

Abstract
As optical reporters and modulators of cellular activity have become increasingly sophisticated, the amount that can be learned about the brain via high-speed cellular imaging has increased dramatically. However, despite fervent innovation, point-scanning two-photon microscopy is facing a fundamental limit in achievable 3D imaging speeds and fields of view. A range of alternative approaches are emerging, some of which are moving away from point-scanning to use axially-extended beams or sheets of light, for example swept confocally aligned planar excitation (SCAPE) microscopy. These methods are proving effective for high-speed volumetric imaging of the nervous system of small organisms such as Drosophila (fruit fly) and D. Rerio (Zebrafish), and are showing promise for imaging activity in the living mammalian brain using both single and two-photon excitation. This article describes these approaches and presents a simple model that demonstrates key advantages of axially-extended illumination over point-scanning strategies for high-speed volumetric imaging, including longer integration times per voxel, improved photon efficiency and reduced photodamage.
Graphical abstract
Introduction
The first microscopes imaged small specimens or thin slices of stained tissue. Physical sectioning removes tissue above and below the plane of interest that would otherwise generate overlapping structures and blurring effects due to light scattering. The breakthrough enabling high-resolution imaging of intact tissues was ‘optical sectioning’ in which only a sub-region of the tissue is illuminated, while light coming from adjacent areas is either not produced, or blocked [1,2]. The two main examples of this approach; confocal and two-photon point-scanning microscopy have had significant impact for a wide range of biomedical applications.
However, the increasing availability of fluorophores capable of reporting cellular function, such as genetically-encoded calcium-sensitive fluorescent proteins (e.g. GCaMP) has heightened the need for faster volumetric imaging speeds over larger fields of view [3]. Higher speed imaging permits neural and other cellular activity to be captured at sufficient temporal resolution to analyze onset timing, amplitudes and response profiles. Volumetric imaging enables parallel recording of ensembles of cells over multiple layers, or throughout the brain of small organisms providing simultaneous evaluation of activity, spontaneous events and system-wide correlations. The increasing use of awake, behaving animals in neuroscience studies places further requirements on motion tolerance, and the ability to track motion, evoke and monitor behavior as well as on simplicity and efficiency of image acquisition.
The pixel-rate limit of point-scanning
Both confocal and two-photon laser scanning microscopes form an image by scanning a tightly focused spot in a 2D or 3D pattern through the sample (Figure 1a-b). Signal detected from this point over time is then assigned to the scanned positions to form a plane or volume image respectively (with 1-photon confocal requiring descanning and a pinhole to reject scattered and out-of-plane light [1]). To increase volumetric imaging speed in point-scanning systems, one approach is to simply increase scanning speed [4–8]. However, as shown in Figure 3a-b, scanning speed is reaching limits, both in terms of available scanners (e.g. up to 24kHz line rate resonant scanning) and integration time per pixel (in the limit, reaching 12 ns per pulse for a standard 80 MHz Ti:Sapphire two-photon laser and 2.7 ns, the fluorescence lifetime of green fluorescent protein (GFP)). Recognizing these physical limits, random-access scanning systems (Figure 1c) have been developed that use elements such as acousto-optic deflectors (AODs) to capture signals from a few hundred pre-determined positions (e.g. neurons) within a 2D or 3D field of view, providing signal from these positions as a function of time [9–12]. However, although effective for some applications, the limits of this technique include the need for a-priori selection of regions of interest, sensitivity to in-vivo movement and low excitation throughput owing to the need for dispersion correction leading to typical all-point sampling rates of around 100 kHz.
Figure 1.

Different in-vivo microscopy configurations to enable high-speed 3D imaging - progressing from point-scanning (a-c) to line (d-e) and multi-point scanning (f) and then light-sheet and axially-extended geometries (g-i) and finally to related two-photon implementations of multi-spot, wide-field and axially extended geometries (j-l). Dark red denotes near infrared two-photon excitation, blue represents single-photon excitation. Fluorescence emission is shown in green for all cases. Black arrows indicate scanning motion in the x-z plane while insets lower right for each case show a top-down (x-y) view of the illumination pattern and scan motion. Configurations modeled in Figure 3 below are indicated by *.
Figure 3.

Model-based comparisons between different volumetric imaging approaches in terms of a) per-voxel integration time, b) scanner line rate, c) total laser power needed for equivalent energy per voxel (based on 0.11 nJ energy deposited per pixel when point-scanning using 0.5mW at 1VPS) and d) predicted photobleaching dynamics for imaging at 10 VPS ignoring the effects of extraneous light during volumetric imaging and e) including photobleaching from extraneous light. See supplemental appendix for all model assumptions and parameters. f) All calculations based on a 300 × 300 × 50 xyz voxel volume. Supplemental figure 1 shows equivalent analysis for 300 × 300 × 300 voxel volume. All results show that axially-extended line and sheet geometries deliver major benefits compared to point scanning.
Lateral beam multiplexing (1-photon)
An alternative approach to very fast scanning of a single point is to increase the size or number of spots illuminating the tissue in parallel. By sampling from multiple locations in parallel, one can reduce the number of sequential samples required to cover a volume, reducing constraints on both scanning speed (which can be slower) and per-pixel integration time (which can be longer).
The commonest approach to this parallelization is to extend illumination in the lateral direction, while maintaining high-NA illumination to restrict axial extent. 1-photon examples of this approach include line-scanning confocal, also called swept-field [13] or scanning laser ophthalmoscopy [14] (Figure 1d) and a variant; theta scanning confocal [15,16] which improves resolution isotropy by creating an angle between the incident beam and the detection path (Figure 1e). In all of these cases, a narrow focused line of illumination (along y) is formed within the tissue and scanned in one dimension (along x) to sample an x-y plane. Confocal descanning enables use of a stationary slit to reject fluorescence from extraneous excitation light, as well as scattered emission light, while a linear detector array can be used to image N pixels in parallel, increasing the effective integration time per pixel by a factor of N for the same frame rate (yielding N times more signal, or N times faster imaging if N times more overall incident power is used). Confocal spinning disk achieves similar integration time benefits by scanning around 500 laterally-distributed points to illuminate a plane in the tissue. The disk also serves to aperture returning light to maintain the confocal condition while building up an image of the illuminated plane on a camera [17] (Figure 1f). The imaging speed and photobleaching benefits of spinning disk confocal over point-scanning have been widely noted for live cell imaging [18].
