

Abstract
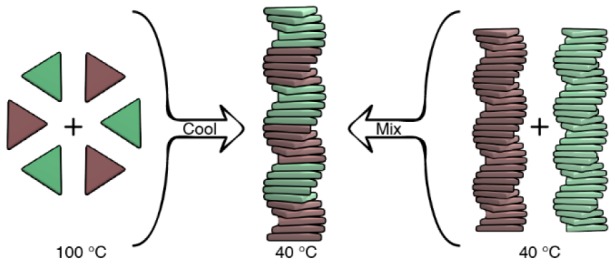
Supramolecular block copolymers are becoming attractive materials in nascent optoelectronic and catalytic technologies. However, their dynamic nature precludes the straightforward tuning and analysis of the polymer’s structure. Here we report the elucidation on the microstructure of triarylamine triamide-based supramolecular block copolymers through a comprehensive battery of spectroscopic, theoretical, and super-resolution microscopic techniques. Via spectroscopic analysis we demonstrate that the direct mixing of preassembled homopolymers and the copolymerization induced by slow cooling of monomers lead to the formation of the same copolymer’s architecture. The small but pronounced deviation of the experimental spectra from the linear combination of the homopolymers’ spectra hints at the formation of block copolymers. A mass balance model is introduced to further unravel the microstructure of the copolymers formed, and it confirms that stable multiblock supramolecular copolymers can be accessed from different routes. The multiblock structure of the supramolecular copolymers originates from the fine balance between favorable hydrogen-bonding interactions and a small mismatch penalty between two different monomers. Finally, we visualized the formation of the supramolecular block copolymers by adapting a recently developed super-resolution microscopy technique, interface point accumulation for imaging in nanoscale topography (iPAINT), for visualizing the architectures formed in organic media. Combining multiple techniques was crucial to unveil the microstructure of these complex dynamic supramolecular systems.
Introduction
The widespread employment of nanotechnologies has stimulated the development of high-performance, nano-ordered materials.1−6 Supramolecular polymers are a compelling platform for introducing diverse functionalities and long-range order.7−10 Their intrinsic self-organizing properties offer the possibility of creating finely tuned dynamic microstructures that are simply not possible with conventional covalent polymers. This potential has motivated rapid progress in developing fundamental principles for designing one-dimensional supramolecular polymers, such as “sergeant and soldiers” chirality amplification11 and supramolecular living polymerization.12−16 Concurrently, theoretical models have been developed to describe supramolecular (co)polymerization17−22 and pathway complexity23−25 of these systems. A crucial step toward competitive functional materials requires control over the sequence of different monomers held together through noncovalent heterointeractions in a supramolecular copolymer. Such control may represent an easy strategy to achieve p–n junctions,26 FRET systems,27 and biosensors.28 Recently, kinetically controlled supramolecular block copolymers have been reported with different microstructures, such as AmBn,26 (ABA)n,29 and 1/2D block nanocrystals.30,31 Nevertheless, the synthesis and characterization of well-defined block structures under thermodynamic control have been elusive.
A promising couple for obtaining functional supramolecular block copolymers is found in triarylamine triamide-based monomers. In the last years, triarylamine-based homopolymers were reported to assemble into potential semiconductive supramolecular fibers under various conditions.32−34 Lately, we have elucidated the mechanism of homopolymerization of S-1 and S-2 (Figure 1, Scheme S1)35 and reported their potential as chiral supramolecular spin filters in water-splitting solar cells.36 Their similar molecular geometry and analogous behavior upon polymerization make the two monomers promising candidates for copolymerization. Additionally, we expect that the small conformational difference35 between the supramolecular homopolymers poly(S-1) and poly(S-2) will result in a modest mismatch penalty. Together with a hydrogen bond directionality in the polymer formed, we anticipate that the copolymer poly[(S-1)x-co-(S-2)(1–x)] can exhibit a multiblock architecture as a result of the balanced H-bonding interactions between preferred homopolymer segments and a limited number of hetero-monomer couplings in the polymer.
Figure 1.
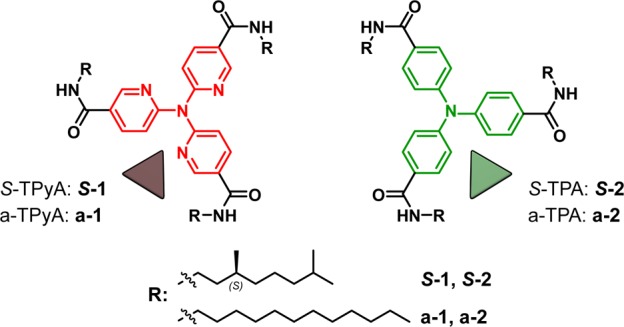
Chemical structures of tri(pyrid-2-yl)amine triamide (1) and triphenylamine triamide (2), with a chiral (S)-3,7-dimethyloctyl chain (S-1, S-2) and achiral dodecyl chain (a-1, a-2).
Herein, we report the noncovalent synthesis of triarylamine triamide-based supramolecular copolymers. Through a combination of spectroscopic, theoretical, and super-resolution microscopic techniques, we unambiguously demonstrate that these copolymers exhibit a stable multiblock architecture.
