

Abstract
Upon different types of stress, the gene encoding the mitosis-promoting phosphatase Cdc25C is transcriptionally repressed by p53, contributing to p53’s enforcement of a G2 cell cycle arrest. In addition, Cdc25C protein stability is also decreased following DNA damage. Mdm2, another p53 target gene, encodes a ubiquitin ligase that negatively regulates p53 levels by ubiquitination. Ablation of Mdm2 by siRNA led to an increase in p53 protein and repression of Cdc25C gene expression. However, Cdc25C protein levels were actually increased following Mdm2 depletion. Mdm2 is shown to negatively regulate Cdc25C protein levels by reducing its half-life independently of the presence of p53. Further, Mdm2 physically interacts with Cdc25C and promotes its degradation through the proteasome in a ubiquitin-independent manner. Either Mdm2 overexpression or Cdc25C downregulation delays cell cycle progression through the G2/M phase. Thus, the repression of the Cdc25C promoter by p53, together with p53-dependent induction of Mdm2 and subsequent degradation of Cdc25C, could provide a dual mechanism by which p53 can enforce and maintain a G2/M cell cycle arrest.
INTRODUCTION
The tumor suppressor p53 is a sequence-specific transcriptional regulator that is expressed at low levels in normal cells. Following DNA damage, hypoxia, oncogene activation and a variety of other stimuli, p53 becomes upregulated resulting in growth arrest, apoptosis and DNA repair, among other responses. These are mediated by multiple factors including p21, 14-3-3σ, Cdc25C, Bax, PUMA and Noxa, whose expression is induced or repressed by p53.1 Consistent with a role in monitoring genomic integrity, p53 is mutated or inactivated in most human cancers, and p53-deficient mice develop early spontaneous tumors.2,3 Owing to its growth-suppressive function, p53 protein levels and transcriptional activity are tightly regulated. Particularly important in this response are two proteins, Mdm2 and Mdm4 (also known as MdmX in humans). Mdm2, another target gene induced by p53, encodes an E3 ligase that ubiquitinates p53 and promotes its proteasome-mediated degradation, creating a negative feedback loop.4 In addition to regulating p53 stability, Mdm2 also inhibits its transcriptional activity by binding to and occluding the p53 transactivation domain, a property shared with Mdm4.5,6 Both Mdm2- and Mdm4-deficient mice die during embryogenesis presumably owing to excessive p53 activity as the lethality can be rescued by deletion of p53.7–9 Mdm2 is found overexpressed in some tumors, also consistent with its role as a negative regulator of p53.10 However, identification of genetic alteration of both p53 and Mdm2 in the same tumor samples indicated that Mdm2 might have p53-independent functions, many of which have been described.11 Finally, several reports have hinted that Mdm2 can in fact induce the opposite effect and have a role in promoting growth arrest.12–17
Cdc25C is a dual specificity phosphatase that promotes entry into mitosis by removing the inhibitory phosphates on cyclin-dependent kinases. Inhibition of Cdc25C activity is critical for the G2 checkpoint and is achieved by several mechanisms. Cdc25C has been shown to become phosphorylated following DNA damage, which results in inhibition of its activity and relocation to the cytoplasm.18 Cdc25C expression is also downregulated in response to DNA damage.19–22 Previous work in our laboratory identified the cdc25C gene as a target for direct transcriptional repression by p53. Cdc25C downregulation was shown to be required for maintenance of the G2 arrest following DNA damage, and overexpression of Cdc25C abrogated this checkpoint following ionizing radiation.23 In this report, evidence for an additional mechanism of inhibition of Cdc25C is presented. We show that Mdm2 interacts with Cdc25C and promotes its degradation through the proteasome in a ubiquitin-independent manner. Moreover, either Mdm2 overexpression or Cdc25C downregulation delays cell cycle progression through the G2/M phase.
RESULTS
Cdc25C protein is downregulated in a p53-dependent manner in response to a variety of stimuli
Previous studies have shown that the cdc25C gene is repressed by multiple transcriptional mechanisms following activation of p53 by DNA damaging agents such as the topoisomerase II poison doxorubicin.19–22,24,25 Cdc25C protein levels were also downregulated following ribosomal stress caused by treatment of HCT116 cells with low doses of actinomycin D (Figure 1a). As is the case with doxorubicin, this decrease in Cdc25C protein was not observed in the p53-null HCT116 isogenic derivative, confirming the p53-dependence of this regulation (Figure 1a). In addition, Cdc25C protein was downregulated by treatment with actinomycin D in U2OS stable clones expressing a control shRNA, but not when p53 expression was ablated by shRNA (Figure 1b). Nutlin-3 is a small molecular weight compound that disrupts the p53-Mdm2 interaction leading to p53 stabilization.26 Treatment with nutlin-3 also decreased Cdc25C protein levels in a p53-dependent manner (Figure 1b). Both actinomycin D and nutlin-3 triggered G1 and G2 arrest of the cell cycle in wild-type p53-expressing U2OS cells, that was not seen in the p53-ablated clone (Figure 1c). These results indicate that Cdc25C repression is p53-dependent. The downregulation of Cdc25C mRNA and protein had been shown to occur in a p21-independent manner in some cell lines but this is not the case for HCT116 cells.23 Treatment of p21-null HCT116 cells with doxorubicin did not affect cdc25C mRNA levels (Figure 1d). However, in this cell line, Cdc25C protein was still significantly downregulated (Figure 1e). This observation led to the hypothesis that there must be an additional mechanism of regulation for Cdc25C protein levels following DNA damage. Indeed, examination of Cdc25C protein stability by cycloheximide chase revealed that Cdc25C becomes destabilized following treatment with doxorubicin (Figure 1f). This result shows that Cdc25C levels are regulated both at the mRNA and protein levels in response to DNA damage and p53 activation.
Figure 1.
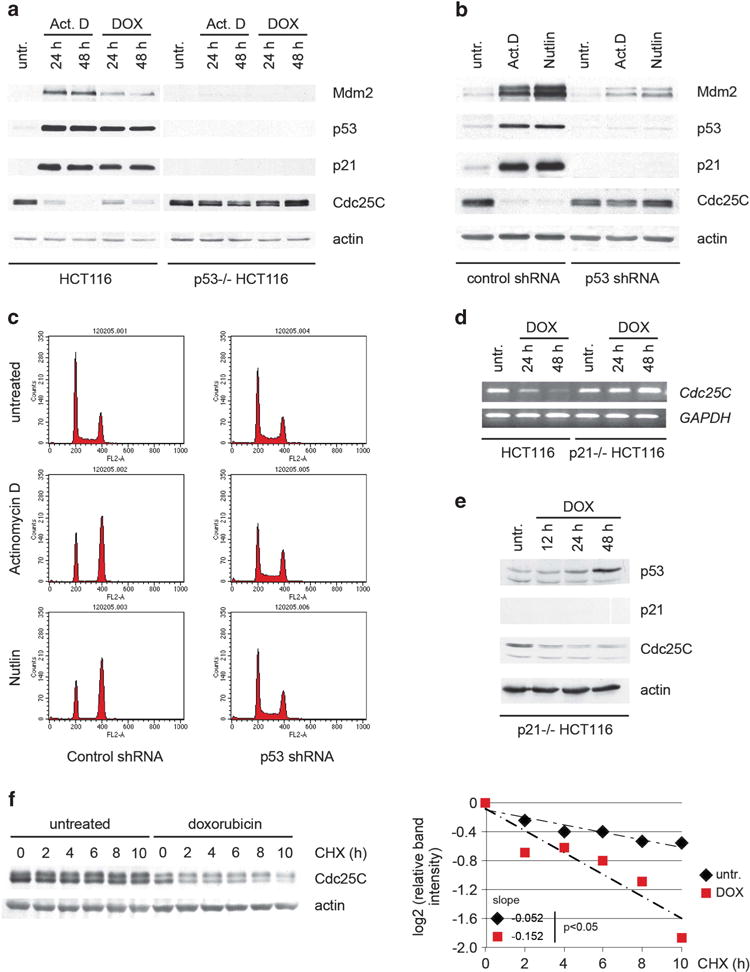
p53 regulates Cdc25C protein stability upon activation. (a) p53 wild-type and p53-null HCT116 cells were treated with 5 nM actinomycin D or 100 ng/ml doxorubicin. Twenty-four and 48 h after treatment, cell lysates were immunoblotted. (b) Stable U2OS clones expressing control or p53 shRNA were treated with 5 nM actinomycin D or 2.5 μM nutlin-3. Forty-eight hours after treatment, cell lysates were immunoblotted. (c) Cells treated as in B were stained and cell cycle profile was analyzed by flow cytometry. (d) Wild-type and p21-null HCT116 cells were treated with 500 ng/ml doxorubicin and subjected to RT–PCR analysis at the indicated time points. (e) p21-null HCT116 cells were treated as in (d). Twelve, 24 and 48 hours after treatment, cell lysates were immunoblotted. (f) HCT116 cells were treated with 100 ng/ml doxorubicin when indicated. Twenty-four hours after treatment, cells were treated with 100 μg/ml cycloheximide and harvested at the indicated time points. Normalized Cdc25C levels quantified by densitometry are shown. Statistical analysis of the slopes using covariance analysis was performed using StatsToDo software (http://www.statstodo.com).
