

Summary
Surgical repair or replacement of the mitral valve is currently the only recommended therapy for severe primary mitral regurgitation. The chronic elevation of wall stress caused by the resulting volume overload leads to structural remodelling of the muscular, vascular and extracellular matrix components of the myocardium. These changes are initially compensatory but in the long term have detrimental effects, which ultimately result in heart failure. Understanding the changes that occur in the myocardium due to volume overload at the molecular and cellular level may lead to medical interventions, which potentially could delay or prevent the adverse left ventricular remodelling associated with primary mitral regurgitation. The pathophysiological changes involved in left ventricular remodelling in response to chronic primary mitral regurgitation and the evidence for potential medical therapy, in particular beta–adrenergic blockers, are the focus of this review.
Keywords: mitral regurgitation, left ventricular remodelling, medical therapy, beta-blocker
Mitral regurgitation (MR) is caused by failure of adequate coaptation of the anterior and posterior mitral leaflets duringleft ventricular contraction, resulting in various degrees ofregurgitation of blood from the left ventricle (LV) into the leftatrium (LA). The result of this regurgitation is twofold. Firstly,there is a reduction in forward stroke volume (FSV) into theaorta, with subsequent reduction in perfusion. Secondly, there isan increase in LA blood volume during ventricular systole, whichresults in an increase in left ventricular preload, the so-called‘volume overloaded’ state.
MR is classified as either primary (organic) or secondary (functional), and acute or chronic.1 Causes of acute MR include infective endocarditis and spontaneous cordal rupture and will not be discussed further in this review. Chronic secondary MR can be ischaemic and/or non-ischaemic in nature and therapies for secondary MR range from medical to surgical.2 By contrast, chronic primary MR is predominantly caused by degenerative disease in developed countries,3 and rheumatic heart disease (RHD) in developing countries.4 RHD is one of the major contributors to the aetiology of heart failure in Africa, where it remains the most common form of acquired cardiovascular disease in children and adults.4
Current therapy for patients with severe chronic primary MR, as recommended by the European Society of Cardiology guidelines,1 comprises surgical repair or replacement in patients who are surgical candidates, or conservative (i.e. palliative) therapy in patients with very poor left ventricular function who are deemed to be poor surgical candidates. At present, there is no recommendation for drug therapy in patients with any degree of chronic primary MR. However, once heart failure develops, angiotensin converting enzyme inhibitors (ACE inhibitors), beta-blockers and spironolactone may be considered.5
Although there have been several recent reviews focusing on ventricular remodelling in ischaemic heart disease, hypertensive heart disease and aortic stenosis, there have been few recent reviews on pathological left ventricular remodelling in patients with primary MR.6-8 In this review we focus in particular on the pathophysiological changes seen in the myocardium of the LV due to volume overload caused by chronic primary MR. We also discuss medical interventions that may attenuate or reverse the adverse changes seen in chronic primary MR, focusing on data related to the use of beta-blockers in these patients.
Pathophysiological changes in the LV in chronic primary MR
Primary MR may present acutely, as a slowly progressive disease, or as chronic progressive MR with sudden deterioration related to acute changes in mitral valve anatomy such as a ruptured cord. Acute MR is usually a medical emergency requiring emergent surgery and is not the focus of this review.
Patients with chronic primary MR are often asymptomatic for long periods of time before presenting at a late stage in heart failure. During this period, there is development of progressive left ventricular dysfunction as the LV is remodelled in an attempt to produce an adequate forward stroke volume.9,10 Five- to 10-year cardiovascular mortality rates vary between 10 and 15%, with a worse prognosis for patients with severe MR.11,12
Alterations in the global structure of the LV in response to primary MR have been reviewed in detail previously.9 Briefly, MR results in increases in LA volume, a reduction in FSV and an increase in left ventricular preload. By mechanisms that are unclear but are discussed in more detail below, the LV responds to the increased preload by eccentric hypertrophy, with a serial increase in myocyte sarcomeres and myofibril slippage (Fig. 1).13-18
Fig. 1.

Left ventricular remodelling in chronic primary mitral regurgitation. A: Normal LV is represented on the left. Wall stress is normal. B: Chronic compensation with eccentric hypertrophy and dilatation. The increase in LV volume is compensated for by the increase in wall thickness. Wall stress appears to be normalised by the eccentric hypertrophy. FSV is normal because of increased LV filling. C: Adversely remodelled LV of decompensated chronic MR. The myocardial wall is thin resulting in an increase in wall stress. The arrow indicates severe MR, which becomes more severe with a dilating LV. LA = left atrium; LV = left ventricle; TSV = total stroke volume; FSV = forward stroke volume; MR = mitral regurgitation.
Eccentric hypertrophy normalises afterload, as estimated by mean systolic stress, compared to patients with aortic regurgitation, leading to a period of so-called ‘compensation’.19 However, the hypertrophy that develops is actually insufficient to fully compensate for the wall stress that develops. This is due to inadequate protein synthesis triggered by MR compared to pressure overload,16,20 and progressive deterioration in myocardial function.21
There is no clear explanation for this phenomenon but it has been proposed that the lower systolic load in the case of MR may result in a reduced hypertrophic response at a time when there is a marked demand for an increased stroke volume.21 Altered cytoskeletal changes, such as microtubular density, may also play a role.22 With time, in the face of inadequate hypertrophy and a dilating LV, systolic wall stress increases [based on the Laplace effect where wall stress (σ) is directly related to the pressure within the ventricle and its radius (Pr), and inversely related to the wall thickness (2h); σ = Pr/2h] due to the increases in LV dimensions and inadequate hypertrophy.21,23,24
Chronic increases in wall stress are detrimental to the myocardium, resulting in activation of a number of complex inflammatory and apoptotic pathways, in a similar manner to heart failure from other causes. Ultimately, there is myocyte loss and sliding displacement of cardiomyocytes, or cell slippage, caused by disruption of the myocardial extracellular matrix (ECM)–integrin linkages.7,25
Various lines of evidence point to time-dependent changes in the up- and downregulation of remodelling pathways in chronic primary MR.26 This process is initiated by diastolic mechanical stretch due to an increase in end-diastolic wall stress, leading to an early increase in reactive oxygen species (ROS) generation, inflammatory cytokine expression and neurohormonal activation, with increases in angiotensin II and catecholamine levels. Early in the remodelling process there is interstitial collagen loss and cell slippage but with time there is myocyte apoptosis and pathological ECM fibrosis.27 Chronic decompensated MR ensues, and the LV resembles end-stage dilated cardiomyopathy.
MR causes mechanical stretch, which triggers mechanoreceptors and activates signal-transduction pathways
Myocardial mechanoreception is currently poorly understood.28-30 There is no evidence that specialised mechanosensory cells exist in the myocardium and the role of stretch-activated channels in sensing stretch is debatable.29 Two systems appear to be particularly important in mechanoreception in the cardiomyocyte: the collagen–integrin–cytoskeleton connections 25,31 and sarcomererelated signalling.30
The contraction–relaxation cycle of the myocyte depends on coordinated interaction between the thin actin filament and the thick myosin filament within the myocyte sarcomere.32 Actin is bound directly to the Z-disc while myosin is bound indirectly to the Z-disc via the giant elastic protein, titin.33,34 In the normal heart, titin is responsible for restoring the stretched sarcomere to its resting length following active contraction.33,34 However, another important role for titin is in mechanoreception and the activation of a number of signal-transduction pathways when there is chronic myocyte stretch.28,29,33,35-37 It does this by changes in the expression of various genes involved in adaptation to the increased load and, ultimately, to the activation of various maladaptive pathways.38-40
Titin complexes with a number of potential ‘signalosomes’ (a mechanosensative signalling complex), including the Z-disclocalised protein MLP (muscle LIM protein), which has been shown to be responsible for hypertrophy and cardiomyopathy in MLP-deficient animals.41,42 MLP, aside from its structural role in the Z-disk and its interaction with signal transduction proteins, is able to translocate to the nucleus and thereby act as a transcription factor modifying gene expression, depending on mechanical stretch.43 MLP may be responsible for control of other transcription factors coordinating alterations in the expression of genes responsible for ventricular remodelling. Another important titin signalosome that controls muscle gene expression is the sarcomere M-band-associated protein titin kinase (TK), which is activated by myocyte stretch.38,40 TK may primarily respond to diastolic stretch,29 which is particularly relevant in the case of pathological volume overload caused by chronic MR (Fig. 2).
Fig. 2.
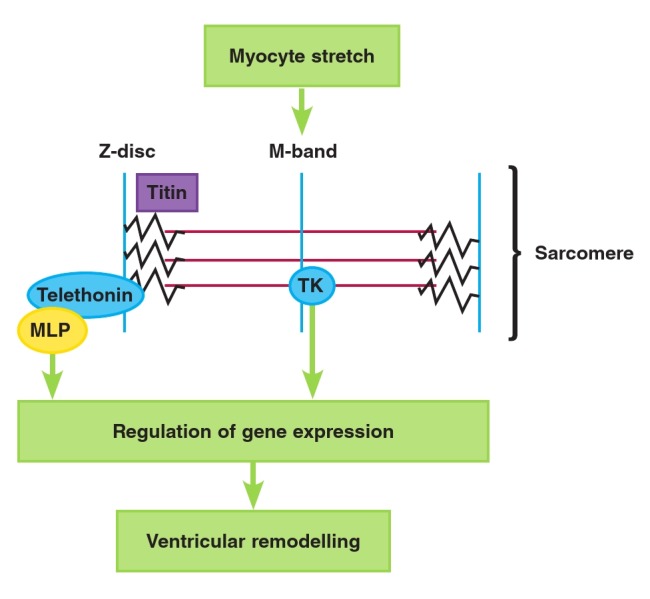
Schematic representation of gene regulation in response to myocyte sarcomere stretch via signal transduction through MLP and TK. MLP = muscle LIM protein; TK = titin kinase. See text for details.
Pathological volume overload-induced mechanical stretch has a number of other effects on the cardiomyocyte. For example, in in vitro44 and in vivo45 rat experiments, TNF-α is produced by cardiomyocytes, resulting in an inflammatory response to stretch, suggesting that TNF-α is an important component in the pathophysiological response of the myocardium to volume overload. Mechanical stretch also results in the local production of angiotensin II46 and ROS,47 which, via transcription factors, such as TRAIL (TNF-related apoptosis-inducing ligand) and NFκB,47 result in local increases in pro-inflammatory cytokines, further contributing to activation of remodelling signaltransduction pathways.48,49 Finally, as discussed in more detail below, mechanical stretch is transmitted through the ECM to cardiomyocyte integrins, which trigger a number of intracellular signal-transduction pathways involved in hypertrophy and apoptosis.31,39
Chronic primary MR increases cardiac reactive oxidative stress
ROS play an important role in signal transduction and physiological regulation in vascular and myocardial cells. However, under pathological conditions, such as excessive myocyte stretch or excessive inflammatory signals, ROS have been shown to activate maladaptive remodelling signal-transduction pathways.50,51 These signal-transduction pathways include (but are not limited to) protein phosphorylation pathways leading to cell growth or apoptosis (depending on ROS levels and other factors); matrix metalloproteinase activation;52 cell cycle protein pathways leading to apoptosis; and pathways leading to the activation of inflammatory transcription factors such as NFκB.53
ROS are increased in patients with congestive heart failure,54 and there is evidence of pathological increases in ROS in patients with chronic isolated MR who still have left ventricular ejection fractions (LVEF) above 60%.55 These data suggest that there is an increase in oxidative stress even before the LV starts to develop systolic dysfunction, which supports the notion that wall stress is present throughout the evolution of left ventricular remodelling in primary MR. This oxidative stress appears to be present as long as the volume overload persists (Fig. 3).56
Fig. 3.
