

SUMMARY
Succinate accumulates during ischemia, and its oxidation at reperfusion drives injury. The mechanism of ischemic succinate accumulation is controversial and is proposed to involve reversal of mitochondrial complex II. Herein, using stable-isotope-resolved metabolomics, we demonstrate that complex II reversal is possible in hypoxic mitochondria but is not the primary succinate source in hypoxic cardiomyocytes or ischemic hearts. Rather, in these intact systems succinate primarily originates from canonical Krebs cycle activity, partly supported by aminotransferase anaplerosis and glycolysis from glycogen. Augmentation of canonical Krebs cycle activity with dimethyl-α-ketoglutarate both increases ischemic succinate accumulation and drives substrate-level phosphorylation by succinyl-CoA synthetase, improving ischemic energetics. Although two-thirds of ischemic succinate accumulation is extracellular, the remaining one-third is metabolized during early reperfusion, wherein acute complex II inhibition is protective. These results highlight a bifunctional role for succinate: its complex-II-independent accumulation being beneficial in ischemia and its complex-II-dependent oxidation being detrimental at reperfusion.
In Brief
Although succinate drives reperfusion injury, its ischemic accumulation mechanism is controversial. Herein, Zhang et al. show that ischemic succinate is generated by canonical Krebs cycle activity, rather than by mitochondrial complex II reversal, and improves ischemic energetics. At reperfusion, most succinate is washed out and may serve a signaling role.
INTRODUCTION
Ischemia reperfusion (IR) injury is driven by mitochondrial metabolism. Specifically, the Krebs cycle metabolite succinate accumulates during ischemia and is rapidly consumed at reperfusion, driving reactive oxygen species (ROS) generation by reverse electron transfer (RET) through mitochondrial complex I (Cx I) (Chouchani et al., 2014). This triggers cell-death mechanisms, such as opening the mitochondrial permeability transition pore, contributing to infarct formation (Bernardi and Di Lisa, 2015; Beutner et al., 2017; Morciano et al., 2015). Despite this detrimental role at reperfusion, ischemic succinate accumulation is conserved across diverse tissues and species (Chouchani et al., 2014; Hochachka and Dressendorfer, 1976; Hochachka et al., 1975), suggesting that the metabolic pathways responsible may serve beneficial roles in ischemia.
One proposed model for ischemic succinate accumulation involves reversal of mitochondrial complex II (Cx I) (Chouchani et al., 2014; Hochachka et al., 1975) (Figure 1, model A). In ischemia, oxygen is not available as the terminal electron acceptor of the respiratory chain. Instead, it is proposed that co-enzyme Q reduction by Cx I drives reversal of Cx II, with fumarate as an electron acceptor yielding succinate. This model has implications for ischemic metabolism: first, NADH would be reoxidized, facilitating glycolysis. Second, NADH oxidation by Cx I is coupled to its proton-pumping activity, which could contribute to maintenance of mitochondrial membrane potential (Pell et al., 2016).
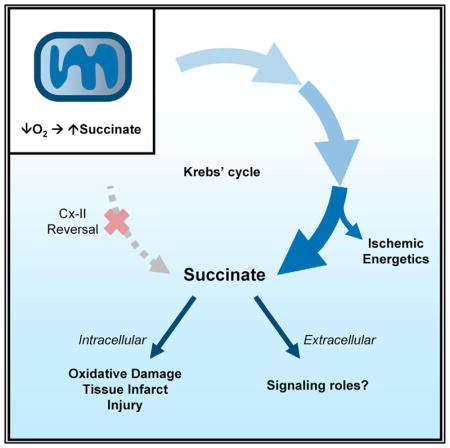
Figure 1. Metabolic Pathways Investigated.

Upper panel: normoxic metabolism. The mitochondrial respiratory chain is shown center left, positioned in the mitochondrial inner membrane, with dashed lines denoting electron flow. Krebs cycle and other metabolites are shown using common abbreviations (e.g., α-KG, α-ketoglutarate; Fum, fumarate). Metabolite transporters (e.g., malate/aspartate shuttle) are shown at right. Dotted lines denote multi-step metabolite interconversions. Inset panels (boxed) show pathways relevant to Figure S1E. Enzyme or transporter inhibitors are shown in red. Lower panels: proposed models for ischemic succinate generation. Model A: Cx II reversal/aspartate model. Model B: Cx II inhibition/canonical Krebs cycle/aminotransferase anaplerosis model. Species shown in color (glucose, orange; palmitate, green; aspartate, blue; glutamine, pink) denote metabolites employed in stable isotope labeling experiments. Dimethyl-α-ketoglutarate (DM-α-KG) is shown in yellow.
Despite its potential significance for ischemic metabolism, Cx II reversal has not been explicitly demonstrated in physiologically relevant complex biological systems. Most evidence for this model comes from isolated Cx II preparations, submitochondrial particles or bacteria (Hirst et al., 1996; Maklashina et al., 1998; Sanadi and Fluharty, 1963; Wilson and Cascarano, 1970). However, the catalytic properties of Cx II do not favor reversal in situ (Sucheta et al., 1992), and early 3H-fumarate tracing experiments suggested that succinate accumulation was insensitive to Cx I inhibition (Hoberman and Prosky, 1967). Furthermore, perfused heart experiments showed that exogenous 14C-fumarate only contributed 15% of ischemic succinate (Laplante et al., 1997), and similar experiments have shown that Cx II reversal contributes only minimally to ischemic cardiac energetics (Cascarano et al., 1968; Hohl et al., 1987; Peuhkurinen et al., 1983).
The metabolic precursor for fumarate in ischemia is thought to be aspartate, originating from both the purine nucleotide cycle (PNC) (Chouchani et al., 2014) and aspartate aminotransferase (AST) (Chouchani et al., 2014, 2016; Hochachka et al., 1975). Although aspartate levels are decreased in ischemia (Pisarenko et al., 1987), the PNC is also likely inactive because of its energy requirement (Idström et al., 1990). In addition, the interpretation of stable-isotope-resolved metabolomics (SIRM) experiments (e.g., with [U-13C]aspartate) (Chouchani et al., 2014) may be confounded within the context of ischemia (see the Results for details).
Herein, we addressed the mechanisms and origin of ischemic succinate accumulation by applying a comprehensive pharmacologic and SIRM-based approach across multiple biological systems. Our in situ studies utilized the Langendorff-perfused mouse heart, affording precise control over delivery of drugs and 13C-labeled substrates without the complication of metabolism by other organs, and permitting rapid tissue sampling without blood contamination (Nadtochiy et al., 2015). We show that while Cx II reversal is possible in isolated hypoxic mitochondria, it is not the primary source of succinate in hypoxic cardiomyocytes or ischemic hearts. Instead, we show that canonical (clockwise) Krebs cycle activity, supported in part by aminotransferase anaplerosis, is the primary source of ischemic succinate under in situ conditions, with substrate-level phosphorylation improving cardiac ischemic energetics (Figure 1, model B). Together with studies on the protective effects of Cx II inhibition at reperfusion, these findings highlight both beneficial and detrimental roles for succinate in IR injury.
RESULTS
A schematic of the metabolic pathways considered in this study during both normoxia and ischemia is shown in Figure 1. The complete dataset for this paper is posted on the open access site FigShare (https://doi.org/10.6084/m9.figshare.6157631).