Cdc25C protein is not downregulated upon induction of p53 after Mdm2 knockdown
Downregulation of Mdm2 by transient transfection of U2OS with siRNA oligonucleotides increased p53 to levels similar to those obtained following treatment with doxorubicin (Figure 2a). Gene expression analysis by quantitative RT–PCR showed that p53 upregulated by Mdm2 siRNA was transcriptionally active, and p53 could both induce and repress the expression of several target genes.27 In particular, p53 downregulated cdc25C mRNA levels, and the extent of the repression was comparable following downregulation of Mdm2 or treatment with doxorubicin (Figure 2b). Doxorubicin also caused downregulation of Cdc25C protein and, consistent with that, Cdc2 was found in its hyperphosphorylated, inactive state, as shown both by its migration pattern in gel electrophoresis and its reduced histone H1 kinase activity (Figures 2a and c). Cyclin B1 levels were also upregulated by doxorubicin, possibly due to the higher proportion of cells in the G2 phase during which Cyclin B1 levels peak28 (Figures 2a and d). Of note, Cyclin B1 is reportedly downregulated at the transcriptional level following persistent DNA damage.22,29 This is also observed under the conditions used here at later time points (data not shown). The downregulation of Cdc25C protein levels by doxorubicin was also observed in two other tumor cell lines, HT1080 and G361, as well as in the normal, diploid WI-38 fibroblasts (Figure 2e and data not shown). However, p53 upregulated by ablation of Mdm2 failed to induce downregulation of Cdc25C protein in all the cell lines tested (Figures 2a and e). Moreover, in U2OS cells, Cdc25C protein levels were, in fact, upregulated following Mdm2 depletion by siRNA (Figures 2a and f). The Cdc2 kinase remained active, showing a predominant band corresponding to its unphosphorylated form (lower band, Figure 2a) and only a slight decrease in its associated kinase activity (Figure 2c). Consistent with the findings involving Cdc25C and Cdc2, treatment with doxorubicin caused a predominant G2 arrest (Figure 2d). In contrast, no significant differences were observed in the cell cycle profile of several cell lines transfected with Mdm2 siRNA, despite their comparable p53 levels (Figure 2d and data not shown). We have previously shown that Mdm2 is required for efficient Cdk2 inhibition by p21, which explains the absence of cell cycle arrest in cells with downregulated Mdm2.27 Taken together, these results revealed a significant disconnect between cdc25C mRNA and Cdc25C protein levels, supporting the previously published observation using p21-null HCT116 cells (Figure 1d and e).23 More importantly, they suggest that Mdm2 has a direct role in the regulation of Cdc25C.
Figure 2.
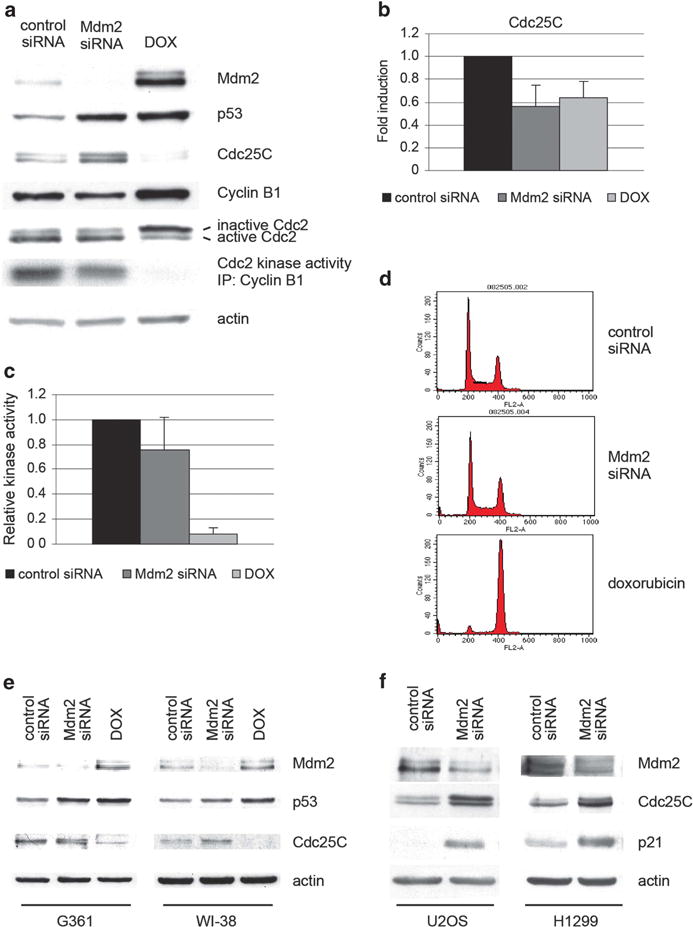
The cdc25C gene is repressed but Cdc25C protein is not downregulated, Cdc2 kinase activity is not inhibited, and cell fails to arrest when p53 is upregulated after Mdm2 siRNA. (a–c) U2OS cells were transfected with control or Mdm2 siRNA or treated with 20 ng/ml doxorubicin. Forty-eight hours after treatment, protein levels in the cell extracts were assayed by immunoblotting (a), total RNA was extracted and Cdc25C mRNA levels were measured by quantitative RT–PCR (b), Cyclin B1 was immunoprecipitated and the associated Cdc2 was assayed for histone H1 kinase activity (c). The indicated values are the average of three independent experiments. Error bars represent s.d. (d) Cell cycle profile of U2OS cells was analyzed by flow cytometry. (e) G361 and WI-38 cells were transfected with control or Mdm2 siRNA or treated with 100 ng/ml doxorubicin. Forty-eight hours after treatment, protein levels in the cells extracts were assayed by immunoblotting. (f) U2OS and H1299 cells were transfected with control or Mdm2 siRNA. Forty-eight hours after treatment, protein levels in the cells extracts were assayed by immunoblotting.
To further explore the effects of Mdm2 on Cdc25C protein levels, wild-type p53-expressing U2OS and p53-null H1299 cells were transfected with a pool of Mdm2 siRNA oligonucleotides. p21 was found upregulated in both cell lines, consistent with previously reported studies showing that Mdm2 regulates p21 stability (Figure 2f, Supplementary Figure S1a). Mdm2 has been shown to negatively regulate p21 protein levels in a p53-independent manner, which can account for this increase in the p53-null H1299 line.30–32 In U2OS cells, upregulation of endogenous p21 is likely to be the result of a combined effect of the increase in p53 levels and transcriptional activity, as well as the increased stability of the p21 protein. Cdc25C levels were found to be upregulated in both cell lines, suggesting that Mdm2 negatively regulates Cdc25C protein levels, and that this effect is not dependent on p53 (Figure 2f). This observation was confirmed using single independent Mdm2 siRNA oligonucleotides (Supplementary Figure S1b). Taken together, these findings point to Mdm2 as a key player in the regulation of Cdc25C protein levels.