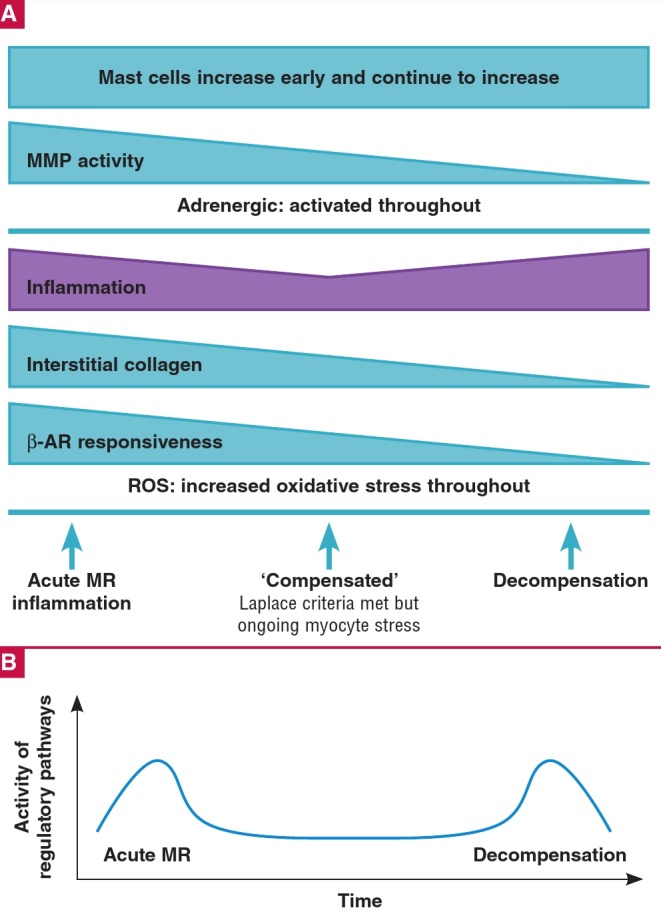
A. Proposed time–dependent changes in various remodelling pathways including changes in measured prevalence of mast cells. B. Proposed overall timedependent changes in remodelling pathway activation. MMP = matrix metalloproteinases; β–AR = β–adrenergic; ROS = reactive oxidative species. See text for details.
Chronic primary MR triggers an inflammatory Response
Tumour necrosis factor (TNF), interleukin-1 (IL-1) and interleukin-6 (IL-6) are produced by all nucleated cells, including cardiac myocytes.57 Cytokines are responsible for beneficial adaptation to short-term stresses, such as haemodynamic overload, within the myocardium. These molecules may play an important role in protecting the heart from oxidative injury and there are several lines of evidence supporting their beneficial role in short-term stress.58 However, the role of cytokines in remodelling is complicated and not easily predictable. For example, TNF can have both a protective and an adverse effect on the myocardium, depending on which TNF receptors are activated.59 Furthermore, prolonged elevation of tissue cytokines has been found to have deleterious effects on the LV.60,61
Chronic elevation of cytokines has an effect on left ventricular remodelling by blunting of β-adrenergic signalling57 and activation of apoptotic pathways.62-64 TNF-α also increases cardiomyocyte apoptosis63,65by activating p38 MAP kinase and NFκB and by down-regulating ERK 1/2 MAP kinase.66 Overexpression of TNF has also been shown to increase tissue matrix metalloproteinase (MMP) activity, with the resultant acute loss in myocardial fibrillar collagen and left ventricular dilatation.67-69 However, with continuing TNF overexpression, there is an increase in tissue inhibitors of metalloproteinase (TIMP-1) expression and reduction in MMP expression, leading to abnormal increases in fibrillar collagen,67,68 suggesting a time-dependent effect of chronic exposure to elevated myocardial TNF.
Cytokines are elevated in patients with heart failure,70 in patients with pressure and volume overload,71 and in other forms of heart disease.72,73Several lines of evidence suggest that the myocardial response to TNFα is similar regardless of aetiology. Gene expression analysis by micro-array suggests that there is a time-dependent inflammatory response to volume overload (Fig. 3).26,45 Very early after the initiation of volume overload in aortocaval fistula rats, there is a marked increase in the expression of inflammatory pathway genes, followed by relative normalisation during the chronic ‘compensated’ period of volume overload.26 This is supported by earlier studies that demonstrate that myocyte stretch induces TNFα secretion from myocytes,44,74 and mast cell-deficient rats with volume overload were protected from TNFα-dependent left ventricular remodelling.69 Furthermore, in humans with compensated chronic primary MR and normal LVEF, there is a down-regulation of inflammatory pathways.56
As the LV becomes dilated and dysfunctional, there is an increase in inflammatory pathway gene expression,26 which is supported by clinical work in patients with severe chronic primary MR71,75 and severe rheumatic aortic regurgitation.76Overall, there appears to be a biphasic elevation in the inflammatory response to mitral regurgitation, with early volume overload activating the expression of numerous inflammatory pathways, and decompensation triggering a second inflammatory response (Fig. 3).
Myocyte loss in chronic primary MR
Apoptosis is activated by several extracellular death signals, including myocyte stretch,77 catecholamines25,78-80 and inflammatory cytokines,57 and various intracellular death signals.81-85 These death signals25 activate transcription factors,86 ultimately resulting in activation of the caspase cascade.87 Loss of myocytes will increase the stress on remaining myocytes. This leads to further increases in ROS,88 cytokine release,44 increases in adrenergic activation,89 perpetuating loss of myocytes in a downward-spiraling process. Time-dependent apoptosis of non-myocyte cells has been described in volume-loaded rats26 and there is evidence that chronic primary MR causes a reduction in contractile elements.21,82,90 Based on this evidence and evidence from studies in myocardial remodelling due to other causes, it is probable that cell loss is an important component in left ventricular dilatation and dysfunction in chronic primary MR.
ECM changes in chronic primary MR
Myocyte arrangement and myocardial integrity is highly organised to enable the continuously moving myocardium to produce coordinated contraction, resulting in stroke volume.91 The structural integrity is provided by the ECM, which comprises a basement membrane, proteoglycans and glycosaminoglycans, and ECM proteins such as type I, III and V collagen, of which approximately 85% is type I collagen.7 This collagen framework serves to maintain cardiac myocyte alignment, without which the myocytes would ‘slip’, altering the shape and size of the cardiac chambers.91
The ECM is a highly dynamic part of the myocardium that changes depending on the degree and type of mechanical stress, neurohormonal activation, inflammation and oxidative stress. These stressors on the ECM result in changes in the expression and activation of the proteins responsible for ECM turnover and, ultimately, alterations in collagen deposition and degradation.
MMPs are a heterogeous family of enzymes responsible for the proteolysis of various protein-based extracellular substances. They include the collagenases (MMP-1, MMP-8 and MMP-13), stromelysins (MMP-3 and MMP-10) and the gelatinases (MMP- 2 and MMP-9). They are expressed and secreted into the extracellular space by a variety of cells, including cardiac myocytes, cardiac fibroblasts and macrophages.92 However, the roles of each MMP and the control of their activity are not yet clearly elucidated and this is an area of on-going research.7 Some studies have demonstrated a correlation between MMP expression and cardiomyopathy phenotypes,93-95and others have demonstrated that serum levels of MMPs have prognostic value in heart failure.96,97
TIMPs are low-molecular-weight proteins that bind to the catalytic domain of active MMPs, preventing substrate binding. There are four species of TIMPs with overlapping functions within the myocardium, which are not restricted to MMP inhibition. Other pleomorphic effects have been described. For example, TIMP-2 increases collagen production by fibroblasts, whereas TIMP-3 is responsible for fibroblast apoptosis.98
Biological and/or mechanical stimuli trigger various signal transduction pathways, resulting in the production of MMP transcription factors and the secretion of these enzymes into the ECM.92,99-101 Mechanical stimuli, such as stretch,102 are transduced through the ECM, which, via collagen–integrin–cytoskeleton connections, are connected to and activate a number of intramyocyte signal-transduction pathways involved in ECM remodelling.31,46,103-106 Local ROS, endothelin-1, angiotensin II and catecholamines, via α- and β-receptors, are also responsible for increases in MMP expression.52,89,107,108
Cytokines, such as TNFα and IL-1, have been found to increase MMP expression,67,102 promoting matrix degradation and ventricular dilatation.7 On the other hand, MMP-9 production can be suppressed by TGF-β-activated NFκB binding in some experiments,109 whereas its expression was up-regulated by angiotensin II-activated NFκB in other experiments.110 Angiotensin II107 and aldosterone both increase ECM remodelling, mainly through TGF-β,111 although the effects of this protein are multiple and often opposing, depending on circumstances.112
TGF-β stimulation induces maturation of fibroblasts to myofibroblasts and enhances ECM protein synthesis via induction of TIMP expression and inhibition of certain MMP expression.111 However, this is dependent on the load on the myocardium and there is clear evidence that volume overload results in reduction in TGF-β level and loss of interstitial collagen,113 whereas pressure overload increases TGF-β.114 The result is increased detection of markers of collagen types I and III turnover in the serum,115 pathological decreases in interstitial collagen15,116,117 and left ventricular dilatation.
In response to different haemodynamic overloads (pressure versus volume), the ECM undergoes different patterns of remodelling.27 Volume overload produces a distinctive loss of collagen fibrils surrounding individual myocytes,15,116,118 with the resultant wall thinning and ventricular dilatation changing the geometrical shape of the LV, whereas excess matrix deposition is observed in pressure overload.119,120 Despite similar fibrotic molecular pathways and cellular effectors, the pathophysiological mechanisms leading to fibrotic remodelling are different, depending on the load on the heart.7 For example, ACE inhibitors reduce remodelling and collagen accumulation in pressure overload,121 but not in chronic MR.15,122 Furthermore, the expression of integrins, which are important in ECM–myocyte connectivity and ECM remodelling, are reduced in MR113 but increased in pressure overload.123 Similarly, profibrotic TGF-β expression was increased in mice with pressure overload114 but was decreased in dogs with experimental MR,113 and expression of PAI-1 was increased in a swine model of early pressure overload124 but decreased in chronic MR.113
There appears to be a time-dependent increase and decrease in MMP activity during the evolution of left ventricular remodelling in response to primary MR (Fig. 3).26,117 Myocardial mast cells have been found to be instrumental in increases in MMP activity in early volume overload,69,117,125,126 and are increased in number in response to volume overload-induced increases in myocardial TNFα.45 In animal models there is an early rise in myocardial MMP levels after the volume-loaded state is created but this seems to normalise after the acute phase.127,128
MMP gene expression in dogs with isolated MR has confirmed that, at four months, there was down-regulation of a number of non-collagen genes important in ECM structure, down-regulation of pro-fibrotic connective tissue growth factor and plasminogen activator, and down-regulation of numerous genes in the TGF-β pathway.113 However, MMP-1 and MMP-9 gene expression was still markedly increased in these dogs with compensated MR compared with controls.113 As the LV started to dilate in dogs with chronic myxomatous mitral valve disease, MMP-9 levels decreased.129
Over time, there are characteristic changes in the MMP/TIMP ratio, enabling the ventricle to initially increase compliance in the acute and compensated phases of MR. However, at some point (the ‘transition’ point) there is excessive degradation of the ECM, leading to the decompensated and dilated LV.27,130 What controls the steady deterioration in the myocardium in response to volume overload is not clear and appears to be complex. In the early stages of volume overload, there are decreases in ECM deposition (which contrasts with the picture in pressure overload),113 but late in the progression of the dilating volumeloaded heart, an increase in perivascular collagen deposition has been noted,26,126 which may reduce ventricular compliance and promote systolic dysfunction.27
Chronic primary MR activates the neurohormonal system: implications for beta-blocker therapy
Patients with chronic primary MR demonstrate LV systolic dysfunction even before a reduction in LVEF occurs.131,132 As with heart failure due to any other cause, chronic MR results in activation of the neurohormonal system and inflammatory cascade at both systemic and local levels.133-135 With neurohormonal activation, myocardial angiotensin II plays an important role in the regulation of cell proliferation, apoptosis, inflammation and production of mediators of remodelling such as platelet-derived growth factor and MMPs.136 Persistent angiotensin receptor-1 activation by angiotensin II not only results in the generation of ROS but also alterations in protein synthesis via tyrosine kinase receptor activation and MAP kinase signalling.137 Furthermore, angiotensin II-activated ROS act as second messengers that also have effects on inflammation and cell growth.138 Angiotensin II also acts on the sympathetic nerve endings in the myocardium to facilitate catecholamine release.139,140
Long-term increases in myocardial angiotensin II levels increase local TGF-β, with the resultant increases in activation of genes involved in ECM production via nuclear translocation of NFκB.110,141 Unlike the pressure-overloaded heart where there is progressive fibrosis,142 the increase in myocardial angiotensin II in volume overload results in an increase in ECM turnover with loss of interstitial ECM.143 Despite the clear link between angiotensin and remodelling in heart failure, to date there has been little clinical evidence to support the role of medical therapy directed against angiotensin in subjects with chronic organic MR.15,144-146 This may be explained by the fact that ACE inhibitors reduce the breakdown of bradykinin, which has been implicated in the initial increase in MMP activity and collagen breakdown seen in volume overload.143
Three types of β-adrenergic receptors (β-ARs) are known to exist in the myocardium: β1, β2 and β3, with an approximate ratio of 80:17:3.147 β1 and β2 are important in the regulation of myocyte excitation–contraction coupling.80 β1-AR is the predominant receptor subtype expressed in the heart and, like other β-ARs, its stimulation results in G-protein-coupled activation of the adenyl cyclase–cAMP–protein kinase A (PKA) signalling cascade. This leads to activation of a number of subcellular pathways important in cardiomyocyte contractile function, including calcium channel activation and troponin I phosphorylation. By contrast, β2-AR signalling has negative effects on adenyl cyclase activation and the subsequent G-protein-activated ionotropic response.80 β3-AR appears to be important in protection from hypertrophic and fibrotic remodelling by preserving NO/cGMP signalling during cardiac stress.148
The β-adrenergic receptor system plays an important role in the pathogenesis of myocardial remodelling and heart failure.84 The exact mechanisms are unclear but it has been known for decades that chronically increased plasma catecholamines can lead to heart failure.149,150 In dogs with chronic primary MR, there is activation of the adrenergic system,89,108 and recent gene array data in chronic primary MR patients with preserved LVEF demonstrate increased expression of genes involved in β-adrenergic signalling.56 This indicates that the adrenergic system is activated during the compensatory phase of MR and supports the concept that blocking these pathways may reduce their adverse consequences. However, with transition to decompensation there is a reduction in adrenergic responsiveness.