Cx II Reversal Is Not the Primary Mechanism of Ischemic Succinate Generation In Situ
Isolated mouse heart mitochondria were subjected to hypoxia in a sealed O2 electrode chamber, with pyruvate and carnitine present to generate NADH in a manner that did not engage the Krebs cycle, i.e., acetyl-coenzyme A (CoA) produced by pyruvate dehydrogenase was shuttled out of mitochondria as acetyl-carnitine (Figure 2A) (Muoio et al., 2012). In this system, addition of fumarate under hypoxia led to succinate generation, and this was sensitive to both the Cx I inhibitor rotenone and the Cx II inhibitor atpenin A5 (AA5) (Figure 2B). Notably, this phenomenon could be recapitulated in normoxia by applying the complex III inhibitor antimycin A (Figure S1A), thus demonstrating that Cx II reversal is possible in this system when the distal respiratory chain is inhibited. Identical results were obtained with malate instead of fumarate (data not shown). To test whether Cx II reversal was sensitive to either ΔpH or Δψm (as reported for Cx I reversal) (Lambert and Brand, 2004), we used nigericin or FCCP to dissipate ΔpH or Δψm, respectively, but observed no effect on hypoxic succinate accumulation (Figure S1B).
Figure 2. Cx II Reversal Is Not the Primary Mechanism of Ischemic Succinate Generation In Situ.

(A) Schematic of isolated mitochondrial experiments. Pyruvate (Pyr) and carnitine were present to generate NADH in a manner that did not engage the Krebs cycle (i.e., acetyl-CoA exits mitochondria as acetyl-carnitine [Ac-Carn]). Rot, rotenone; AA5, atpenin A5; PDH, pyruvate dehydrogenase; Fum, fumarate; Succ, succinate.
(B) Isolated mouse heart mitochondria respired to hypoxia, and then 1 mM fumarate was added, with 1 μM Rot or 1 μM AA5, followed by hypoxic incubation for 20 min. Succinate was measured using HPLC. n = 4–6. *p < 0.05 versus hypoxia with substrates. nd, not detectable.
(C) Perfused hearts were treated with rotenone (1 μM/5 min), AA5 (1 μM/5 min), dimethyl malonate (DMM) (5 mM/10 min), malonate (5 mM/10 min), or thenoyltrifluorocetone (TTFA) (1 mM/5 min) and then immediately subjected to 25 min of ischemia. Hearts were snap-frozen and succinate measured using HPLC. n = 3–8. *p < 0.05 versus untreated baseline (BL). ns, no significant difference between indicated groups.
(D) Isolated primary cardiomyocytes were treated with 1 μM Rot or 1 μM AA5 and subjected to 60 min of hypoxia and then succinate measured using HPLC. n = 3–5. *p < 0.05 versus baseline (BL).
All data are means ± SEM.
See also Figure S1.
Next, we investigated the role of Cx-I-driven Cx II reversal in ischemic succinate accumulation in intact perfused mouse hearts. Although 25 min of ischemia resulted in substantial succinate accumulation, this was insensitive to Cx I inhibition (rotenone) and to several structurally unrelated Cx II inhibitors (AA5, dimethylmalonate [DMM], malonate, and thenoyltrifluoroacetone) (Figure 2C). To confirm inhibition of respiratory complexes in situ, we first assayed Cx I and Cx II activities of inhibitor-treated hearts spectrophotometrically, demonstrating appropriate inhibition (Figures S1C and S1D). Second, the effects of Cx I and Cx II inhibitors on succinate accumulation were investigated in hypoxic isolated primary cardiomyocytes, wherein 60 min of hypoxia led to increased succinate with no effect of rotenone or AA5 (Figure 2D). Similar effects were also seen in cardiomyocytes incubated with antimycin A under normoxia (data not shown).
Overall, while isolated mitochondria can be driven to generate succinate via Cx II reversal, this does not appear to be the primary mechanism for ischemic succinate accumulation in situ. While others have reported that DMM can inhibit cardiac ischemic succinate accumulation in vivo (Chouchani et al., 2014), the interpretation of such results may be complicated by non-cardiac metabolism of DMM.
No Role for PNC, γABA Shunt, ROS, or Autophagy in Ischemic Succinate Accumulation
Previously, aspartate was proposed as a precursor for ischemic succinate via the PNC (Figure 1, model A). However, PNC inhibition with either 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (Swain et al., 1984) or metformin (Ouyang et al., 2011) did not affect ischemic succinate (Figure S1E). Similar to previous reports (Chouchani et al., 2014), the γABA shunt inhibitor vigabatrin also failed to affect ischemic succinate levels.
Although the reaction of α-ketoglutarate (α-KG) with hydrogen peroxide can generate succinate (Fedotcheva et al., 2006), the non-specific thiol antioxidant N-acetylcysteine (NAC) was without effect on ischemic succinate accumulation, suggesting no role for ROS in this system. The autophagy inhibitor chloroquine also had no effect on ischemic succinate accumulation (Figure S1E). Together, these data suggest no role for the PNC, γABA shunt, ROS-mediated α-KG decarboxylation, or autophagy in ischemic succinate accumulation.
Minor Contribution of Aspartate to Ischemic Succinate Accumulation
Consistent with published data (Chouchani et al., 2014; Støttrup et al., 2010), aminooxyacetate (AOA) significantly blunted ischemic succinate accumulation (Figure 3A). Previously, this effect was attributed to AOA inhibition of AST (Figure 1, model A). However, AOA is a non-specific aminotransferase inhibitor (John and Charteris, 1978) and is also reported to react with α-ketoacids, such as pyruvate and α-KG, generating oxime products (Yang et al., 2008). Indeed, AOA delivery to normoxic perfused hearts significantly lowered baseline pyruvate and α-KG levels (Figure 3A), such that it may inhibit ischemic succinate accumulation by depleting metabolic precursors.
Figure 3. Minor Contribution of Aspartate to Ischemic Succinate Accumulation.

(A) Left: perfused hearts were treated with 5 mM aminooxyacetate (AOA) for 5 min and then subjected to 25 min ischemia. Succinate was measured using LC-MS/MS. n = 3–4. *p < 0.05 versus baseline (BL), †p < 0.05 versus ischemia alone. Pyruvate (middle) and α-KG (right) were measured in normoxic perfused hearts treated with 5 mM AOA for 5 min, n = 3–4.
(B) Schematic of SIRM experiments with [U-13C]aspartate delivery for 10 min, followed by sampling immediately or after 25 min of ischemia. Succinate was measured using LC-MS/MS. See also Figure S2 for predicted labeling resulting from [U13-C]aspartate delivery.
(C) Left: abundances of each 13C-labeled succinate species in hearts from (B) are shown, with log-transformed data in the inset. Pictograms below represent labeling status of carbons in succinate (white, unlabeled; blue, labeled). Right: fractional labeling of succinate isotopologues. Pictograms below represent calculated fractions. n = 3. *p < 0.05 versus baseline (BL). ns, no significant difference between indicated groups.
All data are means ± SEM.
To interrogate aspartate as a carbon source for ischemic succinate, a previous study used SIRM (Chouchani et al., 2014). However, following [U-13C]aspartate infusion, hearts were subjected to equal durations of either no-flow ischemia or nor-moxic perfusion with unlabeled aspartate. Although an increase in 13C-succinate was reported at the end of ischemia relative to normoxic perfusion, this relative increment could have been due to [U-13C]aspartate wash out from normoxic hearts (with no washout possible in ischemic hearts).