Mdm2 regulates Cdc25C levels in a p53-independent manner
The studies reported this far showed that ablation of endogenous Mdm2 expression led to an increase in Cdc25C protein. To determine whether increased expression of Mdm2 was sufficient to downregulate Cdc25C, H1299 cells were co-transfected with flag-tagged Cdc25C and increasing amounts of flag-tagged Mdm2 expression constructs. Overexpression of Mdm2 led to a dose-dependent decrease in the level of the transfected Cdc25C protein (Figure 3a), as well as p53 and p21 protein levels (Supplementary Figures S1c and d). GFP expression levels from a co-transfected plasmid were comparable between samples ruling out differences owing to transfection efficiency (Supplementary Figures S1e and f). Infection of U2OS cells with a recombinant adenovirus expressing Mdm2 also caused a decrease in the levels of endogenous Cdc25C, p53 and p21 proteins, compared with infection with a control adenovirus (Supplementary Figure S1g, left). A similar downregulation of endogenous Cdc25C was observed in infected H1299 cells (Supplementary Figure S1g, right).
Figure 3.
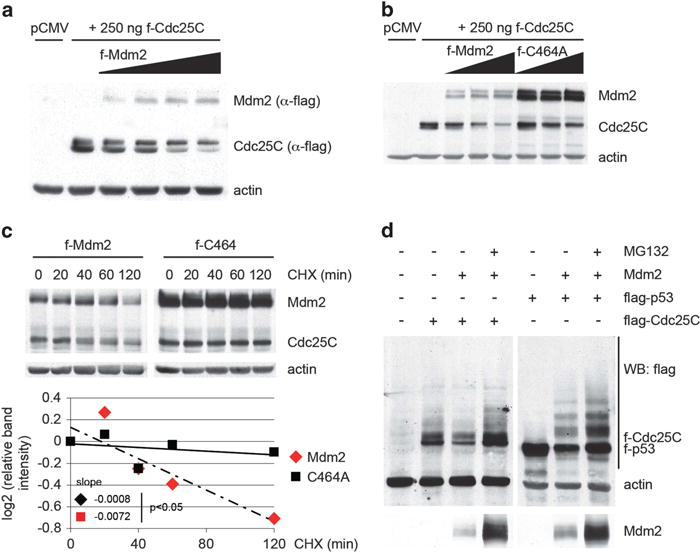
Mdm2 regulates Cdc25C protein stability in a p53-Independent manner. (a) H1299 cells were co-transfected with 250 ng of pCMV-flag-Cdc25C and increasing amounts of pCMV-flag-Mdm2 (0.25, 0.5, 1 and 2 μg) expression plasmids. Twenty-four hours after transfection, cell lysates were immunoblotted. (b) H1299 cells were transfected as in (a) using a C464A-Mdm2 mutant when indicated. (c) H1299 cells were transfected with 250 ng of pCMV-flag-Cdc25C and 2 μg of pCMV-flag-Mdm2 expression plasmids. Twenty-four hours after transfection, cells were treated with 100 μg/ml cycloheximide and harvested at the indicated time points. Normalized Cdc25C levels quantified by densitometry are shown. Statistical analysis of the slopes using covariance analysis was performed using StatsToDo software (http://www.statstodo.com). (d) H1299 cells were transfected with 500 ng of pCMV-flag-Cdc25C or 250 ng of pCMV-flag-p53, 2 μg of pCMV-Mdm2 and treated with 40 μM MG132 for 5 hours before harvesting as indicated. Twenty-four hours after transfection, cell lysates were immunoblotted.
The tumor suppressor p14ARF has been shown to bind to and inhibit Mdm2 through various mechanisms,33,34 prompting an examination of its effect on Cdc25C levels. Transfection of ARF in H1299 cells caused an increase in Cdc25C protein levels (Supplementary Figure S2a), confirming that inhibition of Mdm2 promotes Cdc25c stabilization. In contrast, Nutlin-3, which very specifically inhibits the p53-Mdm2 interaction, while preventing p53 degradation by transfected Mdm2, had no impact on its effect on Cdc25C protein levels (Supplementary Figure S2b).
The C464A mutant of Mdm2 has previously been shown to lack ubiquitin ligase activity.35,36 It was therefore used in a co-transfection study in H1299 cells to assess the requirement for ligase activity on the effect of Mdm2 on Cdc25C. The mutated Mdm2 is unable to promote self-ubiquitination and degradation and thus is expressed at higher levels than wild-type Mdm2 (Figures 3b and c).35 This mutant was also impaired for downregulation of Cdc25C protein levels (Figure 3b). Incubation with cycloheximide showed that the decrease in Cdc25C protein was due to a reduction in its half-life caused by wild-type Mdm2, but not by the C464A-Mdm2 mutant (Figure 3c). Moreover, treatment of the cells with the proteasome inhibitor MG132 prevented the decrease of Cdc25C protein by Mdm2, suggesting that this effect is due to degradation of Cdc25C through the proteasome (Figure 3d). Cdc25C migrates on polyacrylamide gels as a doublet, the upper band of which has been reported to correspond to the form phosphorylated on serine 216.37 Upon checkpoint activation, modification of this residue has been shown to promote binding to 14-3-3 proteins, which in turn relocates Cdc25C to the cytoplasm.37,38 Interestingly, Mdm2 preferentially decreases the lower band of the Cdc25C doublet, whereas its effect on the upper form is comparatively reduced (Figures 3a and b and Supplementary Figures S3b and c). Co-transfection studies using Cdc25C expression constructs bearing deletions and point mutations failed to identify a discrete region or amino acid of the protein as being required for its regulation by Mdm2 (Supplementary Figure S3a). The levels of both ΔN and ΔC truncated proteins, but not the unrelated co-transfected GFP, were decreased by Mdm2. In addition, mutation of serine 216 to alanine or aspartic acid did not prevent the reduction of Cdc25C by Mdm2, suggesting that this process is not affected by the phosphorylation of Cdc25C at serine 216 (Supplementary Figure S3b). Of note, both S216 mutants migrate as a single band with a mobility that corresponds to the lower band of the Cdc25C doublet, supporting the previously reported identity of this band. Finally, a C377S mutant of Cdc25C has been shown to be defective in its phosphatase activity.39 Similarly to wild-type Cdc25C, the levels of the C377S mutant protein were reduced by Mdm2, indicating that the phosphatase activity of Cdc25C does not have a role in Cdc25C degradation (Supplementary Figure S3b). Previous reports have provided evidence for a ubiquitin- and proteasome-dependent degradation of Cdc25C.40,41 In particular, an intact KEN box present in Cdc25C appears to be required for its efficient ubiquitination.40 To gain insight into the mechanism leading to the downregulation of Cdc25C protein levels by Mdm2, a Cdc25C construct with a mutated KEN box (KEN to AAA) was co-transfected with Mdm2 in H1299 cells. Mdm2 was able to decrease the levels of the mutant Cdc25C, suggesting that the KEN box was not involved in this process (Supplementary Figure S3c). Altogether, these results indicate that Mdm2 negatively regulates Cdc25C protein levels by promoting its degradation through the proteasome. Mdm2 appears to target preferentially the serine 216 non-phosphorylated form of Cdc25C and this process is substantially impaired by mutation of the RING domain of Mdm2.
Mdm2 interacts with Cdc25C
In H1299 cells co-transfected with Cdc25C and C464A-Mdm2 expression plasmids, Cdc25C co-immunoprecipitates with Mdm2 and vice versa, indicating that the two proteins physically interact (Figure 4a). The C464A-Mdm2 mutant was used to detect the Cdc25C-Mdm2 complex because of its higher expression level and its impaired downregulation of Cdc25C. Moreover, using cell lines with amplification of the Mdm2 gene and/or treatment with MG132 to stabilize both proteins, we were able to detect a complex between endogenous Cdc25C and Mdm2, further supporting this interaction (Figure 4b, Supplementary Figure S4a).
Figure 4.