In patients with systolic heart failure (HF), several studies in the last three decades show that â1-receptor density151 and its mRNA152,153 are reduced while â2-receptor density remains unchanged.154 Similarly, in animals with HF due to chronic volume overload, â1-AR responsiveness is reduced due to neurohormonal activation (Fig. 3).47,155 These changes in â1-AR expression are caused by sustained adrenergic activity, causing an increase in the expression and activity of GRK 2 (G-proteincoupled receptor kinase; formerly called â-ARK or â-agonist receptor kinase), resulting in â1-AR being phosphorylated and labelled for desensitisation, internalisation and recycling.156 The result is a reduction in the density of â1-ARs and a reduced propensity for myocyte activation by chronic â1-receptor activation, which may protect the myocyte from long-term catecholamine toxicity.80
Beta-blocker therapy improves β1-AR signalling and clinical outcomes in HF
Chronic β1-AR activation causes a number of detrimental effects, ultimately resulting in changes in the ECM and cell loss from necrosis and apoptosis,25 which in turn leads to cardiac dilatation and failure.157 However, the intracellular pathways responsible for these final acts are unclear.25 What is clear is that β1-AR antagonists improve clinical outcomes in patients with systolic heart failure and improve cardiac function and myocardial remodelling.158
Most β-adrenergic blockers are antagonistic to β-ARs (whether β-1, β-2 or β-3) by occupying the receptor and preventing signal transduction via G-protein activation. Importantly, there is an up-regulation of β1-AR expression and improvements in receptor sensitivity, resulting in reversal of cardiac remodelling.147 However, the pharmacological and clinical effects of these agents vary quite considerably. Cardioselective beta-blockers (metoprolol, bisoprolol and atenolol, for example) have a greater affinity for β1-ARs than β2-ARs, whereas carvedilol binds β1-ARs more than β2-ARs and has vasodilatory effects, via nitric oxide and α1-receptor blockade.
There are other important differences between carvedilol and other beta-blockers. For example, metoprolol upregulates cardioprotective β3-AR expression, whereas carvedilol does not.159,160 Carvedilol has antioxidant and antiproliferative properties161-163 and differs from metoprolol in its effects on haemodynamics, left ventricular function and β1-AR expression.164,165 Carvedilol166 and bisoprolol167 have also been shown to improve right ventricular (RV) ejection fraction, attenuate RV dilatation and reduce pulmonary artery hypertension in patients with ischaemic and non-ischaemic dilated cardiomyopathy. Although there are no recent confirmatory studies, these improvements in RV function may be related to reductions in RV afterload and/or improvements in RV contractility.166,167 By contrast, short-term (two-week) metoprolol did not improve RV function in patients with moderate-to-severe degenerative MR.168
Clinical support for β-adrenergic receptor blocker therapy in patients with heart failure is well known,158,169,170 with some data suggesting that patient outcomes are better with carvedilol than the immediate-release form of metoprolol.171
Several mechanisms for the improvement in outcomes with β1-receptor blockade have been proposed,84 including antiarrhythmic properties;172 improved β-adrenergic signalling by cardiac β-AR upregulation;80 free-radical scavenging;161 improvements in calcium cycling by the sarcoplasmic reticulum;83,173 bradycardia reducing myocardial work, mechanical stress,174 and prolonging diastolic calcium uptake and cycling by the sarcoplasmic reticulum; inhibition of the renin– angiotensin–aldosterone system; and there is growing evidence that β-antagonists, in particular carvedilol,162,163 directly reduce apoptosis78,175-177 and collagen loss by MMP activation.178
β1-AR blockade in MR counters adverse adrenergic effects
Since MR leads to a reduction in forward stroke volume, it is hypothesised that the adrenergic system is activated to maintain systemic blood pressure and perfusion, and blockade of the adrenergic system should limit adverse left ventricular remodelling. There is evidence that chronic primary MR results in excessive activation of the sympathetic nervous system, with increases in myocardial catecholamine levels,89,118,134 similar to heart failure from other causes.179
Tallaj et al.118 demonstrated that β-AR blockade with extended release metoprolol succinate attenuated angiotensin II-mediated norepinephrine and epinephrine release in the myocardium of dogs with ‘subacute’ (two to four weeks’ duration) isolated MR. Similarly, Hankes et al.89 demonstrated that norepinephrine release into the cardiac interstitium was significantly higher in dogs with subacute MR, which was reduced by β1-AR blockade. In an earlier study by Tsutsui et al.90 in ‘chronic’ (three months) canine MR, the β1- AR blocker atenolol improved left ventricular function, which was associated with improvement in contractile function of isolated cardiocytes and an increase in the number of contractile elements. This was supported by a similar study by Nemoto et al.,145 which showed that only when a β1-AR blocker was added to an ACE inhibitor did forward stroke volume and cardiac contractility return to normal. Recently, Trappanese et al.180 demonstrated an improvement in β3-AR expression and β3-NO-cGMP coupling with chronic therapy with metoprolol in dogs with primary MR. Since β3-AR is cardioprotective, this may partially explain the potential beneficial effects of β1-AR blockade in primary MR.159
Beta-blockade therapy for chronic primary MR
At present there is no proven medical therapy for chronic primary MR. Surgery is the mainstay of treatment for severe MR1 but carries peri-operative risk, and patients are potentially subjected to a life-time risk of anticoagulation if they undergo mitral valve replacement. Many patients in the developing world are from poor and rural backgrounds where access to regular medication and regular anti-coagulation assessment is difficult. A medication that could limit or even reverse left ventricular dysfunction associated with the volume-loaded state of chronic severe MR would be extremely beneficial to these patients, even if only to delay the need for surgical intervention. This would especially be true in women of child-bearing age. Warfarin is teratogenic and many women with prosthetic valves have complicated pregnancies related to the teratogenic effects of warfarin or the risks related to bleeding during pregnancy.
Persistent, and often worsened, postoperative left ventricular dysfunction is a major cause of morbidity and mortality in these patients,133 although this is not a universal finding, especially when patients are referred for early surgery.181-183 Nevertheless, a means to improve left ventricular function in the peri-operative period might improve the postoperative morbidity and mortality rates associated with left ventricular dysfunction.
Current guidelines1,184 recommend surgery for patients with severe pulmonary hypertension on presentation or a progressively dilating LV, even if they are asymptomatic. However, timing of surgery is uncertain185,186 and there is no clear guideline as to the urgency of the surgery in asymptomatic patients without overt left ventricular systolic dysfunction (LVEF < 60%). Several studies support early surgery for chronic primary MR.187–189 Enriquez–Sarano et al.12 showed that patients with an effective regurgitant orifice area of at least 40 mm2, as assessed by echocardiography, should promptly be considered for cardiac surgery. Barbieri et al.190 found that asymptomatic patients with evidence of pulmonary hypertension ( > 50 mmHg at rest) should undergo prompt surgery. However, there is also evidence that asymptomatic patients with severe MR can be followed until they become symptomatic or demonstrate echocardiographic signs of left ventricular dysfunction.132,191
How these patients should be managed in the interim is unclear but it is important that patients are not left untreated until irreversible left ventricular remodelling has taken place. In resource–limited hospitals, patients who do not need emergency surgery often wait several months before they undergo surgery, by which time there has been progressive advancement of ventricular remodelling, leading to permanent impairment of function. Medical therapy that could reverse or at least attenuate LV remodelling may improve outcomes in these patients.
To date, there is little evidence to support medical therapy in the treatment of patients with organic valve disease,1,119,192 or in the reversal or attenuation of LV remodelling, which may delay the need for surgery in asymptomatic patients.193 Vasodilator therapy, which reduces peripheral vascular resistance and left ventricular afterload,119 has generally not improved outcomes in patients with MR or in experimental MR canine models.15,122,145,194 Although several small studies from the 1970s to the 1990s showed benefit of vasodilator therapy in acute MR,144 small human studies from the same period failed to show long–term benefit.195 One retrospective study, however, demonstrated an improvement in echocardiographic LVEF in patients treated with after load–reducing agents.146 However, there are no large randomised studies assessing long–term vasodilator therapy, including ACE inhibitors, in humans.
There is some clinical evidence to support the concept that β–AR blockade may attenuate remodelling in patients with primary MR (Table 1). Stewart et al.196 recruited 25 patients with moderate or severe degenerative MR and randomly assigned the participants to the β1–AR blocker metoprolol, or placebo for approximately two weeks. Left ventricular function was assessed at baseline and on study completion by cardiac magnetic resonance imaging. They found that the β1–AR blocker resulted in a decrease in left ventricular work and an increase in forward stroke volume.196 Mitral annular dimensions also appeared to improve over the two–week period in the same cohort of patients.197
Table 1. Studies of beta-blocker therapy and left ventricular function in primary MR.
| Authors | Year | Subject | Cause of MR | Number treated with BB | Type of study | Control | Type of BB | Duration of BB | Outcome measures | Favours BB | |
| Tsutsui et al.89 | 1994 | Dog | Experimental chordal rupture | n = 6 | Case Controlled | n = 6 | Atenolol 50mg daily | 3 months | Cardiocyte contractility, myofibrillar density | + | |
| Nemoto et al.144 | 2002 | Dog | Experimental chordal rupture | n = 11 | Longitudinal | NA | Atenolol 100 mg Daily | 3 months | Haemodynamics, LV Function | + | |
| Tallaj et al.117 | 2003 | Dog | Experimental chordal rupture | 2 weeks MR+BB: n = 6 4 weeks MR+BB: n = 8 | Case Controlled | Normal: n = 8 2 weeks MR: n = 8 4 weeks MR: n = 6 | Metoprolol Succinate | 4 weeks | RAAS activation | + | |
| Hankes et al. 88 | 2006 | Dog | Experimental chordal rupture | 4 weeks of MR+BB = 8 | Case Controlled | Normal = 6 Untreated MR = 6 | Metoprolol succinate 100 mg Daily | 4 weeks | NE release into cardiac interstitium | + | |
| Oh et al. 145 | 2007 | Human | 71% degenerative | n = 134 | Retrospective Cohort | NA | Not ascertained | 1–88 Months | Echo LVEF | – | |
| Pat et al. 199 | 2008 | Dog | Experimental chordal rupture | n = 11 | Case Controlled | n = 10 | Metoprolol succinate 100 mg twice daily | 4 months | LV remodelling by MRI and echo; cardiomyocyte function | Improved cardiomyocyte function and BB receptiveness but failure to attenuate remodelling | |
| Sabri et al.107 | 2008 | Dog | Experimental chordal rupture | n = 6 | Case Controlled | Normal = 6 Untreated MR = 6 | Metoprolol succinate 100 mg Daily | 4 weeks | LV remodelling by echo; interstitial collagen quantification; FAK signalling (integrin signalling) | BB reduced FAK tyrosine phosphorylation but no change in remodeling parameters; BB reduced epicardial collagen loss but not endocardial collagen loss | |
| Varadarajan et al.197 | 2008 | Human | LVEF > 55% + ‘severe MR’ | n = 218 | Retrospective observational cohort study | n = 614 | Not stated | 8 years | Mortality | + | |
| Stewart et al. 195 | 2008 | Human | MVP | n = 25 | Double-blind cross-over study | NA | Metoprolol to a maximum 190 mg daily | 14 days | MRI EF | – | |
| LVEDV | – | ||||||||||
| LVESV | – | ||||||||||
| LV ‘work’ (CO) | + | ||||||||||
| Ahmed et al. 198 | 2012 | Human | MVP | n = 19 | RCT | n = 19 | Toprol XL | MRI LVEF | + | ||
| 25–100 mg | MRI LVESV | – | |||||||||
| Daily | LV longitudinal strain rate | – | |||||||||
| Pu et al. 200 | 2013 | Rat | Experimental leaflet disruption | n = 43 ‘Long-term’ BB in 19 | RCT | n = 44 | Carvedilol (1 200 ppm) | 36 weeks | Echo only LV dimensions LVESV and mass index FS and EF Survival probability | – | |
| Trappanese et al.179 | 2015 | Dog | Experimental chordal rupture | n = 8 (MR + BB) | Case Controlled | Normal = 10 | Metoprolol succinate | 4 weeks | Activation of β3AR/ NO-cGMP signalling | + | |
| Untreated MUntreated MR = 8R = 8 | 100 mg Daily | β3-AR expression | + |
BB = beta-blocker; MVP = mitral valve prolapse; RCT = randomised controlled trial; MR = mitral regurgitation; NE = norepinephrine; + indicates that the study favoured BB therapy in primary MR; – indicates that the study did not favour BB therapy in primary MR; LV = left ventricle; LVEF = left ventricular ejection fraction; LVEDV = left ventricular end-diastolic volume; LVESV = left ventricular end-systolic volume; CO = cardiac output; RAAS = renin–angiotensin–aldosterone system; echo = echocardiogram; MRI = magnetic resonance imaging; ppm = parts per million.