Results and Discussion
Spectroscopic Study of the Supramolecular Copolymers
The supramolecular copolymerization between S-1 and S-2 was first evaluated by recording the spectroscopic variations upon mixing preassembled homopolymers, poly(S-2) to poly(S-1). As reported earlier,35 both poly(S-1) and poly(S-2) form via a cooperative mechanism of two assembled states with opposite helicity, state I and state II, as a function of temperature (i.e., both the homopolymers poly(S-1) and poly(S-2) display two states characterized by opposite Cotton effects at λ = 333 nm and λ = 350 nm, respectively) (Figure S1). Recent results revealed that the transition of state I into state II, which occurs at T < 20 °C, is caused by the interaction of supramolecular polymers with codissolved water in alkanes.37 To avoid the complexity that arises from this additional interaction, we performed the copolymerization under thermodynamic control in state I above 20 °C in decalin.
First, we added poly(S-2) to poly(S-1) at 40 °C in a stepwise manner, leading to poly[(S-1)x-co-(S-2)(1–x)], where x and (1–x) are the feed ratios of S-1 and S-2, respectively (Figure 2a). After each addition, followed by equilibration of the solution, we recorded UV–vis, CD, and fluorescence spectra (Figures 2b,c and S2). The resulting CD spectra display a linear transition from poly(S-1) to poly(S-2). The linear combination of the two CD spectra of the homopolymers—calculated assuming no interaction between the two homopolymers—is similar but not identical to the experimental curves. A small but clear deviation at λ = 297 nm (Figures S3, S4) is observed.
Figure 2.

Spectroscopic analysis of the copolymerization achieved via stepwise addition of supramolecular homopolymers. (a) Schematic representation of the experiment performed. CD (b) and fluorescence (c) spectra of poly[(S-1)x-co-(S-2)(1–x)] obtained by stepwise addition of poly(S-2) (green lines) to poly(S-1) (red lines) at 40 °C (decalin, cS-1 = cS-2 = 50 μM). The gray lines represent the different steps performed with different percentage of poly(S-2) added, from poly[(S-1)0.8-co-(S-2)0.2] (lightest gray) to poly[(S-1)0.24-co-(S-2)0.76] (darkest gray). Poly[(S-1)0.53-co-(S-2)0.47] is reported as a light blue line.
To investigate if the deviation between the experimental and the calculated CD spectra of poly[(S-1)0.5-co-(S-2)0.5] is related to an interaction between the two homopolymers, fluorescence (Figures 2c and S2–S4) and 1H NMR experiments (Figures S5–S13) were performed. The fluorescence measurements, performed during the stepwise addition of poly(S-2) to poly(S-1) in decalin at 40 °C, display a sharp change in the emission band already for poly[(S-1)0.8-co-(S-2)0.2] (Figure 2c, lightest gray curve). In this case, the comparison of the measured emission with the linear combination of the emission of the homopolymers reveals the absence of the shoulder at λ = 360 nm (attributable to poly(S-1)) and the dominance of poly(S-2) emission features (Figures 2c and S4). This indicates the presence of supramolecular interactions between the two homopolymers, which affect the electronic levels involved in the emission.
1H NMR spectra were recorded in deuterated chloroform (CDCl3). Because H-bond-driven assembly in chloroform is weaker than in alkane solvents, the experiments were performed at −40 °C. The shift of the aromatic and amide peaks in the copolymer, compared to the ones recorded for separate poly(S-1) and poly(S-2) (Figures S5–S8), is indicative of coaggregation of S-1 and S-2. In addition, the 1H NOE spectra of the mixed S-1 and S-2 solution, acquired under the same conditions, showed the presence of a noncovalent heterointeraction, which is revealed by a negative Overhauser effect of both molecules while irradiating at specific signals of one of the two monomers (Figures S9–S12).39 Since the conditions of 1H NMR are not fully comparable with the ones used for the spectroscopic measurements, we further tested the co-interaction between the two monomers performing a “mixed” sergeant and soldier experiment (Figures S14, S15).40 This time, we mixed achiral poly(a-2) with poly(S-1) in a 1:1 ratio at 40 °C in decalin and recorded the resulting CD spectra. Although the kinetics are slow compared to the S-1:S-2 couple (Figure S15a,b), the mixed sergeant and soldiers experiment reveals chirality transfer from poly(S-1) to poly(a-2). Since S-1 and a-2 have different spectroscopic features, the increase of the CD band related to a-2 is symptomatic of a co-interaction, indicating the presence of both monomers in the same aggregate.
From the results above it becomes clear that upon mixing preassembled homopolymers, a co-interaction occurs excluding the possibility of self-sorting.38 However, its effect on the CD spectra is subtle. This means that the interaction does not significantly interfere with the supramolecular structure of the original homopolymers, excluding as well the possibility of an alternate and random organization.
To further investigate the mechanism of formation of copolymers, we performed the copolymerization via slow cooling of monomers (Figures 3 and S16–S18). S-1 and S-2 were mixed in a 1:1 ratio and monomerically dissolved at 100 °C. Subsequently slow cooling (cooling rate = 15 °C h–1) of the solution induces the supramolecular copolymerization of poly[(S-1)0.50-co-(S-2)0.50] under thermodynamic control (Figure 3a). The variation of the CD value at λ = 341 nm (CD maximum of poly[(S-1)0.50-co-(S-2)0.50] at 40 °C) as a function of temperature permits elucidating the mechanism of copolymerization (Figure 3b).41 During cooling and copolymerization, full UV–vis, fluorescence (Figures S17, S18), and CD spectra are registered every 5 degrees (Figure 3c). This allows an overview of the thermal effect on the CD spectrum of the copolymer.
Figure 3.

