

SUMMARY
Adhesion G-protein coupled receptors (ADGRs) encompass 33 human transmembrane proteins with long N-termini involved in cell-cell and cell-matrix interactions. We show the ADGRB1 gene, which encodes Brain-specific angiogenesis inhibitor 1 (BAI1), is epigenetically silenced in medulloblastomas (MBs) through a methyl-CpG binding protein MBD2-dependent mechanism. Knockout of Adgrb1 in mice augments proliferation of cerebellar granule neuron precursors (GNPs), and leads to accelerated tumor growth in the Ptch1+/− transgenic MB mouse model. BAI1 prevents Mdm2-mediated p53 polyubiquitination, and its loss substantially reduces p53 levels. Reactivation of BAI1/p53 signaling axis by a brain-permeable MBD2 pathway inhibitor suppresses MB growth in vivo. Altogether, our data define BAI1's physiological role in tumorigenesis and directly couple an ADGR to cancer formation.
IN BRIEF
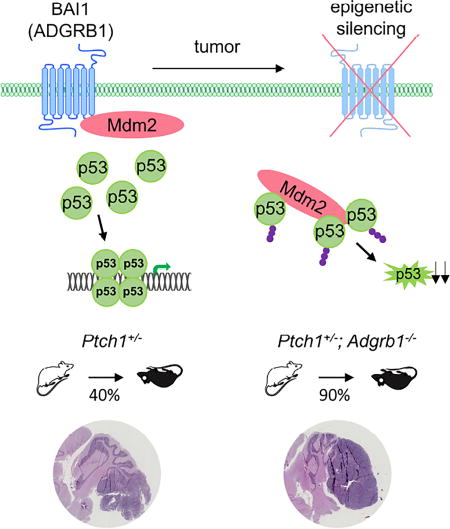
Zhu et al. identify epigenetic silencing of ADGRB1 in medulloblastoma (MB) and show that Adgrb1 loss in a transgenic mouse MB model accelerates tumor growth. ADGRB1 encodes BAI1, which prevents MDM2-mediated p53 polyubiquitination. Rescue of BAI1 expression increases p53 levels and suppresses MB growth.
INTRODUCTION
MB is the most common and aggressive malignant brain tumor in children. Current nonspecific cytotoxic therapies cure only a fraction of patients, and cause life-long neurological, intellectual and physical disabilities (Smoll, 2012), highlighting the urgent need for novel therapeutic approaches. MB has been recently subdivided in four clinically and molecularly distinct subgroups: the wingless (WNT) group, the sonic hedgehog (SHH) group, group 3 and group 4 (Cavalli et al., 2017; Gilbertson and Ellison, 2008; Jones et al., 2012; Northcott et al., 2017). The cellular origin and developmental alterations underlying the formation of each MB subgroup are active areas of investigation. The most studied and best understood is the SHH group. Recent reports suggest that GNPs represent the cells of origin of this subgroup, with genetic alterations in the SHH pathway promoting their transformation (Goodrich et al., 1997; Lee et al., 2010; Schuller et al., 2008). Transgenic mouse models with either Ptch1 deficiency or constitutively active Smo alleles are able to generate MBs that resemble human tumors (Goodrich et al., 1997; Hatton et al., 2008; Lau et al., 2012). However, while these animals are prone to MB formation, the tumor incidence is low and additional genetic defects such as deficiency of either Trp53 or Pten are necessary to increase penetrance and accelerate tumor development (Castellino et al., 2010; Wetmore et al., 2001).
In human MBs, TP53 mutations are frequent in the WNT (16%) and SHH groups (21%), but are rare in group 3 or 4 tumors (Zhukova et al., 2013). However, group 3 tumors display loss of heterozygosity for TP53 as a result of iso17p, and mice with combinations of active c-Myc and Trp53 null or dominant negative alleles are predisposed to group 3 MB (Kawauchi et al., 2012; Pei et al., 2012). Loss of wild-type p53 function can also result from alterations in p53-negative regulators (Castellino et al., 2008; Malek et al., 2011). Yet in many MBs across all 4 subgroups, a clear mechanism underlying p53 pathway inactivation is still missing.
G protein-coupled receptors (GPCRs), the largest family of transmembrane proteins in vertebrates, are the molecular targets for about 50–60% of therapeutic drugs worldwide (Bridges and Lindsley, 2008; Pierce et al., 2002). Many GPCRs are overexpressed in various cancer types and may contribute to tumor growth and metastasis; however, less is known about GPCRs acting as tumor suppressors (Dorsam and Gutkind, 2007). A recent study suggests that GPCR expression patterns can also delineate MB subtypes (Whittier et al., 2013), but whether and how GPCR members contribute to MB tumorigenesis remains unknown.
The BAI proteins (BAI1-3) belong to the adhesion-subgroup of GPCR (Hamann et al., 2015), and are predominantly, although not exclusively, expressed in the brain (Nishimori et al., 1997; Shiratsuchi et al., 1997). They contain several well-defined protein modules in the N-terminus, including thrombospondin type 1 repeats (TSRs), which can regulate angiogenesis (Kaur et al., 2005; Klenotic et al., 2010; Nishimori et al., 1997; Zhu et al., 2011), phagocytosis of apoptotic cells by macrophages (Park et al., 2007) and participate in myogenesis (Hochreiter-Hufford et al., 2013). The BAI1 N-terminus can be cleaved, and when overexpressed, can inhibit angiogenesis and glioma growth (Cork et al., 2012; Kaur et al., 2009; Nishimori et al., 1997; Zhu et al., 2011). Moreover, recent genetic studies have shown that the genes encoding BAI family proteins (ADGRB1-3) are silenced and/or undergo somatic mutations in several cancers, including melanoma, lung, breast, ovarian and brain cancers (Brennan et al., 2013; Kan et al., 2010; Pugh et al., 2012; Robinson et al., 2015; Zhu et al., 2011). However, the significance of these mutations to cancer formation has not been demonstrated. Here, we examined the causal relationship between BAI1 expression and cancer formation, and determined whether it could be targeted for therapeutic gain.
RESULTS
Epigenetic Silencing of the ADGRB1 gene underlies lack of BAI1 expression in MB
We first analyzed ADGRB1 gene expression in frozen tissues of 6 normal cerebella and a cohort of 77 human MBs by RT-PCR. Molecular sub-grouping of all MB samples (Table S1) was performed on a Nanostring instrument as described (Northcott et al., 2012b). ADGRB1 mRNA expression can be readily detected in both fetal and adult normal cerebella; however, its expression is strongly reduced in most MB samples (92%, 71/77, Figures 1A and S1A) and human MB cell lines (Figure S1B). To independently validate our findings, we analyzed a RNA sequencing dataset of 170 MB samples (Northcott et al., 2017) and an expression microarray dataset of 64 MB samples (Remke et al., 2011). Overall down regulation of ADGRB1 expression in all MB subtypes was evidenced in both datasets (Figures 1B and S1C). Examination of a MB tissue array by immunohistochemistry (IHC) evidenced that BAI1 is also reduced at the protein level: 51/60MB specimens (85%) exhibited background levels, while the remaining 9/60 (15%) had weak to moderate expression (Figure 1C). Taken together, these data show that ADGRB1 mRNA and BAI1 protein expression are significantly reduced in MBs.
Figure 1. Epigenetic Silencing of ADGRB1/BAI1 Expression in MB.

(A) RT-PCR analysis of ADGRB1 mRNA expression in patient MB tumors and normal human fetal, infant and adult cerebellum. GAPDH was used as a control.
(B) Analysis of ADGRB1 mRNA levels in 7 normal cerebella (NC) and 170 MB samples by RNA-Seq (Northcott et al., 2017). TPM, Transcripts Per Kilobase Million. Individual data points with the mean ± SEM are shown. *** p<0.0001, ** p<0.01, t test.
(C) IHC staining of BAI1 in a de-identified, formalin-fixed, paraffin-embedded tissue array containing 60 MBs shows low (51 cases; score 0–4) or moderate (9 cases; score 6–9) BAI1 expression. Scale bar, 200 µm. For inset, scale bar, 100 µm.
(D) MS-PCR analysis of DNA methylation in the CpG island of the ADGRB1 promoter. Representative results (n=3) of 1 sample from each MB group and 6 normal cerebella are shown.
(E) β-Gal staining for Adgrb1 promoter activity in Adgrb1-LacZ reporter mice at postnatal day 10 (P10). ML, molecular layer. Scale bar, 100 µm.
See also Figures S1, S2 and Table S1.