A retrospective observational study involving 895 patients in California showed that participants on β1–AR blocker therapy with severe MR and normal left ventricular function had a significantly lowered mortality hazard, regardless of the presence of hypertension or coronary artery disease.198 Ahmed et al.199 published the results of the first randomised, controlled phase IIb trial of beta–blockade in patients with chronic degenerative MR. Thirty–eight asymptomatic patients with moderate–tosevere isolated MR were randomised to either placebo or longacting metoprolol for two years. Cardiac magnetic resonance analysis showed that patients randomised to the β–AR blocker had significantly better LVEFs after two years of therapy.
By contrast, there are several pre–clinical and clinical studies demonstrating that beta–blocker therapy actually worsens left ventricular dimensions and function in chronic primary MR (Table 1).108,146,197,200,201 A recent, longer–term (23 to 35 weeks) study in rats found that echocardiographic measures of left ventricular remodelling were not improved by carvedilol.201 In fact, left ventricular dimensions, ejection fraction and survival were significantly lower with long–term carvedilol use.
Similarly, in a four–month dog model, extended–release metoprolol succinate failed to attenuate the adverse global left ventricular remodelling and ECM loss, but did preserve cardiomyocyte function.200 Interestingly, all dogs treated with β–1–receptor blocker (n = 6) survived to four months, whereas only five out of nine of the untreated dogs survived to four months. Similarly, Sabri et al.108 found that, despite reductions in interstitial collagen degradation and reductions in adverse remodelling–related intracellular signalling, extended–release metoprolol succinate failed to attenuate left ventricular dilatation or decline in left ventricular function.
A retrospective echocardiographic study in 134 human subjects with moderate–to–severe MR (67% degenerative and 20% ‘non–specific thickening’) found that patients exposed to beta–adrenergic blockade developed worsening of their ejection fraction over a mean of 20 months of follow up.146 Finally, despite improvements in left ventricular work and annular dimensions,197 in patients treated over a short period with metoprolol, there were significant increases in left ventricular end–systolic and end–diastolic volume with no significant change in LVEF or regurgitant volume.196
At the present time, there are no recommendations regarding medical therapy in chronic primary MR. Afterload reduction has not consistently been shown to improve long–term outcomes.15,122,145,146,195 Data from heart failure trials158,169,170 as well as from animal models90 and human trials199 suggest a role for beta–blockade in MR, however, other studies do not support this.108,146,197,200,201 The reasons for the discrepancies in these findings are unclear but some explanations can be proposed.
Firstly, the studies have been performed in different experimental models and at different stages in the evolution of MR–related left ventricular remodelling. Many of the experiments performed thus far have been in animal models with controlled formation of volume overload showing that early introduction of beta–blocker therapy89,90,118 may be beneficial, and this is supported to some extent by the work of Ahmed et al.199 in humans. However, there appears to be a time–dependent pattern during remodelling of the LV in chronic primary MR.
Early after the development of MR there is a marked increase in inflammatory and neurohormonal response to the acute volume overload.26,113 A period of compensation and a relatively normal inflammatory response appears to follow until the late decompensated stage is reached, when adverse pathway activation seems to increase.26 Depending on when in this evolution of left ventricular remodelling the studies to date have been performed, there may be discrepancies in the findings with regard to the impact of beta–blockade on left ventricular remodelling. Beta–blockade may have more impressive effects if used early in the evolution of volume overload–related left ventricular remodelling but it may be less effective later on.
Secondly, various beta–adrenergic agents have been tested under different circumstances. Compared to the impressive beneficial results in heart failure patients with the mixed adrenergic blocker, carvedilol,158,202 Pu et al.201 demonstrated that this drug caused worsening left ventricular dimensions and function in animal subjects with primary MR. By contrast, treatment of dogs with the selective beta–blocker atenolol improved left ventricular remodelling,90 whereas treatment with metoprolol in animals and humans have had mixed results.108,180,196,197,199,200
Thirdly, an important question is whether patients presenting in more advanced stages of left ventricular remodelling will respond to anti–remodelling therapy or whether the wall stresses determined by the Laplace law will outweigh any potential beta–blocker effect. In this regard, the findings of Pat et al. 200 are interesting because although there were improvements in cardiomyocyte contractility and beta–receptor responsiveness, left ventricular remodelling was not attenuated by metoprolol. It was postulated that β–AR blockade failed to preserve interstitial collagen loss and therefore failed to prevent ongoing myocyte slippage. There appears to be a strong early adrenergic response to chronic primary MR,56 and beta–blocker therapy before onset of left ventricular dilatation may have more benefit. Lastly, in patients with rheumatic mitral valve disease, there is a possibility that rheumatic fever causing rheumatic carditis may have long–lasting effects on the myocardium, attenuating reverse remodelling by beta–blockers.
Conclusion
Left ventricular remodelling in response to the volume load created by chronic primary MR is a complex process that stems from excessive diastolic stretch of myocytes. Excessive stretch triggers activation of numerous signal transduction pathways, resulting in an initial adaptive remodelling process in the form of eccentric hypertrophy. However, chronic activation of these pathways results in abnormal increases in ROS, catecholamines, angiotensin II and inflammatory cytokines. This is followed by a transition to adverse remodelling involving cardiomyocyte apoptosis and interstitial collagen loss, common to all forms of heart failure.
Limited data do not support the routine long–term use of afterload–reducing agents for the treatment of chronic primary MR. By contrast, there is pre–clinical data demonstrating that β–AR blockade reverses remodelling caused by the volume overload of chronic primary MR, and there are some recent clinical data to support this hypothesis. However, some studies demonstrated worsening left ventricular remodelling with betablocker therapy. Whether these contrasting outcomes are related to differences in beta–blockers, varying experimental models or differences in timing of therapy will need clarification. Ultimately, further studies are required to elucidate the exact mechanisms involved, and large randomised clinical trials are needed to clarify the role of these agents for patients with chronic primary MR.
Acknowledgments
The authors thank Drs Eric Klug and Thomas Kalk for their valuable comments, and Dr Lindsay McCutcheon for help with the artwork.
Contributor Information
McCutcheon Keir, Email: keir_mccutcheon@hotmail.com, Division of Cardiology, Department of Internal Medicine, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg, South Africa.
Manga Pravin, Division of Cardiology, Department of Internal Medicine, Charlotte Maxeke Johannesburg Academic Hospital and University of the Witwatersrand, Johannesburg, South Africa.
References
- 1.Vahanian A, Alfieri O, Andreotti F. et al. Guidelines on the management of valvular heart disease (version 2012): the Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio–Thoracic Surgery (EACTS) Eur J Cardiothorac Surg. 2012;42(4):S1–44. doi: 10.1093/ejcts/ezs455. [DOI] [PubMed] [Google Scholar]
- 2.Ciarka A, van de Veire N. Secondary mitra regurgitation: pathophysiology, diagnosis, and treatment. Heart. 2011;97(12):1012–1023. doi: 10.1136/hrt.2010.219170. [DOI] [PubMed] [Google Scholar]
- 3.Foster E. Clinica practice. Mitra regurgitation due to degenerative mitral–valve disease. N Eng J Med. 2010;363(2):156–165. doi: 10.1056/NEJMcp0906782. [DOI] [PubMed] [Google Scholar]
- 4.Sliwa K, Mocumbi AO. Forgotten cardiovascular diseases in Africa. Clin Res Cardiol. 2010;99(2):65–74. doi: 10.1007/s00392-009-0094-1. [DOI] [PubMed] [Google Scholar]
- 5.McMurray JJ, Adamopoulos S, Anker SD. et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33(14):1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- 6.Burchfield JS, Xie M, Hill JA. Pathologica ventricular remodeling: mechanisms: part 1 of 2. Circulation 2013; 128(4): 388–400. 7. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cel Mo Life Sci. 2014;71(4):549–574. [Google Scholar]
- 7.Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cel Mo Life Sci. 2014;71(4):549–574. doi: 10.1007/s00018-013-1349-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Otto CM, Prendergast B. Aortic–valve stenosis – from patients at risk to severe valve obstruction. N Eng J Med. 2014;371(8):744–756. doi: 10.1056/NEJMra1313875. [DOI] [PubMed] [Google Scholar]
- 9.Carabello BA, Crawford FA. Valvular heart disease. New Eng J Med. 1997;337(1):32–41. doi: 10.1056/NEJM199707033370107. [DOI] [PubMed] [Google Scholar]
- 10.Enriquez–Sarano M, Akins CW, Vahanian A. Mitra regurgitation. Lancet. 2009;373(9672):1382–1394. doi: 10.1016/S0140-6736(09)60692-9. [DOI] [PubMed] [Google Scholar]
- 11.Avierinos JF, Gersh BJ, Melton LJ, 3rd. et al. Natura history of asymptomatic mitra valve prolapse in the community. Circulation. 2002;106(11):1355–1361. doi: 10.1161/01.cir.0000028933.34260.09. [DOI] [PubMed] [Google Scholar]
- 12. Enriquez–Sarano M, Avierinos JF, Messika–Zeitoun D. et al. Quantitative determinants of the outcome of asymptomatic mitra regurgitation. N Eng J Med. 2005;352(9):875–883. doi: 10.1056/NEJMoa041451. [DOI] [PubMed] [Google Scholar]
- 13.Ross J Jr, Sonnenblick EH, Taylor RR, Spotnitz HM, Covell JW. Diastolic geometry and sarcomere lengths in the chronically dilated canine left ventricle. Circ Res. 1971;28(1):49–61. doi: 10.1161/01.res.28.1.49. [DOI] [PubMed] [Google Scholar]
- 14.Grossman W, Jones D, McLaurin LP. Wal stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56(1):56–64. doi: 10.1172/JCI108079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dell’italia LJ, Balcells E, Meng QC. et al. Volume–overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am J Physiol. 1997;273(2 Pt 2):H961–970. doi: 10.1152/ajpheart.1997.273.2.H961. [DOI] [PubMed] [Google Scholar]
- 16.Matsuo T, Carabello BA, Nagatomo Y. et al. Mechanisms of cardiac hypertrophy in canine volume overload. Am J Physiol. 1998;275(1 Pt 2):H65–74. doi: 10.1152/ajpheart.1998.275.1.h65. [DOI] [PubMed] [Google Scholar]
- 17.Kerckhoffs RC, Omens J, McCulloch AD. A single strain–based growth law predicts concentric and eccentric cardiac growth during pressure and volume overload. Mech Res Commun. 2012;42:40–50. doi: 10.1016/j.mechrescom.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spinale FG, Ishihra K, Zile M, DeFryte G, Crawford FA, Carabello BA. Structura basis for changes in left ventricular function and geometry because of chronic mitra regurgitation and after correction of volume overload. J Thorac Cardiovasc Surg. 1993;106(6):1147–1157. [PubMed] [Google Scholar]