To resolve this issue, we perfused hearts for 10 min with [U-13C]aspartate and then sampled either immediately before ischemia or after 25 min of no-flow ischemia (Figure 3B, schematic). This strategy afforded labeling of 15.8% ± 2.2% of the cardiac aspartate pool. As shown schematically in Figure S2, [U-13C]aspartate can contribute labeled carbon to succinate either via AST and Cx II reversal, yielding fully labeled succinate (M+4), or via other pathways in which unlabeled carbons dilute the 13C-label, yielding partially labeled succinate isotopologues. Using this system, consistent with recruitment of Cx II reversal (Chouchani et al., 2014), we observed a significant increase (~35-fold) in M+4 succinate during ischemia. However, this isotopologue only represented ~0.9% of the total ischemic succinate pool (Figure 3C). Since ~16% of cardiac aspartate was labeled by [U-13C]aspartate infusion, these data predict an upper limit on ischemic succinate sourced from aspartate via Cx II reversal of ~5.7% (i.e., 0.9% scaled for 100% aspartate labeling).
The vast majority of ischemic succinate was unlabeled, and although the fractional contribution of partially labeled species to total succinate (sum of M+1, M+2, M+3 = 1.64%) was around double that of the M+4 species, this fraction did not differ between ischemia and normoxia (Figure 3C). This indicates that the indirect metabolism of aspartate to succinate (Figure S2) accounts for ~10.4% of ischemic succinate (i.e., 1.64% scaled for 100% aspartate labeling). Thus, while we demonstrate that aspartate does generate some succinate via Cx II reversal, aspartate metabolism overall is a minor contributor to ischemic succinate.
Glycolysis and Glycogenolysis Facilitate Ischemic Succinate Accumulation
Since upregulation of glycolysis is a hallmark of ischemia (Stanley et al., 2005), we hypothesized it might be important for succinate accumulation. Accordingly, perfusion of hearts with glucose-free media (palmitate as metabolic substrate) blunted ischemic succinate accumulation by 40% (Figure 4A), suggesting glycolysis may play a role. The magnitude of this effect suggests either that glycolysis is not required for the other 60% or that endogenous glucose sources (e.g., glycogen) are recruited in ischemia. Consistent with this, cardiac glycogen content was both diminished (consumed) during ischemia and significantly lower at baseline in hearts pre-perfused for 20 min without glucose (Figure 4A). Thus, glycogen likely serves as a glucose source that facilitates ischemic metabolism.
Figure 4. Glycolysis and Glycogenolysis Facilitate Ischemic Succinate Accumulation.
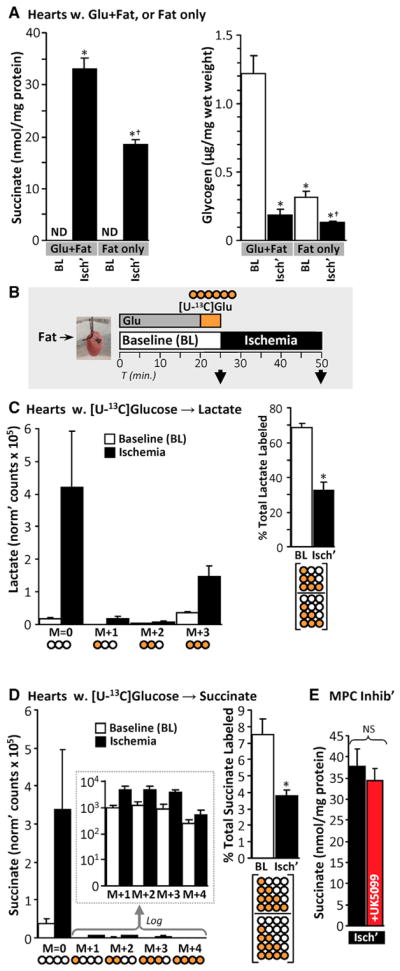
(A) Left: hearts were equilibrated for 20 min with either 5 mM glucose and 100 μM palmitate (Glu+Fat) or palmitate only (Fat only) and sampled either at baseline (BL) or after 25 min of ischemia (Isch’). Succinate was measured using HPLC. n = 3–4. *p < 0.05 versus Glu+Fat baseline (BL), †p < 0.05 versus Glu+Fat ischemia. ND, not detectable. Right: the glycogen content was assayed in the same hearts. n = 3–4. *p < 0.05 versus Glu+Fat baseline (BL), †p < 0.05 versus Fat only ischemia.
(B) Schematic of SIRM experiments with [U-13C]glucose delivery for 5 min, followed by sampling immediately or after 25 min of ischemia.
(C) Left: lactate was measured in hearts from (B) using LC-MS/MS. Abundances of each 13C-labeled species are shown. Pictograms below represent labeling status of carbons in lactate (white, unlabeled; orange, labeled). Right: fractional labeling of all lactate isotopologues. Pictogram below represents fraction calculated. n = 5. *p < 0.05 versus baseline (BL).
(D) Left: succinate was measured in the same hearts as (C) using LC-MS/MS. Abundances of each 13C-labeled succinate species are shown, with log-transformed data in the inset. Pictograms below represent labeling status of carbons in succinate. Right: total fractional labeling of succinate. Pictogram below represents calculated fraction. n = 5. *p < 0.05 versus baseline (BL).
(E) Hearts were perfused with 10 μM of the mitochondrial pyruvate carrier inhibitor UK5099 for 5 min and then subjected to 25 min of ischemia. Succinate was measured using HPLC. n = 3. ns, no significant difference between indicated groups.
All data are means ± SEM.
To investigate further, we perfused hearts with [U-13C]glucose (Figure 4B, schematic), predicting that unlabeled glucose from glycogen would yield a mixed population of unlabeled and fully labeled glycolytic end products (e.g., lactate). Consistent with this, while overall lactate levels were elevated in ischemia, the fractional contribution of labeled lactate species to total lactate was lower (Figure 4C). This indicates recruitment of unlabeled carbon sources to glycolysis during ischemia.
In the same [U-13C]glucose infused hearts, the majority of succinate accumulated in ischemia was unlabeled, and the fractional contribution of all labeled succinate species to total succinate was also lower in ischemia (Figure 4D). These data suggest that, similar to lactate, an unlabeled carbon source is recruited in ischemia to generate succinate.
To test the contribution of glycolytic end products to ischemic succinate accumulation, we employed the mitochondrial pyruvate carrier inhibitor UK5099 (Butcher et al., 1998). Surprisingly, this inhibitor did not affect ischemic succinate accumulation (Figure 4E). Additionally, supplementing cardiac pyruvate levels prior to ischemia with methyl-pyruvate did not further augment ischemic succinate accumulation (data not shown). Overall, these data suggest that while glycogen-derived glucose is important for ischemic succinate accumulation, direct flux of carbon from glycolysis into mitochondria is not required.
Canonical Krebs Cycle Activity, Supported in Part by Aminotransferase Anaplerosis, Is the Primary Source of Ischemic Succinate
In addition to entering mitochondria or being converted to lactate, a third metabolic fate for pyruvate is conversion to alanine via alanine aminotransferase (ALT) (Figure 1). This enzyme is coupled to deamidation of glutamate, yielding α-KG. Alanine is known to accumulate in ischemia (Lewandowski and White, 1995; Pisarenko et al., 1987). Furthermore, ALT is sensitive to inhibition by AOA (John and Charteris, 1978), which we showed inhibits ischemic succinate accumulation (Figure 3A). Together these findings suggest a potential role for ALT in ischemia, both in disposal of pyruvate and provision of a carbon source for succinate.
Although cardiac ALT is thought to operate mainly in the α-KG → glutamate direction (Cohen et al., 2003), its reverse operation (glutamate → α-KG) is speculated to occur in ischemia (Pisarenko et al., 1987) (Figure 1, model B). A potential glutamate source for ALT is glutamine, and to investigate this we perfused hearts with [U-13C,15N]glutamine for 10 min and sampled immediately or following 25 min ischemia (Figure 5A). This strategy yielded 42.0% ± 4.3% labeling of the cardiac glutamine pool at baseline. The total amount of α-KG was depleted in ischemia, but levels of fully labeled α-KG (M+5) were unchanged (Figure 5B). Thus, the resultant increased fractional labeling of α-KG (Figure 5B) demonstrates that anaplerosis from glutamine to α-KG is recruited in ischemia.