Spectroscopic analysis of the copolymerization achieved via slow cooling monomers. (a) Schematic representation of the experiment performed. (b) CD cooling curves (λ = 341 nm, cooling rate = 15 °C h–1) of poly[(S-1)0.5-co-(S-2)0.5] (decalin, ctot = 50 μM) (blue line), poly(S-1) (red line), poly(S-2) (green line), and the linear sum of [poly(S-1) + poly(S-2)] assuming no interaction (decalin, cS-1 = cS-2 = 25 μM) (black dotted line). (c) CD spectra of poly[(S-1)0.5-co-(S-2)0.5] recorded while cooling (cooling rate = 15 °C h–1) and copolymerizing. Spectra acquired every 5 degrees from 100 °C (red line) to 40 °C (blue line).
The cooling curve recorded discloses the formation of the copolymer displaying a cooperative mechanism and the elongation temperature (Te) at 85 °C. Interestingly, the Te of poly[(S-1)0.5-co-(S-2)0.5] (ctot = 50 μM, cS-1 = cS-2 = 25 μM) coincides with the Te of poly(S-1) (cS-1 = 25 μM) (Figure 3b, blue line vs red line). This indicates that the nuclei of poly[(S-1)0.5-co-(S-2)0.5] coincide with the nuclei of poly(S-1). The presence of one single Te is a clear indication of the interaction occurring between S-1 and S-2. Indeed, in the case of independent formation of poly(S-1) and poly(S-2) (estimated by the linear sum of the individual cooling curves of poly(S-1) and poly(S-2) at c = 25 μM), two transitions with different Te would be present in the cooling curve (Figure 3b, black dotted line). The single Te and the coincidence of it with the Te of poly(S-1) reveal that the copolymer nucleates from S-1 nuclei and elongates copolymerizing S-1 with S-2 monomers.
We performed the same cooling experiment on poly[(S-1)0.5-co-(a-2)0.5] (Figure S15c,d). The cooling curve and the Te = 85 °C matched those of poly[(S-1)0.5-co-(S-2)0.5], further supporting the hypothesis that S-1 oligomers act as nuclei for the copolymerization with S-2 or a-2.
To understand whether the copolymerization strategies used play a role in the resulting microstructure, we compared the CD and the fluorescence spectra of poly[(S-1)0.5-co-(S-2)0.5] obtained at 40 °C via addition of homopolymers with the ones measured under thermodynamic control via slow cooling (Figures 4 and S19). Strikingly, both the CD (Figure 4b) and the fluorescence (Figure 4c) spectra perfectly overlap, indicating that the same copolymer can be formed via different pathways, and it is stable over time. The single elongation temperature and the CD spectrum similar but not identical to the linear combination of the homopolymers support the hypothesis of the formation of a block-like copolymer structure. This is analogous to covalent block copolymers, where some spectroscopic features of the homopolymers are conserved and linearly combined in the corresponding block copolymer.42 According to this hypothesis, we speculate that the small deviation observed at λ = 297 nm (Figure 4c) from the CD spectrum of the linear combination is the result of small conformational changes required for the co-interaction of S-1 with S-2.
Figure 4.

Comparison of the spectroscopic features of the copolymers obtained via the two copolymerization strategies (decalin, 40 °C, ctot = 50 μM). (a) Schematic representation of the proposed copolymerization model, (b) CD diagnostic band, and (c) normalized emission spectra. The CD and the emission spectra obtained by addition of homopolymers at 40 °C (light blue lines) coincide with the ones recorded via cooling of monomers (blue lines). The experimental spectra deviate from the linear combination of the homopolymers’ spectra (black dotted lines) in the CD band at 297 nm and in the fluorescence band at 360 nm (linear combination spectra obtained assuming no interaction as [0.5 × poly(S-1) + 0.5 × poly(S-2)] with cS-1 = cS-2 = 50 μM).
To get more insight into how the two monomers are incorporated in the copolymers, we simultaneously analyzed the variation of the CD intensities at multiple wavelengths of poly(S-1), poly(S-2), and poly[(S-1)0.5-co-(S-2)0.5] while cooling (Figure 5a and Figures S20, S21). Following the CD intensities at λ = 333 and 350 nm (i.e., corresponding to the CD maxima of poly(S-1) and poly(S-2), respectively) (Figure 5a top) allows decomposing the CD cooling curve of poly[(S-1)0.5-co-(S-2) 0.5] (Figure 5a bottom and Figure S20) intensities into contributions of poly(S-1) and poly(S-2) and thus calculate the amounts of S-1 and S-2 in the copolymer chain as a function of temperature (Figure 5b, solid dots; see Supporting Information Section 8 for details). As expected, at higher temperatures the copolymer consists primarily of S-1 units, while at 20 °C the effective ratio of the monomers in poly[(S-1)0.5-co-(S-2)0.5] equilibrates to 0.5:0.5, in line with the feed ratio. Additionally, we observe that the incorporation of S-1 occurs rapidly, while S-2 incorporates in a more gradual manner, but it begins to copolymerize at higher temperatures (Figure 5b, green dots) compared to its homopolymer poly(S-2) (Figure 3b, green line). This further provides evidence that S-1 and S-2 do not polymerize independently of each other.
Figure 5.