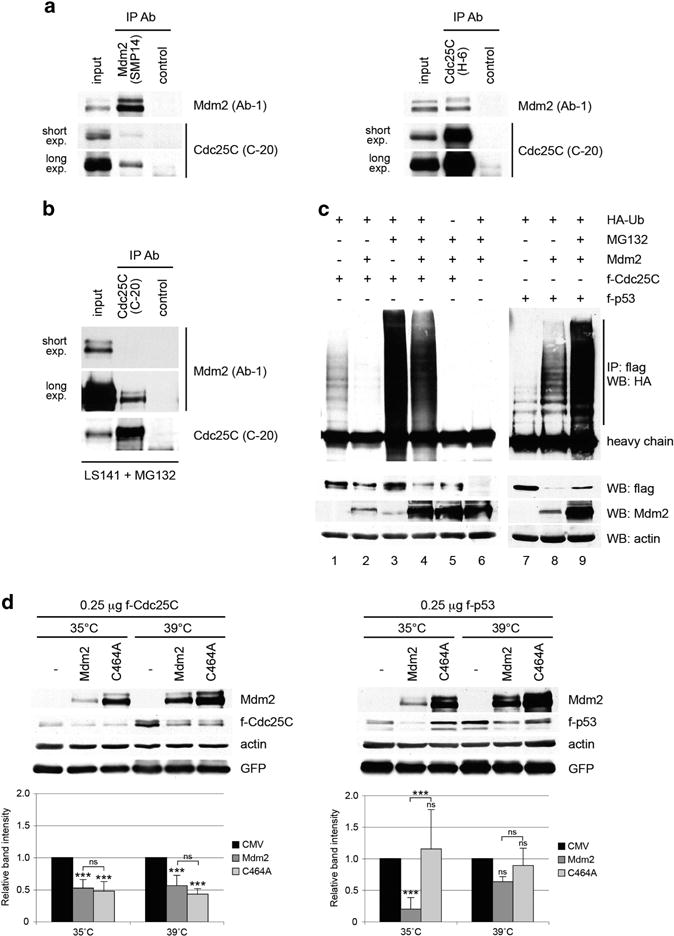
Mdm2 interacts with Cdc25C protein and promotes its ubiquitin-independent degradation. (a) H1299 cells were co-transfected with 1.5 μg of pCMV-flag-Cdc25C and 1.5 μg of pCMV-flag-C464A-Mdm2 mutant. Twenty-four hours after transfection, cell extracts were immunoprecipitated with anti-Mdm2, anti-Cdc25C or a non-related antibody. Mdm2 and Cdc25C in the immunoprecipitates were detected by immunoblotting. (b) LS141 cells were treated with 10 μM MG132 for 4 h and cell extracts were immunoprecipitated with anti-Cdc25C or a non-related antibody. Mdm2 and Cdc25C in the immunoprecipitates were detected by immunoblotting. (c) H1299 cells were co-transfected with the indicated plasmids and treated as described in Materials and Methods. Twenty-four hours after transfection, cell extracts were immunoprecipitated with anti-flag and immunoblotted with anti-HA antibodies. (d) Duplicated dishes of murine ts20 cells were transfected with 250 ng of pCMV-flag-Cdc25C or pCMV-flag-p53, 500 ng of pCMV-Mdm2 and 500 ng of pCMV-GFP expression plasmids. Three hours after transfection, one set of dishes was maintained at 35°C and the other placed at 39 °C for 24 h. Cell extracts were immunoblotted with human specific Cdc25C, p53 and Mdm2 antibodies, as well as actin and GFP antibodies. The indicated values are the average of three to five independent experiments. Error bars represent s.d. Statistical analysis corresponds to two-way ANOVA. Notations above error bars correspond to differences with respect to the CMV control. Differences between Mdm2 and C464A transfected samples are marked with brackets (***P<0.001, ns P>0.05).
Mdm2 regulates Cdc25C in a ubiquitin-independent manner
The observation that the Mdm2 RING domain was needed for efficient Cdc25C degradation suggested that Mdm2 might act as an E3 ligase for Cdc25C and thereby promote its ubiquitination. To address this possibility, H1299 cells were co-transfected with flag-Cdc25C (or flag-p53 as a control), as well as untagged Mdm2 and HA-ubiquitin expression plasmids, in the presence of the proteasome inhibitor MG132. Cells extracts were used for immunoprecipitation using anti-flag antibody followed by immunoblotting with anti-HA antibody, to detect specific ubiquitination of Cdc25C (Figure 4c). Consistent with published studies,40,42 a basal level of ubiquitination of Cdc25C was observed in cells transfected with flag-Cdc25C and HA-Ubiquitin (Figure 4c). Coexpression of Mdm2 and p53 led to a substantial increase in p53 polyubiquitination (Figure 4c, lanes 7–8). In contrast, Mdm2 transfection caused a slight reduction in the ubiquitination of Cdc25C both in the absence and presence of MG132 (Figure 4c, lanes 1–2 and 3–4, Supplementary Figure S4b). This might be due to competition between Mdm2 and endogenous Cdc25C E3 ligase (s) for binding to Cdc25C and for the limited pool of HA-ubiquitin. Thus, Mdm2 would promote the degradation of Cdc25C through a pathway that competes with its basal ubiquitin-dependent processing. Similar results were obtained using a ‘denaturing’ protocol to perform the immunoprecipitations, indicating that the detected HA signal corresponds to ubiquitinated Cdc25C and not to associated unrelated proteins (Supplementary Figure S4c). Finally, results using Mdm2 siRNA transfected U2OS cells, as well as p53− / − and p53/Mdm2− / − mouse embryonic fibroblasts, indicated that Mdm2 does not seem to have a role in Cdc25C basal ubiquitination (Supplementary Figures S4d and e).
The results suggested that Mdm2 promotes the degradation of Cdc25C through the proteasome in an ubiquitin-independent manner. Importantly, there is precedent for this as a similar mechanism has been reported for p21 and pRb.30,43 To confirm this, Cdc25C was co-transfected with Mdm2 in the murine ts20 cells that express a temperature sensitive E1 ubiquitin-activating enzyme.44 These cells have extensively been used to study the ubiquitin-dependence of degradation pathways of several proteins.45–48 At the permissive temperature, overexpression of wild-type but not C464A mutant Mdm2 caused a reduction in the protein levels of co-transfected p53. This downregulation was greatly impaired when ts20 cells were grown at the restrictive temperature (Figure 4d). Of note, the C464A mutant appeared to have a dominant-negative effect on endogenous Mdm2, as p53 levels appear slightly increased at 35°C (Figure 4d, right panel). In contrast, Cdc25C levels were decreased by both Mdm2 constructs and to a similar extent at both temperatures (Figure 4d, left panel). Given the enhanced expression of the C464A mutant, the downregulation of Cdc25C by this protein was significantly less efficient compared with wild-type Mdm2, consistent with previous observations (Figures 3b and c). Taken together, these results suggest that Mdm2 interacts with Cdc25C and promotes its degradation through the proteasome in a ubiquitin-independent manner.
Mdm2 overexpression and Cdc25C downregulation delay cell cycle progression through the G2/M phase
U2OS cells infected with control and Mdm2 expressing adenoviruses, synchronized at the G1/S border by a single thymidine block and, following release, the appearance of cells with 4 N or 2 N DNA content was determined. A greater fraction of cells with a 4 N DNA content followed by a slower (3–4 h) transition from a 4 N to a 2 N DNA content was observed in cells infected with Ad-Mdm2. A consistent delay was observed as cells re-entered the following G1 phase (Figure 5a). Similar results were obtained with U2OS cells transfected with control or Cdc25C siRNA (Figure 6). Cells moved through S-phase at similar rates, as indicated by a nearly simultaneous completion of a 4 N DNA. However, a comparable delay in the decrease from a tetraploid to a diploid state was observed when Cdc25C was downregulated (Figure 6). Taken together, the data indicates that either Mdm2 over-expression or Cdc25C downregulation delays cell cycle progression through the G2/M phase.
Figure 5.