- 19.Wisenbaugh T, Spann JF, Carabello BA. Differences in myocardia performance and load between patients with similar amounts of chronic aortic versus chronic mitra regurgitation. J Am Col Cardiol. 1984;3(4):916–923. doi: 10.1016/s0735-1097(84)80349-6. [DOI] [PubMed] [Google Scholar]
- 20.Imamura T, McDermott PJ, Kent RL, Nagatsu M, Cooper Gt, Carabello BA. Acute changes in myosin heavy chain synthesis rate in pressure versus volume overload. Circ Res. 1994;75(3):418–425. doi: 10.1161/01.res.75.3.418. [DOI] [PubMed] [Google Scholar]
- 21.Urabe Y, Mann DL, Kent RL. et al. Cellular and ventricular contractile dysfunction in experimenta canine mitra regurgitation. Circ Res. 1992;70(1):131–147. doi: 10.1161/01.res.70.1.131. [DOI] [PubMed] [Google Scholar]
- 22.Tsutsui H, Ishihara K, Cooper Gt. Cytoskeleta role in the contractile dysfunction of hypertrophied myocardium. Science. 1993;260(5108):682–687. doi: 10.1126/science.8097594. [DOI] [PubMed] [Google Scholar]
- 23.Carabello BA. Progress in mitra and aortic regurgitation. Prog Cardiovasc Dis. 1988;43(6):457–475. doi: 10.1053/pcad.2001.24597. [DOI] [PubMed] [Google Scholar]
- 24.O’Gara P, Sugeng L, Lang R. et al. The role of imaging in chronic degenerative mitra regurgitation. J Am Col Cardio Cardiovasc Imaging. 2008;1(2):221–237. doi: 10.1016/j.jcmg.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Amin P, Singh M, Singh K. Beta–Adrenergic receptor–stimulated cardiac myocyte apoptosis: role of beta1 integrins. J Signa Transduct. 2011;2011:179057–179057. doi: 10.1155/2011/179057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen YW, Pat B, Gladden JD. et al. Dynamic molecular and histopathologica changes in the extracellular matrix and inflammation in the transition to heart failure in isolated volume overload. Am J Physio Heart Circ Physiol. 2011;300(6):H2251–2260. doi: 10.1152/ajpheart.01104.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchinson KR, Stewart JA Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload–induced heart failure. J Mo Cel Cardiol. 2010;48(3):564–569. doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knoll R, Marston S. On mechanosensation, acto/myosin interaction, and hypertrophy. Trends Cardiovasc Med. 2012;22(1):17–22. doi: 10.1016/j.tcm.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Buyandelger B, Mansfield C, Knoll R. Mechano–signaling in heart failure. Pflugers Arch. 2014;466(6):1093–1099. doi: 10.1007/s00424-014-1468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116(8):1462–1476. doi: 10.1161/CIRCRESAHA.116.304937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Israeli–Rosenberg S, Manso AM, Okada H, Ross RS. Integrins and integrin–associated proteins in the cardiac myocyte. Circ Res. 2014;114(3):572–586. doi: 10.1161/CIRCRESAHA.114.301275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Opie LH BD. Mechanisms of Cardiac Contraction and Relaxation. 10th edn. Philadelphia: Saunders Elsevier, 2015 [Google Scholar]
- 33.Miller MK, Granzier H, Ehler E, Gregorio CC. The sensitive giant: the role of titin–based stretch sensing complexes in the heart. Trends Cel Biol. 2004;14(3):119–126. doi: 10.1016/j.tcb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Zou P, Pinotsis N, Lange S. et al. Palindromic assembly of the giant muscle protein titin in the sarcomeric Z–disk. Nature. 2006;439(7073):229–233. doi: 10.1038/nature04343. [DOI] [PubMed] [Google Scholar]
- 35.Knoll R, Hoshijima M, Hoffman HM. et al. The cardiac mechanica stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell. 2002;111(7):943–955. doi: 10.1016/s0092-8674(02)01226-6. [DOI] [PubMed] [Google Scholar]
- 36.Hishiya A, Kitazawa T, Takayama S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanica stress. Circ Res. 2010;107(10):1220–1231. doi: 10.1161/CIRCRESAHA.110.225649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuwahara K, Kinoshita H, Kuwabara Y. et al. Myocardin–related transcription factor A is a common mediator of mechanica stress– and neurohumora stimulation–induced cardiac hypertrophic signaling leading to activation of brain natriuretic peptide gene expression. Mo Cel Biol. 2010;30(17):4134–4148. doi: 10.1128/MCB.00154-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lange S, Xiang F, Yakovenko A. et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308(5728):1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 39.Wagner MA, Siddiqui MA. The JAK–STAT pathway in hypertrophic stress signaling and genomic stress response. Jakstat. 2012;1(2):131–141. doi: 10.4161/jkst.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Puchner EM, Alexandrovich A, Kho AL. et al. Mechanoenzymatics of titin kinase. Proc Nat Acad Sci USA. 2008;105(36):13385–13390. doi: 10.1073/pnas.0805034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arber S, Hunter JJ, Ross J Jr. et al. MLP–deficient mice exhibit a disruption of cardiac cytoarchitectura organization, dilated cardiomyopathy, and heart failure. Cell. 1997;88(3):393–403. doi: 10.1016/s0092-8674(00)81878-4. [DOI] [PubMed] [Google Scholar]
- 42.Knoll R, Kostin S, Klede S. et al. A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res. 2010;106(4):695–704. doi: 10.1161/CIRCRESAHA.109.206243. [DOI] [PubMed] [Google Scholar]
- 43.Boateng SY, Senyo SE, Qi L, Goldspink PH, Russell B. Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. J Mo Cel Cardiol. 2009;47(4):426–435. doi: 10.1016/j.yjmcc.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang BW, Hung HF, Chang H, Kuan P, Shyu KG. Mechanica stretch enhances the expression of resistin gene in cultured cardiomyocytes via tumor necrosis factor–alpha. Am J Physio Heart Circ Physiol. 2007;293(4):H2305–2312. doi: 10.1152/ajpheart.00361.2007. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Pat B, Zheng J. et al. Tumor necrosis factor–alpha produced in cardiomyocytes mediates a predominant myocardia inflammatory response to stretch in early volume overload. J Mo Cel Cardiol. 2010;49(1):70–78. doi: 10.1016/j.yjmcc.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang TL, Yang YH, Chang H, Hung CR. Angiotensin II signals mechanica stretch–induced cardiac matrix metalloproteinase expression via JAK–STAT pathway. J Mo Cel Cardiol. 2004;37(3):785–794. doi: 10.1016/j.yjmcc.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 47.Gealekman O, Abassi Z, Rubinstein I, Winaver J, Binah O. Role of myocardia inducible nitric oxide synthase in contractile dysfunction and beta–adrenergic hyporesponsiveness in rats with experimenta volumeoverload heart failure. Circulation. 2002;105(2):236–243. doi: 10.1161/hc0202.102015. [DOI] [PubMed] [Google Scholar]
- 48.Baud V, Karin M. Signa transduction by tumor necrosis factor and its relatives. Trends Cel Biol. 2001;11(9):372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 49.Lebrec H, Ponce R, Preston BD, Iles J, Born TL, Hooper M. Tumor necrosis factor, tumor necrosis factor inhibition, and cancer risk. Curr Med Res Opin. 2015;31(3):557–574. doi: 10.1185/03007995.2015.1011778. [DOI] [PubMed] [Google Scholar]
- 50.Griendling KK, Sorescu D, Lassegue B, Ushio–Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20(10):2175–2183. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 51.Grote K, Flach I, Luchtefeld M. et al. Mechanica stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase–2 (MMP–2) via NAD(P)H oxidase–derived reactive oxygen species. Circ Res. 2003;92(11):e80–86. doi: 10.1161/01.RES.0000077044.60138.7C. [DOI] [PubMed] [Google Scholar]
- 52.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physio Cel Physiol. 2001;280(1):C53–60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 53.Birukov KG. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid Redox Signal. 2009;11(7):1651–1667. doi: 10.1089/ars.2008.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keith M, Geranmayegan A, Sole MJ. et al. Increased oxidative stress in patients with congestive heart failure. J Am Col Cardiol. 1998;31(6):1352–1356. doi: 10.1016/s0735-1097(98)00101-6. [DOI] [PubMed] [Google Scholar]
- 55.Ahmed MI, Gladden JD, Litovsky SH. et al. Increased oxidative stress and cardiomyocyte myofibrillar degeneration in patients with chronic isolated mitra regurgitation and ejection fraction > 60% J Am Col Cardiol. 2010;55(7):671–679. doi: 10.1016/j.jacc.2009.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng J, Yancey DM, Ahmed MI. et al. Increased sarcolipin expression and adrenergic drive in humans with preserved left ventricular ejection fraction and chronic isolated mitra regurgitation. Circulation: Heart Failure. 2014;7(1):194–202. doi: 10.1161/CIRCHEARTFAILURE.113.000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116(7):1254–1268. doi: 10.1161/CIRCRESAHA.116.302317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakano M, Knowlton AA, Dibbs Z, Mann DL. Tumor necrosis factoralpha confers resistance to hypoxic injury in the adult mammalian cardiac myocyte. Circulation. 1998;97(14):1392–1400. doi: 10.1161/01.cir.97.14.1392. [DOI] [PubMed] [Google Scholar]
- 59.Hamid T, Gu Y, Ortines RV. et al. Divergent tumor necrosis factor receptor–related remodeling responses in heart failure: role of nuclear factor–kappaB and inflammatory activation. Circulation. 2009;119(10):1386–1397. doi: 10.1161/CIRCULATIONAHA.108.802918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pagani FD, Baker LS, Hsi C, Knox M, Fink MP, Visner MS. Left ventricular systolic and diastolic dysfunction after infusion of tumor necrosis factor–alpha in conscious dogs. J Clin Invest. 1992;90(2):389–398. doi: 10.1172/JCI115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franco F, Thomas GD, Giroir B. et al. Magnetic resonance imaging and invasive evaluation of development of heart failure in transgenic mice with myocardia expression of tumor necrosis factor–alpha. Circulation. 1999;99(3):448–454. doi: 10.1161/01.cir.99.3.448. [DOI] [PubMed] [Google Scholar]
- 62.Krown KA, Page MT, Nguyen C. et al. Tumor necrosis factor alphainduced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cel death. J Clin Invest. 1996;98(12):2854–2865. doi: 10.1172/JCI119114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cel death pathways. J Clin Invest. 2007;117(9):2692–2701. doi: 10.1172/JCI29134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dhingra S, Bagchi AK, Ludke AL, Sharma AK, Singal PK. Akt regulates IL–10 mediated suppression of TNF–alpha–induced cardiomyocyte apoptosis by upregulating Stat3 phosphorylation. PLoS One. 2011;6(9):e25009–e25009. doi: 10.1371/journal.pone.0025009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physio Heart Circ Physiol. 2004;287(3):H1303–1311. doi: 10.1152/ajpheart.00053.2004. [DOI] [PubMed] [Google Scholar]
- 66.Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL–10 attenuates TNF–alpha–induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. 2009;82(1):59–66. doi: 10.1093/cvr/cvp040. [DOI] [PubMed] [Google Scholar]
- 67.Sivasubramanian N, Coker ML, Kurrelmeyer KM. et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;104(7):826–831. doi: 10.1161/hc3401.093154. [DOI] [PubMed] [Google Scholar]