(a, top) CD spectra of poly(S-1) (red line), poly(S-2) (green line), and poly[(S-1)0.5-co-(S-2)0.5] (blue line) at 20 °C and (bottom) CD spectra (gray lines) of poly[(S-1)0.5-co-(S-2)0.5] from 100 to 20 °C (blue line). Vertical dashed lines for λ = 333 and 350 nm (CD maxima of poly(S-1) and poly(S-2), respectively) are wavelengths used for the decomposition of the CD spectra of poly[(S-1)0.5-co-(S-2)0.5] while cooling and polymerizing. (b) Normalized concentration of monomers in the copolymers (computed via CD spectra decomposition) for poly[(S-1)0.5-co-(S-2)0.5] (solid dots) and the simulation for poly[(A)0.5-co-(B)0.5] with ΔHAB = −37 kJ mol–1 (dashed lines). (c) Simulated cooling curves for poly[(A)0.5-co-(B)0.5] (ΔHAA = −53 kJ mol–1, NPA= −40 kJ mol–1, ΔHBB = −50 kJ mol–1, NPB= −20 kJ mol–1, ΔS = −0.06 kJ mol–1, ΔHAB variable, ctot = 50 μM, cA = cB = 25 μM). (d) Evolution of the fraction of A–B bonds in time from a starting homopolymer state (light blue crosses) and for the monomerically dispersed state (blue crosses) for ΔHAB = −37 kJ mol–1 at 50 °C for cA = cB = 25 μM. Both curves level off at 0.15, indicating a block-like structure (in a random copolymer the fraction of A–B bonds would level off at 0.5). (e) Section of multiblock copolymers obtained by stochastic analysis for ΔHAB = −37 kJ mol–1 at 50 °C for cA = cB = 25 μM.
Modeling of Supramolecular Block Copolymer Formation
Recently Das et al.40 showed that theoretical modeling integrated in the study of spectroscopic data helps elucidate the composition in supramolecular copolymers. Aiming to implement the microstructure analysis, we expanded this model to take into account copolymerization of monomers that individually form distinctly cooperative aggregates (Supporting Information Section 9). The main idea of the model is that homobonds (i.e., noncovalent bonds between two equal monomers) in the copolymer behave equal to those in their respective homopolymer and that the copolymerization can thus be fully described by the free energy gain of the formation of a heterobond (i.e., noncovalent bond between two different monomers).
Since it was not possible to reliably fit the CD cooling curve of the homopolymers poly(S-1) and poly(S-2) over a wide concentration range, we used hypothetical polymers poly(A) and poly(B), and we selected thermodynamic parameters (ΔHAA = −53 kJ mol–1, NPA = −40 kJ mol–1, ΔHBB = −50 kJ mol–1, NPB = −20 kJ mol–1, ΔS = −0.06 kJ mol–1), including different cooperativities, which give rise to calculated homopolymerization curves that resemble the experimental curves at 25 μM of poly(S-1) and poly(S-2), respectively (Figure S22). Next, we generated a series of theoretical copolymerization curves by varying the enthalpic interaction between subsequent A and B units (ΔHAB) in the copolymer (Figures 5c and S22) and calculated their number distribution (Figure S23). For a weak A–B interaction (ΔHAB = −25 kJ mol–1), the cooling curve resembles the linear combination of cooling curves of the homopolymers (Figures 3c, 5c, black line), while for relatively strong A–B interaction (ΔHAB = −42 kJ mol–1), the elongation temperature increases significantly (Figure 5c, pink line). For an intermediate A–B interaction of −37 kJ mol–1 the cooling curve shows the typical linear dependence (Figure 5c, blue line) that was also observed experimentally (Figure 3b, blue line).
For this interaction strength, we also plotted the temperature-dependent degree of polymerization, as well as the amounts of A and B in the copolymers as predicted by the model (Figure 5b, dashed lines). This closely matches the composition as extracted by the CD curves (Figure 5b, solid dots). In order to determine the microstructure predicted by the model, we also performed stochastic simulations.43 The simulated microstructure of the copolymer obtained by varying ΔHAB ranged from self-sorted to blocks, to random and alternate copolymers (Figures S24, S25). The enthalpic gain of the co-interaction, ΔHAB, and the cooperativities, related to the values of NPA and NPB, are the main variables that determine the copolymer’s microstructure and the block length, respectively.
The experimental cooling curve of poly[(S-1)0.5-co-(S-2)0.5] (and the relative dependence of monomer composition as a function of temperature) best resembles the simulated curve that exhibits a multiblock structure (Figure 5c,d,e). This match agrees remarkably well with the hypothesis of the formation of supramolecular multiblock copolymers under thermodynamic equilibrium conditions.
In addition, the stochastic simulation allows investigating the evolution of the fraction of A–B bonds (namely, the number of A–B contacts in the copolymer over the total number of contacts) in time starting from different scenarios. We simulated the fraction of A–B bonds during the copolymerization starting either with supramolecular homopolymers or with molecularly dissolved monomers (Figures 5d and S24, S25). The fraction of A–B bonds is shown from t = 0.001 s for computational reasons, and the formation of the first contacts (e.g., A–A, A–B, B–B) is necessary to get a value of the fraction of A–B bonds for the molecularly dissolved case. In line with the spectroscopic data (vide supra), the stochastic simulations highlight the convergence of the curves from two different starting points to the same value of A–B bonds. This occurs for all the different copolymerization scenarios in a reasonable amount of time. For the selected case of ΔHAB = −37 kJ mol–1 the convergence to the value of 0.15 for the fraction of A–B bonds occurred in ∼6 min at 50 °C. This value of 0.15 is indicative of a multiblock copolymer structure, as a value of 0.5 is expected for a random copolymer (Figure S25b). This evidence further supports the possibility of achieving stable supramolecular block architectures under thermodynamic control (Figure 5d,e).