To explore the mechanisms underlying ADGRB1 down regulation, we first considered whether the gene locus is associated with genetic loss in MB. Human ADGRB1 is located on chromosome 8q24, close to the MYC oncogene, a gene amplified in MBs (Bigner et al., 1990; Northcott et al., 2012a; Robinson et al., 2012). Copy number variation analysis in a dataset consisting of 1,087 MB tissues (Northcott et al., 2012a) did not evidence any ADGRB1 loss (Figure S2A), possibly reflecting selection to maintain essential adjacent genes. We then considered epigenetic mechanisms in ADGRB1 silencing. Methylation-specific PCR (MS-PCR) showed extensive methylation in the CpG island of ADGRB1 promoter in all 4 MB groups (Figures 1D, S2B and Table S1), but not in fetal and adult normal cerebella (Figure 1D). Promoter methylation correlated with gene silencing, and the 6/77 tumors that retained ADGRB1 mRNA expression had only partial methylation (Figure S2B). Bisulfite-sequencing on 4 randomly selected tumors (one/group) confirmed dense CpG methylation in 4 sequenced alleles per sample, while cerebellar controls were unmethylated (Figure S2C). Whole genome bisulfite sequencing (WGBS) and DNA methylation array analyses (Hovestadt et al., 2014) revealed that differential DNA methylation between MB and normal cerebellum was confined to the CpG island in the gene promoter (Figures S2D and S2E). Methylation appears to be specific to the ADGRB1 gene as well, as the promoter of another family member (ADGRB2) was unmethylated (Figure S2F). The IRF6 tumor suppressor gene that shows promoter hypermethylation in all four subtypes (Hovestadt et al., 2014) was included as a positive control (Figure S2F). Hypermethylation at the ADGRB1 promoter was associated with reduced gene expression in the same dataset (Figure S2G). Taken together, these data indicate that hypermethylation of the CpG island in the promoter is associated with ADGRB1 gene silencing in MB tissues, even though there are some outliers suggesting additional layers of regulation and/or tumor tissue heterogeneity.
We next determined whether methylation in CpG island at ADGRB1 promoter was a transformation event or was already present in the cells of origin. We focused on SHH-MB as its origin from transformed GNPs during late embryogenesis is widely accepted (Goodrich et al., 1997; Lee et al., 2010; Schuller et al., 2008). During cerebellar development, GNPs originating from a germinal zone in the upper rhombic lip migrate rostrally over the surface of cerebellar anlage to form a mitotically active region named the external granular layer (EGL). Once the GNPs terminally differentiate into cerebellar granule neurons, they migrate to the internal granular layer (IGL), which becomes the granular layer of the mature cerebellum. We used human cerebellum samples from different developmental stages and demonstrated by MS-PCR that there was no evidence for cells with ADGRB1 silencing (Figure 1D) with a detection sensitivity of 1 silenced allele in 10,000 (Figure S2H). The human EGL forms during the second trimester of gestation and is not fully resorbed till 1 year postnatally (Sidman and Rakic, 1973), so our fetal and infant cerebellum samples cover the cell populations from which MB arise in development. To further demonstrate that GNPs actively express Adgrb1, we used transgenic reporter mice carrying a LacZ gene under the control of the endogenous Adgrb1 promoter (Zhu et al., 2015). Abundant β-galactosidase expression was observed in the GNPs present in the transgenic mice EGL during the early post-natal period (Figure 1E). In sum, these data suggest that aberrant DNA methylation at the ADGRB1 promoter is a tumor-specific event.
Adgrb1−/− Mice Show Aberrantly Proliferating GNPs in the EGL
As MB originate from GNPs in the developing cerebellum, and these cells show robust expression of Adgrb1 mRNA (Figure 2A), we examined whether loss of BAI1 expression during early postnatal development may induce pre-neoplastic lesions in Adgrb1−/− mouse cerebellum (Zhu et al., 2015). We observed an overabundance of cells leading to EGL thickening in Adgrb1−/− vs. WT mice (Figure 2B) at postnatal day 14 (P14), a time at which the EGL normally resorbs in mice. MATH1 is a basic helix–loop–helix transcription factor and marker of developing GNPs and SHH-MBs (Northcott et al., 2012b). Crossing Adgrb1−/− mice with Math1-GFP reporter mice (Lumpkin et al., 2003) confirmed that the abnormally thick EGL consisted of aberrant layers of GFP-labeled GNPs (Figure 2C). Consistently, Ki67 staining of the EGL evidenced increased proliferation in Adgrb1−/− vs. WT mice (Figure 2D). Moreover, primary cell cultures of Adgrb1−/− GNPs from postnatal day 5 (P5) had a higher proliferation rate than WT controls (Figure 2E). Jointly, these results support that loss of BAI1 expression in mice leads to excessive growth of GNPs in the EGL.
Figure 2. Adgrb1 Loss Enhances GNP proliferation.

(A) RT-PCR analysis of Adgrb1 mRNA expression in GNPs isolated from WT and Adgrb1−/− mice at postnatal day 5 (P5). Primers in two different gene locations were tested (exons 2 and 3–6).
(B) H&E staining shows the EGL at postnatal day 14 (P14) in WT and Adgrb1−/− mice (KO) cerebellum (arrows). Scale bar, 50 µm. Right, quantification of EGL thickness. ***, p<0.0001, 2-tailed Student’s t-test, n=15.
(C) Fluorescence microscopy of mouse cerebella at P14 shows GFP-positive cells (arrows) in the EGL of Math1-GFP;KO (Adgrb1−/−) mice versus control Math1-GFP;WT (Adgrb1+/+) mice. Scale bar, 50 µm.
(D) IHC and quantification of Ki67 positive cells in EGL at postnatal day 7. **, p<0.001, 2-way ANOVA test, n=8. Scale bar, 30 µm.
(E) In vitro proliferation of primary GNP cultures isolated from P5 WT and Adgrb1−/− (KO) mice. Cycling cells were either stained for Ki67 (green fluorescence) or labeled with BrdU and visualized through the red fluorescence signal. Hoechst 33342 counterstaining (blue) reveals the nuclei. Scale bar, 200 µm. Histograms show quantification of BrdU and Ki67 positive cells, ***, p<0.0001, 2-way ANOVA test, n=5. All histogram data represent mean ± SEM.
Deletion of Adgrb1 in Mice Accelerates SHH-MB Formation
To determine whether loss of BAI1 expression might predispose the developing cerebellum to transformation, we aged Adgrb1−/− mice and monitored spontaneous brain tumor formation. We did not observe any cancer incidence for at least one and a half years (Figure 3A and data not shown); a small number of mice had to be euthanized during this period due to fighting wounds. Since SHH-MBs are known to originate from GNP transformation, we next examined whether reduced BAI1 expression might synergize with PTCH1 loss in the Ptch1+/− transgenic mice (Goodrich et al., 1997; Oliver et al., 2005). Ptch1−/− mice are embryonic lethal, but heterozygotes are viable and develop SHH-MB from spontaneous transformation of GNPs in the EGL, albeit with low penetrance (Goodrich et al., 1997; Oliver et al., 2005; Wechsler-Reya and Scott, 2001). Consistent with the literature, Kaplan-Meier survival curves demonstrated that ~38% of Ptch1+/− mice died of MB by 12 months of age (Figure 3A). In sharp contrast, ~90% of the Ptch1+/− mice had died by 4 and 5 months of age on the Adgrb1−/− and Adgrb1+/− backgrounds, respectively, showing dramatic acceleration of the age-of-onset and incidence of MB (p<0.0001; log-rank test). Upon histological examination, Ptch1+/− mice brains displayed large intracranial tumors in the posterior fossa harboring typical MB features with strong staining of GFAP and MATH1 (Figures 3B and S3), and a higher proliferation rate was observed in the tumor cells from the Ptch1+/−;Adgrb1−/− mice by Ki67 staining (Figure 3B).
Figure 3. Adgrb1 Loss Increases Penetrance and Accelerates Tumor Growth in the Ptch1+/− SHH-MB Mouse Model.

(A) Kaplan-Meier curves show the survival of Ptch1+/− mice in the presence of hemi- or homozygous deletion of Adgrb1 (p<0.0001; log-rank test).
(B) H&E staining of 5-month-old Ptch1+/− and Ptch1+/−;Adgrb1−/− mouse cerebella with tumor. IHC shows Ki67 staining in tumor cells and quantification is shown by histogram, p=0.005, t test, n=6. Scale bar, 30 µm.
(C) RT-PCR analysis of mRNA expression of Adgrb1, Ptch1 and SHH pathway markers (Gli1 and Ccnd1) in adult normal cerebellum and MB from mice with indicated genotypes.