- 68.Diwan A, Dibbs Z, Nemoto S. et al. Targeted overexpression of noncleavable and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes. Circulation. 2004;109(2):262–268. doi: 10.1161/01.CIR.0000109642.27985.FA. [DOI] [PubMed] [Google Scholar]
- 69.Levick SP, Gardner JD, Holland M, Hauer–Jensen M, Janicki JS, Brower GL. Protection from adverse myocardia remodeling secondary to chronic volume overload in mast cel deficient rats. J Mo Cel Cardiol. 2008;45(1):56–61. doi: 10.1016/j.yjmcc.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Eng J Med. 1990;323(4):236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 71.Kapadia SR, Yakoob K, Nader S, Thomas JD, Mann DL, Griffin BP. Elevated circulating levels of serum tumor necrosis factor–alpha in patients with hemodynamically significant pressure and volume overload. J Am Col Cardiol. 2000;36(1):208–212. doi: 10.1016/s0735-1097(00)00721-x. [DOI] [PubMed] [Google Scholar]
- 72.Sliwa K, Skudicky D, Bergemann A, Candy G, Puren A, Sareli P. Peripartum cardiomyopathy: analysis of clinica outcome, left ventricular function, plasma levels of cytokines and Fas/APO–1. J Am Col Cardiol. 2000;35(3):701–705. doi: 10.1016/s0735-1097(99)00624-5. [DOI] [PubMed] [Google Scholar]
- 73.Bujak M, Frangogiannis NG. The role of IL–1 in the pathogenesis of heart disease. Arch Immuno Ther Exp (Warsz) 2009;57(3):165–176. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palmieri EA, Benincasa G, Di Rella F. et al. Differentia expression of TNF–alpha, IL–6, and IGF–1 by graded mechanica stress in norma rat myocardium. Am J Physio Heart Circ Physiol. 2002;282(3):H926–934. doi: 10.1152/ajpheart.00436.2001. [DOI] [PubMed] [Google Scholar]
- 75.Oral H, Sivasubramanian N, Dyke DB. et al. Myocardia proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitra regurgitation. Circulation. 2003;107(6):831–837. doi: 10.1161/01.cir.0000049745.38594.6d. [DOI] [PubMed] [Google Scholar]
- 76.Spina GS, Tarasoutchi F, Sampaio RO. et al. Neurohormona profile of rheumatic patients with significant chronic aortic regurgitation. Arq Bras Cardiol. 2009;92(2):143–56. doi: 10.1590/s0066-782x2009000200012. [DOI] [PubMed] [Google Scholar]
- 77.Cheng W, Li B, Kajstura J. et al. Stretch–induced programmed myocyte cel death. J Clin Invest. 1995;96(5):2247–2259. doi: 10.1172/JCI118280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Communal C, Singh K, Pimentel DR, Colucci WS. Norepinephrine stimulates apoptosis in adult rat ventricular myocytes by activation of the beta–adrenergic pathway. Circulation. 1998;98(13):1329–1334. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 79.Geng YJ, Ishikawa Y, Vatner DE. et al. Apoptosis of cardiac myocytes in Gsalpha transgenic mice. Circ Res. 1999;84(1):34–42. doi: 10.1161/01.res.84.1.34. [DOI] [PubMed] [Google Scholar]
- 80.Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113(6):739–753. doi: 10.1161/CIRCRESAHA.113.300308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Orrenius S, McConkey DJ, Bellomo G, Nicotera P. Role of Ca2+ in toxic cel killing. Trends Pharmaco Sci. 1989;10(7):281–285. doi: 10.1016/0165-6147(89)90029-1. [DOI] [PubMed] [Google Scholar]
- 82.Mulieri LA, Tischler MD, Martin BJ. et al. Regiona differences in the force–frequency relation of human left ventricular myocardium in mitra regurgitation: implications for ventricular shape. Am J Physio Heart Circ Physiol. 2005;288(5):H2185–2191. doi: 10.1152/ajpheart.00905.2003. [DOI] [PubMed] [Google Scholar]
- 83.Gupta RC, Mishra S, Mishima T, Goldstein S, Sabbah HN. Reduced sarcoplasmic reticulum Ca(2+)–uptake and expression of phospholamban in left ventricular myocardium of dogs with heart failure. J Mo Cel Cardiol. 1999;31(7):1381–1389. doi: 10.1006/jmcc.1999.0970. [DOI] [PubMed] [Google Scholar]
- 84.Sabbah HN. Biologic rationale for the use of beta–blockers in the treatment of heart failure. Heart Fai Rev. 2004;9(2):91–97. doi: 10.1023/B:HREV.0000046363.59374.23. [DOI] [PubMed] [Google Scholar]
- 85.Leszek P, Korewicki J, Klisiewicz A. et al. Reduced myocardia expression of calcium handling protein in patients with severe chronic mitra regurgitation. Eur J Cardiothorac Surg. 2006;30(5):737–743. doi: 10.1016/j.ejcts.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 86.Dorn GW, 2nd. Apoptotic and non–apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res. 2009;81(3):465–473. doi: 10.1093/cvr/cvn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Foo RS, Mani K, Kitsis RN. Death begets failure in the heart. J Clin Invest. 2005;115(3):565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Remondino A, Kwon SH, Communal C. et al. Beta–adrenergic receptorstimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c–Jun NH2–termina kinase–dependent activation of the mitochondria pathway. Circ Res. 2003;92(2):136–8. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 89.Hankes GH, Ardell JL, Tallaj J. et al. Beta1–adrenoceptor blockade mitigates excessive norepinephrine release into cardiac interstitium in mitra regurgitation in dog. Am J Physio Heart Circ Physiol. 2006;291(1):H147–151. doi: 10.1152/ajpheart.00951.2005. [DOI] [PubMed] [Google Scholar]
- 90.Tsutsui H, Spinale FG, Nagatsu M. et al. Effects of chronic beta–adrenergic blockade on the left ventricular and cardiocyte abnormalities of chronic canine mitra regurgitation. J Clin Invest. 1994;93(6):2639–2648. doi: 10.1172/JCI117277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Janicki JS, Brower GL. The role of myocardia fibrillar collagen in ventricular remodeling and function. J Card Fail. 2002;8(6 Suppl):S319–325. doi: 10.1054/jcaf.2002.129260. [DOI] [PubMed] [Google Scholar]
- 92.Spinale FG. Myocardia matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physio Rev. 2007;87(4):1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 93.Spinale FG, Coker ML, Heung LJ. et al. A matrix metalloproteinase induction/activation system exists in the human left ventricular myocardium and is upregulated in heart failure. Circulation. 2000;102(16):1944–1949. doi: 10.1161/01.cir.102.16.1944. [DOI] [PubMed] [Google Scholar]
- 94.King MK, Coker ML, Goldberg A. et al. Selective matrix metalloproteinase inhibition with developing heart failure: effects on left ventricular function and structure. Circ Res. 2003;92(2):177–185. doi: 10.1161/01.res.0000052312.41419.55. [DOI] [PubMed] [Google Scholar]
- 95.Ahmed SH, Clark LL, Pennington WR. et al. Matrix metalloproteinases/ tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinica manifestations of hypertensive heart disease. Circulation. 2006;113(17):2089–2096. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 96.George J, Patal S, Wexler D, Roth A, Sheps D, Keren G. Circulating matrix metalloproteinase–2 but not matrix metalloproteinase–3, matrix metalloproteinase–9, or tissue inhibitor of metalloproteinase–1 predicts outcome in patients with congestive heart failure. Am Heart J. 2005;150(3):484–487. doi: 10.1016/j.ahj.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 97.Buralli S, Dini FL, Ballo P. et al. Circulating matrix metalloproteinase– 3 and metalloproteinase–9 and tissue Doppler measures of diastolic dysfunction to risk stratify patients with systolic heart failure. Am J Cardiol. 2010;105(6):853–856. doi: 10.1016/j.amjcard.2009.11.038. [DOI] [PubMed] [Google Scholar]
- 98.Lovelock JD, Baker AH, Gao F. et al. Heterogeneous effects of tissue inhibitors of matrix metalloproteinases on cardiac fibroblasts. Am J Physio Heart Circ Physiol. 2005;288(2):H461–468. doi: 10.1152/ajpheart.00402.2004. [DOI] [PubMed] [Google Scholar]
- 99.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mo Cel Biochem. 2003;253(1–2):269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 100.Vincenti MP, Brinckerhoff CE. Signa transduction and cell–type specific regulation of matrix metalloproteinase gene expression: can MMPs be good for you? J Cel Physiol. 2007;213(2):355–364. doi: 10.1002/jcp.21208. [DOI] [PubMed] [Google Scholar]
- 101.Klein T, Bischoff R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids. 2011;41(2):271–290. doi: 10.1007/s00726-010-0689-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Siwik DA, Chang DL, Colucci WS. Interleukin–1beta and tumor necrosis factor–alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86(12):1259–1265. doi: 10.1161/01.res.86.12.1259. [DOI] [PubMed] [Google Scholar]
- 103.MacKenna DA, Dolfi F, Vuori K, Ruoslahti E. Extracellular signalregulated kinase and c–Jun NH2–termina kinase activation by mechanica stretch is integrin–dependent and matrix–specific in rat cardiac fibroblasts. J Clin Invest. 1998;101(2):301–310. doi: 10.1172/JCI1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.MacKenna D, Summerour SR, Villarrea FJ. Role of mechanica factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res. 2000;46(2):257–263. doi: 10.1016/s0008-6363(00)00030-4. [DOI] [PubMed] [Google Scholar]
- 105.Manso AM, Elsherif L, Kang SM, Ross RS. Integrins, membrane–type matrix metalloproteinases and ADAMs: potentia implications for cardiac remodeling. Cardiovasc Res. 2006;69(3):574–584. doi: 10.1016/j.cardiores.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 106.Ross RS. he extracellular connections: the role of integrins in myocardia remodeling. T. J Card Fai. 2002;8(6 Suppl):S326–331. doi: 10.1054/jcaf.2002.129263. [DOI] [PubMed] [Google Scholar]
- 107.Coker ML, Jolly JR, Joffs C. et al. Matrix metalloproteinase expression and activity in isolated myocytes after neurohormona stimulation. Am J Physio Heart Circ Physio. 2001;281(2):H543–551. doi: 10.1152/ajpheart.2001.281.2.H543. [DOI] [PubMed] [Google Scholar]
- 108.Sabri A, Rafiq K, Seqqat R, Kolpakov MA, Dillon R, Dell’italia LJ. Sympathetic activation causes foca adhesion signaling alteration in early compensated volume overload attributable to isolated mitra regurgitation in the dog. Circ Res. 2008;102(9):1127–1136. doi: 10.1161/CIRCRESAHA.107.163642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ogawa K, Chen F, Kuang C, Chen Y. Suppression of matrix metalloproteinase– 9 transcription by transforming growth factor–beta is mediated by a nuclear factor–kappaB site. Biochem J. 2004;381(Pt 2):413–422. doi: 10.1042/BJ20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rouet–Benzineb P, Gontero B, Dreyfus P, Lafuma C. Angiotensin II induces nuclear factor– kappa B activation in cultured neonata rat cardiomyocytes through protein kinase C signaling pathway. J Mo Cel Cardio. 2000;32(10):1767–1778. doi: 10.1006/jmcc.2000.1211. [DOI] [PubMed] [Google Scholar]
- 111.Seeland U, Haeuseler C, Hinrichs R. et al. Myocardia fibrosis in transforming growth factor–beta(1) (TGF–beta(1)) transgenic mice is associated with inhibition of interstitia collagenase. Eur J Clin Invest. 2002;32(5):295–303. doi: 10.1046/j.1365-2362.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- 112.Bujak M, Frangogiannis NG. The role of TGF–beta signaling in myocardia infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zheng J, Chen Y, Pat B. et al. Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitra regurgitation in the dog. Circulation. 2009;119(15):2086–2095. doi: 10.1161/CIRCULATIONAHA.108.826230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang YM, Bo J, Taffet GE. et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. 2006;20(7):916–125. doi: 10.1096/fj.05-5129com. [DOI] [PubMed] [Google Scholar]