Figure 5. Canonical Krebs cycle Activity, Supported in Part by Aminotransferase Anaplerosis, Is the Primary Source of Ischemic Succinate.
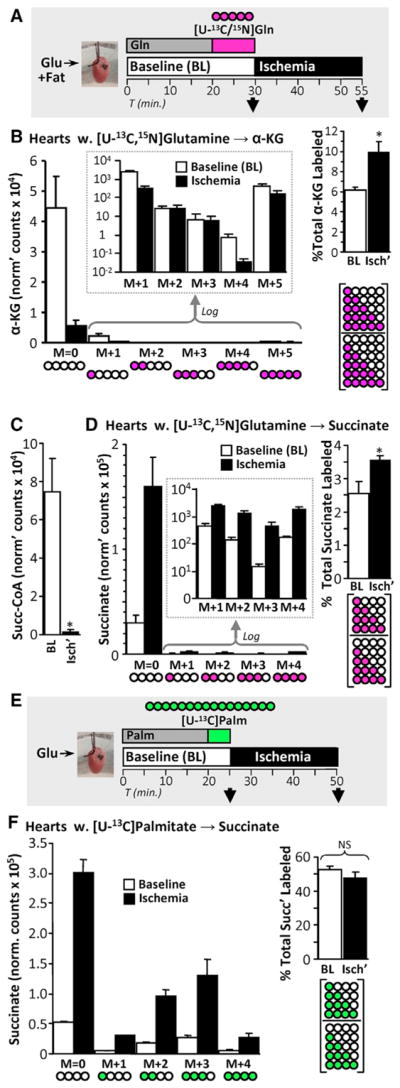
(A) Schematic of SIRM experiments with [U-13C,15N]glutamine delivery for 10 min, followed by sampling immediately or after 25 min of ischemia. α-KG and succinate were measured using LC-MS/MS.
(B) Left: abundances of each 13C-labeled α-KG species in hearts from (A) are shown, with log-transformed data in the inset. Pictograms below represent labeling status of carbons in α-KG (white, unlabeled; pink, labeled) Right: total fractional labeling of α-KG. Pictogram below represents fraction calculated. n = 6. *p < 0.05 versus baseline (BL).
(C) Perfused hearts were sampled either at normoxic baseline (BL) or following 25 min of ischemia (Isch’), and total succinyl-CoA levels were measured using LC-MS/MS. n = 3. *p < 0.05 versus baseline.
(D) Left: abundances of each 13C-labeled succinate species in hearts from (A) are shown, with log-transformed data in the inset. Pictograms below represent labeling status of carbons in succinate. Right: total fractional labeling of succinate. Pictogram below represents fraction calculated. n = 6. *p < 0.05 versus baseline (BL).
(E) Schematic of steady-state SIRM experiments, with [U-13C]palmitate delivery for 5 min, achieving steady-state labeling of Krebs cycle metabolites. Hearts were sampled immediately or after 25 min of ischemia. Succinate was measured using LC-MS/MS.
(F) Left: abundances of each 13C-labeled succinate species in hearts from (E) are shown. Pictograms below represent the labeling status of carbons in succinate (white, unlabeled; green, labeled). Right: total fractional labeling of succinate. Pictogram below represents fraction calculated. n = 3. ns, no significant difference between indicated groups.
All data are means ± SEM.
A route from α-KG to succinate is the canonical Krebs cycle via α-KG dehydrogenase and succinyl-CoA synthetase (Figure 1, model B), and ischemic succinate accumulation may utilize existing Krebs cycle metabolite pools. In support of this, we observed ischemic depletion of not only α-KG (Figure 5B, see above) but also succinyl-CoA (Figure 5C). Additionally, in hearts perfused with [U-13C,15N]glutamine, while the majority of ischemic succinate was unlabeled, the fractional contribution of all labeled succinate species to the total succinate pool rose significantly, from 2.6% at baseline to 3.5% in ischemia (Figure 5D). Correcting for labeling efficiency of the cardiac glutamine pool, these data predict that ~8.4% of ischemic succinate is sourced from glutamine anaplerosis (i.e., 3.5% scaled for 100% glutamine labeling).
The hypothesis that ischemic succinate accumulation may consume existing Krebs cycle intermediates can be further tested using steady-state isotope labeling of metabolite pools. Fatty acid oxidation is the primary cardiac fuel source (Stanley et al., 2005), and consistent with this [U-13C]palmitate rapidly and robustly labels the Krebs cycle, reaching steady-state labeling within 5 min (Nadtochiy et al., 2015). As shown in Figures 5E and 5F, steady-state pre-labeling with [U-13C]palmitate yielded ~50% fractional saturation of the succinate pool at baseline (normoxia). However, despite a multiple-fold elevation of succinate in ischemia, fractional succinate labeling was unchanged. Furthermore, acknowledging the recent discovery that circulating lactate is a carbon source for the cardiac Krebs cycle (Hui et al., 2017), steady-state pre-labeling with [U-13C]lactate yielded 42% saturation of the succinate pool at baseline, and this fraction was unchanged by ischemia (Figure S3). Together, these data indicate that ischemic succinate accumulation primarily consumes pre-existing Krebs cycle metabolites.
Summarizing isotope labeling studies, 5.7% of ischemic succinate accumulation derives from aspartate via Cx II reversal, and 8.4% derives from glutamine anaplerosis into the canonical Krebs cycle. Glucose, and specifically pyruvate entry into mitochondria, does not contribute carbon to ischemic succinate, and steady-state pre-labeling of Krebs cycle intermediates suggests that the source of most ischemic succinate is the existing Krebs cycle metabolite pool. Overall, canonical Krebs cycle activity supported in part by aminotransferase anaplerosis is the source of ischemic succinate (Figure 1, model B).
Ischemic Succinate Generation Enhances Cardiac Energetics
Since α-KG was depleted in ischemia (Figure 5B), we hypothesized α-KG may be limiting, so increasing its availability may boost ischemic succinate generation. Consistent with this, supplementing perfused hearts with cell-permeable dimethyl-α-ketoglutarate (DM-α-KG) significantly increased ischemic succinate accumulation (Figure 6A). Furthermore, the inhibition of ischemic succinate accumulation by aminotransferase inhibition (AOA) was bypassed by DM-α-KG supplementation (Figures 1, model B, and 6B). Notably, the ~4.5-fold increase in ischemic succinate induced by DM-α-KG (versus ischemia alone) (Figure 6A), was significantly blunted (to 1.25-fold) by AOA (Figure 6B). This is likely due to the direct reaction between AOA and α-ketoacids as noted earlier (Figure 3A) (Yang et al., 2008). Nevertheless, even in the presence of AOA ischemic succinate was elevated 5.4-fold by addition of DM-α-KG.
Figure 6. Ischemic Succinate Generation Enhances Cardiac Energetics.