(D) Representative images show IHC staining for endothelial cell markers CD31 (top) and CD144 (VE-cadherin, bottom) in 5-month-old mice MB. Scale bar, 100 µm.
All histogram data represent mean ± SEM. See also Figure S3.
To gain further insight into the tumorigenic process, we isolated tumors and examined Adgrb1 expression and found it was remarkably reduced in all tumors regardless of Adgrb1 allele status (Figure 3C). This suggests that tumor formation in Adgrb1+/− mice selects for loss of expression of the remaining WT allele. Moreover, it indicates that spontaneous formation of MB in Ptch1+/− mice also selects for loss of BAI1 expression. As expected, expression of Gli1, a transcriptional driver of an activated SHH pathway, and its target Ccnd1, are substantially induced in the tumor tissues (Figure 3C). IHC with vascular markers (CD31 and CD144) showed comparable microvascular density (CD31, p=0.60 and CD144, p=0.765, respectively, 2-tailed t-test, n=6) in tumors from Ptch1+/− and Ptch1+/−;Adgrb1−/− mice (Figure 3D). Taken together, these results demonstrate that deletion of Adgrb1 in mice is insufficient to trigger spontaneous tumor formation per se, but dramatically increases the penetrance and growth of SHH-MB in the Ptch1+/− mice, likely in an angiogenesis-independent fashion.
Expression of p53 is dramatically down regulated in the Adgrb1−/− mice cerebellum
To explore the mechanisms underlying increased penetrance of tumor initiation due to Adgrb1 absence, we examined the early postnatal mouse cerebellum for putative oncogenic signaling. Examination of proliferation-related proteins at P14 revealed a substantial reduction of p53, along with its target, the p21 cell cycle inhibitor (Figure 4A). This reduction was specific to Adgrb1−/− mice, as p53 was not decreased in Ptch1+/−;Adgrb1+/+ mice. Interestingly, the drop in p53 levels selectively impacted downstream signaling, with reduced gene expression of cell cycle regulators Cdkn1a and Gadd45a, but not other p53 target genes involved in apoptosis or metabolism (Figure 4B). This target gene selectivity may reflect the partial acetylation status of p53 in these cells (Kruse and Gu, 2009). We next explored the mechanisms underlying p53 reduction. There were no alterations in Trp53 mRNA levels at P14 (Figure 4B), suggesting a post-transcriptional mechanism. To test BAI1’s effects on p53 stability, we first generated human D556 MB cells in which BAI1 expression can be induced by doxycycline treatment. We treated D556+BAI1 (Tet-on) cells with cycloheximide (CHX), a protein synthesis blocker, and compared the kinetics of p53 degradation with or without BAI1 induction by doxycycline (Figure 4C). In the presence of BAI1, p53 half-life was significantly increased; after 3 hr of protein synthesis inhibition, there was minimal p53 degradation, while p53 protein was markedly degraded in control cells. In sum, these findings reveal a reduction in p53 tumor suppressor levels in the Adgrb1−/− cerebellum, and demonstrate that BAI1 expression can impact p53 stability in human MB cells.
Figure 4. BAI1 blocks Mdm2-mediated p53 Polyubiquitination and Degradation.

(A) Representative immunoblots (n=3) show indicated protein expression in WT and Adgrb1−/− mouse cerebellum at P14.
(B) qRT-PCR analysis of p53 target gene expression in WT and Adgrb1−/− cerebellum at P14. Histogram data represent mean ± SEM.
(C) Western blot showing p53 expression in the D556+BAI1 (Tet-on) MB cells +/− doxycycline (Dox) treatment. CHX (50 µg/ml) was used and normalized fraction of remaining p53 levels after 1–3 hr is indicated.
(D) Left panel, IP was performed with anti–p53 antibody, and polyubiquitination of p53 was determined by anti-ubiquitin antibody with or without Dox treatment. Right panel, ONS-76 cells were transiently transfected with BAI1 and HA-ubiquitin expression vectors. IP was performed with anti–p53 antibody, followed by IB detection of ubiquitin chains (anti-HA antibody) or Mdm2.
(E) IP with BAI1 antibodies in D556 and ONS-76 human MB cells followed by IB of Mdm2.
(F) Representative confocal images of PLA in D556-BAI1 tet-on cells (+/− 1 µg/ml Dox, 48 hr). Green spots are regions of signal amplification denoting BAI1-Mdm2 interaction. Nuclear stain (DAPI) is blue. Scale bars, 10 µm.
(G) Top, schematic diagram shows myc-tagged BAI1 truncated constructs. NT=N-terminus, ΔNT=N-terminal truncated, CT=C-terminus. Bottom, IP with Mdm2 antibodies in HEK293 cells followed by IB of myc tag.
(H) Cell fractionation shows distribution of 90-kDa Mdm2 and p53 in D556+BAI1 (tet-on) cells and transiently transfected ONS-76 cells. Cytoplasmic (C) and nuclear (N) fractions are shown. All blots show representative images from 3 independent experiments with similar results.
(I) Analysis of p53 and Mdm2 nuclear localization by immunofluorescence upon BAI1 induction in D556+BAI1 (tet-on) cells. Scale bar, 10 µm.
(J) D556+BAI1 (tet-on) cell surface proteins were biotinylated and collected by streptavidin-bead pulled-down. IP was then performed with BAI1 antibodies on purified cell surface proteins followed by IB of Mdm2.
See also Figure S4.
Association of BAI1 with Mdm2 Inhibits p53 Polyubiquitination and Degradation
We then considered whether BAI1 might act as a negative regulator of Mdm2, the E3 ubiquitin ligase that regulates p53 stability (Kubbutat et al., 1997). Although a slight decrease in Mdm2 levels was observed in human MB cells upon BAI1 induction (Figure 4C), reduced p53 stability in Adgrb1−/− cerebellum was not related to changes in the expression of either total Mdm2 or its phosphorylated form at serine 166 (Figure 4A), which can activate p53 degradation (Abe et al., 2008). Next, we examined whether BAI1 could block Mdm2-mediated p53 ubiquitination. In BAI1-inducible D556 cells, co-immunoprecipitation (co-IP) experiments evidenced that p53 polyubiquitination from endogenous ubiquitin was remarkably inhibited, which stabilized p53 (Figure 4D, left panel). Similar results were obtained upon transient transfection of BAI1-silent human SHH-MB ONS-76 cells (Ivanov et al., 2016) with expression vectors for BAI1 and HA-tagged ubiquitin. Co-IP showed exogenous BAI1 disrupted p53-Mdm2 interaction, reduced p53 polyubiquitination and stabilized p53 levels (Figure 4D, right panel). To probe for a mechanism, we hypothesized that BAI1 might directly interact with and inhibit Mdm2’s E3 ligase activity towards p53. Co-IP experiments revealed an interaction between Mdm2 and BAI1 in MB cells (Figure 4E). The interaction is BAI1-specific as no interactions between Mdm2 and BAI2 or BAI3 were detected (Figure S4A), and neither BAI2 nor BAI3 can stabilize p53 in MB cells (Figure S4B). To independently confirm the Mdm2-BAI1 interaction, we used in situ proximity ligation assay (PLA) (Soderberg et al., 2008), an assay that can visualize protein interaction in close proximity (<40 nm) in whole cells. Upon BAI1 induction in D556 cells, fluorescent PLA spots were easily detected, while they were inexistent in non-induced cells (Figure 4F) or in parental cells with or without dox (data not shown).
To map the Mdm2 binding region on BAI1, we performed co-IP experiments with transfected myc-tagged truncated BAI1 constructs in HEK293 cells, and these revealed that the seven-transmembrane domain (ΔNT-construct) is required for Mdm2-BAI1 interaction (Figure 4G). Consistently, only the ΔNT-construct enhanced endogenous p53 stability in ONS-76 cells (Figure S4C). To identify a putative Mdm2 binding site in BAI1’s 7TM, we compared the 3 cytoplasmic loops of BAI1-3 (Figure S4D). This alignment evidenced a unique VSV motif in BAI1 loop 1, while loops 2 and 3 were more conserved. Co-IP with a BAI1 VSV-deletion mutant evidenced a strong reduction in Mdm2 binding, while a loop 2 mutant had no effect (Figure S4E). Consistently, the ΔVSV mutant failed to stabilize p53 in human MB cells (Figure S4F). Taken together, these results support a direct cause-effect relationship between BAI1-Mdm2 interaction and p53 stability.
