

Abstract
We studied the formation of supraparticles from nanocrystals confined in slowly evaporating oil droplets in an oil-in-water emulsion. The nanocrystals consist of an FeO core, a CoFe2O4 shell, and oleate capping ligands, with an overall diameter of 12.5 nm. We performed in situ small- and wide-angle X-ray scattering experiments during the entire period of solvent evaporation and colloidal crystallization. We observed a slow increase in the volume fraction of nanocrystals inside the oil droplets up to 20%, at which a sudden crystallization occurs. Our computer simulations show that crystallization at such a low volume fraction is only possible if attractive interactions between colloidal nanocrystals are taken into account in the model as well. The spherical supraparticles have a diameter of about 700 nm and consist of a few crystalline face-centered cubic domains. Nanocrystal supraparticles bear importance for magnetic and optoelectronic applications, such as color tunable biolabels, color tunable phosphors in LEDs, and miniaturized lasers.
Keywords: Colloidal nanocrystals, self-assembly, supraparticles, in situ small-/wide-angle X-ray scattering, spherical confinement
The self-assembly of colloids into larger periodic structures is a phenomenon commonly observed in nature. For example, opals form by colloidal crystallization of highly monodisperse spherical particles with diameters in the 100–1000 nm range.1−3 In the last two decades, there has been more and more focus on colloids with sizes in the nanometer range.4 Assembly of colloidal nanocrystals (NCs) dispersed in nonpolar solutions has been studied extensively.5−9 Single- and binary-component NC superlattices have been realized as three-dimensional crystals,6,10 thin films,9 and in some cases even as two-dimensional sheets with a large variety of NCs.8,9,11−15 In this field of nanoscience, there is a quest to design superstructures with tunable optical, magnetic, and electric properties that emerge from the properties of the building blocks, and the quantum mechanical and dipolar interactions between them.6,7
Recently, several groups realized that a versatile route to obtain hierarchical nanocrystal superstructures is through the self-assembly of so-called supraparticles (SPs):16 particles built up from nanoparticles. Several of us used the slow drying of emulsion droplets as the way to arrive at SPs that are thus self-assembled in a spherical confinement.17−19 These supraparticles are nearly spherical with diameters in the 100 to 10000 nm range; they may show a periodic ordering of the NC building blocks and crystalline facets, if the constituent NC building blocks have a sufficiently low polydispersity (<10%). Designer SPs based on CdSe/(Cd,Zn)S core–shell building blocks have attracted interest for their tunable emission, from pure colors to white light,18 and for the appearance of whispering gallery modes.19
Despite the broad applicability of NC assembly in spherical confinement and the obvious scientific and technologic interest, the study of the kinetic and mechanistic aspects has been addressed in only a few reports so far.17,20−24 However, the growth of nanocrystal superstructures as thin films or even two-dimensional sheets got much more attention with time-resolved, in situ synchrotron X-ray scattering methods.25−27
Here, we report a thorough real-time, in situ study of the colloidal crystallization of nanocrystals in spherical confinement. Using time-resolved wide-angle and small-angle X-ray scattering data (WAXS and SAXS), we followed the crystallization process over a wave vector (q) range of 5 × 10–3 to 34 nm–1 and thus, in a large spatial domain, obtaining information ranging from the shrinking oil droplets to the position of the NCs inside the droplets. This study allowed us to propose a mechanism for the colloidal crystallization of our NCs in the spherical confinement of an oil droplet. The model system chosen for this study is FeO/CoFe2O4 core/shell NCs, as they have been shown to easily form crystalline SPs17 and because the shape of these NCs can be tuned from spherical to cubic.28 By comparison of our data with simulations on the crystallization mechanism of hard spheres,17 it is clear that in the confined assembly of FeO/CoFe2O4 core/shell NCs of more than 10 nm in diameter, attractions between the NCs must be taken into account.