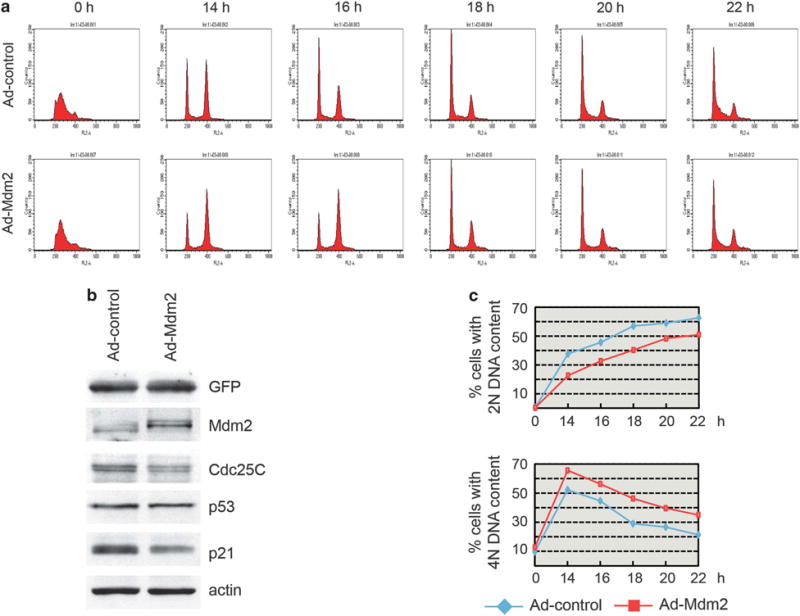
Mdm2 overexpression delays cell cycle progression through G2/M. (a–c) U2OS cells were transduced with control or Mdm2 expressing adenoviruses. Progression through the cell cycle of synchronized cells collected at 14–22 h after release from thymidine block was analyzed by flow cytometry. Representative cell cycle profiles (a), protein levels in the cells extracts (b) and the average of three independent experiments (c) are shown.
Figure 6.
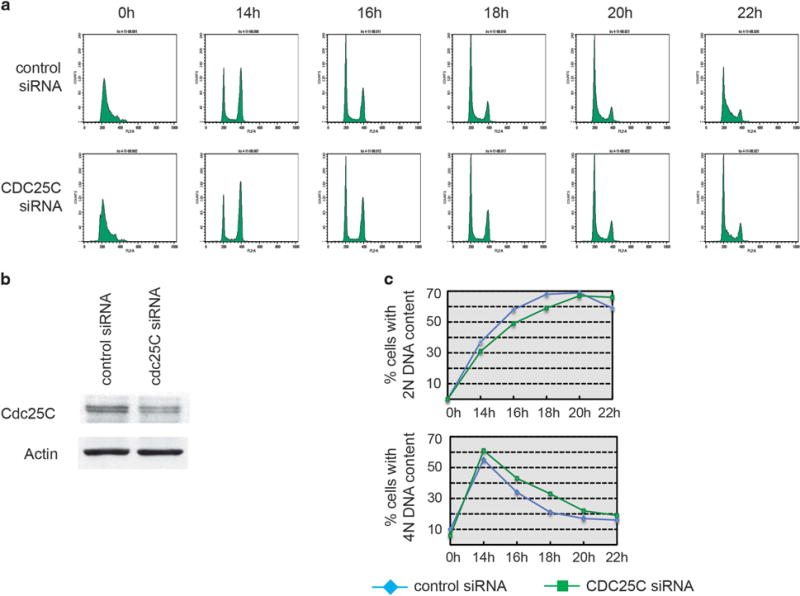
Cdc25C downregulation delays cell cycle progression through G2/M. (a–c) U2OS cells were transfected with control or Cdc25C siRNA. Progression through the cell cycle of synchronized cells collected at 14–22 h after release from thymidine block was analyzed by flow cytometry. Representative cell cycle profiles (a), protein levels in the cells extracts (b) and the average of three independent experiments (c) are shown.
Cdc25C is downregulated through redundant mechanisms during the DNA damage response
The results presented so far showed that Mdm2 can promote the degradation of transfected or endogenous Cdc25C in basal conditions (in transfected cells, in the absence of DNA damage or other sources of stress). Moreover, Cdc25C protein stability is decreased following activation of p53 by doxorubicin. It was therefore sensible to reason that Mdm2 induced during the p53 response to stress could contribute to the downregulation of Cdc25C and the cell cycle arrest. Although it is well know that degradation of p53 by Mdm2 is prevented after DNA damage through post-translational modification of both proteins, it has also been shown that MdmX degradation by Mdm2 is induced following DNA damage.49 To test whether a similar scenario could apply to Cdc25C, U2OS cells were simultaneously transfected with control or Mdm2 siRNA and treated with doxorubicin or actinomycin D. In unstressed cells, two independent duplexes of oligonucleotides targeting Mdm2 caused an upregulation of Cdc25C, as previously shown. However, the reduction in Cdc25C protein levels by doxorubicin or actinomycin D was as (or more) efficient in cells with downregulated Mdm2, compared with the control transfected cells (Supplementary Figures S5a and b). Importantly, ablation of Mdm2 resulted in significantly higher levels of p21 following stress. p21 is a strong component in the downregulation of Cdc25C during the DNA damage response23 and the increased p21, as well as other possible redundant mechanisms, likely compensated for the decreased Mdm2 levels, leading to the reduced Cdc25C levels observed. Indeed, p21 ablation in U2OS cells markedly impaired this effect (data not shown).
DISCUSSION
Cdc25C is a member of the family of phosphatases that remove inhibitory phosphates from cyclin-dependent kinases at Thr14 and Tyr15, a rate-limiting step in the activation of Cdc2 kinase and progression into mitosis. Being an important player at the G2/M transition, Cdc25C is a target of checkpoint pathways and its activity is regulated in different ways, including transcriptional repression, post-translational modification, subcellular localization and protein destabilization.50
The cdc25C gene is a target of the p53 tumor suppressor and has been shown to be transcriptionally repressed following treatment with DNA damaging agents in a p53-dependent manner.19–24 Moreover, its repression by p53 has been shown to be required for a proper G2 arrest.23 Here, Cdc25C protein levels were downregulated in a p53-dependent manner in multiple cell lines in response to different stimuli. This is distinct from Cdc25A, which is degraded after DNA damage in a p53-independent manner.51 Treatment of p21-null HCT116 cells with doxorubicin caused a decrease in Cdc25C protein levels without changes in its mRNA levels23 (Figure 1d and e). This revealed a distinction between Cdc25C mRNA and protein levels, and suggested the existence of an additional p53-dependent mechanism for the regulation of Cdc25C protein. Examination of Cdc25C half-life in DNA damaged cells showed that the p53-dependent downregulation of Cdc25C by doxorubicin, in addition to the previously described transcriptional repression, involves another mechanism that affects Cdc25C protein stability (Figure 1 f). These results are consistent with a previously published report where Cdc25C was shown to be destabilized in U2OS cells following irradiation.52
After p53 stabilization by siRNA-mediated downregulation of Mdm2 in U2OS cells, cdc25C mRNA was repressed but Cdc25C protein was not downregulated (Figure 2a and b). One explanation is that there is increased stability of Cdc25C protein under these conditions that compensated for the reduction in mRNA levels. Cdc25C was found also upregulated in the p53-null cells H1299, showing that Mdm2 regulates Cdc25C protein levels independently of p53 (Figure 2f). Of note, consistent with the fact that in untreated cells p53 levels are maintained low and the protein is transcriptionally inactive, and that basal Mdm2 levels in wild-type and p53-null cells are comparable, basal Cdc25C levels were also similar in the tested cell lines, regardless of their p53 status.
Co-transfection studies in H1299 cells showed that Mdm2 was capable of decreasing Cdc25C stability and reducing Cdc25C protein levels (Figures 3a and c). In addition, Cdc25C can be found in cells forming a complex to Mdm2 (Figures 4a and b and Supplementary Figure S4a). The upper band of the Cdc25C doublet has been reported to correspond to the serine 216 phosphorylated form of Cdc25C that can be retained in the cytoplasm through binding to 14-3-3 proteins.37,38 Chk1 has been proposed to be one of the kinases that modify this residue following DNA damage and G2 checkpoint activation.53 Interestingly, Mdm2 preferentially reduces the lower band of the Cdc25C doublet (Figures 3a and b and Supplementary Figures S3b and c). This observation is consistent with the nuclear localization of Mdm2 and suggests that Mdm2 could therefore target the 216 unphosphorylated Cdc25C that has failed to be relocated to the cytoplasm.