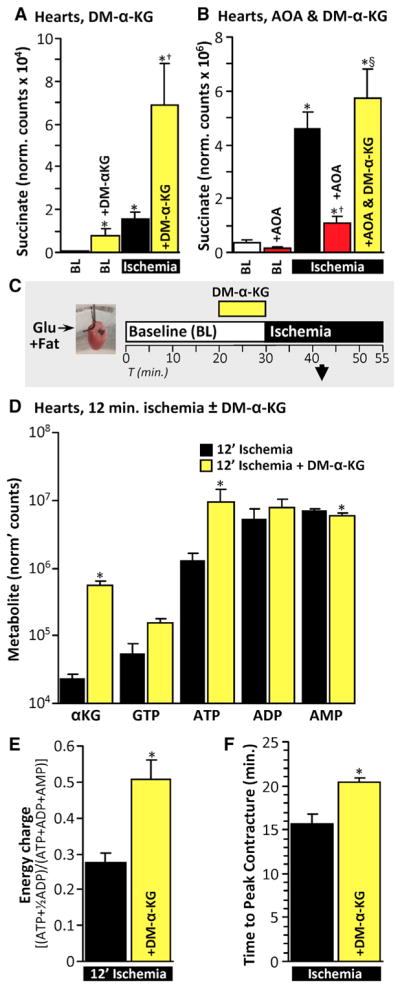
(A) Hearts were perfused with 5 mM dimethyl-α-KG (DM-α-KG) for 10 min and sampled immediately or following 25 min of ischemia. Succinate was measured using LC-MS/MS. n = 3–5. *p < 0.05 versus untreated baseline (BL), †p < 0.05 versus untreated ischemia.
(B) Hearts were perfused with 5 mM DM-α-KG for 10 min and/or 5 mM AOA for 5 min and sampled either at baseline (BL) or after 25 min of ischemia. Succinate was measured using LC-MS/MS. n = 3–6. *p < 0.05 versus untreated baseline, †p < 0.05 versus untreated ischemia, §p < 0.05 versus ischemia +AOA.
(C) Schematic of experiments investigating mid-point ischemic energetics. Hearts were perfused with 5 mM DM-α-KG for 10 min and sampled after 12 min of ischemia. Metabolites were detected using LC-MS/MS.
(D) Metabolites of hearts from (C). n = 5–6. *p < 0.05 versus untreated (with Bonferroni correction for multiple testing).
(E) Energy charge was calculated from (D). n = 5–6. *p < 0.05 versus untreated.
(F) Time to peak ischemic contracture was measured in hearts perfused with 5 mM DM-α-KG for 10 min and then subjected to ischemia. n = 6. *p < 0.05 versus untreated.
All data are means ± SEM.
If ischemic succinate is generated via canonical Krebs cycle activity, substrate-level phosphorylation by succinyl-CoA synthetase could benefit ischemic energetics. To investigate this, we measured nucleotide levels in hearts perfused with DM-α-KG. Preliminary experiments showed that ATP was mostly depleted by 25 min ischemia, even with DM-α-KG supplementation (data not shown). However, at 12 min of ischemia (Figure 6C), DM-α-KG supplemented hearts had significantly less ATP depletion versus non-supplemented ischemic controls (Figure 6D), and their energy charge was also significantly higher (Figure 6E). A major consequence of ischemic ATP depletion is ischemic contracture, and consistent with improved energetics the time to reach peak ischemic contracture was extended by DM-α-KG supplementation (Figure 6F). Thus, confounding the portrayal of succinate as a solely detrimental metabolite in ischemia, boosting ischemic succinate accumulation affords beneficial effects on cardiac energetics. This may partially explain the evolutionary conservation of ischemic succinate accumulation (Chouchani et al., 2014; Hochachka and Dressendorfer, 1976; Hochachka et al., 1975).
Rapid Succinate Oxidation in Early Reperfusion Mediates IR Injury
At reperfusion, rapid succinate oxidation by Cx II is thought to drive pathologic ROS generation via Cx I RET (Chouchani et al., 2014). In agreement with this we found that myocardial succinate levels returned to baseline within 5 min of reperfusion (Figures 7A and 7B). However, consistent with previous proposals that ischemic succinate may accumulate in the extracellular space (Andrienko et al., 2017; Støttrup et al., 2010), we also found that approximately two-thirds of succinate accumulated in ischemic hearts was washed into the perfusate within 5 min of reperfusion. While the greater portion of ischemic succinate was lost to washout, nevertheless approximately one-third was unaccounted for (i.e., was metabolized). Furthermore, in agreement with the concept that succinate consumption by Cx II at reperfusion drives cardiac injury, we found that acute delivery of the Cx II inhibitor AA5 at the onset of reperfusion was cardioprotective (Figures 7C and 7D).
Figure 7. Rapid Succinate Oxidation in Early Reperfusion Mediates IR Injury.
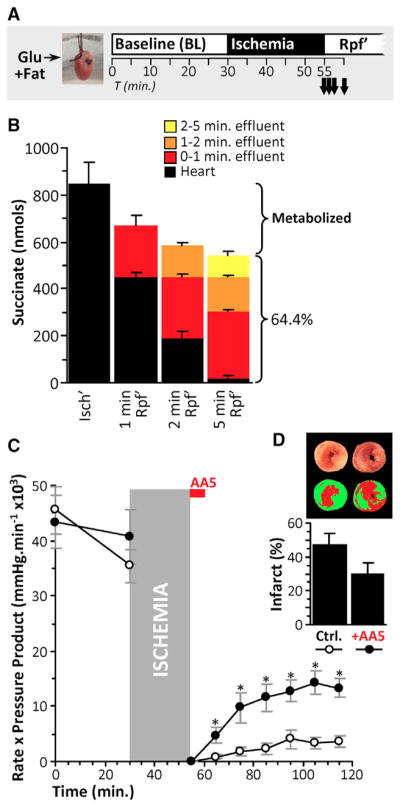
(A) Schematic of experiments measuring succinate consumption during early reperfusion (Rpf’).
(B) Hearts were subjected to 25 min ischemia then reperfused for 0, 1, 2, and 5 min. Succinate was measured in hearts (black bars). Effluent was collected during reperfusion, binned into 0–1 min (red), 1–2 min (orange), and 2–5 min (yellow). No succinate was detected in the effluent after 5 min reperfusion. Succinate was measured using HPLC. n = 3–4. Brackets to the right of graph show the fraction of total cardiac succinate at the end of ischemia (i.e., leftmost black bar) that was released into effluent, with the remainder presumed to be metabolized.
(C) Hearts were subjected to 25 min ischemia and 60 min reperfusion. 100 nM AA5 was infused for 5 min at the onset of reperfusion where indicated. Cardiac functional recovery (rate x pressure product) was monitored. n = 6–8. *p < 0.05 versus control (ctrl)
(D) Infarct sizes as percent of area-at-risk (i.e., total heart area) in hearts from (C). n = 6–8. All data are means ± SEM.
DISCUSSION
Succinate accumulation in hypoxia/ischemia is a universal phenomenon in vertebrates (Chouchani et al., 2014; Hochachka and Dressendorfer, 1976; Hochachka et al., 1975), for which two models have been proposed (Figure 1). Model A posits mitochondrial Cx II reversal, reducing fumarate to succinate, with fumarate originating from aspartate via AST (Chouchani et al., 2014; Sanadi and Fluharty, 1963). Model B posits Cx II inhibition, with canonical Krebs cycle activity partly supported by aminotransferase anaplerosis (Hochachka et al., 1975). Evidence for model A comes in part from in vivo studies, interpretation of which is complicated by the presence of other organs (Chouchani et al., 2014), and from artificial systems such as isolated mitochondria (Sanadi and Fluharty, 1963; Wilson and Cascarano, 1970) (Figure 2B). Although the perfused heart preparation is not without limitations (e.g., edema with prolonged perfusion) it is the model of choice for studying metabolism in situ, since it affords precise control over metabolic substrate delivery and sampling. Using this system our data indicate that ischemic succinate primarily originates from canonical Krebs cycle activity using existing metabolites, with only minor contributions from Cx II reversal and aminotransferase anaplerosis (model B). As such, proposals to target Cx II reversal to elicit cardioprotection in IR injury (Chouchani et al., 2014, 2016; Pell et al., 2016; Valls-Lacalle et al., 2016) may require reevaluation.