In Situ X-ray Scattering
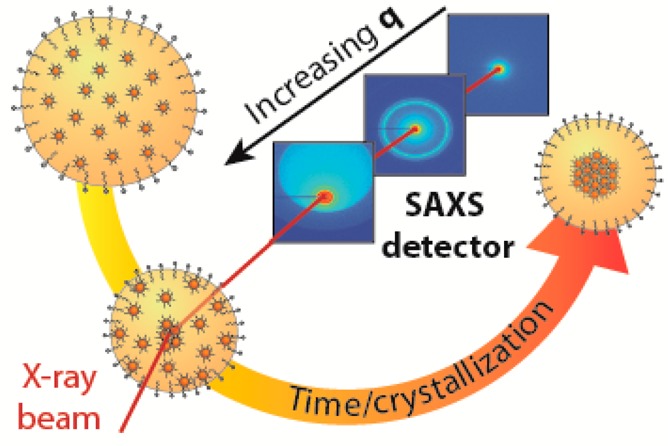
We performed in situ WAXS and SAXS measurements at the ID02 beamline of ESRF in Grenoble. A scheme of the experimental setup is shown in Figure 1. The “oil droplets” of the oil-in-water emulsion form, in fact, a suspension themselves: they consist of FeO/CoFe2O4 core/shell NCs of 10.5 ± 1.2 nm overall diameter (Figures 1a and S20–21) dispersed in cyclohexane, the initial NC volume fraction being 0.0077 ± 0.0012 (see SI for details about droplets formation). The CoFe2O4 shell around the core has a thickness of approximately 1.5 nm.29 Colloidal crystallization and the formation of SPs in the confined volume of the oil droplets were initiated by increasing the NC concentration upon evaporation of the cyclohexane. The emulsion was, therefore, heated to 68 °C in a reaction vessel (left part of Figure 1b). During this process, the oil-in-water suspension was pumped through a capillary through which the collimated X-ray beam passed. The scattered beam was detected by a WAXS detector that collected the atomic diffraction from the system. Detection at small angles (SAXS) occurred using three different sample-to-detector distances: 1, 5, and 30 m (see Figure 1). In such a way, the scattered signal could be detected over a broad wave vector region (5 × 10–3 to 34 nm–1), and structural information on the nanocrystal organization in the oil droplet was obtained on length scales spanning from ∼2.5 μm to 0.1 nm. Hence, we were able to follow the fate of the oil droplets and that of the nanocrystals inside each droplet, in situ, and in real time (Figure S19).
Figure 1.

Scheme of the experimental setup and of the experimental conditions in which the self-assembly was investigated. (a) Representative TEM image of FeO/CoFe2O4 NCs used in these measurements. (b) Emulsion, while heated and stirred in a vial in order to evaporate the nonpolar phase, is pumped through a capillary using a peristaltic pump. The emulsion is then probed, while in the capillary, by X-rays. The experiment has been performed three times with the SAXS detector at three different distances, 1, 5, and 30 m, in order to probe the full q range: from the region where the NCs scatter up to the region where the droplets and, later, the SPs scatter. (c) Representative SE-STEM image of a SP made of FeO/CoFe2O4 NCs as obtained from this experiment.
Evolution of the X-ray Scattering Patterns over a Broad Wave Vector Range
The time-resolved X-ray scattering signal during solvent evaporation and NC colloidal crystallization is presented in Figure 2. Panel (a) displays the time-resolved scattering patterns over the entire q range. In the high q range, the signal is modulated by the form factor of the spherical NCs. From the position and the depth of the minima, we can quantify the size of the NCs and their polydispersity: the NCs are 10.7 nm in diameter with a polydispersity of ∼9% (Figure S21). This is in good agreement with the values of 10.5 ± 1.2 nm obtained from transmission electron microscopy (TEM) images (Figures 1a and S20). In the small q region, the scattering pattern is modulated by the form factor of the cyclohexane/NCs droplets in the water medium (see arrows in Figure 2a). We can extract the initial size of the droplets being ∼3.2 μm, with a polydispersity of ∼21%, in good agreement with ex situ optical microscopy measurements of the oil droplets (Figure S23 in the SI).
Figure 2.

Time-resolved X-ray scattering patterns of the self-assembly. (a) Scattering patterns of the emulsion over time; the crystallization in ordered SPs is confirmed by the appearance of structure factor peaks in the region associated with NC scattering. The arrows indicate the first minima of the form factor relative to the scattering from the droplets/SPs. (b) Droplet size as a function of the reaction time. The error bars in dark blue show the error in the determination of the size relative to each point. (c) Volume fraction of NCs in the droplet as a function of time. The error bars in light red show the error in the determination of the volume fraction relative to each point. (d) Percentage of NCs not assembled in an SP over time; the crystallization is highlighted by the sharp change in the slope of the curve.
