACUC)-approved protocol 804226. For syngeneic experiments, the mice were all female, and for experiments with NSG mice, there was a mix of male and female mice. The mice were approximately 8 weeks old at the beginning of each study.
CRISPR/Cas9 Gene Disruption
Primary donor T cells were engineered with CRISPR/Cas9 as reported previously (Ren et al., 2017). TCR-deficient T cells were achieved with single guide RNA (sgRNA) targeting GGAGAATGACGAGTGGACCC within the TCRB region. For the IL-18R knockout, we identified an sgRNA targeting IL-18Rα (GTACAAAAGCAGTGGATCAC) with high efficiency.
More details are available in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Augmented proliferation of synthetic IL-18-expressing human T cells
rIL-18 augments IFN-γ secretion and proliferation of anti-CD3 activated T cells
IL-18-secreting CD4+ T cells promote CD8+ T cells through a helper effect
IL-18 CAR T cells have superior proliferation and antitumor activity in mouse models
Acknowledgments
This work was funded by NIH 2R01CA120409 (to C.H.J. and Y.Z.), the Novartis Alliance, and NIH T32 CA009140 (to B.H.). We thank Patricia Tsao and Yinan Lu from the Human Immunology Core. We also thank Dr. Taku Kambayashi for providing the vector containing the 1D3 scFV fragment recognizing murine CD19. B.H., C.H.J., and Y.Z. have a pending patent application based on this work.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, C.H.J. and B.H.; Study Design, B.H.; CRISPR/Cas9 Gene Editing, J.R., B.H., and Y.Z.; Animal Procedures, B.H. and Y.L.; Scientific Resources, J.S.; Drafting and Editing of the Manuscript, B.H., C.H.J., B.K., and R.M.Y.
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.09.002.
References
- Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RG, Carpenito C, Shan X, Danet-Desnoyers G, Liu R, Jiang S, Albelda SM, Golovina T, Coukos G, Riley JL, et al. Distinct effects of IL-18 on the engraftment and function of human effector CD8 T cells and regulatory T cells. PLoS ONE. 2008;3:e3289. doi: 10.1371/journal.pone.0003289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257:83–90. doi: 10.1111/imr.12125. [DOI] [PubMed] [Google Scholar]
- Cohen J. IL-12 deaths: explanation and a puzzle. Science. 1995;270:908. doi: 10.1126/science.270.5238.908a. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD, Jr, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, Lin AA, Schlom J, June CH, Sherwin SA. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22. doi: 10.1186/s40425-017-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008;358:2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med. 1998;188:2357–2368. doi: 10.1084/jem.188.12.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, Olivares S, Rabinovich B, Huls H, Forget MA, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci USA. 2016;113:E7788–E7797. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, Wilson WH, Rosenberg SA. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrecque N, Whitfield LS, Obst R, Waltzinger C, Benoist C, Mathis D. How much TCR does a T cell need? Immunity. 2001;15:71–82. doi: 10.1016/s1074-7613(01)00170-4. [DOI] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, Samanta M, Lakhal M, Gloss B, Danet-Desnoyers G, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montufar-Solis D, Wang HC, Klein JR. Stimulatory and costimulatory effects of IL-18 directed to different small intestinal CD43 T cell subsets. J Leukoc Biol. 2007;82:1166–1173. doi: 10.1189/jlb.0207108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Okamura H, Wada M, Nagata K, Tamura T. Endotoxin-induced serum factor that stimulates gamma interferon production. Infect Immun. 1989;57:590–595. doi: 10.1128/iai.57.2.590-595.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Okamura H, Nagata K, Komatsu T, Tamura T. Purification of a factor which provides a costimulatory signal for gamma interferon production. Infect Immun. 1993;61:64–70. doi: 10.1128/iai.61.1.64-70.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, Bautista C, Chang WC, Ostberg JR, Jensen MC. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, Kathman S, Pandite LN, Oei C, Kirby LC, et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res. 2006;12:4265–4273. doi: 10.1158/1078-0432.CCR-06-0121. [DOI] [PubMed] [Google Scholar]
- Tanyi JL, Stashwick C, Plesa G, Morgan MA, Porter D, Maus MV, June CH. Possible Compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T cells. J Immunother. 2017;40:104–107. doi: 10.1097/CJI.0000000000000160. [DOI] [PubMed] [Google Scholar]
- Zhang L, Feldman SA, Zheng Z, Chinnasamy N, Xu H, Nahvi AV, Dudley ME, Rosenberg SA, Morgan RA. Evaluation of γ-retroviral vectors that mediate the inducible expression of IL-12 for clinical application. J Immunother. 2012;35:430–439. doi: 10.1097/CJI.0b013e31825898e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, Nahvi AV, Ngo LT, Sherry RM, Phan GQ, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding inter-leukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M. structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.