Using ts20 cells, a functional E1 ubiquitin-activating enzyme was shown to be dispensable for the effect of Mdm2 on Cdc25C, indicating that this regulation occurs in a ubiquitin-independent manner (Figure 4d). The C464A ligase-defective mutant of Mdm2 was able to interact with Cdc25C and reduce its ubiquitination. Compared with wild-type Mdm2, the C464A mutant was less efficient at decreasing the half-life and promoting the degradation of Cdc25C, specially considering its much higher expression levels (Figures 3b, c and 4d, and Supplementary Figure S1e). This could be due to the C464A mutant itself retaining partial ability to degrade Cdc25C, or to an indirect stabilizing effect on endogenous Mdm2. A similar scenario where integrity of the Mdm2 RING domain was required for efficient ubiquitin-independent degradation of a substrate was described in the case of pRb. In that report, this observation was attributed to the fact that C464A-Mdm2 was impaired in, but not completely devoid of, its ability to interact with the C8 subunit of the proteasome.43 Mdm2 has also been shown to induce a conformational change in p21 that increases its interaction with the C8 subunit.32
Several studies have reported regulation of Cdc25C protein stability in response to a variety of agents that induce G2 arrest such as arsenite,40 isothiocyanates like sulforaphane,31 PEITC,41 vanadate54 and infection with adeno-associated virus.55 p14ARF was shown to induce a p53-independent cell cycle arrest at the G2 phase by a mechanism that involved ERK1/2 activation and Cdc25C protein degradation.56 Uncapped telomeres activated the ATM/ATR-Chk1/Chk2 pathway and promoted Cdc25C degradation, preventing mitotic entry.57 BRCA1 overexpression in p53 wild-type MCF-7 cells led to a G2 arrest that was accompanied by activation of ERK1/2 and a decrease in Cdc25C protein. Here, the BRCA1/BARD1 complex was identified as an E3 ligase for Cdc25C.58,59 In all of these studies, the decrease in Cdc25C protein levels was mediated by the proteasome, and some of these reports provided evidence of ubiquitination of Cdc25C.40,56,58
The results presented here describe a new mechanism of degradation for Cdc25C through a ubiquitin-independent pathway and identify Mdm2 as a key player in this process. Of note, the observations made in U2OS and HCT116 cells on Cdc25C stability are p14ARF-independent, since these cells lines do not express p14ARF.60,61 However, as ARF is known to inhibit Mdm2, its overexpression caused upregulation of Cdc25C (Supplementary Figure 2a).
Mdm2 is overexpressed in several types of tumors, and its overexpression or amplification has been hypothesized to inactivate the p53 pathway in these cancers. Mdm2 also has p53-independent activities and promotes the degradation of other proteins involved in cell proliferation including pRb and E2F-1.30,43,62–67 In contrast, other studies have suggested a growth-suppressive role for Mdm2 or some of its splice variants.11 Overexpression of Mdm2 has been shown to induce G1 arrest in some cell lines.12,13,17 Stable overexpression of Mdm2 in H1299 cells containing an inducible p53 improved the G2 arrest of these cells following induction of p53.16 Mdm2 splice variants have been shown to act in a dominant-negative manner resulting in activation of p53.68,69 Contradictory results have been reported about the effect of Mdm2 on the p53 family member p63.70–72 On the other hand, Mdm2 has been reported to stabilize another family member p73 and to enhance its activity.73
In the same way Mdm2 exhibits both growth promoting and suppressive activities, the biological significance of the negative regulation of Cdc25C by Mdm2 is potentially double. First, it is attractive to envision Mdm2 as an effector of p53, contributing, together with p21, to the downregulation of Cdc25C through a new mechanism during the DNA damage response. Many reports have provided evidence that Mdm2 cooperates with p53 and is not just one of its negative regulators. Mdm2 functions as a ubiquitin ligase of the IGF-R1, a target of repression by p53.74 Mdm2 also targets MdmX for proteasomal-dependent degradation following DNA damage.49,75 MdmX, like Mdm2, can inhibit p53 transcriptional activity by binding to its transactivation domain, and its degradation by Mdm2 was suggested to ensure p53 optimal activation.49 More recently, several studies have reported new activities of Mdm2 that might have a role in the p53 decision-making process, notably growth arrest vs apoptosis.27,76–78 Although not ruling out the possibility, the results reported here failed to demonstrate an involvement of Mdm2 in the downregulation of Cdc25C in the DNA damage response. Unfortunately, the multiplicity, different timing and likely redundancy of mechanisms, as well as the complex interplay between the factors involved, make it extremely difficult to assess the exact contribution of each one of them to this process. The dual p53- and p21-dependent transcriptional repression of Cdc25C, the regulatory effect of Mdm2 on p21, p53-independent regulatory mechanisms and possibly the Mdm2-mediated degradation of Cdc25C create an intricate scenario hard to dissect. Furthermore, the Cdh1-APC E3 ligase has been shown to become activated after X-irradiation and to have a crucial role in the maintenance of the G2 arrest.79 As APC has already been implicated in the degradation of Cdc25C through its KEN box, this could constitute yet another mechanism of regulation of Cdc25C following DNA damage.
Another potential implication of the regulation of Cdc25C by Mdm2 is related to Mdm2’s role as an oncogene. Work by Vecchione and colleagues identified the Cdc2/Cdc25C complex as a target of the tumor suppressor Fez1/Lzts1. Destabilization of Cdc25C in the absence of Lzts1 was associated to reduced Cdc2 activity, accelerated mitotic progression, improper chromosome segregation and increased tumor susceptibility in mice.42 The results presented in this study suggest that increased degradation of Cdc25C by aberrant expression of Mdm2 could similarly contribute to tumor development. Supporting this idea, targeted overexpression of Mdm2 to breast epithelial cells of transgenic mice inhibited normal development by arresting cells in S-phase, but also caused polyploidy in a fraction of cells.15 Interestingly, a mathematical model has been generated in an attempt to elucidate the mechanisms by which Mdm2 overexpression leads to uncoupling of DNA synthesis and mitosis, resulting in polyploidy. This led to the prediction that Mdm2 could cause polyploidy by interfering with Cdc25C activity.80 Consistent with this, megakaryocytes downregulate Cdc25C expression as they become polyploid.81
In conclusion, the results presented in this study show Cdc25C to be a target of degradation by Mdm2 through a ubiquitin-independent pathway. Due to its ability to promote Cdc25C degradation, p53-induced Mdm2 may cooperate with p53 to regulate cellular Cdc25C levels following DNA damage. Furthermore, Mdm2 could ensure Cdc25C downregulation even in cases where p53 repression of the gene may be impaired owing to cell-type specific conditions. On the other hand, uncontrolled degradation of Cdc25C resulting from Mdm2 amplification and overexpression could constitute one of the mechanisms by which it functions as an oncogene.
MATERIALS AND METHODS
Cells and drugs
U2OS, HCT116, G361, WI-38, H1299, LS141 and SW872 cell lines were purchased from ATCC. p53- and p21-null HCT116 cell lines and p53− / − and p53/Mdm2− / − mouse embryonic fibroblasts were generous gifts from B Vogelstein and G Lozano, respectively. ts20 cells were kindly provided by H Ozer. Cells were grown in Dulbecco’s modified Eagle’s medium, containing 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin. Control and p53 shRNA stable cell lines were generated by co-transfection of either TranSilent control or p53 shRNA vectors (Panomics, Santa Clara, CA, USA) and pBabe-Puro. Doxorubicin, actinomycin D, cycloheximide, MG132 and N-ethylmaleimide were purchased from Sigma (Billerica, MA, USA). Nutlin-3 (racemic mix) was purchased from Sigma.
Plasmids
pEF-HA-Ub encodes HA-tagged ubiquitin under control of the eEF2 promoter and was a gift from Z Pan. All other expression plasmids were driven by the CMV promoter and encoded the corresponding human proteins. The pCMV-flag-Mdm2 and pCMV-Mdm2 plasmids, expressing flag-tagged and untagged human Mdm2, respectively, were a gift from Z Ronai. The pCMV-flag-C46A-Mdm2 and pCMV-flag-Cdc25C derived mutant plasmids were generated by site-directed mutagenesis based on the protocol of the QuickChange Site-Directed Mutagenesis Kit (Stratagene-Agilent, Santa Clara, CA, USA) or previously described.82
siRNA and plasmid transfections
For siRNA transfections, Mdm2 and Cdc25C pools or individual RNA oligonucleotides were purchased from Dharmacon (Lafayette, CO, USA) and used according to the manufacturer’s instructions at a final concentration of 100 nM. U2OS and WI-38 cells were transfected using Oligofectamine, and G361 and H1299 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. For plasmid DNA, H1299 cells were transfected using Lipofectamine Reagent (Invitrogen).