Axial beam multiplexing (1-photon)
An alternative to distributing the excitation pattern in a lateral direction is extending it in an axial direction. Axially-extended beams have already been used extensively in conventional light sheet microscopy, wherein a sheet of light is generated across the tissue, only exciting fluorescence in a single plane [19,20] (Figure 1g). An orthogonally positioned camera, aligned to focus at the plane of the light sheet can then create an optically sectioned image whose point spread function (PSF) corresponds to the product of the orthogonal excitation and emission PSFs [16,21]. A volumetric image can be created if the object is translated relative to this co-aligned plane. Although conventional, dual-objective light sheet methods have been applied to functional imaging of the larval zebrafish brain [22], alternative approaches such as objective coupled planar excitation microscopy (OCPI) [23] (Figure 1h), oblique plane microscopy (OPM) [24], axial plane optical microscopy [25], and swept confocally aligned planar excitation (SCAPE) microscopy (Figure 1i) [26] leverage the use of light sheets, but use simpler single-objective geometries that can accommodate more diverse neuroimaging applications while in some cases enabling higher speed, translationless volumetric imaging. A further recent example of axial-beam imaging includes ‘extended depth of field’ two-photon microscopy approaches, discussed further below [27–29] (Figure 1l).
While still achieving optical sectioning, axially-extending illumination yields several additional benefits compared to lateral-beam multiplexing as depicted in Figure 2:
The most recognized benefit of light sheet approaches is that they selectively illuminate only the plane being imaged, significantly reducing the light reaching parts of the sample that are not being imaged at that moment in time. This effect can considerably reduce photobleaching and phototoxicity compared to high NA confocal methods which illuminate tissue above and below the focal plane and must reject light from these regions using apertures.
A second major benefit of axially extended beams is that photons within the light sheet have multiple opportunities to interact with fluorophores along their propagation direction (Figure 2). A photon that is not absorbed by a fluorophore at the start of its path will continue to have a probability of being absorbed by a fluorophore as it continues along its path. Axial illumination strategies can thus produce a much higher fluorescent yield per photon entering the tissue compared to multi-spot, lateral line or spinning disc confocal approaches that only utilize signal from photon-interactions occurring at a single location along their path (e.g. the excitation focal plane). To provide equivalent irradiance of all voxels within a plane, a thin light sheet propagating along z, of size Ny × Nz could thus require up to 1/Nx less total power compared to forming an en-face x-y plane of size Nx × Ny using light propagating along z; a geometry equivalent to spinning disk confocal and ‘wide-field’ two-photon with temporal focusing [30]. Although axial beams and sheets will experience some attenuation as they propagate, this same attenuation affects point-scanning and lateral multiplexing approaches and is typically compensated for by utilizing even higher illumination powers for deeper planes.
The third benefit (shared with multi-spot and en-face methods) is that parallel detection of light from an illuminated plane of size Nx by Ny (or Ny by Nz) enables integration time per pixel to increase by a factor of Nx × Ny (or Ny × Nz) for the same volumetric imaging rate (Figure 3a). This benefit means that equivalent signal at each location can be generated with much lower per-pixel incident power compared to point-scanning, where shorter per-pixel integration times require much higher incident powers that can approach fluorophore saturation limits [18], while also worsening photobleaching as examined further below. Longer integration times also enable the use of array (e.g. camera-based) detectors which operate at higher noise equivalent powers compared to the very high speed detectors required for point-scanning applications (such as photomultiplier tubes and Gallium arsenide-based detectors). Parallelization also reduces the speed at which illumination patterns need to be moved during volumetric imaging compared to single point-scanning (Figure 3b).
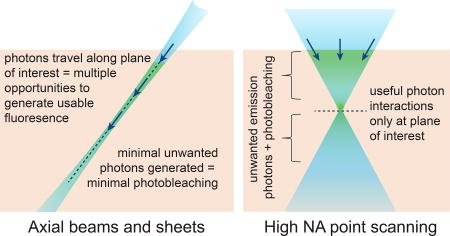
Figure 2.

Comparing high-NA point-scanning with low-NA light sheet or axial beam illumination. High NA confocal microscopy illuminates tissue above and below the focal plane causing photodamage to regions that are not being imaged, while requiring a pinhole to reject light originating from these regions. Axial illumination restricts light to only the plane / line being imaged, reducing both background and photodamage to regions that are not being imaged. However, a further benefit of axial beams is that the beam or sheet is composed of photons traveling in their direction of propagation. As a result, photons forming the sheet have repeated opportunities to interact with fluorophores along their path, whereas high NA confocal imaging only detects interactions occurring at the focal plane. This effect makes axially-extended beams more efficient – generating more usable fluorescent photons per photon entering the tissue.
Modeling the trade-offs between point-scanning and multiplexed imaging
A mathematical model is needed to weigh these factors against each other to properly compare each imaging approach for equivalent imaging speeds and signal generated. Figure 3 shows modeling results that compare scanning speed, per-pixel integration time, total power required and estimate photobleaching dynamics for a range of image-formation methodology. A conservative volume of 300 × 300 × 50 (x-y-z) voxels, each 1 micron3 in size, is assumed. Full model details and additional calculations for a 300 × 300 × 300 (x-y-z) volume are provided in a supplemental appendix and supplemental figure 1.
Results shown in Figure 3a-b depict integration times and scanning speeds for the 1- and 2-photon methods shown in Figure 1a,j,l,d,f,i (in order). These results easily confirm that any increase in parallelization will yield scanning speed and integration time benefits, while demonstrating that point scanning methods are reaching fundamental limits (and exceeding limits for larger volumes, Supplemental Figure 1a-b).
Figure 3c computes the total amount of incident power required for equivalent signal for 1-photon embodiments of each method. Calculations assume that fluorescence generated will be proportional to energy incident at a voxel (energy = power × time), and used 0.11 nJ, corresponding to point-scanning at 1 volume per second (VPS) with 0.5 mW at the sample. Axially-extended beam and sheet illumination is assumed to provide photons with repeated opportunities for interactions along their path, as described above, an effect which combines with increased integration times to yield the lowest required total incident powers at the sample for axially-extended methods (Figure 3c). As explained further below, calculations for the oblique sheet (SCAPE) method include a conservative 50% detection efficiency.