During cyclohexane evaporation, taking ∼4.5–5 h, the droplets shrink, increasing the NC concentration. This shrinkage can be followed over time by the change in the position of the form factor minima (see arrows in Figure 2a). The diameter of the droplets decreases linearly with time (Figure 2b), with an average value of 6.5 nm/min, indicating that the rate of solvent evaporation is determined by the droplet surface area, and that the intrinsic evaporation rate per unit of area is constant, independent of the droplet size (see SI for details). After ∼270–290 min, we observe a sudden “apparent” decrease in the droplet size from ∼1040 to ∼690 nm, which would correspond to a 70% decrease in the droplet volume. We remark that such a sudden and strong shrinkage of the suspension droplets is unlikely, and therefore, we name this an “apparent” decrease and not a true decrease in droplet size. We believe that the sudden shrinkage is due to X-ray scattering on an object with high electron density, which suddenly emerges from the NC dispersion. The simplest interpretation is that this object is an NC SP with a considerably larger NC density formed by colloidal crystallization (Figures S9–S14). In the SI, we show that alternative explanations based on simulated scattering patterns cannot explain the results. In particular, while we cannot fully exclude that a particle monolayer is adsorbed at the interface (as considerable adsorption energies have been predicted) from the scattering pattern (see Figure S15), this is clearly not related to the nucleation of the SPs that are observed in our results. Our interpretation that colloidal crystallization has started in the bulk of the droplet is corroborated by the concomitant appearance of structure factor peaks in the high q region of the scattering pattern (Figure 2a).
Knowing the size of the droplets and the initial NC volume fraction in the dispersion, we quantified the NC volume fraction in the dispersion in the droplets during solvent evaporation (Figure 2c). The NC volume fraction steadily increases over time from the initial value of 0.0077 ± 0.0012. At the moment of the crystallization, the volume fraction has a value of 0.20, and we observe an apparent increase in the volume fraction up to the value of 0.74, typical for a colloidal crystal with a face-centered-cubic (FCC) packing. According to the crystallization model for hard spheres, crystallization should occur at ∼0.5 NC volume fraction.30 In our case, crystallization occurs at a much lower volume fraction, indicating the existence of attractive forces between the NCs (further discussion follows below).
The WAXS signal shows reflections of the atomic planes of the FeO and CoFe2O4 crystals (Figure S16), in agreement with the TEM data (Figure S20). Since we are sampling over many SPs we have no information on possible alignment of the NCs in one SP (see SI for details).
Analysis of the Structure Factor Peaks
By integration of the scattering intensity of the structure factor peaks (see SI for details) in the high q range (5 × 10–1 to 6 nm–1), we are able to estimate the percentage of NCs dispersed, compared to those present in the SPs, over time during oil evaporation (Figure 2d). We assume that at time zero all the NCs were dispersed, and that, upon the shrinkage of the droplets indicating the crystallization, all the NCs are present in the SPs. We can then fit the data in Figure 2d (for t > 270 min) with a single exponential (see Figure S5 in the SI), yielding an average time constant for the NC crystallization process in a droplet of 7.5 min (see SI for details).
Now, we focus on the mechanism of NC crystallization into SPs by the analysis of the evolution of the structure factor peaks over time (Figure 3a). The first peak arises at 0.51 nm–1, corresponding to the reflection of the {111} planes in an FCC NC lattice. The peak can be fitted with a Lorentzian (Figure S2). The distance between adjacent NCs in the SP can be obtained from the position of the peak (Figure 3b,c), and the spatial extent of the periodic order from the full-width at half-maximum (fwhm) (Figure 3b,c and see SI for details on the methods). After a sharp change in the initial stage of the crystallization, the fwhm stabilizes to a constant value. We derive that the average crystalline domain size increases to a value of 260 nm, i.e., roughly 2.5 times smaller than the average SP size observed from the form factor scattering of the SP. Hence, each SP consists of 10–15 crystalline domains on average. The peak position shifts steadily toward higher q values (Figure 3b). The derived average distance between adjacent NCs in the SP is initially 14.3 nm, roughly corresponding to the size of two NCs with extended ligands (oleate) around them. This distance steadily decreases to 13.6 nm and then slowly to 13.3 nm. This decrease in the NC–NC distance must reflect interpenetration of the oleate ligands of the two adjacent ligand shells.