Although glycolysis supported by cardiac glycogen appears to play a role in ischemic succinate accumulation (Figure 4A), mitochondrial pyruvate entry is not required (Figure 4E), indicating that glucose is not a major carbon source. Furthermore, the ability of AOA to blunt ischemic succinate accumulation (Figure 3A) suggests a requirement for aminotransferases. Since it is known that alanine accumulates in ischemia (Lewandowski and White, 1995; Pisarenko et al., 1987), glycolysis and pyruvate are likely required for deamidation of glutamate by ALT to generate α-KG for Krebs cycle anaplerosis (Pyr + Glu →Ala + α-KG) (Figure 1, model B). Notably in cancer, glutamine anaplerosis into the Krebs cycle, followed by reductive carboxylation of α-KG to citrate, is important for tumor metabolism (Mullen et al., 2014; Smith et al., 2016). Since the latter reaction consumes reducing equivalents and is stimulated in hypoxia, it may facilitate α-KG oxidation to succinate.
Although in silico analysis has been used to predict a role for cytosolic AST and Cx II reversal in ischemic succinate accumulation (Chouchani et al., 2014) (Figure 1, model A), this only accounts for ~6% of ischemic succinate (Figure 3C). A further 10.4% is accounted for by aspartate anaplerosis via AST into canonical Krebs cycle activity (Figure 3C), and another 8.4% from glutamine anaplerosis via ALT (Figure 5D). Combined, these reactions account for one-fourth of ischemic succinate (6% + 10% + 8%), with steady-state labeling experiments ([U-13C]palmitate or [U-13C]lactate) (Figures 5E, 5F, and S3) suggesting that the bulk of ischemic succinate originates from pre-existing Krebs cycle metabolites (Figure 1, model B). In support of this, the immediate precursors for succinate generation in the canonical Krebs cycle are succinyl-CoA and α-KG, both of which were depleted by ischemia (Figures 5B and 5C). Accordingly, α-KG repletion with cell-permeable DM-α-KG boosted ischemic succinate generation (Figure 6A) and also improved cardiac energetics, likely due to stimulation of substrate-level phosphorylation by succinyl-CoA synthetase (Figures 6D–6F). Extension of the time to ischemic contracture in mice may translate to a significant therapeutic benefit in human cardiac ischemia.
A notable feature of ischemic succinate identified herein was its largely extracellular nature, with approximately two-thirds of ischemic succinate released into the perfusate during early reperfusion (Figure 7B). Succinate is thought to exit cells via the monocarboxylate transporter MCT1 under low pH conditions (Andrienko et al., 2017), and it is known that plasma succinate levels are elevated in response to exercise (Hochachka and Dressendorfer, 1976) or hypoxia (Hochachka et al., 1975). Plasma succinate regulates vascular tone (Leite et al., 2016) and cardiac hypertrophy (Yang et al., 2016) via the succinate receptor SUCNR1 (formerly GPR91) (Gilissen et al., 2016) and is also an inflammatory mediator (Mills et al., 2016; Tannahill et al., 2013). As such, extracellular succinate released at reperfusion may signal ischemia to the whole organism, akin to a “mitokine.”
There are further similarities among succinate, lactate, and alanine in whole animal hypoxic physiology. Lactate and alanine are key intermediates in the Cori and Cahill cycles, respectively (Cori, 1931; Felig et al., 1970), wherein they are released into blood for regeneration into glucose by hepatic gluconeogenesis. Plasma succinate may participate in a similar cycle, perhaps with glutamine/glutamate as metabolites returning to hypoxic tissue. In agreement with this, a recent study on plasma metabolites in the coronary sinus of ischemic human myocardium identified elevated succinate and lower glutamate (Kohlhauer et al., 2018).
Despite washout of approximately two-thirds of ischemic succinate upon reperfusion, approximately one-third appears to be metabolized by the heart, potentially facilitating IR injury (Figure 7B) (Chouchani et al., 2014). Accordingly, it has been shown that inhibition of ischemic succinate accumulation with AOA confers cardioprotection (Nielsen et al., 2011; Støttrup et al., 2010). In addition there is pervasive interest in Cx II inhibition as a cardioprotective strategy (Chouchani et al., 2014; Valls-Lacalle et al., 2016, 2018; Wojtovich and Brookes, 2009). However, the optimal time-window for Cx II inhibition during IR is not yet clear.
Ischemic succinate accumulation via model A (Cx II reversal) may afford sustenance of cardiac energetics via continued proton pumping by Cx I (Pell et al., 2016). As such, inhibiting Cx II activity during ischemia may be detrimental. Furthermore, ischemic succinate accumulation via model B (canonical Krebs cycle with anaplerosis) can also afford sustenance of cardiac energetics via substrate-level phosphorylation. In this case, inhibiting Cx II in ischemia may be redundant, since the model itself invokes Cx II inhibition (Figure 1). Thus overall, Cx II inhibition in ischemia may have detrimental effects on cardiac energetics.
In contrast, the finding that one-third of ischemic succinate is oxidized during the first 5 min of reperfusion (and 59% of this within the first minute) (Figure 7B) indicates early reperfusion is a critical time window for therapeutic targeting of Cx II. Consistent with this, AA5 delivery at the moment of reperfusion elicited significant improvement in functional recovery of perfused hearts (Figure 7C), with a trend toward lower infarct size (Figure 7D). The reported ability of Cx II inhibitors to afford protection when delivered prior to ischemia (Chouchani et al., 2014; Wojtovich and Brookes, 2009) may therefore be due in-part to their residual presence at reperfusion inhibiting succinate oxidation.
In conclusion, our data support a model in which cardiac ischemic succinate is primarily derived from canonical Krebs cycle activity, supported in part by aminotransferase anaplerosis. Succinate accumulation plays a multifunctional role in IR, being beneficial for ischemic energetics, a source of damage at reperfusion, and a potential mitokine signaling hypoxia/ischemia to other tissues.
EXPERIMENTAL PROCEDURES
More details can be found in the Supplemental Experimental Procedures.
Animals and Reagents
Experimental procedures complied with the NIH Guide for Care and Use of Laboratory Animals and were approved by the University of Rochester Committee on Animal Resources (protocol number 2007-087). Male C57BL/6J mice, 2–4 months old, were housed in a pathogen-free vivarium with 12 hr light-dark cycles and food and water ad libitum. Anesthesia was induced with 250 mg/kg i.p. 2,2,2-tribromoethanol (Avertin). Unless otherwise stated (see the Supplemental Experimental Procedures), reagents were obtained from Sigma (St. Louis, MO, USA).
Hypoxic Mitochondria
Mouse heart mitochondria were isolated via differential centrifugation in sucrose media (Zhang et al., 2016). Mitochondrial protein (0.5 mg) was added to 0.5 mL of buffer containing (in millimolars): KCl (120), sucrose (25), MgCl2 (5.0), KH2PO4 (5.0), EGTA (1.0), HEPES (10), and 0.1% (w/v) fat-free BSA (pH 7.3) at 37°C in an O2 electrode chamber (YSI, Yellow Springs, OH, USA). After baseline measurements, 2 mM pyruvate, 5 mM carnitine, and 100 μM ADP were added sequentially. This yielded a respiratory control ratio of 3.6 ± 0.8 (mean ± SD). Once hypoxic (< 2% O2), 1 μM rotenone or 1 μM atpenin A5 (AA5) and 1 mM fumarate were added. After 20 min of hypoxia, 8% perchloric acid was added, and samples were frozen at −80°C until metabolite analysis.