Photobleaching dynamics shown in Figure 3d-e were modeled for single-photon point-scanning, 500 point spinning disk confocal, an x-y scanned axial-beam, and oblique sheet (SCAPE) geometries. Calculations are based on imaging at 10 VPS, assuming equivalent signal per voxel for each method, here 63pJ per voxel based on using 0.5 mW in a resonant scanning confocal acquiring 512×512 frames at 30 fps. The model incorporates recently characterized properties of fluorescent proteins which suggest a supralinear relationship with incident power (P−α where α ~ 1.23 for GFP [31]). This relationship implies that higher power illumination for a shorter period will cause more photobleaching than lower power illumination for a longer period of time (with equivalent overall energy = power × time) delivered. This effect alone predicts lower photobleaching for more parallelized methods as demonstrated in Figure 3d, which shows how signal would decay within a single plane of the volume, ignoring additional bleaching from extraneous light during volumetric imaging. Supplemental figure 2a(i) confirms that these methods would have equivalent single-plane bleaching rates if α = 1. This supralinear effect also explains why imaging with a full light sheet (lower irradiance, longer integration), rather than a scanning axial beam (higher irradiance, shorter integration) provides less photobleaching (Figure 3d), an effect also compared for conventional 90° light-sheet geometries in supplemental figure 2b.
Figure 3e add the effects of extraneous illumination on all parts of the tissue during volumetric imaging to the model. For point-scanning and spinning disk, every voxel in a given plane will be illuminated during the acquisition of every other depth-plane during volumetric imaging. In contrast, axial sheet and beam methods selectively illuminate the plane being imaged. These effects lead to dramatically reduced photobleaching compared to point scanning for equivalent volume rates and fluorescence yield. Supplemental figures 1d-e and 2b(ii), c(ii) and d(ii) illustrate that these benefits are consistent at different volumetric imaging speeds (2 VPS), for larger volumes and irrespective of α.
The net consequence of these favorable photobleaching effects is that axially-extended imaging methods can either utilize higher average power levels to improve signal to noise ratios (SNR) for high-speed imaging (for lower or equivalent photodamage), or that these methods can be used to image light-sensitive samples such as developing C. Elegans embryos [32], perfused tissues, visual circuits and possibly samples expressing channel rhodopsins, all of which are susceptible to being damaged or perturbed by imaging.
Implementations of single-photon axial-sheet imaging
As a result of these advantages, light-sheet microscopy has found many applications for imaging biological processes including the early-stage development of small, low-scattering organisms such as Drosophila embryos and larval zebrafish. However, conventional implementations of light sheet utilize an orthogonal arrangement of two objective lenses at the sample: one to generate the light sheet, and a second to focus onto the sheet and relay the image to a camera [20,22,33,34] (Figure 1g). This need for co-alignment makes formation of a volumetric image challenging, with the fastest implementations scanning the light sheet through the excitation objective in synchrony with piezo-actuated translation of the detection objective. Although electric-lens implementations have also been demonstrated as alternatives to piezo scanning [35], the need for synchrony of both movements impose limitations on achievable volumetric imaging speeds (under 2 VPS) for isotropic sampling to date) [36]. The sample geometry in such systems is also severely restricted by the positioning of dual (or 3 or 4) [34,37] orthogonal objective lenses (Figure 1g). These configurations constrain the size of sample that can be imaged while limiting working distances, immersion media and generally requiring rigid mounting of the sample, preventing free behavior. Mounting further restricts ancillary modulations and measurements such as electrode recording and stimulation, perfusion of agents and behavioral monitoring.
OCPI, OPM and SCAPE are variants of light-sheet microscopy that illuminate the tissue with an axially-extended light sheet entering the tissue at an oblique angle. Another variant, Axial plane optical microscopy (APOM) both illuminates and images an axial plane through a single high NA objective [25]. APOM and OPM have been primarily applied to in-vitro cellular imaging. However, OCPI was developed specifically for imaging functional activity in olfactory tissues and generates the light sheet using a fiber optic component attached to a high numerical aperture objective lens, positioned such that the light sheet is rigidly aligned with the detection plane (Figure 1h) [23,38]. To generate a volumetric image, the system is translated relative to the sample. Although the need for physical motion can limit achievable speeds, this method has been foundational in establishing the use of light sheet imaging in intact neural tissues.
SCAPE microscopy (Figure 1i) is well-suited to brain imaging owing to its ability to capture large 3D fields of view at high-speeds without requiring translation of the sample or objective lens [26]. SCAPE achieves this translationless high-speed imaging by combining oblique light sheet illumination with confocal scanning and descanning principles. Figure 4a shows a SCAPE system layout, demonstrating how SCAPE’s oblique light sheet (along y-z’) is formed through the primary objective lens, while fluorescence light excited by this sheet is collected back through the same objective lens. In order to acquire a focused image of the oblique light sheet, two telescopes are used to relay the 3D object space at the sample to a 3D intermediate image space. An obliquely aligned camera is then focused onto the oblique image of the light sheet in this intermediate image space. This configuration achieves the equivalent of a light sheet imaging condition at the sample, yet retains a simple sample geometry similar to a standard upright (or inverted) two-photon or confocal microscope.
Figure 4.