Visualization of Supramolecular Block Copolymers by iPAINT
To confirm the proposal (based on spectroscopy and simulations) of a multiblock structure, we employed a recently developed super-resolution microscopy technique, interface point accumulation for imaging in nanoscale topography (iPAINT),44−46 to visualize the copolymer obtained. iPAINT allows imaging in organic solvent by single-molecule localization with a spatial resolution on the order of ∼20 nm. Exploiting the spontaneous physisorption of the dyes to the 1D supramolecular polymers,46 iPAINT does not require the synthesis of ad-hoc dye-functionalized monomers. Although the measurements are performed in conditions that render a direct comparison with the spectroscopic data difficult, this microscopy technique can provide visual evidence for the blocky character of the copolymer.
Hence, we stained individually the supramolecular homopolymers assembled in methylcyclohexane (MCH) with photoactivatable caged dyes: Cage-635 for poly(S-1) giving poly(S-1)Cage-635, and Cage-552 for poly(S-2) giving poly(S-2)Cage-552. Successively, the prestained homopolymers poly(S-1)Cage-635 and poly(S-2)Cage-552 are mixed in a 1:1 ratio at 40 °C (Figure 6a). The low solubility of the dyes in MCH ensures the dye–polymers correspondence imposed by prestaining—which is fundamental for the experiment—without hampering the intrinsic dynamicity of the supramolecular fibers.45 The solution is then injected in the sample chamber, and the imaging is performed once the fibers are physisorbed on the coverslip. At this point the dynamicity is suppressed and no further exchange occurs.
Figure 6.

(a) Schematic representation of staining (poly(S-1) with Cage-635 giving poly(S-1)Cage-635 and poly(S-2) with Cage-552 giving poly(S-2)Cage-552) and mixing at 40 °C (cS-1 = cS-2 = 200 μM in MCH, 0.5% v/v Cage-dye c = 10 mM in DMSO, 1% i-PrOH). (b) iPAINT images of supramolecular copolymers, reconstructed images of merged channels, and single Cage-635 and Cage-552 channels.
Remarkably, iPAINT revealed the presence of red and green bi- and triblock fibers, confirming our hypothesis of formation of supramolecular block copolymers. Differently from what we observed when mixing the same homopolymer oppositely stained (e.g., poly(S-1)Cage-552 + poly(S-1)Cage-635), in which a full reconstruction of the fiber in the two channels is obtained,45 the addition of poly(S-2)Cage-552 to poly(S-1)Cage-635 confirmed the formation of supramolecular block copolymers poly[(S-1)Cage-6350.5-co-(S-2)Cage-5520.5] in alkane solvents (Figure 6b) and the capability to discern between random aggregates and block architectures.45
We noticed that the block length observed in the iPAINT experiments differs from the one estimated by the model. This discrepancy likely results from the different conditions used between spectroscopic and microscopy experiments. iPAINT is performed in more concentrated conditions and in the presence of 1% 2-propanol (i-PrOH), 0.5% of dimethyl sulfoxide (DMSO), and the dye. The imaging is then acquired on the fibers physisorbed on a glass coverslip, which is fundamental to achieve reconstructed images with high spatial resolution.47
To evaluate the impact of i-PrOH and DMSO on the fiber assembly, we performed CD spectroscopy on the supramolecular polymers in the same conditions as used for iPAINT imaging (c = 200 μM in MCH, 0.5% DMSO, 1% i-PrOH v/v) (Figure S26). For poly(S-1) and poly(S-2) the shape of the CD spectrum (Figure S26a,b) was unaltered, although the CD intensity was reduced. This indicates partial denaturation of the assemblies upon addition of the polar solvents.41,48
The stepwise addition of poly(S-2) to poly(S-1) in iPAINT conditions performed at 40 °C was also evaluated by CD spectroscopy, and poly[(S-1)0.5-co-(S-2)0.5] displayed the same CD features as those obtained in pure MCH (Figure S26c). It is worth noticing that the presence of i-PrOH seems to affect the copolymer more than the homopolymers. The spectrum recorded in the absence of polar solvents displays a CD maximum at the same wavelength (λ = 341 nm) at the intensity of the crossing point of the homopolymers’ CD curves as expected (Figures 2b, S26a). Conversely, the copolymer’s CD spectrum recorded in the presence of DMSO and i-PrOH is partially decreased in intensity (Figure S26b). This indicates that the denaturing effect of i-PrOH/DMSO is enhanced in the copolymer when compared to the relative homopolymers. This result is consistent with the data obtained with the mass balance model, where the enthalpic gain of heterointeractions (ΔHAB) is less favorable than the homointeractions (ΔHAA and ΔHBB) (Figures 5c and S22). This means that the A–B contacts are easier to break than A–A or B–B and more affected by i-PrOH and DMSO. On the basis of this, we speculate that the addition of 0.5% DMSO and 1% i-PrOH (v/v) partially favors the homointeractions making bi- or triblock copolymers more stable than multiblock copolymers. Despite the differences induced by the denaturing effect, the system is still stable enough to copolymerize, demonstrating once more the reproducible tendency of the system to form block copolymers under different conditions.
Kinetic Spectroscopic and iPAINT Studies
Finally, we explored the kinetics involved in the formation of poly[(S-1)0.5-co-(S-2)0.5] by adding poly(S-2) to poly(S-1) at 20 °C (Figure 7a). The addition at this temperature resulted in a CD spectrum perfectly superimposable with the linear combination of the homopolymers, also in the diagnostic band (Figure 7b, left). Subsequent heating to 40 °C led to a transition visible in the CD diagnostic band as a flattening (Figure 7b, right) and in the heating curve as an additional transition at ∼27 °C (Figure 7c). Subsequently, in the emission spectra a loss of the shoulder at λ = 360 nm and an increase in emission intensity were observed (Figure S27).