Figure 3.

Crystallization of the NCs in FCC SPs. (a) SAXS patterns at high q at different times during the crystallization. (b) Variation of the peak position (dots) and relative peak width (lines) of the first structure factor peak (0.5 nm–1) over time. (c) Variation of the NC–NC distance (blue) and average crystalline domain size (red) over time. (d) Indexing of the first 21 reflections of an FCC lattice in the scattering pattern of the SPs.
All structure factor peaks originate from reflections from different crystal planes in the NC superlattice, which allowed us to identify the crystal structure of the SP as FCC (Figure 3d). We were able to index as many as 21 reflections, which landmarks the excellent crystallinity of our SPs. The position of the structure factor peaks was also compared to those of the random hexagonal-close-packed (RHCP) and hexagonal-close-packed (HCP) structures (Figure S22). Our analysis, however, unambiguously showed that the structure factor peaks can only be indexed as originating from an FCC colloidal crystal.
Mechanism of Colloidal Crystallization in Spherical Confinement
Recently, the colloidal crystallization of nanocrystals in spherical confinement has been simulated by assuming that the nanocolloids act as hard spheres.17 It was found that crystallization sets in at a NC volume fraction of ∼0.5; crystal nuclei that have a NC volume fraction of 0.55 are formed by heterogeneous nucleation at the surface of the oil droplets. In our case, crystallization starts at a much lower volume fraction of ∼0.20. This suggests that NC–NC attractions should be incorporated in the model, as also supported by computer simulations presented below. It has been previously shown that in nearly ideal dispersions, the crystallization of small (<8 nm in diameter) semiconductor NCs can be seen as hard-sphere crystallization.14,31 However, in many other cases the formation of NC superlattices by self-assembly was understood on the basis of a subtle balance of entropy arguments and NC–NC attractions.7,9 Analysis of the state of a dispersion of nanocrystals in organic solvents indicated that the NC–NC attraction corresponds to a pair potential of a few times the thermal energy at room temperature, increasing with the size of the nanocrystals.32,33 This was also corroborated by calculations of the effective NC interactions (see SI).14
We propose a mechanism of crystallization that is schematically shown in Figure 4. In the initial stage, the NCs are homogeneously distributed in the oil droplets. Regarding the X-ray scattering, such NC/oil droplets can be assumed as having a homogeneous electron density, with a scattering length density somewhere between that of the cyclohexane and the NC solid. Over time the scattering contrast increases due to the densification of the NCs in the oil droplets. This temporal regime shows a gradual and constant decrease in the droplet size with time. We then observe a critical time regime of a few minutes in which the scattering pattern changes dramatically. This is attributed to the NC crystallization in the oil droplets occurring at a volume fraction of ∼0.20, which is corroborated by the sudden appearance of FCC structure factor peaks. One or more crystalline SP(s) start(s) to form via homogeneous nucleation inside each oil droplet, growing by NC addition and, possibly, aggregation of smaller SPs into a larger one. The scattered X-ray photons in the low-q region are now mainly due to the growing SP, the SP being considerably smaller than the oil droplet itself. In fact, the droplets can be seen as consisting of a strongly scattering core (the NC SP) and a weakly scattering shell of the remaining cyclohexane. The scattering length density difference, i.e., the contrast, of the SP compared to the cyclohexane is ∼350 times stronger. This explains the sudden “apparent” decrease of the size of the oil droplet. In the final stage, the remaining cyclohexane evaporates, leaving the SP covered by a layer of surfactants, which will enable its dispersion in water.
Figure 4.