Hypoxic Mouse Cardiomyocytes
Adult mouse cardiomyocytes were isolated by enzymatic digestion (Zhang et al., 2016), yielding −8 × 105 rod-shaped cells per heart with >80% viability. Cells were seeded per well onto laminin-coated 12-well plates (1 × 105 cells/well) with unbuffered DMEM containing (in millimolars) glucose (5.5), palmitate (0.1), and pyruvate (0.1). Following equilibration, 10 μM rote-none and 1 μM AA5 were added, and the plate was placed into a hypoxic chamber (< 0.1% O2, Coy, Grass Lake, MI, USA) at 37°C. After 1 hr, 8% perchloric acid was added, and samples were frozen at −80°C until metabolite analysis.
Perfused Mouse Hearts
Mouse hearts were excised and perfused with 0.22-μm filtered Krebs-Henseleit buffer (KH) containing (in millimolars): NaCl (118), KCl (4.7), MgSO4 (1.2), NaHCO3 (25), KH2PO4 (1.2), CaCl2 (2.5), glucose (5), pyruvate (0.2), lactate (1.2), and palmitate (0.1), with 95% O2 and 5% CO2 at 37°C (Nadtochiy et al., 2015). Cardiac function was monitored throughout with a baseline rate × pressure product of 43,884 ± 7,209 mmHg·min−1 (mean ± SD).
For metabolic studies, hearts were infused with substrates or inhibitors (see the Supplemental Experimental Procedures for concentrations and durations) and either immediately frozen in liquid N2 or subjected to 25 min of global no-flow ischemia and then frozen. Frozen hearts were pulverized and stored at −80°C until metabolite analysis. For IR injury studies, hearts were subjected to 25 min of global no-flow ischemia and then 60 min of reperfusion. AA5 (100 nM) was infused for 5 min immediately at reperfusion. Infarct was assessed following staining with triphenyltetrazolium chloride. For reperfusion washout studies, hearts were subjected to 25 min of global no-flow ischemia and then reperfused for 1, 2, or 5 min before sampling. In addition, where feasible, effluent from reperfusion was collected and treated with 8% perchloric acid. Samples were stored at −80°C until metabolite analysis.
Metabolite Analysis by High-Performance Liquid Chromatography
Heart powder was serially extracted in 8% perchloric acid. Metabolites were resolved on high-performance liquid chromatography (HPLC) (Shimadzu Prominence 20 System) with photodiode array detection, quantified via bracket-calibration with a standard curve (see the Supplemental Experimental Procedures for details), and normalized to protein.
Metabolite Analysis by Liquid Chromatography-Tandem Mass Spectrometry
Heart powder was serially extracted in 80% methanol, evaporated under N2, and resuspended in 50% methanol. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was essentially as previously described (Nadtochiy et al., 2015). Metabolites were resolved by HPLC (Shimadzu Prominence 20 System) and identified by single-reaction monitoring on a triple-quadrupole mass spectrometer (Thermo Quantum TSQ) (see the Supplemental Experimental Procedures for details). Data were analyzed using MzRock (https://code.google.com/archive/p/mzrock/). Selected metabolites were also analyzed using XCalibur Qual Browser (Thermo Scientific). Data were normalized to protein content.
Quantification and Statistical Analysis
Comparisons between groups were made using ANOVA and with a post hoc Student’s t test applied where appropriate. Data are shown as means ± SEM unless otherwise noted. Numbers of replicates (n) are noted in the figure legends. Significance was set at α = 0.05.
DATA AND SOFTWARE AVAILABILITY
All raw data and statistical analyses are available in the original dataset on Figshare: 10.6084/m9.figshare.6157631.
Supplementary Material
Highlights.
Canonical Krebs cycle, rather than complex II reversal, generates ischemic succinate
Ischemic succinate accumulation may improve ischemic energetics
At reperfusion, succinate is primarily washed out rather than oxidized
Acknowledgments
Work in the lab of P.S.B. was funded by an NIH grant (R01-HL071158). J.Z. is funded by grants from AHA (17PRE33410360) and NIH (F30-HL134203). We thank Mike Murphy (University of Cambridge) and Edward Chouchani (Harvard Medical School) for helpful discussions.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.104.
AUTHOR CONTRIBUTIONS
J.Z. and P.S.B. conceived of and designed the study. J.Z. performed the experiments and analyzed the results. P.S.B. prepared the figures. Y.T.W., J.H.M., M.M.D., and J.C.M. assisted with the experiments. J.Z. and P.S.B. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP. The role of succinate and ROS in reperfusion injury - A critical appraisal. J Mol Cell Cardiol. 2017;110:1–14. doi: 10.1016/j.yjmcc.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutner G, Alavian KN, Jonas EA, Porter GA., Jr The mitochondrial permeability transition pore and ATP synthase. Handb Exp Pharmacol. 2017;240:21–46. doi: 10.1007/164_2016_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher L, Coates A, Martin KL, Rutherford AJ, Leese HJ. Metabolism of pyruvate by the early human embryo. Biol Reprod. 1998;58:1054–1056. doi: 10.1095/biolreprod58.4.1054. [DOI] [PubMed] [Google Scholar]
- Cascarano J, Chick WL, Seidman I. Anaerobic rat heart: effect of glucose and Krebs cycle metabolites on rate of beating. Proc Soc Exp Biol Med. 1968;127:25–30. doi: 10.3181/00379727-127-32612. [DOI] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23:254–263. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- Cohen DM, Guthrie PH, Gao X, Sakai R, Taegtmeyer H. Glutamine cycling in isolated working rat heart. Am J Physiol Endocrinol Metab. 2003;285:E1312–E1316. doi: 10.1152/ajpendo.00539.2002. [DOI] [PubMed] [Google Scholar]
- Cori CF. Mammalian carbohydrate metabolism. Physiol Rev. 1931;11:143–275. [Google Scholar]
- Fedotcheva NI, Sokolov AP, Kondrashova MN. Nonezymatic formation of succinate in mitochondria under oxidative stress. Free Radic Biol Med. 2006;41:56–64. doi: 10.1016/j.freeradbiomed.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Felig P, Pozefsky T, Marliss E, Cahill GF., Jr Alanine: key role in gluconeogenesis. Science. 1970;167:1003–1004. doi: 10.1126/science.167.3920.1003. [DOI] [PubMed] [Google Scholar]
- Gilissen J, Jouret F, Pirotte B, Hanson J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol Ther. 2016;159:56–65. doi: 10.1016/j.pharmthera.2016.01.008. [DOI] [PubMed] [Google Scholar]
- Hirst J, Sucheta A, Ackrell B, Armstrong F. Electrocatalytic voltammetry of succinate dehydrogenase: direct quantification of the catalytic properties of a complex electron-transport enzyme. J Am Chem Soc. 1996;118:5031–5038. [Google Scholar]
- Hoberman HD, Prosky L. Evidence of reduction of fumarate to succinate in perfused rat liver under conditions of reduced O2 tension. Bio-chim Biophys Acta. 1967;148:392–399. doi: 10.1016/0304-4165(67)90135-3. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Dressendorfer RH. Succinate accumulation in man during exercise. Eur J Appl Physiol Occup Physiol. 1976;35:235–242. doi: 10.1007/BF00423282. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Owen TG, Allen JF, Whittow GC. Multiple end products of anaerobiosis in diving vertebrates. Comp Biochem Physiol B. 1975;50:17–22. doi: 10.1016/0305-0491(75)90292-8. [DOI] [PubMed] [Google Scholar]