Immunoblotting
Cells were washed and harvested in phosphate-buffered saline (PBS), then lysed in 50 mM Hepes, pH 7.5, 1% Triton X-100, 150 mM NaCl, 1 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin and 50 μg/ml aprotinin. Equal amounts of proteins were electrophoresed on SDS–PAGE, transferred to nitrocellulose membranes (BioRad, Hercules, CA, USA) and blotted with anti-p53 (DO-1), anti-p21 (C-19), anti-Cdc25C (H-6, C-20), anti-cdc2 (17), anti-cyclin B1 (GNS1) from Santa Cruz Biotechnology Inc (Dallas, TX, USA). Anti-Mdm2 (Ab-1) and anti-actin (Ab-1) were from Calbiochem (Billerica, MA, USA), and anti-flag (M2) was from Sigma (St. Louis, MO, USA). Secondary anti-mouse, and anti-rabbit antibodies were from ICN Biomedicals Inc (Irvine, CA, USA). The signal was detected using the enhanced chemiluminescence detection reagent from Amersham Life Sciences (Pittsburgh, PA, USA).
Protein half-life determination
Cells were treated with doxorubicin or transfected as indicated. Twenty-four hours later, cells were treated with 100 μg/ml cycloheximide and harvested at the indicated time points. Cells were lysed and protein levels assayed as described for immunoblotting. Bands were quantitated by densitometry using a GS-800 Calibrated Densitometer and Quantity One software (Biorad).
Cdc2 kinase assays
Method is as previously described27 using 200–500 μg of proteins that were immunoprecipitated with 5–10 μl of anti-cyclin B1 (GNS1) antibodies and 40–50 μl of 50% slurry of protein A-Sepharose (Amersham BioSciences, Little Chalfont, UK).
RNA extraction and RT–PCR
RNA was extracted using RNeasy Kit (Qiagen, Hilden, Germany). Immediately after extraction, 1–2 μg of RNA were used for cDNA synthesis with SuperScript II (Invitrogen) according to the manufacturer’s instruction. Semi-quantitative PCR for cdc25C and GAPDH was performed as described.23 Quantitative real-time PCR was done using the following PCR primers on an ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA)
cdc25C: 5′-GGCCAAGGAAAGCTCAGG-3′/5′-GCCCCTGGTTAGAATCTTCC-3′;
GAPDH: 5′-CAATGACCCCTTCATTGACC-3′/5′-GATCTCGCTCCTGGAAGATG-3′.
The reactions were performed in quadruplicates. The median of the cdc25C replicates was normalized using the median of the GAPDH replicates. Fold-induction was calculated as 2(GAPDHmedian−cdc25Cmedian)treated+(cdc25Cmedian−GAPDHmedian)control.
Flow cytometry
Cells were fixed in 70% ethanol, washed and then incubated in PBS in the presence of 20 μg/ml propidium iodide and 1 mg/ml RNase A. Cell cycle distribution was analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) using CellQuest software (BD Biosciences, Franklin Lakes, NJ, USA). When indicated, cells were synchronized at G1/S by incubation with 2.5 mM thymidine for 36 h. Cells were then washed twice with PBS and released by addition of growth medium.
Immunoprecipitations
In total, 100 mm dishes of H1299 cells were transfected with 1.5 μg of pCMV-flag-Cdc25C and 1.5 μg of pCMV-flag-C464A-Mdm2 expression vectors using Lipofectamine Reagent (Invitrogen) for 24 h. A total of 150 mm dishes of LS141 cells were treated with 10 μM MG132 for 4–5 h. Cells were harvested in PBS and lysed as described for immunoblotting, with addition of phosphatase inhibitors (15 mM PNPP, and 0.1 mM sodium orthovanadate, 0.1 M sodium fluoride). Protein extracts (1–3 mg) were incubated with 4 μl of anti-Mdm2 (SMP14, Santa Cruz), anti-Cdc25C (H-6 or C-20) or a control antibody and 40 μl of 50% slurry of protein G Sepharose 4 Fast Flow beads (Amersham Biosciences) in a final volume of 600 μl of lysis buffer, and rocked at 4 °C for 2 h. Beads were washed three times with 25 mM Tris-HCl pH 7.8, 50 mM NaCl, 0.5% sodium deoxycholate, 0.2% NP-40, resuspended in 40 μl of 1 × protein sample buffer, and heated to 95 °C for 5 min. Immunocomplexes were resolved in 10% polyacrylamide gels and analyzed by immunoblotting using anti-Mdm2 (Ab-1) and anti-Cdc25C (C-20).
Detection of ubiquitinated Cdc25C in cells
150 mm dishes of H1299 cells were transfected with 2.5 μg of pCMV-flag-Cdc25C, 0.615 μg of pCMV-flag-p53, 5 μg of pCMV-Mdm2, 10 μg of pEF-HA-Ub expression vectors, and total amount of DNA was normalized using a pCMV empty vector. After 24 h of transfection, indicated dishes were treated with 40 μM MG132 for 5 h prior to harvesting. Cells were harvested in PBS and lysed in radioimmunoprecipitation assay buffer (5 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate, 5 mM ethylenediaminetetraacetic acid) with protease inhibitors as described for immunoblotting and 10 mM N-ethylmaleimide. Protein extracts (250–750 μg) were incubated with 5 μg of anti-flag antibody (M2) and rocked overnight at 4 °C. In total, 30 μl of 50% slurry of protein G Sepharose 4 Fast Flow beads were added and the samples were rocked for 2 h. Beads were collected by a brief spin and washed three times with 1 ml of RIPA buffer, and three times with 1 ml of NET-gel buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.25% gelatin and 0.1% NP-40) for 5 min. Beads were finally resuspended in 30 μl of 2 × protein sample buffer, and heated to 95 °C for 5 min. Samples were resolved in 8% polyacrylamide gels and analyzed by immunoblotting using anti-HA (clone 12CA5, Roche, Basel, Switzerland). Input was analyzed in separate gels and probed with anti-flag, anti-HA and anti-Mdm2 antibodies.