SCAPE microscopy for high-speed 3D neuroimaging. a) Optical layout of a typical SCAPE system permitting oblique sheet scanning through a stationary, single objective lens. Insets show imaging geometries for brain imaging in zebrafish larvae (left) and awake, behaving mouse brain (right). b(i) shows SCAPE data acquired at 10 VPS (140 × 750 × 149 x-y-z voxels = 372 × 1032 × 174 micron field of view (FOV)) in an awake, behaving mouse capturing spontaneous activity in apical dendrites of layer 5 neurons in whisker barrel cortex via GCaMP6f (AAV9.Syn.GCaMP6f) using methods similar to [26], but with improved resolution and SNR compared to our first demonstration. Maximum intensity views from the top (x-y) and side (y-z) are shown for activity occurring between 22 and 28 seconds (with colors denoting time). Raw fluorescence time-courses from the regions of interest indicated are shown as raw data, showing excellent SNR, minimal photobleaching over 60 seconds and the ability to probe firing dynamics along individual dendrites during a single spontaneous event. A time-color merge of all dendritic activity for the full 60 seconds is shown in supplemental figure 3. c) shows imaging of spontaneous activity in the whole brain of larval zebrafish: Data was acquired at 6 VPS over a 820 × 380 × 260 µm FOV (i) shows a volume rendering of a time-maximum intensity projection taken over all 360 time points over a 1 minute run. Inset shows time series extracted from 6 neurons at 6 depth planes within the fish as indicated. (ii) Shows a time-encoded color projection of 3 depth planes showing spontaneous activity over a range of brain regions. Insets show ~2× close ups of indicated regions. 7 day past fertilization HuC:H2B-GCaMP6f fish obtained from Janelia Farm.
SCAPE’s unique way of acquiring a volumetric image is then to sweep the light sheet across the sample in the × direction using a galvanometer mirror. However, in addition to scanning the sheet within the sample, this moving mirror also descans the returning fluorescence light such that the intermediate image remains stationary, yet always stays aligned with the plane being illuminated by the moving light sheet [26]. As a result, the camera also stays focused on the light sheet, even as it scans across the volume in the x-direction, and can thus capture a sequence of y-z’ planes that can be stacked to form a 3D image. The galvanometer mirror is the only moving part of the system, and only needs to move at the volume rate (e.g. 20 Hz for 20 VPS). The primary limit on SCAPE’s volumetric imaging speed (and/or x-direction sampling density) is thus the read-out rate of available cameras and fluorophore brightness. Current standard sCMOS cameras can read-out 100 rows (z’ depths) across 2000 columns (y-pixels) at over 2,000 frames per second, enabling 10 VPS imaging of 200 × 2000 × 100 (xyz) voxel volumes.
One essential part of all single-objective light sheet approaches, including SCAPE, is need to form an image of an oblique or axial image plane. This image rotation can be achieved in a number of ways, but as characterized by Dunsby [24,39], all will generally lose some fraction of the light emitted from the sample. For this reason, modeling in Figure 3c-e incorporates a 50% loss in detection efficiency to account for this effect. Despite this factor, oblique illumination (SCAPE) still exhibits major benefits in all parameters modeled.
Examples of high-speed functional imaging using SCAPE are shown in Figure 4b-c. Figure 4b shows spontaneous calcium (GCaMP6f) activity in apical dendrites from layer 5 neurons in the awake mouse barrel cortex acquired at 10 VPS to a depth of around 150 microns using 488 nm excitation. Raw fluorescence intensity data extracted from 2×2×2 voxel regions along individual dendrites is also shown. This dataset highlights SCAPE’s ability to capture all activity in a 3D field of view over 1 mm wide, to resolve fine structures in the dendritic branches, as well as the excellent signal to noise and minimal photobleaching observed over 1 minute of acquisition. This data demonstrates a significant improvement over imaging of the same preparation using our first SCAPE prototype as presented here [26].
Figure 4c shows SCAPE imaging of nuclear-localized GCaMP6f in neurons in a zebrafish larva, capturing spontaneous activity throughout the brain. SCAPE permits imaging without needing to translate the objective lens or the sample, reducing disturbance. The fact that SCAPE’s oblique light sheet does not need to pass through the fish’s eye is also a major advantage, while the simple mounting of the fish permits simple presentation of stimuli such as visual patterns, as well as surveillance of the animal including recording swim-efforts or simultaneous electrode recording. In this sample, data was acquired at 6 VPS for over 1 hour without evidence phototoxicity (such as marked decreases in neural activity).
These examples demonstrate that SCAPE delivers the significant benefits of light-sheet imaging in terms of reduced photodamage and improved SNR, combined with the ability acquire volumetric images of unmounted, intact, behaving or freely moving samples at high enough speeds to visualize cellular-level functional activity over large fields of view.
Extending analysis to two-photon implementations
The above analysis focused primarily on single-photon excitation, yet similar principles can hold for two-photon excitation, which can enable deeper imaging into scattering tissues such as the mammalian brain. However, for two-photon implementations two additional factors need to be considered - the first is limitations on laser power, and the second is the need for spatially-resolved detection.
In terms of laser power, most point-scanning two-photon systems use a standard (yet costly) Ti:Sapphire laser generating up to 1 W of 690-1040 nm near infrared light consisting of ~100 fs pulses at 80 MHz. Depending on system losses, it is common to use 10–100% of this available power for high-speed point-scanning in the mammalian brain. As such, generating multiple excitation spots [40], lines or planes of two-photon excitation from a standard Ti:Sapphire laser would result in reduced laser power per unit area if the maximum available laser power limit is reached. Since two-photon emission scales as power2 splitting a beam into 4 equal beams, for example, would reduce signal at each point to 1/16th of its original value. Although 4× longer integration time could be used for the same imaging rate, the net detected signal would decrease by a factor of 4. Even if higher average power lasers were available, recent studies have suggested that >200 mW of average power at the sample will cause excessive heating [41], while saturation of typical in-vivo fluorophores is expected to occur at around 2nJ per pulse per micron2 (corresponding to 160 mW at 80 MHz).
Achieving parallelized two-photon excitation thus requires careful navigation of these limits, but can also enable alternative strategies. For example, consider that for the same average power, if a laser’s pulse repetition rate is decreased from 80 MHz to 1 MHz, pulse peak power can be increased by a factor of 80. This increase should yield 802 more two-photon signal per pulse. Accounting for the fact that there will be 1/80 fewer pulses, this lower repetition rate could still yield an 80× improvement in signal (per unit time) without exceeding average power limits. High-speed point-scanning cannot benefit from this advantage, since it is already facing 80 MHz as a pixel-rate limit as shown in Figure 3a. However, this analysis shows that a 1 MHz laser beam could be split into 80 parallel spots, which could be measured in parallel at 1 MHz and should yield equivalent signal to 80 MHz sequential point scanning for the same volume rate, average power and incident pulse energy per unit area. This approach has been employed for temporal focusing two-photon microscopy (Figure 1k) to enable parallel two-photon excitation of a lateral x-y plane at a particular depth [30,42,43,44].