- Hohl C, Oestreich R, Rösen P, Wiesner R, Grieshaber M. Evidence for succinate production by reduction of fumarate during hypoxia in isolated adult rat heart cells. Arch Biochem Biophys. 1987;259:527–535. doi: 10.1016/0003-9861(87)90519-4. [DOI] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Zhan Yanxiang Guo J, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idström JP, Soussi B, Elander A, Bylund-Fellenius AC. Purine metabolism after in vivo ischemia and reperfusion in rat skeletal muscle. Am J Physiol. 1990;258:H1668–H1673. doi: 10.1152/ajpheart.1990.258.6.H1668. [DOI] [PubMed] [Google Scholar]
- John RA, Charteris A. The reaction of amino-oxyacetate with pyridoxal phosphate-dependent enzymes. Biochem J. 1978;171:771–779. doi: 10.1042/bj1710771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhauer M, Dawkins S, Costa ASH, Lee R, Young T, Pell VR, Choudhury RP, Banning AP, Kharbanda RK, Saeb-Parsy K, et al. Oxford Acute Myocardial Infarction (OxAMI) Study. Metabolomic profiling in acute ST-segment-elevation myocardial infarction identifies succinate as an early marker of human ischemia-reperfusion injury. J Am Heart Assoc. 2018;7:e007546. doi: 10.1161/JAHA.117.007546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante A, Vincent G, Poirier M, Des Rosiers C. Effects and metabolism of fumarate in the perfused rat heart. A 13C mass isotopomer study. Am J Physiol. 1997;272:E74–E82. doi: 10.1152/ajpendo.1997.272.1.E74. [DOI] [PubMed] [Google Scholar]
- Leite LN, Gonzaga NA, Simplicio JA, do Vale GT, Carballido JM, Alves-Filho JC, Tirapelli CR. Pharmacological characterization of the mechanisms underlying the vascular effects of succinate. Eur J Pharmacol. 2016;789:334–343. doi: 10.1016/j.ejphar.2016.07.045. [DOI] [PubMed] [Google Scholar]
- Lewandowski ED, White LT. Pyruvate dehydrogenase influences postischemic heart function. Circulation. 1995;91:2071–2079. doi: 10.1161/01.cir.91.7.2071. [DOI] [PubMed] [Google Scholar]
- Maklashina E, Berthold DA, Cecchini G. Anaerobic expression of Escherichia coli succinate dehydrogenase: functional replacement of fumarate reductase in the respiratory chain during anaerobic growth. J Bacteriol. 1998;180:5989–5996. doi: 10.1128/jb.180.22.5989-5996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470. e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, Campo G, Pinton P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol. 2015;78:142–153. doi: 10.1016/j.yjmcc.2014.08.015. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadtochiy SM, Urciuoli W, Zhang J, Schafer X, Munger J, Brookes PS. Metabolomic profiling of the heart during acute ischemic preconditioning reveals a role for SIRT1 in rapid cardioprotective metabolic adaptation. J Mol Cell Cardiol. 2015;88:64–72. doi: 10.1016/j.yjmcc.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TT, Støttrup NB, Løfgren B, Bøtker HE. Metabolic fingerprint of ischaemic cardioprotection: importance of the malate-aspartate shuttle. Cardiovasc Res. 2011;91:382–391. doi: 10.1093/cvr/cvr051. [DOI] [PubMed] [Google Scholar]
- Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286:1–11. doi: 10.1074/jbc.M110.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. Moving forwards by blocking back-flow: the yin and yang of MI therapy. Circ Res. 2016;118:898–906. doi: 10.1161/CIRCRESAHA.115.306569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuhkurinen KJ, Takala TE, Nuutinen EM, Hassinen IE. Tricarboxylic acid cycle metabolites during ischemia in isolated perfused rat heart. Am J Physiol. 1983;244:H281–H288. doi: 10.1152/ajpheart.1983.244.2.H281. [DOI] [PubMed] [Google Scholar]
- Pisarenko OI, Studneva IM, Portnoy VF, Arapov AD, Korostylev AM. Ischemic heart arrest: nitrogenous metabolism in energy-depleted human myocardium. Eur Surg Res. 1987;19:329–336. doi: 10.1159/000128718. [DOI] [PubMed] [Google Scholar]
- Sanadi DR, Fluharty AL. On the mechanism of oxidative phosphorylation: the energy-requiring reduction of pyridine nucleotide by succinate and the energy-yielding oxidation of reduced pyridine nucleotide by fumarate. Biochemistry. 1963;2:523–528. doi: 10.1021/bi00903a023. [DOI] [PubMed] [Google Scholar]
- Smith B, Schafer XL, Ambeskovic A, Spencer CM, Land H, Munger J. Addiction to coupling of the Warburg effect with glutamine catabolism in cancer cells. Cell Rep. 2016;17:821–836. doi: 10.1016/j.celrep.2016.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Støttrup NB, Løfgren B, Birkler RD, Nielsen JM, Wang L, Caldarone CA, Kristiansen SB, Contractor H, Johannsen M, Bøtker HE, Nielsen TT. Inhibition of the malate-aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Res. 2010;88:257–266. doi: 10.1093/cvr/cvq205. [DOI] [PubMed] [Google Scholar]
- Sucheta A, Ackrell BA, Cochran B, Armstrong FA. Diode-like behaviour of a mitochondrial electron-transport enzyme. Nature. 1992;356:361–362. doi: 10.1038/356361a0. [DOI] [PubMed] [Google Scholar]
- Swain JL, Hines JJ, Sabina RL, Harbury OL, Holmes EW. Disruption of the purine nucleotide cycle by inhibition of adenylosuccinate lyase produces skeletal muscle dysfunction. J Clin Invest. 1984;74:1422–1427. doi: 10.1172/JCI111553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls-Lacalle L, Barba I, Miró-Casas E, Alburquerque-Béjar JJ, Ruiz-Meana M, Fuertes-Agudo M, Rodríguez-Sinovas A, García-Dorado D. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res. 2016;109:374–384. doi: 10.1093/cvr/cvv279. [DOI] [PubMed] [Google Scholar]
- Valls-Lacalle L, Barba I, Miró-Casas E, Ruiz-Meana M, Rodríguez-Sinovas A, García-Dorado D. Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep. 2018;8:2442. doi: 10.1038/s41598-018-20866-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Cascarano J. The energy-yielding oxidation of NADH by fumarate in submitochondrial particles of rat tissues. Biochim Biophys Acta. 1970;216:54–62. doi: 10.1016/0005-2728(70)90158-1. [DOI] [PubMed] [Google Scholar]
- Wojtovich AP, Brookes PS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. 2009;104:121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Kombu RS, Kasumov T, Zhu SH, Cendrowski AV, David F, Anderson VE, Kelleher JK, Brunengraber H. Metabolomic and mass isotopomer analysis of liver gluconeogenesis and citric acid cycle. I. Interrelation between gluconeogenesis and cataplerosis; formation of methoxamates from aminooxyacetate and ketoacids. J Biol Chem. 2008;283:21978–21987. doi: 10.1074/jbc.M803454200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Yu D, Mo R, Zhang J, Hua H, Hu L, Feng Y, Wang S, Zhang WY, Yin N, Mo XM. The succinate receptor GPR91 is involved in pressure overload-induced ventricular hypertrophy. PLoS ONE. 2016;11:e0147597. doi: 10.1371/journal.pone.0147597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nadtochiy SM, Urciuoli WR, Brookes PS. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am J Physiol Heart Circ Physiol. 2016;310:H29–H38. doi: 10.1152/ajpheart.00926.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.