To address the mechanism underlying BAI1 inhibition of Mdm2-mediated p53 degradation, we hypothesized that BAI1 binding may impede Mdm2 nuclear localization, as PLA results suggested the majority of Mdm2-BAI1 interaction spots occurred outside the nuclear compartment (Figure 4F). Analysis of cytoplasmic and nuclear fractions in MB cells showed BAI1 expression reduced nuclear 90 kDa Mdm2 (Figures 4H and 4I), the main isoform that controls p53 stability (Cheng and Cohen, 2007). As expected, BAI1 expression induced a remarkable increase of p53 protein in the nucleus (Figures 4H and 4I). To examine whether BAI1 traps Mdm2 at the cell surface, we used biotin to label all surface proteins (Zhu et al., 2008) followed by streptavidin bead pull down and BAI1 immunoprecipitation. Examination of co-precipitated Mdm2 showed that BAI1 recruits at least part of intracellular Mdm2 to the cell surface membrane (Figure 4J). Altogether, these data show that BAI1 protects p53 from Mdm2-mediated degradation by sequestering Mdm2 outside the nucleus.
Restoration of BAI1 in MB Cells Inhibits Tumor Growth in vitro and in vivo
Stable restoration of BAI1 expression in BAI1-silent human MB cells reduced cell proliferation by ~2-fold compared to controls, and this effect was p53-dependent (Figure 5A). Genetic knockout of TP53 in colon cancer HCT116 cells also abrogated BAI1’s growth suppressive effects (Figure S5). These results demonstrate that BAI1 inhibits in vitro tumor cell growth in a p53-dependent manner. Consistently, Mdm2-binding construct BAI1-ΔNT could stabilize p53 (Figure S4C) and inhibit cell proliferation, while the BAI1-ΔVSV mutant did not bind Mdm2 and had no inhibitory effect on MB cell growth (Figure 5B). To examine BAI1’s effects on in vivo MB growth, we established orthotopic xenografts by stereotaxically injecting MB cells (D556 and ONS-76) into the cerebella of athymic nude mice. Kaplan-Meier survival curves show that restoration of BAI1 expression had dramatic anti-tumor effects in both models. 90% of mice with ONS-76 xenografts experienced long-term survival (Figure 5C) (p=0.0003, log-rank test), while median survival was extended from 28 to 50 days (p=0.0002) in mice with D556 xenografts (Figure 5D). Remarkably, the therapeutic effect was lost upon stable shRNA-mediated p53 knockdown in both models. These data further establish BAI1 as a tumor suppressor in human MB, and indicate p53 is a key mediator of its downstream suppressive signaling.
Figure 5. BAI1 inhibits MB Growth in vitro and in vivo in a p53-dependent manner.

(A) Left, cell proliferation of D556+BAI1 (tet-on) cells with stable p53 knockdown upon BAI1 induction. Fold is the ratio of live cells on day 3/day 0. Right, cell proliferation of ONS-76 MB cells with stable transfection of BAI1 +/− p53 shRNA lentivirus was measured for 5 days. Efficiency of shRNA-mediated knockdown was evaluated by Western blots (insets). ***, p<0.0001, 2-way ANOVA, n=3.
(B) Effect of BAI1 WT and mutants on ONS-76 cell proliferation. Fold is the ratio of live cells on day 3/day 0. ***, p<0.0001, 2-way ANOVA, n=5.
(C) Kaplan-Meier curves show the survival of mice with intracranial xenografts of ONS-76 cells infected by lentiviral-expressed BAI1 +/− stable p53 knockdown (p=0.0003; log-rank test).
(D) Kaplan-Meier curves show the survival of mice with intracranial xenografts of D556 cells infected by lentiviral-expressed BAI1 +/− stable p53 knockdown (p=0.0002; log-rank test). Right, representative images show H&E staining of MB sections, and arrows show tumor. Scale bar, 1 mm.
All histogram data represent mean ± SEM. See also Figure S5.
Methylated DNA binding protein MBD2 participates in ADGRB1 gene silencing
Next, we wanted to gain a better understanding of the molecular mechanism underlying ADGRB1 gene silencing in human MB cells with the hope of identifying opportunities for therapeutic reactivation. We performed chromatin immunoprecipitation (ChIP) in BAI1-silent MB cells, and found strong tri-methylation of histone H3 at lysine 9 (H3K9me3), a marker of condensed (silenced) chromatin, and little evidence for histone H3 acetylated at lysine 9 (H3K9ac), a marker of active chromatin (Figure 6A), confirming gene silencing. Interestingly, promoter silencing was accompanied by the binding of MBD2 (Figure 6A), a methyl-CpG–binding domain protein that binds methylated DNA and suppresses gene expression (Ballestar and Wolffe, 2001; Wade, 2001). To test whether there was a functional interaction, we stably knocked down MBD2 expression in MB cells and observed reactivation of BAI1 expression (Figure 6B). Taken together, these results suggest that MBD2 plays a direct role in ADGRB1 silencing.
Figure 6. KCC-07 distributes to the Brain and Inhibits Tumor Growth in vitro.

(A) Analysis of histone H3 marks and MBD2 binding on ADGRB1 promoter in human MB cells by ChIP assay.
(B) WB analysis of BAI1 protein expression in human MB cells with MBD2-shRNAs.
(C) Left panel, structure of MBD2 pathway inhibitor KCC-07. Right, concentration of KCC-07 in the plasma (PL), cerebral cortex (CC) and cerebellum (CB) one hr after i.p. injection (100 mg/kg).
(D) ChIP assay shows the effect of KCC-07 treatment (10 µM, 48 hr) on the binding of MBD2 to ADGRB1 promoter in MB cells.
(E) RT-PCR analysis of ADGRB1 expression in MB cell lines with KCC-07 treatment.
(F) Western blot shows the effect of KCC-07 on BAI1/p53/p21 protein expression in MB cells. All blots show representative images (n=3).
(G) Effect of KCC-07 treatment (10 µM, 3 days) on the growth of MB cell lines in vitro. ***, p<0.0001, 2-way ANOVA test, n=3. Histogram data represent mean ± SEM.
Small Molecule MBD2 Pathway Inhibitor Suppresses MB Growth
We then explored the feasibility of pharmacologically reactivating the endogenously silenced ADGRB1 alleles in the tumor toward therapeutic gain. We used KCC-07, a small molecule that prevents binding of MBD2 to methylated DNA (Reichert, 2010; Wyhs et al., 2014). We then evaluated KCC-07 for its ability to reactivate ADGRB1 expression and permeate across the blood-brain barrier. KCC-07 had excellent brain distribution as more than 2-fold higher drug concentration in the brain tissue than in the plasma (Figure 6C). Moreover, treatment of BAI1-silent MB cells with KCC-07 largely abrogated MBD2 binding to the ADGRB1 promoter (Figure 6D) and restored BAI1 mRNA and protein expression (Figures 6E and 6F) at low concentrations. KCC-07 also clearly inhibited MB cell growth in vitro (Figure 6G), consistent with induction of anti-proliferative BAI1/p53/p21 signaling (Figure 6F).
We next tested KCC-07’s effect on two orthotopic human MB xenograft models (D556 and D425). Fourteen days after MB cell implantation in the cerebellum, mice were treated intraperitoneally (i.p.) with KCC-07. The drug increased median survival in both models: from 22.5 to 29 days (p<0.0001) in D556 xenografts (Figure 7A), and 25.5 to 30 days (p=0.0054) in D425 xenografts (Figure S6A). Tumor size was reduced, and reactivation of BAI1 expression along with augmented nuclear p53 was detected in the KCC-07 group (Figure 7B). Since MBD2 pathway inhibition may reprogram the expression of multiple genes, we considered whether the anti-tumor effect of KCC-07 was BAI1-dependent. We infected D556 cells with lentiviruses expressing shRNAs targeting BAI1, which neutralized the reactivation of BAI1 expression by KCC-07 (Figure 7C). We then tested the therapeutic effect of KCC-07 in mice implanted with the D556-BAI1-shRNA cells. Remarkably, the silencing of BAI1 completely abrogated the survival advantage conferred by KCC-07 (p=0.16, log-rank test) (Figure 7C), demonstrating that the anti-tumor effects observed were BAI1-dependent. We next determined whether the anti-tumor effect of KCC-07 was p53-dependent. Knockdown of p53 blocked the beneficial effect of KCC-07 both in vitro (Figure S6B) and in vivo (Figure 7D). In summary, these data show that KCC-07 is able to inhibit MB growth in vivo by reactivating the BAI1-p53 tumor suppressor axis and, thereby, represents a promising chemical scaffold for further drug development.
Figure 7. KCC-07 Inhibits Orthotopic MB Growth in mice xenografts.