If we now consider the photon-efficiency benefits of axially-extended beams and light sheets (Figure 2), we can consider forming an oblique light sheet instead of a lateral line of 80 points along the tissue surface as described above. If this line continued as a low NA sheet into the tissue along z’, to a depth of perhaps 300 microns, the integration time per measurement could increase by a factor of 300 (for the same volume rate). In fact, measurement rates would permit laser repetition rates at or below 100 kHz, providing further available peak power for the same average power. These factors provide significant excess signal to account for the lower NA illumination, formation of a wider light sheet or acquisition and higher volumetric imaging speeds. These basic relationships are described by the following equation,
where Tint1 = integration time per pixel for single-point scanning at a given volume rate, Ndot is the number of lateral spots illuminated (in the limit a line formed at the surface as the axial sheet enters the tissue), Nz is the number of depth-planes acquired in parallel, Rrep = laser repetition rate, Tp is the laser pulse width and Pave is the laser average power (derivation provided in Supplemental appendix 2). This equation does not include the additional benefit that photons in an axially-extended beam will all travel similar paths, reducing group dispersion compared to a high NA focus, potentially providing improved two-photon excitation efficiency [45].
Although the analysis above demonstrates the feasibility of achieving multiplexed two-photon excitation, another challenge is capturing and disambiguating emitted fluorescent light. A major advantage of single-point scanning in two-photon microscopy is that the non-linearity of the two-photon process restricts excitation to a single focal point. As a result, all fluorescence light detected from the tissue can be unambiguously assigned to the location of the focal point at any given moment in time, without the need for descanning or pinholes [2]. However, this condition breaks down once multiple points, lines or planes are excited in parallel. One solution is to encode signal from multiple spots in some way while still using single-channel detectors to collect all of the light emitted. Examples include using two different excitation wavelengths and a mixture of two fluorophores [46], encoding via modulation of each beam at a different temporal frequency [47] or encoding via time-delay of excitation pulses [48]. Each of these methods has been used for simultaneous multi-depth imaging in the living brain, but all are limited in scalability to <5 beams; limits imposed by complexity, cost, imaging speed and fluorescence lifetime.
Another approach, is to use a single detector to acquire all fluorescence signal generated in the sample while an axial Bessel beam is scanned in an x-y raster pattern Figure 1l. Collecting all signal generated by this axial beam at any point in time generates a depth-averaged sum image of the volume sampled [27,29]. This method thus does not provide true 3D imaging, but can provide significant advantages in sparsely labeled samples, samples with modest axial motion, and samples that don’t require the 3D location of objects to be tracked. Fast ‘volumetric’ imaging speeds can be achieved since a single x-y raster scan samples the entire volume, rather than requiring a full x-y scan per plane in addition to axial scanning. Meanwhile the method shares the integration time benefits of parallel illumination, and the efficiency of axial illumination as detailed in Figure 3 and above, providing good SNR even at high speeds. High enough resolution to resolve dendritic spines can be achieved if excitation is sufficiently constrained to a narrow Bessell beam.
Besides these ‘summation’ methods, true 3D volumetric two-photon imaging using multiple spots [40], lines [45] or planes [30,42,43,44] requires detection of a spatially resolved image of the illuminated regions, for example using a camera. Although near infrared light can penetrate more deeply than the visible light typically used for 1-photon excitation, emitted fluorescence light will likely be in the visible range, and will thus experience scatter, attenuation and distortion as it emerges from the tissue, representing a current limit to achievable penetration depths for parallelized two-photon. Despite these limitations, implementations of two-photon light sheet to date, although slow owing to their use of standard Ti: Sapphire lasers, demonstrate expected improved penetration, optical sectioning and reduced background compared to single-photon implementations [45]. The clear advantages of axially-extended illumination, as well as the ability to leverage modern variable repetition rate lasers suggest that high-speed two-photon light sheet is a promising direction for high-speed volumetric imaging deeper into the mammalian brain.
Conclusions
An emerging direction in high-speed volumetric microscopy is to extend illumination axially, rather than laterally. Compared to point-scanning and lateral multiplexing, axial beams and sheets deliver reduced photodamage and can thus yield improved SNR. Fast volumetric imaging speeds can be achieved without high-speed beam scanning, and with no axial scanning of the objective lens, eliminating both inertia concerns and physical disturbance of sensitive in-vivo samples.
The SCAPE imaging examples shown here highlighted the method’s ability to capture large fields of view at high speeds, and did not explore or compare limits of spatial resolution. Although obtaining resolution equivalent to high NA confocal point scanning may be challenging using axial beams and sheets, such performance may be attainable with optical designs optimized for specific applications. Moreover, many strategies already developed for conventional light-sheet imaging can be applied to SCAPE and related approaches to improve resolution and penetration depth, including beam shaping, structured illumination and wavefront optimization, spatiotemporal unmixing as well as the potential to extend to 2-photon implementations [49–54].
The high SNR, high volumetric imaging speeds, and the ability to achieve optical sectioning without two-photon excitation of axial methods also offer promise for imaging diverse new optical reporters of neural activity including red and near-infrared calcium and voltage indicators. Some of these indicators are incompatible with two-photon excitation, while some require ultrafast detection [55–57]. Newer intensified cameras that can stream pixels at over 1.2 GHz, promising single-plane rates of 10,000 FPS, volume rates exceeding 100 VPS, or 3D fields of view extending several millimeters in x and y. The lower illumination powers needed for light sheet and axial-beam imaging methods (Figure 3c), as well as the ability to image volumetrically without axial translation of the objective lens makes these methods well suited for combination with modern optogenetic activation studies [58].
Against other emerging volumetric imaging methods such as light field microscopy [59,60], axially-extended approaches bring the major benefit of not needing complex computational reconstruction in order to visualize 3D dynamics, while also having optical sectioning advantages and the ability to implement two-photon excitation. These features combine to make axially-extended illumination strategies for in-vivo imaging of brain function a promising route towards holistic, real-time volumetric imaging of neural activity.