Figure 7.

(a) Addition at 20 °C, kinetic trap of self-sorted homopolymers and consequent evolution to poly[(S-1)0.5-co-(S-2)0.5] by heating. (b) CD diagnostic band of (left) self-sorted trapped state at 20 °C and evolution to poly[(S-1)0.5-co-(S-2)0.5] (right) at 40 °C (decalin ctot = 50 μM). Comparison of the experimental results (light blue lines) with the linear combination spectra (black dotted lines) (c) CD heating curves followed at λ = 341 nm for the originally self-sorted solution [0.5 × poly(S-1) + 0.5 × poly(S-2)] (light blue curve) and for the supramolecular copolymer poly[(S-1)0.5-co-(S-2)0.5] (blue curve).
The CD spectrum recorded at 40 °C perfectly overlaps with the one recorded via slow cooling of monomers. We attribute this to the possibility to kinetically trap the “self-sorted” homopolymers by mixing at 20 °C. Subsequently, giving thermal energy to the system or equilibrating (Figure S28), the metastable mixed state [0.5 × poly(S-1) + 0.5 × poly(S-2)] transforms into poly[(S-1)0.5-co-(S-2)0.5] block copolymers.
To confirm this hypothesis, we imaged with iPAINT the evolution of the mixture prepared at 20 °C at different time lapses. By mixing the stained supramolecular homopolymer at 20 °C (Figure 8a) and directly imaging them, we verified the self-sorting at low temperatures (Figure 8b). By repeating the imaging on the mixed solution equilibrated for 1 h at 20 °C we confirmed that copolymerization occurred (Figure 8c).
Figure 8.
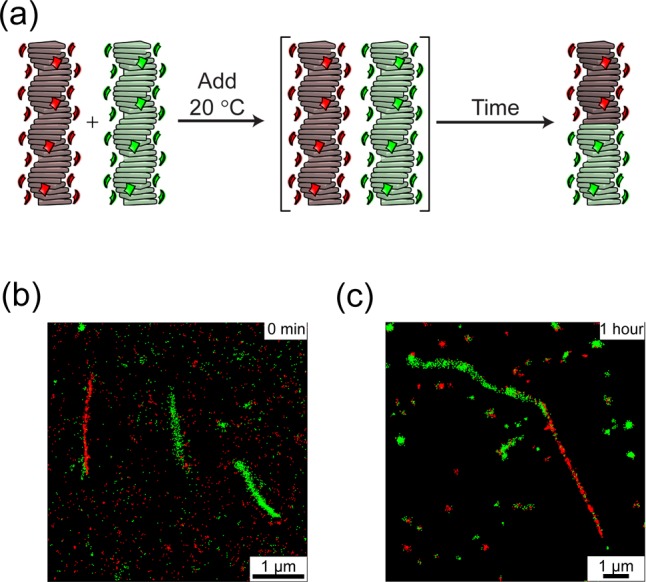
(a) Addition at 20 °C, kinetic trap of self-sorted homopolymers, and consequent evolution to poly[(S-1)Cage-6350.5-co-(S-2)Cage-5520.5] by equilibration at 20 °C. iPAINT image at (b) t = 0 min and (c) 1 h after the addition (cS-1 = cS-2 = 200 μM in MCH, 0.5% v/v Cage-dye c = 10 mM in DMSO, 1% v/v i-PrOH).
Conclusions
Taken all together, the results presented above clearly indicate the formation of supramolecular block copolymers under thermodynamic equilibrium conditions. The strategy applied here is generally applicable to create supramolecular block copolymers. This approach takes advantage of the balance between the mismatch penalty that hampers the complete mixing and the enthalpic cost of the chain-ends that drives co-interactions. In a similar way that propagation of errors in olefin polymerization occurs,49 blocky structures can be formed.
On the basis of the above, we can list (some of) the requirements needed for monomer couples to obtain multiblock supramolecular copolymers. Besides the symmetry prerequisite needed to achieve successful interactions among the different monomers, such as H-bonding, the monomer couple has to display a similar cooperative homopolymerization and a moderately small enthalpic gain. This last factor, unique for every couple, is a combination of multiple energies involved, for example, the mismatch penalties paid for the conformational change required to interact with the other type of monomers as well as positive contributions (e.g., H-bonding formation, donor–acceptor interactions). The enthalpic gain defines the formation of a certain microstructure (from alternate to self-sorted). The more negative the value, the more favorable the interaction, which leads to random or alternated structure. Oppositely, multiblocks and block copolymers are formed when ΔHAB is much less negative than the enthalpy for the relative homopolymers (|ΔHAB| ≪ |ΔHAA| and |ΔHAB| ≪ |ΔHBB|). Moreover, since the cooperativity plays a fundamental role in the homopolymer and copolymer growth, the difference between the two cooperativities will define the length of the respective blocks.
The application of this strategy allows the formation of stable noncovalent block copolymers. The use of coassembling monomers with diverse optoelectronic properties would lead to new, unexpected functionalities in the field of supramolecular electronics.
Acknowledgments
We thank Dr. Nathan J. Van Zee, Eindhoven University of Technology, for fruitful discussions and Prof. Dr. David Leigh and Adrian Haertsch, University of Manchester, for providing the instrumentation for the NMR experiments. This research was supported by the Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035), the European Union (ERC-2014-StG Contract No. 635928), and the Dutch Science Foundation (NWO ECHO Grant No. 712.016.002).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b02706.