(A) Kaplan-Meier survival curves show effect of KCC-07 (100 mg/kg i.p.; 5 days/week) on the survival of mice harboring D556 intracranial xenografts. p<0.0001, log-rank test.
(B) Effect of KCC-07 on tumor size (H&E), BAI1 and p53 expression (IHC) in tumors from (A). Scale bar in H&E images, 1 mm. Scale bar in IHC images, 25 µm.
(C) Kaplan-Meier survival curves shows effect of KCC-07 on the survival of mice harboring intracranial xenografts of D556 cells with lentiviral shRNA-mediated abrogation of BAI1 (p=0.16, log-rank test).
(D) Kaplan-Meier survival curves shows effect of KCC-07 on the survival of mice harboring intracranial xenografts of D556 cells with lentiviral shRNA-mediated abrogation of p53 (p=0.23, log-rank test).
See also Figure S6.
DISCUSSION
BAI1 is an ADGR highly expressed in the brain and we now show that its expression is lost in all four subtypes of human MB, the most aggressive pediatric brain tumor. We demonstrate a role for BAI1 in regulating proper cerebellar development and preventing transformation of neural progenitors during EGL expansion. Our findings suggest BAI1 is a tumor suppressor in MB, define it as an upstream protector of p53, and thereby reveal that alteration in ADGRB1/BAI1 represents a point of vulnerability in cancer.
When crossing Adgrb1−/− mice with several mutant mouse lines that are predisposed to cancer, we found a dramatic decrease in survival rate in the Ptch1+/− mice, a SHH-MB mouse model. Histology of the mouse brain evidenced MB in the cerebellum, showing that physiological deficiency of BAI1 contributes to cerebellar transformation. Adgrb1 loss per se did not lead to spontaneous tumor formation, nor did it generally augment tumorigenesis in the mouse brain, as crossings with Pten+/−, Ink4a−/−, or Gfap-Cre;Trp53LoxP/LoxP mice yielded no tumors (data not shown). This indicates that deletion of Adgrb1 alone is not sufficient to initiate malignant transformation, or that other Bai family proteins may compensate for BAI1 loss. Unexpectedly, Adgrb1 loss did not alter the vascular phenotype of spontaneous mouse MB, in contrast to BAI1’s effect in overexpression studies (Cork et al., 2012; Kaur et al., 2009). This suggests that under physiological conditions, BAI1 suppresses tumor growth independently of angiogenesis, which is consistent with the lack of vascular defects in Adgrb1−/− mice brains (Zhu et al., 2015).
Importantly, we identified a physiological function for BAI1 as a protector of the p53 tumor suppressor. This is consistent with prior findings showing that p53 loss increases penetrance and accelerates tumor formation in mouse MB models (Lau et al., 2012; Momota et al., 2008; Uziel et al., 2005; Wetmore et al., 2001). Mechanistically, we showed that BAI1 suppresses tumor formation by binding Mdm2, the p53 E3 ubiquitin ligase, thereby reducing its nuclear levels and stabilizing p53. Taken together, these results define a BAI1-p53 signaling axis and show its importance in brain tumorigenesis. Although the majority of MBs lack p53 mutations, about 40% show dysfunctional p53 target gene expression (de Bont et al., 2008), suggesting that additional mechanisms eliminate the tumor surveillance activity of p53. Our findings now demonstrate that loss of BAI1 expression leads to an alternative way to reduce p53 activity across all four groups of MB. The reduction in p53 expression is partial, so we do not exclude that in some tumors this might precede further inactivation of the p53 pathway through subsequent mutations. Further studies are warranted to define the role of BAI1/p53 signaling loss in other cancers. Somatic mutations in ADGRB1 have been found in many cancers including melanoma, lung, breast, ovarian and brain tumors (Brennan et al., 2013; Kan et al., 2010; Pugh et al., 2012; Robinson et al., 2015), but whether these alter BAI1 function or are passenger mutations is unknown.
We found that the extensive loss of expression of BAI1 in MB was at least in part related to epigenetic alterations of the ADGRB1 gene, and that the silencing was not present in fetal and post-natal cerebellum, including GNPs of the EGL, a cell population at the origin of some MB subtypes. ADGRB1 promoter methylation was associated with the binding of MBD2, a methyl-CpG binding protein believed to play an active role in gene silencing by establishing a repressive chromatin environment (Ballestar and Wolffe, 2001; Le Guezennec et al., 2006; Wade, 2001). The presence of MBD2 on the chromatin was necessary to maintain epigenetic silencing, as we were able to reactivate ADGRB1 expression through targeting MBD2.
MB is the most common malignant brain tumor in children and there is hope that rational and targeted therapies can be developed to treat patients with MB (Huse and Holland, 2010). To address this need, we sought pharmacological means to target epigenetic silencing events at the ADGRB1 gene. We showed that KCC-07, an inhibitor of MBD2 binding to methylated DNA (Reichert, 2010), could reactivate ADGRB1 expression. KCC-07 was bio-available, depleted MBD2 from the ADGRB1 promoter, led to induction of BAI1 and p53 expression, and reduced human MB growth in culture and in MB mouse models in a p53 and BAI1-dependent way. Since dysfunction of the p53 pathway plays a key role in resistance to MB chemotherapy (Tabori et al., 2010; Weeraratne et al., 2011), p53 reactivation through epigenetic restoration of ADGRB1/BAI1 expression represents an important therapeutic goal in this disease. Importantly, KCC-07 showed excellent brain distribution and significantly extended the survival of MB xenografts in vivo. KCC-07 was well tolerated, consistent with the finding that MBD2-knockout mice are viable with only mild phenotypic changes (Hendrich et al., 2001). The pharmacological targeting of MBD2 in cancer has not been realized to date, highlighting the importance of our study. Prior studies investigating epigenetic approaches to cancer therapy have mainly targeted either DNA methyltransferases (DNMTs) or the histone deacetylases (HDACs) (Foulks et al., 2012; Mahindra et al., 2012). Our data demonstrate that pharmacological targeting of MBD2 is possible in the brain, and represents an epigenetic therapy for MB. Further optimization of this scaffold holds promise for the development of a clinical candidate drug.
In summary, we found that Adgrb1 deficiency leads to developmental defects in the mouse cerebellum, and significantly accelerates MB tumorigenesis. These results advance our understanding of the diversity and complexity of signaling pathways that can engender MB disease, and establish a mouse model for therapeutic testing. Combined with the silencing of ADGRB1 gene expression in patient-derived MB samples, these data suggest that adhesion GPCR BAI1 is a tumor suppressor in the brain. Our data further identify a hitherto unknown interaction between BAI1 and Mdm2, which is a regulatory mechanism for p53 stability; and unveils a point of vulnerability of the p53 pathway through silencing or mutation of ADGRB1 in cancer. Our demonstration that a brain-permeable small-molecule MBD2 inhibitor potently antagonizes MB growth provides proof-of-concept for its further development towards a first in class therapeutic. Altogether, our findings provide insight into the neurobiological mechanisms underlying cerebellum development and neoplastic transformation, and establish BAI1 and the associated signaling pathways as important therapeutic targets for MB treatment.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Erwin G. Van Meir (evanmei@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All protocols for mouse experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at Emory University. Adgrb1−/− mice (Zhu et al., 2015) and Ptch1+/− mice (Goodrich et al., 1997; Oliver et al., 2005) were initially on a mixed C57/Bl6/129Sv background and were backcrossed with WT C57/Bl6 mice for more than 10 generations before any experiments in this paper. Adgrb1+/− mice were crossed with Ptch1+/− mice to generate cohorts for MB experiments. Mice were aged and humanely euthanized upon signs of morbidity. Math1-GFP mice (Lumpkin et al., 2003) were bred with Adgrb1−/− mice to generate pups for quantification of GFP-positive cells in the EGL.
Xenografts in nude mice
Outbred athymic nude mice (Hsd:Athymic Nude-Foxn1nu 069, Envigo/Harlan, females; 8–10 weeks old) were anesthetized, an incision was made in the midline of the scalp over the cerebellum, and a small hole was made in the skull (1 mm lateral to midline) using a beveled (sharp point) 18G needle. 5×105 cells in a 5 µL volume were injected with a 24 G Hamilton syringe mounted on a micromanipulator at a 30° angle to the surface of the cerebellum, at a depth of 1 mm.
Cell culture
Human MB cell lines (ONS-76, D556, D425 and Daoy) were maintained in DMEM with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in an atmosphere of 5% CO2. STR profiling to authenticate the cell lines was performed by the Cancer Center of Texas Tech University Health Sciences Center.