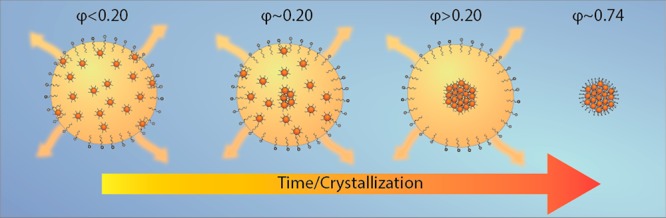
Crystallization mechanism. In the initial state (volume fraction, φ < 0.20), the NCs are homogeneously distributed in the droplet; when the volume fraction of NCs becomes critical (φ ≈ 0.20), a nucleation event starts inside the droplet. After a certain time, the NCs are nearly all assembled in the SP, which is still inside the droplet. Upon complete evaporation of the cyclohexane, the SP is covered by a layer of surfactants and suspended in water.
Computer Simulations
In order to support our hypothesis on the crystallization mechanism, we perform event-driven Brownian dynamics (EDBD) simulations of 100 000 NCs interacting with an attractive square-well potential in a slowly shrinking droplet as modeled by a spherical hard wall (see SI for details). As the confinement shrinks, the volume fraction of the nanoparticles in the droplet increases. At volume fraction (η) ≈ 0.11, we observe the formation of multiple crystalline nuclei (Figure 5a), which are identified using a bond-orientational order parameter and a cluster criterion (see SI for further details). We clearly observe from Figure 5a that the crystallization proceeds via homogeneous nucleation of multiple crystallites in the bulk and is not induced by crystallization at the wall as in the case of pure hard spheres (animations S1 and S2 in SI).17 With further increase in volume fraction, we observe that the nuclei merge and form a single crystalline domain that becomes eventually spherical due to the shrinking confinement. Remarkably, we observe that the surface of our SP assembled from attractive NCs shows ledges and facets indicating that the crystallization is not surface-induced. This should be contrasted to the case of SPs formed from hard spheres, which show a much smoother surface (Figure S24). In order to investigate the evolution of the crystallization mechanism, we plot in Figure 5b the fraction of the crystalline particles as a function of the volume fraction (time) for attractive NCs and for pure hard spheres. Figure 5b shows that (i) the onset of the crystallization for attractive NCs is at a much lower volume fraction (η ≈ 0.11) than for hard-sphere NCs (η ≈ 0.5) and that (ii) the crystallization of attractive NCs occurs via homogeneous nucleation, while it proceeds through heterogeneous nucleation for hard-sphere NCs, which can also be appreciated from the corresponding thumbnails of Figure 5b, showing a cross-section of the attractive and hard-sphere NC SPs after crystallization has set in. We remark that a more quantitative agreement, between simulations and experiments, in the estimated volume fraction at which there is an onset of crystallization, could be obtained by tuning the strength of the attraction between the NCs. Furthermore, the increase in the fraction of crystalline particles with volume fraction is very steep, indicating a very rapid crystallization process, which is in qualitative agreement with the experimental observations. Finally, we show in Figure 5c (i) the number of crystalline particles in the largest crystal domain as a function of volume fraction (time) (in green), which increases in time until ∼83% of the NCs constitute the main crystal, and (ii) the number of crystal nuclei (in purple), which shows a peak during the shrinking process and finally decays to one corresponding to the formation of a single cluster. Both trends support our proposed crystallization mechanism.
Figure 5.

Computer simulations. (a) Cross-section (through the center) of the SP at different times (t/τMD) in a slowly shrinking spherical confinement. The crystallization proceeds through homogeneous nucleation of multiple crystal nuclei, which merge together at long simulation times to form a single crystalline domain. Colors are assigned randomly to distinct different nuclei in every time snapshot (cyan always denotes disordered particles). (b) Number of crystalline particles as a function of volume fraction for attractive NCs and hard-sphere NCs in a shrinking spherical confinement. The onset of the crystallization for attractive NCs is at a much lower volume fraction (η ≈ 0.11) than for hard-sphere NCs (η ≈ 0.5). The thumbnails show a cross-section of the attractive and hard-sphere NC SPs after crystallization has set in, thereby demonstrating that the crystallization of attractive NCs occurs via homogeneous nucleation, while it proceeds through heterogeneous nucleation for hard-sphere NCs. (c) Size of the largest cluster and the number of differently oriented clusters as a function of volume fraction (time). Eventually there is only one main crystalline domain.