Supplementary Material
Highlights.
Neuroscience needs faster in-vivo volumetric microscopy approaches.
Axially-extended beams and light sheets provide alternatives to point scanning.
The performance and photobleaching benefits of axially extended illumination are modeled.
SCAPE microscopy leverages axial benefits for high-speed 3D imaging of neural activity.
SCAPE imaging in awake mouse brain and larval zebrafish brain is demonstrated.
Acknowledgments
The authors acknowledge the foundational contributions of Matthew Bouchard to the development of SCAPE as well as the contributions of many collaborators in developing and refining SCAPE and associated imaging principles for neuroscience and in-vivo applications including Wesley Grueber, Richard Mann, Cesar Mendes, Neeli Mishra, Evan Schaffer, Rebecca Vaadia, David Schoppik, Richard Axel, Randy Bruno, Clay Lacefield, Ed Boyden, Clare Wyart, Peter Canoll. Data in figure 4c was acquired in collaboration with Jeremy Ullmann, with fish provided by Misha Ahrens and Janelia Farm. We thank David Boas, Martin Booth, David Piston and Tim Holy for helpful discussions. Funding for this work was provided by National Institutes of Health BRAIN initiative grant 5U01NS094296 and R01HL13143801A1, the National Science Foundation (graduate fellowships to Bouchard and Patel and CAREER CBET-0954796 to Hillman), the Simons Foundation Collaboration on the Global Brain, Department of Defense MURI W911NF-12-1-0594, the Kavli Institute for Brain Science, the Columbia-Coulter Translational Research Partnership and Coulter Foundation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest
SCAPE intellectual property is licensed to Leica Microsystems for commercial development. All authors have a potential financial conflict of interest relating to SCAPE microscopy.
References
- 1.White JG, Amos WB, Fordham M. An evaluation of confocal versus conventional imaging of biological structures by fluorescence light microscopy. J Cell Biol. 1987;105:41–48. doi: 10.1083/jcb.105.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 3.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gobel W, Kampa BM, Helmchen F. Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nature Methods. 2007;4:73–79. doi: 10.1038/nmeth989. [DOI] [PubMed] [Google Scholar]
- 5.Grewe BF, Voigt FF, van’t Hoff M, Helmchen F. Fast two-layer two-photon imaging of neuronal cell populations using an electrically tunable lens. Biomed. Opt. Express. 2011;2:2035–2046. doi: 10.1364/BOE.2.002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6*.Sofroniew NJ, Flickinger D, King J, Svoboda K. A large field of view two-photon mesoscope with subcellular resolution for in vivo imaging. Elife. 2016;5 doi: 10.7554/eLife.14472. This paper describes and demonstrates a state of the art design for large field of view point-scanning two-photon microscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7*.Stirman JN, Smith IT, Kudenov MW, Smith SL. Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain. Nat Biotechnol. 2016;34:857–862. doi: 10.1038/nbt.3594. This paper describes and demonstrates a state of the art design for large field of view point-scanning two-photon microscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Botcherby EJ, Smith CW, Kohl MM, Debarre D, Booth MJ, Juskaitis R, Paulsen O, Wilson T. Aberration-free three-dimensional multiphoton imaging of neuronal activity at kHz rates. Proc Natl Acad Sci U S A. 2012;109:2919–2924. doi: 10.1073/pnas.1111662109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salomé R, Kremer Y, Dieudonné S, Léger; J-F, Krichevsky O, Wyart C, Chatenay D, Bourdieu L. Ultrafast random-access scanning in two-photon microscopy using acousto-optic deflectors. Journal of Neuroscience Methods. 2006;154:161–174. doi: 10.1016/j.jneumeth.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Katona G, Szalay G, Maak P, Kaszas A, Veress M, Hillier D, Chiovini B, Vizi ES, Roska B, Rozsa B. Fast two-photon in vivo imaging with three-dimensional random-access scanning in large tissue volumes. Nat Methods. 2012;9:201–208. doi: 10.1038/nmeth.1851. [DOI] [PubMed] [Google Scholar]
- 11*.Szalay G, Judak L, Katona G, Ocsai K, Juhasz G, Veress M, Szadai Z, Feher A, Tompa T, Chiovini B, Maak P, Rozsa B. Fast 3D Imaging of Spine, Dendritic, and Neuronal Assemblies in Behaving Animals. Neuron. 2016;92:723–738. doi: 10.1016/j.neuron.2016.10.002. Paper demonstrating fast random-access scanning approach to two-photon microscopy in mouse brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Nadella KM, Ros H, Baragli C, Griffiths VA, Konstantinou G, Koimtzis T, Evans GJ, Kirkby PA, Silver RA. Random-access scanning microscopy for 3D imaging in awake behaving animals. Nat Methods. 2016;13:1001–1004. doi: 10.1038/nmeth.4033. Paper demonstrating fast random-access scanning approach to two-photon microscopy in mouse brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castellano-Munoz M, Peng AW, Salles FT, Ricci AJ. Swept field laser confocal microscopy for enhanced spatial and temporal resolution in live-cell imaging. Microsc Microanal. 2012;18:753–760. doi: 10.1017/S1431927612000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webb RH, Hughes GW, Delori FC. Confocal scanning laser ophthalmoscope. Appl Opt. 1987;26:1492–1499. doi: 10.1364/AO.26.001492. [DOI] [PubMed] [Google Scholar]
- 15.Dwyer PJ, DiMarzio CA, Zavislan JM, Fox WJ, Rajadhyaksha M. Confocal reflectance theta line scanning microscope for imaging human skin in vivo. Opt Lett. 2006;31:942–944. doi: 10.1364/ol.31.000942. [DOI] [PubMed] [Google Scholar]
- 16.Lindek S, Stelzer EH. Resolution improvement by nonconfocal theta microscopy. Opt Lett. 1999;24:1505–1507. doi: 10.1364/ol.24.001505. [DOI] [PubMed] [Google Scholar]