METHOD DETAILS
RT-PCR
Total RNA was isolated with Trizol reagent (Thermo Fisher Scientific) and the first-strand cDNA was prepared using an AMV RNA PCR kit (TaKaRa) from 1 µg total RNA. PCR amplification for ADGRB1 mRNA detection was carried out with an initial denaturing step at 95°C for 5 min, then 30 (or 35) cycles of PCR (95°C for 30 s, 52°C for 45 s, and 72°C for 45 s) and a further extension at 72°C for 10 min. Quantitative PCR was performed in triplicate using SYBR green (Thermo Fisher Scientific) and a 7500 Fast Real-Time PCR machine (Applied Biosystems). Specific primer sequences are listed in Table S2.
Transfection of cells
Transient transfection of MB cells was performed with lipofectamine 3000 according to the manufacturer’s instruction (Thermo Fisher Scientific) with minor modifications. Cells were incubated with serum free DMEM containing plasmids/lipofectamine 3000 reagent mixture for at least 8 hr before being switched to DMEM+10% serum for 2–3 days. The transfection efficiency could reach ~90%. For stable MB cell clones with either BAI1- or p53-knockdown, MB cells were infected with BAI1- or p53-specific shRNA-expressing lentiviral particles in the presence of polybrene (5 µg/ml) (Santa Cruz). Two days later, cells were selected with puromycin (1 µg/ml) (Santa Cruz) for 2 weeks. Nuclear and cytoplasmic extract preparation was performed with the nuclear extract kit (Active Motif).
Immunohistochemical Analysis
For the tissue array study, 60 MB tumor specimens were sectioned and mounted on one slide. Sections were deparaffinized and subjected to antigen retrieval by boiling (20 min, 100°C) in 0.01 M Tris HCL (pH 10). Slides were then incubated with a 1:200 dilution of BAI1 antibody. Immunostaining was detected with the avidin-biotin complex method, using diaminobenzidine as the chromogen (Abcam). Slides were scanned with a Nanozoomer 2.0 HT (Hamamatsu) and the MetaMorph Premier software was used to quantify staining intensity (Five fields/tumor). BAI1 status was assessed based on relative staining intensity unit [absent (0), weak (1) (units, 1–75), moderate (2) (units, 76–150), strong (3) (units, 151–225)] and percentage of positive tumor cells [0% (0), <10% (1), 10–50% (2), 51–80% (3), 81–100% (4)]. Immunoreactivity scores (IHC scores) were determined through multiplying the staining score by the percentage score to give a maximum of 12 (Zhu et al., 2011). Quantification of Ki67-positive cells was performed with NIH ImageJ software with IHC toolbox plugin.
Immunoprecipitation and immunoblotting
Cells were rinsed with ice-cold PBS and solubilized for 15 min on ice in radioimmuno-precipitation assay (RIPA) buffer supplemented with protease inhibitors. Lysates were then centrifuged in a microfuge at 12,000 rpm for 10 min, and the supernatants were immunoprecipitated at 4°C with antibodies (5 µg/ml) for 4 hr followed by protein A/G beads (Santa Cruz) overnight. After centrifugation, beads were washed five times with RIPA buffer. Bound proteins were eluted with 2× SDS sample buffer and subjected to SDS-PAGE. Proteins resolved on SDS-PAGE were transferred to nitrocellulose membranes (Bio-Rad). Nitrocellulose blots were incubated at room temperature for 1 hr in blocking buffer (PBS with 0.1% Tween 20 and 5% milk), followed by incubation with indicated antibodies at 4°C overnight. After three 15-min washes with PBS containing 0.1% Tween 20, the blots were incubated with horseradish peroxidase-conjugated secondary antibody (Thermo Fisher Scientific). Immunoreactive bands were visualized using enhanced chemiluminescence substrate (Thermo Fisher Scientific). All immunoblotting was repeated at least three times. For quantitative analysis, autoradiographic films were scanned with an Epson 1680 scanner, and the captured image was analyzed with NIH ImageJ software.
X-Gal staining
The cerebellum was removed and fixed in fresh 4% paraformaldehye/PBS (pH 7.4) for 1 hr at 4°C. The tissue was then rinsed (3×15 min) with washing buffer (100 mM sodium phosphate, pH 7.4, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40) at room temperature. Staining was performed overnight in washing buffer plus 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and 1 mg/ml X-gal (Thermo Fisher Scientific). The tissue was then washed once and post-fixed overnight in 10% formalin at 4°C before sending to the Winship Cancer Institute Cancer Tissue and Pathology shared resource for sectioning.
DNA methylation analysis of the ADGRB1 gene core promoter
Genomic DNA was isolated from the tissues using the DNeasy tissue kit (Qiagen). The bisulfite reaction was carried out on 1 µg of genomic DNA with the EZ DNA Methylation-Direct kit (Zymo). For bisulfite-sequencing, PCR amplification was performed on bisulfite-modified DNA (see Table S2 for primers) and PCR products containing 30 CpG sites within the CpG island of the ADGRB1 gene core promoter were purified by electrophoresis on 1% agarose gels using the Qiaquick™ gel extraction kit (Qiagen), ligated into pGEM-T vectors (Promega) and then introduced into TOP10 competent bacteria (Thermo Fisher Scientific). Four randomly selected bacterial colonies containing individual alleles from each sample were sequenced.
Primary GNP culture
Cerebella from P5 mice were dissected aseptically, cut into small pieces, and digested in 0.05% trypsin at 37°C for 5 min. The tissue was then triturated using pipettes to obtain a single-cell suspension. The cell suspension was added on top of a step gradient of 35% and 65% Percoll and centrifuged in a table top microfuge at high speed (12,000 rpm) for 15 min at room temperature. GNPs were harvested from the 35/65% interface. For staining of bromodeoxyuridine (BrdU) in GNP cultures, cells were pulsed with 10 µM BrdU (Sigma) for the final 4 hr of culture. At the end of culture, cells were fixed with 4% paraformaldehyde for 15 min and incubated with 2N hydrochloric acid (HCl) for 10 min to denature DNA. The acid was neutralized with 0.1 M sodium tetraborate (pH 8.5), and cells were washed in PBS and subsequently stained overnight with anti-BrdU antibodies.
Chromatin Immunoprecipitation Assay (ChIP)
Cells were washed with PBS for 3 times and incubated with 1% formaldehyde for 10 min. After cross-linking, the cells were lysed in FA buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8, 1% Triton X-100, 0.1% Sodium deoxycholate, 0.1% SDS, plus protease inhibitors) and sonicated using a Misonix sonicator MX2020 (setting 8, 15 s for 3 times). Sonicated lysates were centrifuged in a microfuge at 14,000 rpm at 4°C for 15 min to remove insoluble fractions. An aliquot of the chromatin preparation was set aside and designated as input fraction. The cleared chromatin (200 µg aliquot) was immunoprecipitated with 5 µg of antibodies and incubated overnight at 4°C with rotation. Protein A/G agarose slurry preabsorbed with salmon sperm DNA was then added and rocked overnight at 4°C. Protein A/G immune complexes were collected by brief centrifugation at 3,000 rpm and washed. Immune complexes were eluted with buffer (100 mM NaHCO3 and 1% SDS) and DNA was recovered by DNA purification columns (Qiagen), and analyzed by PCR.
Proximity Ligation Assay (PLA)
PLA was performed using the Duolink in situ PLA Detection Kit (Sigma). Briefly, D556+BAI1 (Tet-on) cells were treated with doxycycline (1 µg/ml) for 2 days to induce BAI1 expression. Cells without Dox treatment were used as negative controls. Cells were fixed with 4% paraformaldehyde for 15 min, permeabilized in 0.1% Triton-X-100 for 15 min followed by blocking with 5% goat serum for 1 hr. Cells were then incubated overnight with rabbit anti-BAI1 polyclonal antibody and mouse anti-Mdm2 monoclonal antibody. The proximity ligation reaction and visualization of signals were performed as per the manufacturer’s protocol. Hoechst staining was performed as the last step to detect cell nuclei. PLA signals, recognized as green fluorescent dots, were visualized and images were taken by confocal microscopy (Leica TCS SP8).
Biotin labeling of surface proteins
To label surface proteins, cells were washed with cold PBS containing 1 mM MgCl2 and 0.1 mM CaCl2 and incubated with sulfo-NHS-SS-biotin (0.5 mg/ml) (Thermo Fisher Scientific) in the same buffer at 4°C for 30 min. The labeling reaction was quenched by incubation with 0.1 M glycine for 10 min. Cells were washed 3 times with cold PBS and harvested in RIPA buffer containing protease inhibitors. Lysates were incubated with streptavidin agarose resins (Thermo Fisher Scientific) overnight at 4°C to isolate biotinylated proteins.