We studied the mechanism of the self-assembly of colloidal NCs into NC SPs in a NC dispersion being confined to an oil droplet dispersed in water. We performed in situ small- and wide-angle X-ray scattering experiments at the ID02 beamline at ESRF, Grenoble. Our results showed that colloidal crystallization sets in at a NC volume fraction of ∼0.20; this indicates that attractive interactions between the NCs are important as a driving force for the self-assembly. The completion of the crystallization of the SPs takes on average about 7.5 min to incorporate all NCs. The SPs themselves consist of a few FCC crystalline domains. SPs bear importance for optoelectronic applications, as color tunable biolabels, phosphors in LEDS, and possibly also miniaturized lasers.
Acknowledgments
The authors thank F. T. Rabouw and W. van der Stam for very fruitful discussions, Jan Ilavsky for support in the use of IRENA, Jacques Gorini for assistance during the scattering measurements, and G. Fiorucci, M. Chiappini, and G. Coli for support in calculating crystal reflections. This work was supported by the European Commission via the Marie-Sklodowska Curie action Phonsi (H2020-MSCA-ITN-642656). D.V. wishes to thank the Dutch FOM (program DDC13), NWO–CW (Toppunt 718.015.002), and the European Research Council under HORIZON 2020 (Grant 692691 FIRSTSTEP) for financial support. J.J.G. gratefully acknowledges financial support from the joined UU and ESRF graduate program (NWO grant number 022.004.016).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b00809.
Synthesis of FeO/CoFe2O4 NCs, synthesis of the SPs, experimental setup, analysis of the SAXS and WAXS data, determination of the volume fraction, determination of the crystallization mechanism, analysis of the crystal structure of the SPs, EDBD simulations (PDF)
Square-well attraction of 2kT (cross-section through center) animation (AVI)
Hard spheres animation (AVI)
The authors declare no competing financial interest.
Supplementary Material
References
- Sanders J. V. Colour of Precious Opal. Nature 1964, 204, 1151–1153. 10.1038/2041151a0. [DOI] [Google Scholar]
- Iler R. K. Formation of Precious Opal. Nature 1965, 207, 472–473. 10.1038/207472a0. [DOI] [Google Scholar]
- Darragh P. J.; Gaskin A. J.; Terrell B. C.; Sanders J. V. Origin of Precious Opal. Nature 1966, 209, 13–16. 10.1038/209013a0. [DOI] [Google Scholar]
- Cheng Z. Colloidal Crystallization. Fluids, Colloids Soft Mater. An Introd. to Soft Matter Phys. 2016, 203–248. 10.1002/9781119220510.ch12. [DOI] [Google Scholar]
- Murray C. B.; Kagan C. R.; Bawendi M. G. Self-Organization of CdSe Nanocrystallites into Three-Dimensional Quantum Dot Superlattices. Science 1995, 270, 1335–1338. 10.1126/science.270.5240.1335. [DOI] [Google Scholar]
- Vanmaekelbergh D. Self-Assembly of Colloidal Nanocrystals as Route to Novel Classes of Nanostructured Materials. Nano Today 2011, 6, 419–437. 10.1016/j.nantod.2011.06.005. [DOI] [Google Scholar]
- Talapin D. V.; Lee J. S.; Kovalenko M. V.; Shevchenko E. V. Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications. Chem. Rev. 2010, 110, 389–458. 10.1021/cr900137k. [DOI] [PubMed] [Google Scholar]
- Redl F. X.; Cho K.-S.; Murray C. B.; O’Brien S. Three-Dimensional Binary Superlattices of Magnetic Nanocrystals and Semiconductor Quantum Dots. Nature 2003, 423, 968–971. 10.1038/nature01702. [DOI] [PubMed] [Google Scholar]