Experimental details, CD, UV, fluorescence spectroscopy, 1H NMR, model analysis, and details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Schumer J. Nat. Nanotechnol. 2016, 11 (10), 828–834. 10.1038/nnano.2016.232. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Zhong X.; Pavlica E.; Li S.; Klekachev A.; Bratina G.; Ebbesen T. W.; Orgiu E.; Samorì P. Nat. Nanotechnol. 2016, 11 (10), 900–906. 10.1038/nnano.2016.125. [DOI] [PubMed] [Google Scholar]
- Baroncini M.; d’Agostino S.; Bergamini G.; Ceroni P.; Comotti A.; Sozzani P.; Bassanetti I.; Grepioni F.; Hernandez T. M.; Silvi S.; Venturi M.; Credi A. Nat. Chem. 2015, 7, 634. 10.1038/nchem.2304. [DOI] [PubMed] [Google Scholar]
- Borges J.; Rodrigues L. C.; Reis R. L.; Mano J. F. Adv. Funct. Mater. 2014, 24 (36), 5624–5648. 10.1002/adfm.201401050. [DOI] [Google Scholar]
- Guo P.; Weimer M. S.; Emery J. D.; Diroll B. T.; Chen X.; Hock A. S.; Chang R. P. H.; Martinson A. B. F.; Schaller R. D. ACS Nano 2017, 11 (1), 693–701. 10.1021/acsnano.6b07042. [DOI] [PubMed] [Google Scholar]
- Chen F.; Zhu Y.-J. ACS Nano 2016, 10 (12), 11483–11495. 10.1021/acsnano.6b07239. [DOI] [PubMed] [Google Scholar]
- Aida T.; Meijer E. W.; Stupp S. I. Science 2012, 335 (6070), 813–817. 10.1126/science.1205962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Würthner F.; Saha-Möller C. R.; Fimmel B.; Ogi S.; Leowanawat P.; Schmidt D. Chem. Rev. 2016, 116 (3), 962–1052. 10.1021/acs.chemrev.5b00188. [DOI] [PubMed] [Google Scholar]
- López-Andarias J.; Rodriguez M. J.; Atienza C.; López J. L.; Mikie T.; Casado S.; Seki S.; Carrascosa J. L.; Martín N. J. Am. Chem. Soc. 2015, 137 (2), 893–897. 10.1021/ja510946c. [DOI] [PubMed] [Google Scholar]
- Bakker M. H.; Lee C. C.; Meijer E. W.; Dankers P. Y. W.; Albertazzi L. ACS Nano 2016, 10 (2), 1845–1852. 10.1021/acsnano.5b05383. [DOI] [PubMed] [Google Scholar]
- Smulders M. M. J.; Schenning A. P. H. J.; Meijer E. W. J. Am. Chem. Soc. 2008, 130 (2), 606–611. 10.1021/ja075987k. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay R. D.; Ajayaghosh A. Science 2015, 349 (6245), 241–242. 10.1126/science.aac7422. [DOI] [PubMed] [Google Scholar]
- Kang J.; Miyajima D.; Mori T.; Inoue Y.; Itoh Y.; Aida T. Science 2015, 347 (6222), 646–651. 10.1126/science.aaa4249. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Sugiyasu K.; Manna S.; Samitsu S.; Takeuchi M. Nat. Chem. 2014, 6 (3), 188–195. 10.1038/nchem.1849. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Stepanenko V.; Sugiyasu K.; Takeuchi M.; Würthner F. J. Am. Chem. Soc. 2015, 137 (9), 3300–3307. 10.1021/ja511952c. [DOI] [PubMed] [Google Scholar]
- Aliprandi A.; Mauro M.; De Cola L. Nat. Chem. 2016, 8 (1), 10–15. 10.1038/nchem.2383. [DOI] [PubMed] [Google Scholar]
- Markvoort A. J.; ten Eikelder H. M. M.; Hilbers P. A. J.; de Greef T. F. A.; Meijer E. W. Nat. Commun. 2011, 2, 509. 10.1038/ncomms1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korevaar P. A.; Grenier C.; Markvoort A. J.; Schenning A. P. H. J.; de Greef T. F. A.; Meijer E. W. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (43), 17205–17210. 10.1073/pnas.1310092110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbari-Farouji S.; van der Schoot P. J. Chem. Phys. 2012, 137 (6), 64906. 10.1063/1.4742192. [DOI] [PubMed] [Google Scholar]
- Gestel J. v.; van der Schoot P.; Michels M. A. J. J. Chem. Phys. 2004, 120 (17), 8253–8261. 10.1063/1.1689645. [DOI] [PubMed] [Google Scholar]
- Buchelnikov A. S.; Evstigneev V. P.; Evstigneev M. P. Chem. Phys. 2013, 421, 77–83. 10.1016/j.chemphys.2013.06.006. [DOI] [Google Scholar]
- ten Eikelder H. M. M.; Markvoort A. J.; de Greef T. F. A.; Hilbers P. A. J. J. Phys. Chem. B 2012, 116 (17), 5291–5301. 10.1021/jp300622m. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; George S. J.; Markvoort A. J.; Smulders M. M. J.; Hilbers P. A. J.; Schenning A. P. H. J.; De Greef T. F. A.; Meijer E. W. Nature 2012, 481 (7382), 492–496. 10.1038/nature10720. [DOI] [PubMed] [Google Scholar]
- Ogi S.; Fukui T.; Jue M. L.; Takeuchi M.; Sugiyasu K. Angew. Chem., Int. Ed. 2014, 53 (52), 14363–14367. 10.1002/anie.201407302. [DOI] [PubMed] [Google Scholar]