Molecular subgrouping of MB tissues
MB tumor sample subgroup affiliation was determined using 22 MB subgroup-specific gene expression profiles. 100 ng total RNA was hybridized to oligonucleotide probes and nCounter Elements TagSet (NanoString Technologies, Inc, Seattle, WA) at 67°C for 16 hr. Procedures of mRNA quantification were carried out according to the protocol from NanoString nCounter System (NanoString Technologies, Inc).
Analysis of array datasets
For RNA-seq analysis of ADGRB1 expression in 7 control cerebella and 170 primary MBs (Figure 1B), the TPM (Transcripts Per Kilobase Million) value of each sample (kindly provided by Dr. Peter Lichter and colleagues) from the (Northcott et al, 2017) dataset was plotted to make a chart with software Graphpad Prism 5.
For analysis of ADGRB1 mRNA expression in human MBs (Figure S1C), dataset GSE28245 (Remke et al., 2011) was used. It contains 64 MB samples from 4 subtypes with a pool of 24 normal cerebellum tissues as controls. The probe ID for ADGRB1 is A_23_P125056. The log2 value of each sample/normal control was obtained from the array directly and plotted for all 4 subtypes by Graphpad Prism 5.
For analysis of DNA methylation pattern in ADGRB1 gene by whole-genome bisulfite-sequencing (Figure S2D), the methylation value (kindly provided by Dr. Peter Lichter and colleagues) for 3,141 informative CpG sites across human chromosome 8:143,530,799–143,626,259 (GRCh37 coordinates) was plotted with the Spotfire software package (TIBCO Software, Palo Alto, CA).
For analysis of DNA methylation pattern in ADGRB1 gene by methylation array (Figure S2E), the β value [methylated/(methylated+unmethylated)] from 109 probes across ADGRB1 gene (29 upstream of TSS, 6 in the 5’UTR/1st exon, 72 in the gene body and 2 in the 3’UTR) was obtained from the dataset GSE54880 (Hovestadt et al., 2014). The Spotfire software package was used to generate a heatmap with sample values.
DNA methylation at 3 gene core promoter (Figure S2F) was analyzed in the DNA methylation dataset GSE54880 (Hovestadt et al., 2014). Three probes within the 5’UTR/1st exon region were randomly chosen for each gene analyzed. ADGRB1 1st exon is designated as ch8:143,545,341–143,546,343. For ADGRB1, probes were cg03924566 (5'UTR/1st exon), cg09968723 (1st exon) and cg14695492 (1st exon). For ADGRB2, probes were cg07187863 (5'UTR/1st exon), cg10773309 (5'UTR) and cg15469507 (5'UTR). For IRF6, probes were cg09509183 (TSS200), CG10074409 (5’UTR/1st exon), and cg22029157 (TSS200).
For analysis of DNA methylation/expression of ADGRB1 gene (Figure S2G), gene expression value from RNA-seq dataset (Northcott et al., 2017) and β value from DNA methylation dataset (Hovestadt et al., 2014) were used. Methylation pattern was shown in CpG island in ADGRB1 exon 1 (5 probes) in 5 normal cerebella and 169 MB samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
Results were analyzed using either two-tailed Student's t test or 2-way analysis of variance (ANOVA) in Graphpad Prism 5.0 software to assess statistical significance. p values <0.05 were considered to be statistically significant. All histogram data represent mean ± SEM (standard error of the mean).
Supplementary Material
SIGNIFICANCE.
The demonstration that BAI1 is a tumor suppressor in MB reveals a direct crosstalk between ADGR and p53 signaling, and provides a causal relationship between ADGRs and cancer. The discovery of an upstream regulator of the p53 tumor suppressor is highly significant because of this pathway’s involvement in many cancers. Alteration of this pathway through BAI1 silencing reveals a vulnerability in cancer, and provides an opportunity for therapeutic exploitation through epigenetic reactivation. We provide proof-of-principle that this can be achieved with a chemical scaffold targeting the MBD2 pathway, and this lead molecule is actionable for translation into a first-in-class therapeutic intervention against MB, and possibly other cancers.
HIGHLIGHTS.
Adgrb1 gene deficiency dramatically enhances tumor growth in a mouse MB model
BAI1 protects p53 from Mdm2-mediated degradation
ADGRB1 is silenced in human MB and can be reactivated by a MBD2 pathway inhibitor
Epigenetic reactivation of BAI1 stabilizes p53 and blocks MB growth in mice
Acknowledgments
We thank Dr. Tracy-Ann Read for providing Math1-GFP and Ptch1+/− transgenic mice, and Drs. Roger Abounader, Craig C. Castellino and Bert Vogelstein for providing human tumor cell lines and RNA samples. We thank Dr. Oskar Laur from the Emory Custom Cloning Core Facility for generation of BAI1 mutant constructs. We thank Dr. Tomoko Shofuda and Ms. Ema Yoshioka for technical help with molecular subgrouping of MBs. We thank Drs. Peter Lichter, Ivo Buchhalter, Volker Hovestadt, and Marc Zapatka for providing access to prior published MB datasets. Research reported here was supported in part by Emory University Integrated Genomics Core (EIGC) Shared Resource of the Winship Cancer Institute. This work was supported by grants from the NIH CA086335, CA163722 and NS096236 (to EGVM), CA138292 (to Winship Cancer Institute), NS055077 (to Center for Neurodegenerative Disease), CA151129 (to G.Y.G.) and the CURE Childhood Cancer Foundation, Southeastern Brain Tumor Foundation, St. Baldrick’s Foundation, and the Emory Pediatric Research Center (to EGVM). MB samples were obtained from the Pediatric Brain Tumor Repository at Children’s Healthcare of Atlanta, the University of Florida Brain Tumor Tissue Bank, the University of Alabama at Birmingham Patient Biospecimen Core and the NIH NeuroBioBank at the University of Maryland Brain and Tissue Bank.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
D.Z. and E.G.V.M. initiated the project and D.Z. performed most of the experiments. Z.Z. and J.J.O. performed the stereotactic neurosurgery. S.O. and D.Z. performed the genomic analyses on published MB datasets. Z.R. and W.N. performed the screen that identified KCC-07. L.Y. and Z.Z. established the tet-on BAI1-inducible D556-Med cells. Y.J. and C.T. performed KCC-07 PK analysis in the brain. Y.K. and R.N. provided MB samples and performed the molecular subgrouping of MBs. D.Z., N.S.D., D.B., H.Z. and S.Y. performed transgenic mice crossings and maintained the mouse colonies. T.M. provided the MB tissue array. Y.G. and S.T. provided MB samples. D.Z. and E.G.V.M. wrote the manuscript. All authors provided advice and comments on the manuscript.
DECLARATION OF INTERESTS
The authors have no competing financial interests.