- Dong A.; Chen J.; Vora P. M.; Kikkawa J. M.; Murray C. B. Binary Nanocrystal Superlattice Membranes Self-Assembled at the Liquid-Air Interface. Nature 2010, 466, 474–477. 10.1038/nature09188. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Altantzis T.; Zanaga D.; Bals S.; Van Tendeloo G.; Pileni M.-P. Supracrystalline Colloidal Eggs: Epitaxial Growth and Freestanding Three-Dimensional Supracrystals in Nanoscaled Colloidosomes. J. Am. Chem. Soc. 2016, 138, 3493–3500. 10.1021/jacs.5b13235. [DOI] [PubMed] [Google Scholar]
- Ye X.; Chen J.; Engel M.; Millan J. a; Li W.; Qi L.; Xing G.; Collins J. E.; Kagan C. R.; Li J.; et al. Competition of Shape and Interaction Patchiness for Self-Assembling Nanoplates. Nat. Chem. 2013, 5, 466–473. 10.1038/nchem.1651. [DOI] [PubMed] [Google Scholar]
- Bodnarchuk M. I.; Li L.; Fok A.; Nachtergaele S.; Ismagilov R. F.; Talapin D. V. Three-Dimensional Nanocrystal Superlattices Grown in Nanoliter Microfluidic Plugs. J. Am. Chem. Soc. 2011, 133, 8956–8960. 10.1021/ja201129n. [DOI] [PubMed] [Google Scholar]
- Cargnello M.; Johnston-Peck A. C.; Diroll B. T.; Wong E.; Datta B.; Damodhar D.; Doan-Nguyen V. V. T.; Herzing A. a; Kagan C. R.; Murray C. B. Substitutional Doping in Nanocrystal Superlattices. Nature 2015, 524, 450–453. 10.1038/nature14872. [DOI] [PubMed] [Google Scholar]
- Evers W. H.; De Nijs B.; Filion L.; Castillo S.; Dijkstra M.; Vanmaekelbergh D. Entropy-Driven Formation of Binary Semiconductor-Nanocrystal Superlattices. Nano Lett. 2010, 10, 4235–4241. 10.1021/nl102705p. [DOI] [PubMed] [Google Scholar]
- Boneschanscher M. P.; Evers W. H.; Geuchies J. J.; Altantzis T.; Goris B.; Rabouw F. T.; Van Rossum S. A. P.; Van Der Zant H. S. J.; Siebbeles L. D. A.; Van Tendeloo G.; et al. Long-Range Orientation and Atomic Attachment of Nanocrystals in 2D Honeycomb Superlattices. Science 2014, 344, 1377–1380. 10.1126/science.1252642. [DOI] [PubMed] [Google Scholar]
- Wang T.; LaMontagne D.; Lynch J.; Zhuang J.; Cao Y. C. Colloidal Superparticles from Nanoparticle Assembly. Chem. Soc. Rev. 2013, 42, 2804–2823. 10.1039/C2CS35318K. [DOI] [PubMed] [Google Scholar]
- de Nijs B.; Dussi S.; Smallenburg F.; Meeldijk J. D.; Groenendijk D. J.; Filion L.; Imhof A.; Dijkstra M.; Van Blaaderen A. Entropy-Driven Formation of Large Icosahedral Colloidal Clusters by Spherical Confinement. Nat. Mater. 2015, 14, 56–60. 10.1038/nmat4072. [DOI] [PubMed] [Google Scholar]
- Montanarella F.; Altantzis T.; Zanaga D.; Rabouw F. T.; Bals S.; Baesjou P.; Vanmaekelbergh D.; van Blaaderen A. Composite Supraparticles with Tunable Light Emission. ACS Nano 2017, 11, 9136–9142. 10.1021/acsnano.7b03975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmaekelbergh D.; van Vugt L. K.; Bakker H.; Rabouw F. T.; de Nijs B.; van Dijk-Moes R.; Beasjou P.; van Blaaderen A. Shape-Dependent Multiexciton Emission and Whispering Gallery Modes in Supraparticles of CdSe/Multishell Quantum Dots. ACS Nano 2015, 9, 3942–3950. 10.1021/nn507310f. [DOI] [PubMed] [Google Scholar]
- Lacava J.; Ouali A.-A.; Raillard B.; Kraus T. On the Behaviour of Nanoparticles in Oil-in-Water Emulsions with Different Surfactants. Soft Matter 2014, 10, 1696. 10.1039/c3sm52949e. [DOI] [PubMed] [Google Scholar]