- 17.Graf R, Rietdorf J, Zimmermann T. Live cell spinning disk microscopy. Adv Biochem Eng Biotechnol. 2005;95:57–75. doi: 10.1007/b102210. [DOI] [PubMed] [Google Scholar]
- 18.Wang E, Babbey CM, Dunn KW. Performance comparison between the high-speed Yokogawa spinning disc confocal system and single-point scanning confocal systems. J Microsc. 2005;218:148–159. doi: 10.1111/j.1365-2818.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- 19.Keller PJ, Dodt HU. Light sheet microscopy of living or cleared specimens. Curr Opin Neurobiol. 2012;22:138–143. doi: 10.1016/j.conb.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EHK. Optical Sectioning Deep Inside Live Embryos by Selective Plane Illumination Microscopy. Science. 2004;305:1007–1009. doi: 10.1126/science.1100035. [DOI] [PubMed] [Google Scholar]
- 21.Engelbrecht CJ, Stelzer EH. Resolution enhancement in a light-sheet-based microscope (SPIM) Opt Lett. 2006;31:1477–1479. doi: 10.1364/ol.31.001477. [DOI] [PubMed] [Google Scholar]
- 22**.Ahrens MB, Orger MB, Robson DN, Li JM, Keller PJ. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Meth. 2013;10:413–420. doi: 10.1038/nmeth.2434. Paper presenting the first whole-brain imaging of neural activity in zebrafish larvae with conventional light sheet microscopy. [DOI] [PubMed] [Google Scholar]
- 23**.Holekamp TF, Turaga D, Holy TE. Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy. Neuron. 2008;57:661–672. doi: 10.1016/j.neuron.2008.01.011. Paper first demonstrating OCPI in which an oblique light sheet is used to provide optical sectioning in intact mouse olfactory bulb. [DOI] [PubMed] [Google Scholar]
- 24**.Dunsby C. Optically sectioned imaging by oblique plane microscopy. Opt Express. 2008;16:20306–20316. doi: 10.1364/oe.16.020306. Paper first describing oblique plane microscopy and characterizing performance parameters. [DOI] [PubMed] [Google Scholar]
- 25.Li T, Ota S, Kim J, Wong ZJ, Wang Y, Yin X, Zhang X. Axial plane optical microscopy. Sci Rep. 2014;4:7253. doi: 10.1038/srep07253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26**.Bouchard MB, Voleti V, Mendes CS, Lacefield C, Grueber WB, Mann RS, Bruno RM, Hillman EM. Swept confocally-aligned planar excitation (SCAPE) microscopy for high speed volumetric imaging of behaving organisms. Nat Photonics. 2015;9:113–119. doi: 10.1038/nphoton.2014.323. light sheet microscopy for high-speed volumetric imaging for in-vivo samples including awake mouse brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27**.Lu RW, Sun WZ, Liang YJ, Kerlin A, Bierfeld J, Seelig JD, Wilson DE, Scholl B, Mohar B, Tanimoto M, Koyama M, Fitzpatrick D, Orger MB, Ji N. Video-rate volumetric functional imaging of the brain at synaptic resolution. Nature Neuroscience. 2017;20 doi: 10.1038/nn.4516. 620-+. Demonstration and reduction to practice of extended depth of field two-photon microscopy using an axial Bessel beam for depth-summed imaging. (also Theriault et al). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dufour P, Piche M, De Koninck Y, McCarthy N. Two-photon excitation fluorescence microscopy with a high depth of field using an axicon. Appl Opt. 2006;45:9246–9252. doi: 10.1364/ao.45.009246. [DOI] [PubMed] [Google Scholar]
- 29**.Theriault G, De Koninck Y, McCarthy N. Extended depth of field microscopy for rapid volumetric two-photon imaging. Opt Express. 2013;21:10095–10104. doi: 10.1364/OE.21.010095. Demonstration and reduction to practice of extended depth of field two-photon microscopy using an axial Bessel beam for depth-summed imaging. (also Lu et al) [DOI] [PubMed] [Google Scholar]
- 30.Vaziri A, Shank CV. Ultrafast widefield optical sectioning microscopy by multifocal temporal focusing. Opt. Express. 2010;18:19645–19655. doi: 10.1364/OE.18.019645. [DOI] [PubMed] [Google Scholar]
- 31**.Cranfill PJ, Sell BR, Baird MA, Allen JR, Lavagnino Z, de Gruiter HM, Kremers GJ, Davidson MW, Ustione A, Piston DW. Quantitative assessment of fluorescent proteins. Nat Methods. 2016;13:557–562. doi: 10.1038/nmeth.3891. Paper detailing characterization of the photobleaching dynamics of modern fluorescent proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Ghitani A, Christensen R, Santella A, Du Z, Rondeau G, Bao Z, Colon-Ramos D, Shroff H. Inverted selective plane illumination microscopy (iSPIM) enables coupled cell identity lineaging and neurodevelopmental imaging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2011;108:17708–17713. doi: 10.1073/pnas.1108494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dodt H-U, Leischner U, Schierloh A, Jahrling N, Mauch CP, Deininger K, Deussing JM, Eder M, Zieglgansberger W, Becker K. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nature Methods. 2007;4:331–336. doi: 10.1038/nmeth1036. [DOI] [PubMed] [Google Scholar]
- 34.Tomer R, Khairy K, Amat F, Keller PJ. Quantitative high-speed imaging of entire developing embryos with simultaneous multiview light-sheet microscopy. Nat Methods. 2012;9:755–763. doi: 10.1038/nmeth.2062. [DOI] [PubMed] [Google Scholar]
- 35.Fahrbach FO, Voigt FF, Schmid B, Helmchen F, Huisken J. Rapid 3D light-sheet microscopy with a tunable lens. Opt Express. 2013;21:21010–21026. doi: 10.1364/OE.21.021010. [DOI] [PubMed] [Google Scholar]
- 36.Lemon WC, Pulver SR, Hockendorf B, McDole K, Branson K, Freeman J, Keller PJ. Whole-central nervous system functional imaging in larval Drosophila. Nat Commun. 2015;6:7924. doi: 10.1038/ncomms8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chhetri RK, Amat F, Wan Y, Hockendorf B, Lemon WC, Keller PJ. Whole-animal functional and developmental imaging with isotropic spatial resolution. Nat Methods. 2015;12:1171–1178. doi: 10.1038/nmeth.3632. [DOI] [PubMed] [Google Scholar]