- 115.Jellis C, Martin J, Narula J, Marwick TH. Assessment of nonischemic myocardia fibrosis. J Am Col Cardio. 2010;56(2):89–97. doi: 10.1016/j.jacc.2010.02.047. [DOI] [PubMed] [Google Scholar]
- 116.Dell’Italia LJ, Meng QC, Balcells E. et al. Increased ACE and chymaselike activity in cardiac tissue of dogs with chronic mitra regurgitation. Am J Physio. 1995;269(6 Pt 2):H2065–2073. doi: 10.1152/ajpheart.1995.269.6.H2065. [DOI] [PubMed] [Google Scholar]
- 117.Stewart JA, Jr, Wei CC, Brower GL. et al. Cardiac mast cell– and chymase–mediated matrix metalloproteinase activity and left ventricular remodeling in mitra regurgitation in the dog. J Mo Cel Cardio. 2003;35(3):311–319. doi: 10.1016/s0022-2828(03)00013-0. [DOI] [PubMed] [Google Scholar]
- 118.Tallaj J, Wei CC, Hankes GH. et al. Beta1–adrenergic receptor blockade attenuates angiotensin II–mediated catecholamine release into the cardiac interstitium in mitra regurgitation. Circulation. 2003;108(2):225–230. doi: 10.1161/01.CIR.0000079226.48637.5A. [DOI] [PubMed] [Google Scholar]
- 119.Lin A, Stewart R. Medica treatment of asymptomatic chronic aortic regurgitation. Expert Rev Cardiovasc Ther. 2011;9(9):1249–1254. doi: 10.1586/erc.11.97. [DOI] [PubMed] [Google Scholar]
- 120.Weidemann F, Herrmann S, Stork S. et al. Impact of myocardia fibrosis in patients with symptomatic severe aortic stenosis. Circulation. 2009;120(7):577–584. doi: 10.1161/CIRCULATIONAHA.108.847772. [DOI] [PubMed] [Google Scholar]
- 121.Weinberg EO, Schoen FJ, George D. et al. Angiotensin–converting enzyme inhibition prolongs surviva and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis. Circulation. 1994;90(3):1410–1422. doi: 10.1161/01.cir.90.3.1410. [DOI] [PubMed] [Google Scholar]
- 122.Perry GJ, Wei CC, Hankes GH. et al. Angiotensin II receptor blockade does not improve left ventricular function and remodeling in subacute mitra regurgitation in the dog. J Am Col Cardio. 2002;39(8):1374–1379. doi: 10.1016/s0735-1097(02)01763-1. [DOI] [PubMed] [Google Scholar]
- 123.Babbitt CJ, Shai SY, Harpf AE, Pham CG, Ross RS. Modulation of integrins and integrin signaling molecules in the pressure–loaded murine ventricle. Histochem Cel Bio. 2002;118(6):431–439. doi: 10.1007/s00418-002-0476-1. [DOI] [PubMed] [Google Scholar]
- 124.Bloor CM, Nimmo L, McKirnan MD, Zhang Y, White FC. Increased gene expression of plasminogen activators and inhibitors in left ventricular hypertrophy. Mo Cel Biochem. 1997;176(1–2):265–71. [PubMed] [Google Scholar]
- 125.Brower GL, Chancey AL, Thanigaraj S, Matsubara BB, Janicki JS. Cause and effect relationship between myocardia mast cel number and matrix metalloproteinase activity. Am J Physio Heart Circ Physio. 2002;283(2):H518–525. doi: 10.1152/ajpheart.00218.2000. [DOI] [PubMed] [Google Scholar]
- 126.Janicki JS, Brower GL, Gardner JD. et al. Cardiac mast cel regulation of matrix metalloproteinase–related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res. 2006;69(3):657–665. doi: 10.1016/j.cardiores.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 127.Nagatomo Y, Carabello BA, Coker ML. et al. Differentia effects of pressure or volume overload on myocardia MMP levels and inhibitory control. Am J Physio Heart Circ Physio. 2000;278(1):H151–161. doi: 10.1152/ajpheart.2000.278.1.H151. [DOI] [PubMed] [Google Scholar]
- 128.Chancey AL, Brower GL, Peterson JT, Janicki JS. Effects of matrix metalloproteinase inhibition on ventricular remodeling due to volume overload. Circulation. 2002;105(16):1983–1988. doi: 10.1161/01.cir.0000014686.73212.da. [DOI] [PubMed] [Google Scholar]
- 129.Ljungval I, Rajamaki MM, Crosara S. et al. Evaluation of plasma activity of matrix metalloproteinase–2 and –9 in dogs with myxomatous mitra valve disease. Am J Vet Res. 2011;72(8):1022–1028. doi: 10.2460/ajvr.72.8.1022. [DOI] [PubMed] [Google Scholar]
- 130.Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J Am Col Cardio. 2002;39(8):1384–1391. doi: 10.1016/s0735-1097(02)01756-4. [DOI] [PubMed] [Google Scholar]
- 131.Moustafa SE, Kansa M, Alharthi M, Deng Y, Chandrasekaran K, Mookadam F. Prediction of incipient left ventricular dysfunction in patients with chronic primary mitra regurgitation: a velocity vector imaging study. Eur J Echocardiogr. 2011;12(4):291–298. doi: 10.1093/ejechocard/jer003. [DOI] [PubMed] [Google Scholar]
- 132.Magne J, Mahjoub H, Dulgheru R, Pibarot P, Pierard LA, Lancellotti P. Left ventricular contractile reserve in asymptomatic primary mitra regurgitation. Eur Heart J. 2014;35(24):1608–1616. doi: 10.1093/eurheartj/eht345. [DOI] [PubMed] [Google Scholar]
- 133.Le Tourneau T, de Groote P, Millaire A. et al. Effect of mitra valve surgery on exercise capacity, ventricular ejection fraction and neurohormona activation in patients with severe mitra regurgitation. J Am Col Cardio. 2000;36(7):2263–2269. doi: 10.1016/s0735-1097(00)01015-9. [DOI] [PubMed] [Google Scholar]
- 134.Mehta RH, Supiano MA, Ora H. et al. Compared with contro subjects, the systemic sympathetic nervous system is activated in patients with mitra regurgitation. Am Heart J. 2003;145(6):1078–1085. doi: 10.1016/S0002-8703(03)00111-X. [DOI] [PubMed] [Google Scholar]
- 135.Nagatsu M, Zile MR, Tsutsui H. et al. Native beta–adrenergic support for left ventricular dysfunction in experimenta mitra regurgitation normalizes indexes of pump and contractile function. Circulation. 1994;89(2):818–826. doi: 10.1161/01.cir.89.2.818. [DOI] [PubMed] [Google Scholar]
- 136.Touyz RM, Schiffrin EL. Signa transduction mechanisms mediating the physiologica and pathophysiologica actions of angiotensin II in vascular smooth muscle cells. Pharmaco Rev. 2000;52(4):639–672. [PubMed] [Google Scholar]
- 137.Schieffer B, Bernstein KE, Marrero MB. The role of tyrosine phosphorylation in angiotensin II mediated intracellular signaling and cel growth. J Mo Med (Berl) 1996;74(2):85–91. doi: 10.1007/BF00196783. [DOI] [PubMed] [Google Scholar]
- 138.Abe J, Berk BC. Reactive oxygen species as mediators of signa transduction in cardiovascular disease. Trends Cardiovasc Med. 1998;8(2):59–64. doi: 10.1016/S1050-1738(97)00133-3. [DOI] [PubMed] [Google Scholar]
- 139.Brasch H, Sieroslawski L, Dominiak P. Angiotensin II increases norepinephrine release from atria by acting on angiotensin subtype 1 receptors. Hypertension. 1993;22(5):699–704. doi: 10.1161/01.hyp.22.5.699. [DOI] [PubMed] [Google Scholar]
- 140.Farrel DM, Wei CC, Tallaj J. et al. Angiotensin II modulates catecholamine release into interstitia fluid of canine myocardium in vivo. Am J Physio Heart Circ Physio. 2001;281(2):H813–822. doi: 10.1152/ajpheart.2001.281.2.H813. [DOI] [PubMed] [Google Scholar]
- 141.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor–beta expression in rat glomerular mesangia cells. J Clin Invest. 1994;93(6):2431–2437. doi: 10.1172/JCI117251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goldsmith EC, Bradshaw AD, Spinale FG. Cellular mechanisms of tissue fibrosis. 2. Contributory pathways leading to myocardia fibrosis: moving beyond collagen expression. Am J Physio Cel Physio. 2013;304(5):C393–402. doi: 10.1152/ajpcell.00347.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ryan TD, Rothstein EC, Aban I. et al. Left ventricular eccentric remodeling and matrix loss are mediated by bradykinin and precede cardiomyocyte elongation in rats with volume overload. J Am Col Cardio. 2007;49(7):811–821. doi: 10.1016/j.jacc.2006.06.083. [DOI] [PubMed] [Google Scholar]
- 144.Levine HJ, Gaasch WH. Vasoactive drugs in chronic regurgitant lesions of the mitra and aortic valves. J Am Col Cardio. 1996;28(5):1083–1091. doi: 10.1016/S0735-1097(96)00288-4. [DOI] [PubMed] [Google Scholar]
- 145.Nemoto S, Hamawaki M, De Freitas G, Carabello BA. Differentia effects of the angiotensin–converting enzyme inhibitor lisinopri versus the beta–adrenergic receptor blocker atenolo on hemodynamics and left ventricular contractile function in experimenta mitra regurgitation. J Am Col Cardio. 2002;40(1):149–154. doi: 10.1016/s0735-1097(02)01926-5. [DOI] [PubMed] [Google Scholar]
- 146.Oh SH, Meyers DG. Afterload reduction may halt and beta–adrenergic blockade may worsen progression of left ventricular dysfunction in patients with chronic compensated mitra regurgitation: a retrospective cohort study. Angiology. 2007;58(2):196–202. doi: 10.1177/0003319707300357. [DOI] [PubMed] [Google Scholar]
- 147.Woo AY, Xiao RP. Beta–adrenergic receptor subtype signaling in heart: from bench to bedside. Acta Pharmaco Sin. 2012;33(3):335–341. doi: 10.1038/aps.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belge C, Hammond J, Dubois–Deruy E. et al. Enhanced expression of β3–adrenoceptors in cardiac myocytes attenuates neurohormone– induced hypertrophic remodeling through nitric oxide synthase. Circulation. 2014;129(4):451–462. doi: 10.1161/CIRCULATIONAHA.113.004940. [DOI] [PubMed] [Google Scholar]
- 149.Chidsey CA, Braunwald E, Morrow AG, Mason DT. Myocardia norepinephrine concentration in man. Effects of reserpine and of congestive heart failure. N Eng J Med. 1963;269:653–658. doi: 10.1056/NEJM196309262691302. [DOI] [PubMed] [Google Scholar]
- 150.Fowler MB, Bristow MR. Rationale for beta–adrenergic blocking drugs in cardiomyopathy. Am J Cardio. 1985;55(10):120d–124d. doi: 10.1016/0002-9149(85)91066-5. [DOI] [PubMed] [Google Scholar]
- 151.Bristow MR, Ginsburg R, Minobe W. et al. Decreased catecholamine sensitivity and beta–adrenergic–receptor density in failing human hearts. N Eng J Med. 1982;307(4):205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 152.Lowes BD, Minobe W, Abraham WT. et al. Changes in gene expression in the intact human heart. Downregulation of alpha–myosin heavy chain in hypertrophied, failing ventricular myocardium. J Clin Invest. 1997;100(9):2315–2324. doi: 10.1172/JCI119770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lowes BD, Gilbert EM, Abraham WT. et al. Myocardia gene expression in dilated cardiomyopathy treated with beta–blocking agents. N Eng J Med. 2002;346(18):1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 154.Engelhardt S, Bohm M, Erdmann E, Lohse MJ. Analysis of beta–adrenergic receptor mRNA levels in human ventricular biopsy specimens by quantitative polymerase chain reactions: progressive reduction of beta 1–adrenergic receptor mRNA in heart failure. J Am Col Cardio. 1996;27(1):146–154. doi: 10.1016/0735-1097(95)00425-4. [DOI] [PubMed] [Google Scholar]
- 155.Hammond HK, Roth DA, Inse PA. et al. Myocardia beta–adrenergic receptor expression and signa transduction after chronic volumeoverload hypertrophy and circulatory congestion. Circulation. 1992;85(1):269–80. doi: 10.1161/01.cir.85.1.269. [DOI] [PubMed] [Google Scholar]
- 156.Choi DJ, Koch WJ, Hunter JJ, Rockman HA. Mechanism of betaadrenergic receptor desensitization in cardiac hypertrophy is increased beta–adrenergic receptor kinase. J Bio Chem. 1997;272(27):17223–17229. doi: 10.1074/jbc.272.27.17223. [DOI] [PubMed] [Google Scholar]
- 157.Osadchii OE, Norton GR, McKechnie R, Deftereos D, Woodiwiss AJ. Cardiac dilatation and pump dysfunction without intrinsic myocardia systolic failure following chronic beta–adrenoreceptor activation. Am J Physio Heart Circ Physio. 2007;292(4):H1898–1905. doi: 10.1152/ajpheart.00740.2006. [DOI] [PubMed] [Google Scholar]
- 158.Packer M, Fowler MB, Roecker EB. et al. Effect of carvedilo on the morbidity of patients with severe chronic heart failure: results of the carvedilo prospective randomized cumulative surviva (COPERNICUS) study. Circulation. 2002;106(17):2194–2199. doi: 10.1161/01.cir.0000035653.72855.bf. [DOI] [PubMed] [Google Scholar]