- Lacava J.; Born P.; Kraus T. Nanoparticle Clusters with Lennard-Jones Geometries. Nano Lett. 2012, 12, 3279–3282. 10.1021/nl3013659. [DOI] [PubMed] [Google Scholar]
- Schmitt J.; Hajiw S.; Lecchi A.; Degrouard J.; Salonen A.; Impéror-Clerc M.; Pansu B. Formation of Superlattices of Gold Nanoparticles Using Ostwald Ripening in Emulsions: Transition from Fcc to Bcc Structure. J. Phys. Chem. B 2016, 120, 5759–5766. 10.1021/acs.jpcb.6b03287. [DOI] [PubMed] [Google Scholar]
- Kister T.; Mravlak M.; Schilling T.; Kraus T. Pressure-Controlled Formation of Crystalline, Janus, and Core–shell Supraparticles. Nanoscale 2016, 8, 13377–13384. 10.1039/C6NR01940D. [DOI] [PubMed] [Google Scholar]
- Yang P. W.; Thoka S.; Lin P. C.; Su C. J.; Sheu H. S.; Huang M. H.; Jeng U. S. Tracing the Surfactant-Mediated Nucleation, Growth, and Superpacking of Gold Supercrystals Using Time and Spatially Resolved X-Ray Scattering. Langmuir 2017, 33, 3253–3261. 10.1021/acs.langmuir.6b04319. [DOI] [PubMed] [Google Scholar]
- Weidman M. C.; Smilgies D.-M.; Tisdale W. A. Kinetics of the Self-Assembly of Nanocrystal Superlattices Measured by Real-Time in Situ X-Ray Scattering. Nat. Mater. 2016, 15, 775–781. 10.1038/nmat4600. [DOI] [PubMed] [Google Scholar]
- Geuchies J. J.; van Overbeek C.; Evers W. H.; Goris B.; de Backer A.; Gantapara A. P.; Rabouw F. T.; Hilhorst J.; Peters J. L.; Konovalov O.; et al. In Situ Study of the Formation Mechanism of Two-Dimensional Superlattices from PbSe Nanocrystals. Nat. Mater. 2016, 1, 1–8. 10.1038/nmat4746. [DOI] [PubMed] [Google Scholar]
- Wu L.; Willis J. J.; McKay I. S.; Diroll B. T.; Qin J.; Cargnello M.; Tassone C. J. High-Temperature Crystallization of Nanocrystals into Three-Dimensional Superlattices. Nature 2017, 548, 197–201. 10.1038/nature23308. [DOI] [PubMed] [Google Scholar]
- Wang D.; Hermes M.; Kotni R.; Wu Y.; Tasios N.; Liu Y.; de Nijs B.; van der Wee E. B.; Murray C. B.; Dijkstra M.; et al. Submitted 2018. [DOI] [PMC free article] [PubMed]
- Bodnarchuk M. I.; Kovalenko M. V.; Groiss H.; Resel R.; Reissner M.; Hesser G.; Lechner R. T.; Steiner W.; Schaffler F.; Heiss W. Exchange-Coupled Bimagnetic Wustite/metal Ferrite Core/shell Nanocrystals: Size, Shape, and Compositional Control. Small 2009, 5, 2247–2252. 10.1002/smll.200900635. [DOI] [PubMed] [Google Scholar]
- Hoover W. G.; Ree F. H. Melting Transition and Communal Entropy for Hard Spheres. J. Chem. Phys. 1968, 49, 3609–3617. 10.1063/1.1670641. [DOI] [Google Scholar]
- Bodnarchuk M. I.; Kovalenko M. V.; Heiss W.; Talapin D. V. Energetic and Entropic Contributions to Self-Assembly of Binary Nanocrystal Superlattices: Temperature as the Structure-Directing Factor. J. Am. Chem. Soc. 2010, 132, 11967–11977. 10.1021/ja103083q. [DOI] [PubMed] [Google Scholar]
- Klokkenburg M.; Dullens R. P. A.; Kegel W. K.; Erné B. H.; Philipse A. P. Quantitative Real-Space Analysis of Self-Assembled Structures of Magnetic Dipolar Colloids. Phys. Rev. Lett. 2006, 96, 1–4. 10.1103/PhysRevLett.96.037203. [DOI] [PubMed] [Google Scholar]
- van Rijssel J.; Erné B. H.; Meeldijk J. D.; Casavola M.; Vanmaekelbergh D.; Meijerink A.; Philipse A. P. Enthalpy and Entropy of Nanoparticle Association from Temperature-Dependent Cryo-TEM. Phys. Chem. Chem. Phys. 2011, 13, 12770–12774. 10.1039/c1cp20297a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.