References
- Abe Y, Oda-Sato E, Tobiume K, Kawauchi K, Taya Y, Okamoto K, Oren M, Tanaka N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc Natl Acad Sci U S A. 2008;105:4838–4843. doi: 10.1073/pnas.0712216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem. 2001;268:1–6. doi: 10.1046/j.1432-1327.2001.01869.x. [DOI] [PubMed] [Google Scholar]
- Bigner SH, Friedman HS, Vogelstein B, Oakes WJ, Bigner DD. Amplification of the c-myc gene in human medulloblastoma cell lines and xenografts. Cancer Res. 1990;50:2347–2350. [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- Castellino RC, Barwick BG, Schniederjan M, Buss MC, Becher O, Hambardzumyan D, Macdonald TJ, Brat DJ, Durden DL. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS One. 2010;5:e10849. doi: 10.1371/journal.pone.0010849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellino RC, De Bortoli M, Lu X, Moon SH, Nguyen TA, Shepard MA, Rao PH, Donehower LA, Kim JY. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245–256. doi: 10.1007/s11060-007-9470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017;31:737–754 e736. doi: 10.1016/j.ccell.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Cohen SN. Human MDM2 isoforms translated differentially on constitutive versus p53-regulated transcripts have distinct functions in the p53/MDM2 and TSG101/MDM2 feedback control loops. Mol Cell Biol. 2007;27:111–119. doi: 10.1128/MCB.00235-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork SM, Kaur B, Devi NS, Cooper L, Saltz JH, Sandberg EM, Kaluz S, Van Meir EG. A proprotein convertase/MMP-14 proteolytic cascade releases a novel 40 kDa vasculostatin from tumor suppressor BAI1. Oncogene. 2012;31:5144–5152. doi: 10.1038/onc.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bont JM, Packer RJ, Michiels EM, den Boer ML, Pieters R. Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro Oncol. 2008;10:1040–1060. doi: 10.1215/15228517-2008-059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7:79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Foulks JM, Parnell KM, Nix RN, Chau S, Swierczek K, Saunders M, Wright K, Hendrickson TF, Ho KK, McCullar MV, et al. Epigenetic drug discovery: targeting DNA methyltransferases. J Biomol Screen. 2012;17:2–17. doi: 10.1177/1087057111421212. [DOI] [PubMed] [Google Scholar]
- Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Hamann J, Aust G, Arac D, Engel FB, Formstone C, Fredriksson R, Hall RA, Harty BL, Kirchhoff C, Knapp B, et al. International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev. 2015;67:338–367. doi: 10.1124/pr.114.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68:1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, Klibanov AL, Yan Z, Mandell JW, Ravichandran KS. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature. 2013;497:263–267. doi: 10.1038/nature12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510:537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10:319–331. doi: 10.1038/nrc2818. [DOI] [PubMed] [Google Scholar]
- Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: Choosing the right tool for the job. J Biotechnol. 2016;236:10–25. doi: 10.1016/j.jbiotec.2016.07.028. [DOI] [PubMed] [Google Scholar]
- Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- Kaur B, Brat DJ, Devi NS, Van Meir EG. Vasculostatin, a proteolytic fragment of brain angiogenesis inhibitor 1, is an antiangiogenic and antitumorigenic factor. Oncogene. 2005;24:3632–3642. doi: 10.1038/sj.onc.1208317. [DOI] [PubMed] [Google Scholar]
- Kaur B, Cork SM, Sandberg EM, Devi NS, Zhang Z, Klenotic PA, Febbraio M, Shim H, Mao H, Tucker-Burden C, et al. Vasculostatin inhibits intracranial glioma growth and negatively regulates in vivo angiogenesis through a CD36-dependent mechanism. Cancer Res. 2009;69:1212–1220. doi: 10.1158/0008-5472.CAN-08-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21:168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenotic PA, Huang P, Palomo J, Kaur B, Van Meir EG, Vogelbaum MA, Febbraio M, Gladson CL, Silverstein RL. Histidine-rich glycoprotein modulates the anti-angiogenic effects of vasculostatin. Am J Pathol. 2010;176:2039–2050. doi: 10.2353/ajpath.2010.090782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lau J, Schmidt C, Markant SL, Taylor MD, Wechsler-Reya RJ, Weiss WA. Matching mice to malignancy: molecular subgroups and models of medulloblastoma. Childs Nerv Syst. 2012;28:521–532. doi: 10.1007/s00381-012-1704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, Stunnenberg HG. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. 2006;26:843–851. doi: 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EY, Ji H, Ouyang Z, Zhou B, Ma W, Vokes SA, McMahon AP, Wong WH, Scott MP. Hedgehog pathway-regulated gene networks in cerebellum development and tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:9736–9741. doi: 10.1073/pnas.1004602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumpkin EA, Collisson T, Parab P, Omer-Abdalla A, Haeberle H, Chen P, Doetzlhofer A, White P, Groves A, Segil N, et al. Math1-driven GFP expression in the developing nervous system of transgenic mice. Gene Expr Patterns. 2003;3:389–395. doi: 10.1016/s1567-133x(03)00089-9. [DOI] [PubMed] [Google Scholar]
- Mahindra A, Laubach J, Raje N, Munshi N, Richardson PG, Anderson K. Latest advances and current challenges in the treatment of multiple myeloma. Nat Rev Clin Oncol. 2012;9:135–143. doi: 10.1038/nrclinonc.2012.15. [DOI] [PubMed] [Google Scholar]
- Malek R, Matta J, Taylor N, Perry ME, Mendrysa SM. The p53 inhibitor MDM2 facilitates Sonic Hedgehog-mediated tumorigenesis and influences cerebellar foliation. PLoS One. 2011;6:e17884. doi: 10.1371/journal.pone.0017884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momota H, Shih AH, Edgar MA, Holland EC. c-Myc and beta-catenin cooperate with loss of p53 to generate multiple members of the primitive neuroectodermal tumor family in mice. Oncogene. 2008;27:4392–4401. doi: 10.1038/onc.2008.81. [DOI] [PubMed] [Google Scholar]
- Nishimori H, Shiratsuchi T, Urano T, Kimura Y, Kiyono K, Tatsumi K, Yoshida S, Ono M, Kuwano M, Nakamura Y, et al. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene. 1997;15:2145–2150. doi: 10.1038/sj.onc.1201542. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Grobner S, Segura-Wang M, Zichner T, Rudneva VA, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–317. doi: 10.1038/nature22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012a;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C, Eberhart CG, Dubuc A, Guettouche T, Cardentey Y, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012b;123:615–626. doi: 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Read TA, Kessler JD, Mehmeti A, Wells JF, Huynh TT, Lin SM, Wechsler-Reya RJ. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development. 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012;21:155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert Z. Targeting epigenetic repression by interfering with methyl -binding domain protein function. Dissertation Advisor: Dr William G Nelson; 2010. [Google Scholar]
- Remke M, Hielscher T, Korshunov A, Northcott PA, Bender S, Kool M, Westermann F, Benner A, Cin H, Ryzhova M, et al. FSTL5 is a marker of poor prognosis in non-WNT/non-SHH medulloblastoma. J Clin Oncol. 2011;29:3852–3861. doi: 10.1200/JCO.2011.36.2798. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;162:454. doi: 10.1016/j.cell.2015.06.053. [DOI] [PubMed] [Google Scholar]
- Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratsuchi T, Nishimori H, Ichise H, Nakamura Y, Tokino T. Cloning and characterization of BAI2 and BAI3, novel genes homologous to brain-specific angiogenesis inhibitor 1 (BAI1) Cytogenet Cell Genet. 1997;79:103–108. doi: 10.1159/000134693. [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- Smoll NR. Relative survival of childhood and adult medulloblastomas and primitive neuroectodermal tumors (PNETs) Cancer. 2012;118:1313–1322. doi: 10.1002/cncr.26387. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Tabori U, Baskin B, Shago M, Alon N, Taylor MD, Ray PN, Bouffet E, Malkin D, Hawkins C. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28:1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]
- Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade PA. Methyl CpG binding proteins: coupling chromatin architecture to gene regulation. Oncogene. 2001;20:3166–3173. doi: 10.1038/sj.onc.1204340. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci. 2001;24:385–428. doi: 10.1146/annurev.neuro.24.1.385. [DOI] [PubMed] [Google Scholar]
- Weeraratne SD, Amani V, Neiss A, Teider N, Scott DK, Pomeroy SL, Cho YJ. miR-34a confers chemosensitivity through modulation of MAGE-A and p53 in medulloblastoma. Neuro Oncol. 2011;13:165–175. doi: 10.1093/neuonc/noq179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513–516. [PubMed] [Google Scholar]
- Whittier KL, Boese EA, Gibson-Corley KN, Kirby PA, Darbro BW, Qian Q, Ingram WJ, Robertson T, Remke M, Taylor MD, et al. G-protein coupled receptor expression patterns delineate medulloblastoma subgroups. Acta Neuropathol Commun. 2013;1:66. doi: 10.1186/2051-5960-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyhs N, Walker D, Giovinazzo H, Yegnasubramanian S, Nelson WG. Time-Resolved Fluorescence Resonance Energy Transfer Assay for Discovery of Small-Molecule Inhibitors of Methyl-CpG Binding Domain Protein 2. J Biomol Screen. 2014;19:1060–1069. doi: 10.1177/1087057114526433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Hunter SB, Vertino PM, Van Meir EG. Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res. 2011;71:5859–5870. doi: 10.1158/0008-5472.CAN-11-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Li C, Swanson AM, Villalba RM, Guo J, Zhang Z, Matheny S, Murakami T, Stephenson JR, Daniel S, et al. BAI1 regulates spatial learning and synaptic plasticity in the hippocampus. J Clin Invest. 2015;125:1497–1508. doi: 10.1172/JCI74603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Yang Z, Luo Z, Luo S, Xiong WC, Mei L. Muscle-specific receptor tyrosine kinase endocytosis in acetylcholine receptor clustering in response to agrin. J Neurosci. 2008;28:1688–1696. doi: 10.1523/JNEUROSCI.4130-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC, Castelo-Branco P, Baskin B, Ray PN, Bouffet E, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31:2927–2935. doi: 10.1200/JCO.2012.48.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.