- 159.Kohout TA, Takaoka H, McDonald PH. et al. Augmentation of cardiac contractility mediated by the human beta(3)–adrenergic receptor overexpressed in the hearts of transgenic mice. Circulation. 2001;104(20):2485–2491. doi: 10.1161/hc4501.098933. [DOI] [PubMed] [Google Scholar]
- 160.Zhao Q, Wu TG, Jiang ZF, Chen GW, Lin Y, Wang LX. Effect of beta–blockers on beta3–adrenoceptor expression in chronic heart failure. Cardiovasc Drugs Ther. 2007;21(2):85–90. doi: 10.1007/s10557-007-6016-4. [DOI] [PubMed] [Google Scholar]
- 161.Yue TL, Cheng HY, Lysko PG. et al. Carvedilol, a new vasodilator and beta adrenoceptor antagonist, is an antioxidant and free radica scavenger. J Pharmaco Exp Ther. 1992;263(1):92–98. [PubMed] [Google Scholar]
- 162.Ohlstein EH, Douglas SA, Sung CP. et al. Carvedilol, a cardiovascular drug, prevents vascular smooth muscle cel proliferation, migration, and neointima formation following vascular injury. Proc Nat Acad Sci USA. 1993;90(13):6189–6193. doi: 10.1073/pnas.90.13.6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wisler JW, DeWire SM, Whalen EJ. et al. A unique mechanism of betablocker action: carvedilo stimulates beta–arrestin signaling. Proc Nat Acad Sci USA. 2007;104(42):16657–16662. doi: 10.1073/pnas.0707936104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gilbert EM, Abraham WT, Olsen S. et al. Comparative hemodynamic, left ventricular functional, and antiadrenergic effects of chronic treatment with metoprolo versus carvedilo in the failing heart. Circulation. 1996;94(11):2817–2825. doi: 10.1161/01.cir.94.11.2817. [DOI] [PubMed] [Google Scholar]
- 165.Stoschitzky K, Koshucharova G, Zweiker R. et al. Differing betablocking effects of carvedilo and metoprolol. Eur J Heart Fai. 2001;3(3):343–349. doi: 10.1016/s1388-9842(01)00126-x. [DOI] [PubMed] [Google Scholar]
- 166.Quaife RA, Christian PE, Gilbert EM, Datz FL, Volkman K, Bristow MR. Effects of carvedilo on right ventricular function in chronic heart failure. Am J Cardio. 1998;81(2):247–250. doi: 10.1016/s0002-9149(97)00874-6. [DOI] [PubMed] [Google Scholar]
- 167.Beck–da–Silva L, de Bold A, Davies R. et al. Effect of bisoprolo on right ventricular function and brain natriuretic peptide in patients with heart failure. Congest Heart Fai. 2004;10(3):127–132. doi: 10.1111/j.1527-5299.2004.03316.x. [DOI] [PubMed] [Google Scholar]
- 168.Hongning Y, Stewart RA, Whalley GA. The impact of beta–blockade on right ventricular function in mitra regurgitation. Heart Lung Circ. 2014;23(4):378–380. doi: 10.1016/j.hlc.2013.10.090. [DOI] [PubMed] [Google Scholar]
- 169.The Cardiac Insufficiency Bisoprolo Study II (CIBIS–II): a randomised trial. Lancet. 1999;353(9146):9–13. [PubMed] [Google Scholar]
- 170.Effect of metoprolo CR/X in chronic heart failure: Metoprolo CR/X Randomised Intervention Tria in Congestive Heart Failure (MERIT–HF) Lancet. 1999;353(9169):2001–2007. [PubMed] [Google Scholar]
- 171.Poole–Wilson PA, Swedberg K, Cleland JG. et al. Comparison of carvedilo and metoprolo on clinica outcomes in patients with chronic heart failure in the Carvedilo Or Metoprolo European Tria (COMET): randomised controlled trial. Lancet. 2003;362(9377):7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- 172.Naccarelli GV, Lukas MA. Carvedilol’s antiarrhythmic properties: therapeutic implications in patients with left ventricular dysfunction. Clin Cardio. 2005;28(4):165–173. doi: 10.1002/clc.4960280403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bristow MR. beta–adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101(5):558–569. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- 174.Nagatsu M, Spinale FG, Koide M. et al. Bradycardia and the role of beta–blockade in the amelioration of left ventricular dysfunction. Circulation. 2000;101(6):653–659. doi: 10.1161/01.cir.101.6.653. [DOI] [PubMed] [Google Scholar]
- 175.Communa C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)– and beta(2)–adrenergic receptors on cardiac myocyte apoptosis: role of a pertussis toxin–sensitive G protein. Circulation. 1999;100(22):2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 176.Mason RP, Giles TD, Sowers JR. Evolving mechanisms of action of beta blockers: focus on nebivolol. J Cardiovasc Pharmaco. 2009;54(2):123–128. doi: 10.1097/FJC.0b013e3181ad207b. [DOI] [PubMed] [Google Scholar]
- 177.Saini–Chohan HK, Hatch GM. Biologica actions and metabolism of currently used pharmacologica agents for the treatment of congestive heart failure. Curr Drug Metab. 2009;10(3):206–219. doi: 10.2174/138920009787846314. [DOI] [PubMed] [Google Scholar]
- 178.Wu TC, Chen YH, Leu HB. et al. Carvedilol, a pharmacologica antioxidant, inhibits neointima matrix metalloproteinase–2 and –9 in experimenta atherosclerosis. Free Radic Bio Med. 2007;43(11):1508–1522. doi: 10.1016/j.freeradbiomed.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 179.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of betaadrenergic signaling in heart failure? Circ Res. 2003;93(10):896–906. doi: 10.1161/01.RES.0000102042.83024.CA. [DOI] [PubMed] [Google Scholar]
- 180.Trappanese DM, Liu Y, McCormick RC. et al. Chronic beta1–adrenergic blockade enhances myocardia beta3–adrenergic coupling with nitric oxide–cGMP signaling in a canine mode of chronic volume overload: new insight into mechanisms of cardiac benefit with selective beta1– blocker therapy. Basic Res Cardio. 2015;110(1):456–456. doi: 10.1007/s00395-014-0456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Starling MR. Effects of valve surgery on left ventricular contractile function in patients with long–term mitra regurgitation. Circulation. 1995;92(4):811–818. doi: 10.1161/01.cir.92.4.811. [DOI] [PubMed] [Google Scholar]
- 182.Witkowski TG, Thomas JD, Delgado V. et al. Changes in left ventricular function after mitra valve repair for severe organic mitra regurgitation. Ann Thorac Surg. 2012;93(3):754–760. doi: 10.1016/j.athoracsur.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 183.Kainuma S, Taniguchi K, Toda K. et al. B–type natriuretic peptide response and reverse left ventricular remodeling after surgica correction of functiona mitra regurgitation in patients with advanced cardiomyopathy. J Cardio. 2015;66(4):279–285. doi: 10.1016/j.jjcc.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 184.Bonow RO, Carabello BA, Chatterjee K. et al. 2008 focused update incorporated into the ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1998 guidelines for the management of patients with valvular heart disease). Endorsed by the Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Col Cardio. 2008;52(13):e1–142. doi: 10.1016/j.jacc.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 185.Hetzer R, Dande M. Early detection of left ventricular dysfunction in patients with mitra regurgitation due to flai leaflet is stil a challenge. Eur Heart J. 2011;32(6):665–667. doi: 10.1093/eurheartj/ehq399. [DOI] [PubMed] [Google Scholar]
- 186.Carabello BA. Beta–blockade for mitra regurgitation: could the management of valvular heart disease actually be moving into the 21st century? J Am Col Cardio. 2012;60(9):839–840. doi: 10.1016/j.jacc.2012.04.028. [DOI] [PubMed] [Google Scholar]
- 187.Samad Z, Kau P, Shaw LK. et al. Impact of early surgery on surviva of patients with severe mitra regurgitation. Heart. 2011;97(3):221–224. doi: 10.1136/hrt.2010.202432. [DOI] [PubMed] [Google Scholar]
- 188.Kang DH, Park SJ, Sun BJ. et al. Early surgery versus conventiona treatment for asymptomatic severe mitra regurgitation: a propensity analysis. J Am Col Cardio. 2014;63(22):2398–2407. doi: 10.1016/j.jacc.2014.02.577. [DOI] [PubMed] [Google Scholar]
- 189.Yazdchi F, Koch CG, Mihaljevic T. et al. Increasing disadvantage of ‘watchfu waiting’ for repairing degenerative mitra valve disease. Ann Thorac Surg. 2015;99(6):1992–2000. doi: 10.1016/j.athoracsur.2015.01.065. [DOI] [PubMed] [Google Scholar]
- 190.Barbieri A, Bursi F, Grigioni F. et al. Prognostic and therapeutic implications of pulmonary hypertension complicating degenerative mitra regurgitation due to flai leaflet: a multicenter long–term internationa study. Eur Heart J. 2011;32(6):751–759. doi: 10.1093/eurheartj/ehq294. [DOI] [PubMed] [Google Scholar]
- 191.Rosenhek R, Rader F, Klaar U. et al. Outcome of watchfu waiting in asymptomatic severe mitra regurgitation. Circulation. 2006;113(18):2238–2244. doi: 10.1161/CIRCULATIONAHA.105.599175. [DOI] [PubMed] [Google Scholar]
- 192.Eskesen K, Olsen NT, Dimaano VL, Fritz–Hansen T, Sogaard P, Abraham TP. Effects of early and late–onset treatment with carvedilo in an experimenta mode of aortic regurgitation. Springerplus. 2015;4:52–52. doi: 10.1186/s40064-015-0829-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Carabello BA. The current therapy for mitra regurgitation. J Am Col Cardio. 2008;52(5):319–326. doi: 10.1016/j.jacc.2008.02.084. [DOI] [PubMed] [Google Scholar]
- 194.Wisenbaugh T. Vasoactive medications in the treatment of severe mitra regurgitation. Coron Artery Dis. 2000;11(1):19–22. doi: 10.1097/00019501-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 195.Wisenbaugh T, Sinovich V, Dullabh A, Sareli P. Six month pilot study of captopri for mildly symptomatic, severe isolated mitra and isolated aortic regurgitation. J Heart Valve Dis. 1994;3(2):197–204. [PubMed] [Google Scholar]
- 196.Stewart RA, Raffe OC, Kerr AJ. et al. Pilot study to assess the influence of beta–blockade on mitra regurgitant volume and left ventricular work in degenerative mitra valve disease. Circulation. 2008;118(10):1041–1046. doi: 10.1161/CIRCULATIONAHA.108.770438. [DOI] [PubMed] [Google Scholar]
- 197.Ennis DB, Rudd–Barnard GR, Li B. et al. Changes in mitra annular geometry and dynamics with ss–blockade in patients with degenerative mitra valve disease. Circ Cardiovasc Imaging. 2010;3(6):687–693. doi: 10.1161/CIRCIMAGING.110.959171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Varadarajan P, Joshi N, Appe D, Duvvuri L, Pai RG. Effect of Betablocker therapy on surviva in patients with severe mitra regurgitation and norma left ventricular ejection fraction. Am J Cardio. 2008;102(5):611–615. doi: 10.1016/j.amjcard.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 199.Ahmed MI, Aban I, Lloyd SG. et al. A Randomized controlled phase IIb tria of beta1–receptor blockade for chronic degenerative mitra regurgitation. J Am Col Cardio. 2012;60(9):833–838. doi: 10.1016/j.jacc.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pat B, Killingsworth C, Denney T. et al. Dissociation between cardiomyocyte function and remodeling with beta–adrenergic receptor blockade in isolated canine mitra regurgitation. Am J Physio Heart Circ Physio. 2008;295(6):H2321–2327. doi: 10.1152/ajpheart.00746.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Pu M, Gao Z, Pu DK, Davidson WR Jr. Effects of early, late, and longterm nonselective beta–blockade on left ventricular remodeling, function, and surviva in chronic organic mitra regurgitation. Circ Heart Fai. 2013;6(4):756–762. doi: 10.1161/CIRCHEARTFAILURE.112.000196. [DOI] [PubMed] [Google Scholar]
- 202.Capomolla S, Febo O, Gnemmi M. et al. Beta–blockade therapy in chronic heart failure: diastolic function and mitra regurgitation improvement by carvedilol. Am Heart J. 2000;139(4):596–608. doi: 10.1016/s0002-8703(00)90036-x. [DOI] [PubMed] [Google Scholar]