- 38.Liang X, Holy TE, Taghert PH. Synchronous Drosophila circadian pacemakers display nonsynchronous Ca(2)(+) rhythms in vivo. Science. 2016;351:976–981. doi: 10.1126/science.aad3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Botcherby EJ, Juskaitis R, Booth MJ, Wilson T. Aberration-free optical refocusing in high numerical aperture microscopy. Opt Lett. 2007;32:2007–2009. doi: 10.1364/ol.32.002007. [DOI] [PubMed] [Google Scholar]
- 40.Watson BO, Nikolenko V, Yuste R. Two-photon imaging with diffractive optical elements. Front Neural Circuits. 2009;3:6. doi: 10.3389/neuro.04.006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podgorski K, Ranganathan G. Brain heating induced by near-infrared lasers during multiphoton microscopy. J Neurophysiol. 2016;116:1012–1023. doi: 10.1152/jn.00275.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi H, Yew EY, Hallacoglu B, Fantini S, Sheppard CJ, So PT. Improvement of axial resolution and contrast in temporally focused widefield two-photon microscopy with structured light illumination. Biomed Opt Express. 2013;4:995–1005. doi: 10.1364/BOE.4.000995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowlands CJ, Bruns OT, Bawendi MG, So PT. Objective, comparative assessment of the penetration depth of temporal-focusing microscopy for imaging various organs. J Biomed Opt. 2015;20:61107. doi: 10.1117/1.JBO.20.6.061107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dana H, Marom A, Paluch S, Dvorkin R, Brosh I, Shoham S. Hybrid multiphoton volumetric functional imaging of large-scale bioengineered neuronal networks. Nature Communications. 2014;5 doi: 10.1038/ncomms4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45*.Truong TV, Supatto W, Koos DS, Choi JM, Fraser SE. Deep and fast live imaging with two-photon scanned light-sheet microscopy. Nat Methods. 2011;8:757–760. doi: 10.1038/nmeth.1652. Paper describing and demonstrating a system for two-photon light sheet microscopy. [DOI] [PubMed] [Google Scholar]
- 46.Grosberg LE, Chen BR, Hillman EM. Simultaneous multiplane in vivo nonlinear microscopy using spectral encoding. Opt Lett. 2012;37:2967–2969. doi: 10.1364/OL.37.002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ducros M, Goulam Houssen Y, Bradley J, de Sars V, Charpak S. Encoded multisite two-photon microscopy. Proc Natl Acad Sci U S A. 2013;110:13138–13143. doi: 10.1073/pnas.1307818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng A, Goncalves JT, Golshani P, Arisaka K, Portera-Cailliau C. Simultaneous two-photon calcium imaging at different depths with spatiotemporal multiplexing. Nat Methods. 2011;8:139–142. doi: 10.1038/nmeth.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalgarno HIC, Cizmar T, Vettenburg T, Nylk J, Gunn-Moore FJ, Dholakia K. Wavefront corrected light sheet microscopy in turbid media. Applied Physics Letters. 2012;100 [Google Scholar]
- 50.Mertz J, Kim J. Scanning light-sheet microscopy in the whole mouse brain with HiLo background rejection. J Biomed Opt. 2010;15:016027. doi: 10.1117/1.3324890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olarte OE, Licea-Rodriguez J, Palero JA, Gualda EJ, Artigas D, Mayer J, Swoger J, Sharpe J, Rocha-Mendoza I, Rangel-Rojo R, Loza-Alvarez P. Image formation by linear and nonlinear digital scanned light-sheet fluorescence microscopy with Gaussian and Bessel beam profiles. Biomed Opt Express. 2012;3:1492–1505. doi: 10.1364/BOE.3.001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilding D, Pozzi P, Soloviev O, Vdovin G, Verhaegen M. Adaptive illumination based on direct wavefront sensing in a light-sheet fluorescence microscope. Opt Express. 2016;24:24896–24906. doi: 10.1364/OE.24.024896. [DOI] [PubMed] [Google Scholar]
- 53.Wilding D, Pozzi P, Soloviev O, Vdovin G, Sheppard CJ, Verhaegen M. Pupil filters for extending the field-of-view in light-sheet microscopy. Opt Lett. 2016;41:1205–1208. doi: 10.1364/OL.41.001205. [DOI] [PubMed] [Google Scholar]
- 54.Nobauer T, Skocek O, Pernia-Andrade AJ, Weilguny L, Traub FM, Molodtsov MI, Vaziri A. Video rate volumetric Ca2+ imaging across cortex using seeded iterative demixing (SID) microscopy. Nature Methods. 2017;14 doi: 10.1038/nmeth.4341. 811-+ [DOI] [PubMed] [Google Scholar]
- 55.Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. Sensitive red protein calcium indicators for imaging neural activity. Elife. 2016;5:e12727. doi: 10.7554/eLife.12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grimm JB, Muthusamy AK, Liang Y, Brown TA, Lemon WC, Patel R, Lu R, Macklin JJ, Keller PJ, Ji N, Lavis LD. A general method to fine-tune fluorophores for live-cell and in vivo imaging. Nat Methods. 2017;14:987–994. doi: 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gong Y, Wagner MJ, Zhong Li J, Schnitzer MJ. Imaging neural spiking in brain tissue using FRET-opsin protein voltage sensors. Nat Commun. 2014;5:3674. doi: 10.1038/ncomms4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neuroscience. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 59.Prevedel R, Yoon YG, Hoffmann M, Pak N, Wetzstein G, Kato S, Schrodel T, Raskar R, Zimmer M, Boyden ES, Vaziri A. Simultaneous whole-animal 3D imaging of neuronal activity using light-field microscopy. Nat Methods. 2014;11:727–730. doi: 10.1038/nmeth.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60*.Cong L, Wang Z, Chai Y, Hang W, Shang C, Yang W, Bai L, Du J, Wang K, Wen Q. Rapid whole brain imaging of neural activity in freely behaving larval zebrafish (Danio rerio) Elife. 2017;6 doi: 10.7554/eLife.28158. Paper demonstrating high-speed imaging of calcium activity in freely moving zebrafish larvae using light-field microscopy. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.