- van der Zwaag D.; Pieters P. A.; Korevaar P. A.; Markvoort A. J.; Spiering A. J. H.; de Greef T. F. A.; Meijer E. W. J. Am. Chem. Soc. 2015, 137 (39), 12677–12688. 10.1021/jacs.5b08138. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Jin W.; Fukushima T.; Saeki A.; Seki S.; Aida T. Science 2011, 334 (6054), 340–343. 10.1126/science.1210369. [DOI] [PubMed] [Google Scholar]
- Ajayaghosh A.; Vijayakumar C.; Praveen V. K.; Babu S. S.; Varghese R. J. Am. Chem. Soc. 2006, 128 (22), 7174–7175. 10.1021/ja0621905. [DOI] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Gazit E. Chem. Soc. Rev. 2014, 43 (20), 6881–6893. 10.1039/C4CS00164H. [DOI] [PubMed] [Google Scholar]
- Görl D.; Zhang X.; Stepanenko V.; Würthner F. Nat. Commun. 2015, 6, 7009. 10.1038/ncomms8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupar P. A.; Chabanne L.; Winnik M. A.; Manners I. Science 2012, 337 (6094), 559–562. 10.1126/science.1221206. [DOI] [PubMed] [Google Scholar]
- Qiu H.; Hudson Z. M.; Winnik M. A.; Manners I. Science 2015, 347 (6228), 1329–1332. 10.1126/science.1261816. [DOI] [PubMed] [Google Scholar]
- Moulin E.; Niess F.; Maaloum M.; Buhler E.; Nyrkova I.; Giuseppone N. Angew. Chem., Int. Ed. 2010, 49, 6974–6978. 10.1002/anie.201001833. [DOI] [PubMed] [Google Scholar]
- Faramarzi V.; Niess F.; Moulin E.; Maaloum M.; Dayen J.-F.; Beaufrand J.-B.; Zanettini S.; Doudin B.; Giuseppone N. Nat. Chem. 2012, 4, 485–490. 10.1038/nchem.1332. [DOI] [PubMed] [Google Scholar]
- Haedler A. T.; Kreger K.; Issac A.; Wittmann B.; Kivala M.; Hammer N.; Köhler J.; Schmidt H.-W.; Hildner R. Nature 2015, 523, 196. 10.1038/nature14570. [DOI] [PubMed] [Google Scholar]
- Adelizzi B.; Filot I. A. W.; Palmans A. R. A.; Meijer E. W. Chem. - Eur. J. 2017, 23 (25), 6103–6110. 10.1002/chem.201603938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtangi W.; Tassinari F.; Vankayala K.; Vargas Jentzsch A.; Adelizzi B.; Palmans A. R. A.; Fontanesi C.; Meijer E. W.; Naaman R. J. Am. Chem. Soc. 2017, 139 (7), 2794–2798. 10.1021/jacs.6b12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unpublished results.
- Onogi S.; Shigemitsu H.; Yoshii T.; Tanida T.; Ikeda M.; Kubota R.; Hamachi I. Nat. Chem. 2016, 8 (8), 743–752. 10.1038/nchem.2526. [DOI] [PubMed] [Google Scholar]
- Claridge T. D. W.; Claridge T. D. W. High-Resolution NMR Techniques in Organic Chemistry 2016, 315–380. 10.1016/B978-0-08-099986-9.00009-9. [DOI] [Google Scholar]
- Das A.; Vantomme G.; Markvoort A. J.; ten Eikelder H. M. M.; Garcia-Iglesias M.; Palmans A. R. A.; Meijer E. W. J. Am. Chem. Soc. 2017, 139 (20), 7036–7044. 10.1021/jacs.7b02835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Greef T. F. A.; Smulders M. M. J.; Wolffs M.; Schenning A. P. H. J.; Sijbesma R. P.; Meijer E. W. Chem. Rev. 2009, 109 (11), 5687–5754. 10.1021/cr900181u. [DOI] [PubMed] [Google Scholar]
- Lee I.; Bates F. S. Macromolecules 2013, 46 (11), 4529–4539. 10.1021/ma400479b. [DOI] [Google Scholar]
- Markvoort A. J.; Eikelder H. M. M. t.; Hilbers P. A. J.; de Greef T. F. A. ACS Cent. Sci. 2016, 2 (4), 232–241. 10.1021/acscentsci.6b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloi A.; Vargas Jentzsch A.; Vilanova N.; Albertazzi L.; Meijer E. W.; Voets I. K. J. Am. Chem. Soc. 2016, 138 (9), 2953–2956. 10.1021/jacs.5b13585. [DOI] [PubMed] [Google Scholar]
- Adelizzi B., Aloi A., Van Zee N. J., Palmans A. R. A., Meijer E. W., Voets I. K.. ACS Nano 2018, DOI: 10.1021/acsnano.8b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloi A.; Vilanova N.; Albertazzi L.; Voets I. K. Nanoscale 2016, 8, 8712–8716. 10.1039/C6NR00445H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertazzi L.; van der Zwaag D.; Leenders C. M. A.; Fitzner R.; van der Hofstad R. W.; Meijer E. W. Science 2014, 344 (6183), 491–495. 10.1126/science.1250945. [DOI] [PubMed] [Google Scholar]
- Korevaar P. A.; Schaefer C.; de Greef T. F. A.; Meijer E. W. J. Am. Chem. Soc. 2012, 134 (32), 13482–13491. 10.1021/ja305512g. [DOI] [PubMed] [Google Scholar]
- Coates G. W. Chem. Rev. 2000, 100 (4), 1223–1252. 10.1021/cr990286u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.