Supplementary Material
Acknowledgments
We are grateful to Matthew O’Connell and Zhen-Qiang Pan for their technical advice, helpful discussions and sharing reagents. Present and past members of the Manfredi laboratory: Dana Lukin, Anthony Mastropietro, Wendy Liu, Sejal Patel, Shohreh Varmeh-Ziaie, Pierre-Jacques Hamard and Kester Haye are thanked for their help and support. Quantitative PCR was performed at the Mount Sinai shared research facility. These studies were supported by grants from the National Cancer Institute F31CA150539 to LAC, R03CA216466 to JJM, and developmental support from P30CA196521. Grant support: National Cancer Institute F31CA150539 to LAC, R03CA216466 to JJM, and developmental support from P30CA196521.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 2.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 5.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 6.Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. Embo J. 1996;15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- 7.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 8.Migliorini D, Denchi EL, Danovi D, Jochemsen A, Capillo M, Gobbi A, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–5538. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 10.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 11.Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010;24:1580–1589. doi: 10.1101/gad.1941710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown DR, Thomas CA, Deb SP. The human oncoprotein MDM2 arrests the cell cycle: elimination of its cell-cycle-inhibitory function induces tumorigenesis. Embo J. 1998;17:2513–2525. doi: 10.1093/emboj/17.9.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang J, Kuo ML, Eischen CM, Stepanova L, Sherr CJ, Roussel MF. The RING domain of Mdm2 can inhibit cell proliferation. Cancer Res. 2002;62:1222–1230. [PubMed] [Google Scholar]
- 14.Folberg-Blum A, Sapir A, Shilo BZ, Oren M. Overexpression of mouse Mdm2 induces developmental phenotypes in Drosophila. Oncogene. 2002;21:2413–2417. doi: 10.1038/sj.onc.1205305. [DOI] [PubMed] [Google Scholar]
- 15.Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, Barfield CR, et al. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11:714–725. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- 16.Ohkubo S, Tanaka T, Taya Y, Kitazato K, Prives C. Excess HDM2 impacts cell cycle and apoptosis and has a selective effect on p53 dependent transcription. J Biol Chem. 2006;281:16943–16950. doi: 10.1074/jbc.M601388200. [DOI] [PubMed] [Google Scholar]
- 17.Zhou R, Frum R, Deb S, Deb SP. The growth arrest function of the human oncoprotein mouse double minute-2 is disabled by downstream mutation in cancer cells. Cancer Res. 2005;65:1839–1848. doi: 10.1158/0008-5472.CAN-03-3755. [DOI] [PubMed] [Google Scholar]
- 18.Piwnica-Worms H. Cell cycle. Fools rush. Nature. 1999;401:537. doi: 10.1038/44029. [DOI] [PubMed] [Google Scholar]
- 19.Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M, et al. Direct p53 Transcriptional Repression: In Vivo Analysis of CCAAT-Containing G2/M Promoters. Mol Cell Biol. 2005;25:3737–3751. doi: 10.1128/MCB.25.9.3737-3751.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Gac G, Esteve PO, Ferec C, Pradhan S. DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J Biol Chem. 2006;281:24161–24170. doi: 10.1074/jbc.M603724200. [DOI] [PubMed] [Google Scholar]
- 21.Lohr K, Moritz C, Contente A, Dobbelstein M. p21/CDKN1A Mediates Negative Regulation of Transcription by p53. J Biol Chem. 2003;278:32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 22.Manni I, Mazzaro G, Gurtner A, Mantovani R, Haugwitz U, Krause K, et al. NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25C promoters upon induced G2 arrest. J Biol Chem. 2001;276:5570–5576. doi: 10.1074/jbc.M006052200. [DOI] [PubMed] [Google Scholar]
- 23.St Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, et al. DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell. 2004;16:725–736. doi: 10.1016/j.molcel.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 24.Badie C, Itzhaki JE, Sullivan MJ, Carpenter AJ, Porter AC. Repression of CDK1 and other genes with CDE and CHR promoter elements during DNA damage-induced G(2)/M arrest in human cells. Mol Cell Biol. 2000;20:2358–2366. doi: 10.1128/mcb.20.7.2358-2366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.St Clair S, Manfredi JJ. The dual specificity phosphatase Cdc25C is a direct target for transcriptional repression by the tumor suppressor p53. Cell Cycle. 2006;5:709–713. doi: 10.4161/cc.5.7.2628. [DOI] [PubMed] [Google Scholar]
- 26.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 27.Giono LE, Manfredi JJ. Mdm2 is required for inhibition of Cdk2 activity by p21, thereby contributing to p53-dependent cell cycle arrest. Mol Cell Biol. 2007;27:4166–4178. doi: 10.1128/MCB.01967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pines J, Hunter T. Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell. 1989;58:833–846. doi: 10.1016/0092-8674(89)90936-7. [DOI] [PubMed] [Google Scholar]
- 29.Innocente SA, Abrahamson JL, Cogswell JP, Lee JM. p53 regulates a G2 checkpoint through cyclin B1. Proc Natl Acad Sci USA. 1999;96:2147–2152. doi: 10.1073/pnas.96.5.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jin Y, Lee H, Zeng SX, Dai MS, Lu H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. Embo J. 2003;22:6365–6377. doi: 10.1093/emboj/cdg600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, et al. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004;279:25813–25822. doi: 10.1074/jbc.M313538200. [DOI] [PubMed] [Google Scholar]
- 32.Xu H, Zhang Z, Li M, Zhang R. MDM2 promotes proteasomal degradation of p21Waf1 via a conformation change. J Biol Chem. 2010;285:18407–18414. doi: 10.1074/jbc.M109.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chin L, Pomerantz J, DePinho RA. The INK4a/ARF tumor suppressor: one gene–two products–two pathways. Trends Biochem Sci. 1998;23:291–296. doi: 10.1016/s0968-0004(98)01236-5. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 35.Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–8951. doi: 10.1074/jbc.275.12.8945. [DOI] [PubMed] [Google Scholar]
- 36.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 37.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 38.Graves PR, Lovly CM, Uy GL, Piwnica-Worms H. Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene. 2001;20:1839–1851. doi: 10.1038/sj.onc.1204259. [DOI] [PubMed] [Google Scholar]
- 39.Turowski P, Franckhauser C, Morris MC, Vaglio P, Fernandez A, Lamb NJ. Functional cdc25C dual-specificity phosphatase is required for S-phase entry in human cells. Mol Biol Cell. 2003;14:2984–2998. doi: 10.1091/mbc.E02-08-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen F, Zhang Z, Bower J, Lu Y, Leonard SS, Ding M, et al. Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 2002;99:1990–1995. doi: 10.1073/pnas.032428899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao D, Johnson CS, Trump DL, Singh SV. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol Cancer Ther. 2004;3:567–575. [PubMed] [Google Scholar]
- 42.Vecchione A, Baldassarre G, Ishii H, Nicoloso MS, Belletti B, Petrocca F, et al. Fez1/ Lzts1 absence impairs Cdk1/Cdc25C interaction during mitosis and predisposes mice to cancer development. Cancer Cell. 2007;11:275–289. doi: 10.1016/j.ccr.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 44.Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Asher G, Lotem J, Sachs L, Kahana C, Shaul Y. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci USA. 2002;99:13125–13130. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Chi Y, Bloecher A, Aebersold R, Clurman BE, Roberts JM. N-acetylation and ubiquitin-independent proteasomal degradation of p21(Cip1) Mol Cell. 2004;16:839–847. doi: 10.1016/j.molcel.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol. 2006;26:2019–2028. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003;278:45946–45953. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- 50.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, et al. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 52.Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, et al. Involvement of the MKK6-p38gamma cascade in gamma-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O’Connor PM, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–5605. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- 54.Liu TT, Liu YJ, Wang Q, Yang XG, Wang K. Reactive-oxygen-species-mediated Cdc25C degradation results in differential antiproliferative activities of vanadate, tungstate, and molybdate in the PC-3 human prostate cancer cell line. J Biol Inorg Chem. 2012;17:311–320. doi: 10.1007/s00775-011-0852-1. [DOI] [PubMed] [Google Scholar]
- 55.Raj K, Ogston P, Beard P. Virus-mediated killing of cells that lack p53 activity. Nature. 2001;412:914–917. doi: 10.1038/35091082. [DOI] [PubMed] [Google Scholar]
- 56.Eymin B, Claverie P, Salon C, Brambilla C, Brambilla E, Gazzeri S. p14ARF triggers G2 arrest through ERK-mediated Cdc25C phosphorylation, ubiquitination and proteasomal degradation. Cell Cycle. 2006;5:759–765. doi: 10.4161/cc.5.7.2625. [DOI] [PubMed] [Google Scholar]
- 57.Thanasoula M, Escandell JM, Suwaki N, Tarsounas M. ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. Embo J. 2012;31:3398–3410. doi: 10.1038/emboj.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shabbeer S, Omer D, Berneman D, Weitzman O, Alpaugh A, Pietraszkiewicz A, et al. BRCA1 targets G2/M cell cycle proteins for ubiquitination and proteasomal degradation. Oncogene. 2013;32:5005–5016. doi: 10.1038/onc.2012.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene. 2005;24:3285–3296. doi: 10.1038/sj.onc.1208492. [DOI] [PubMed] [Google Scholar]
- 60.Burri N, Shaw P, Bouzourene H, Sordat I, Sordat B, Gillet M, et al. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab Invest. 2001;81:217–229. doi: 10.1038/labinvest.3780230. [DOI] [PubMed] [Google Scholar]
- 61.Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyd MT, Vlatkovic N, Haines DS. A novel cellular protein (MTBP) binds to MDM2 and induces a G1 arrest that is suppressed by MDM2. J Biol Chem. 2000;275:31883–31890. doi: 10.1074/jbc.M004252200. [DOI] [PubMed] [Google Scholar]
- 63.Ciznadija D, Zhu XH, Koff A. Hdm2- and proteasome-dependent turnover limits p21 accumulation during S phase. Cell Cycle. 2011;10:2714–2723. doi: 10.4161/cc.10.16.16725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loughran O, La Thangue NB. Apoptotic and growth-promoting activity of E2F modulated by MDM2. Mol Cell Biol. 2000;20:2186–2197. doi: 10.1128/mcb.20.6.2186-2197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Uchida C, Miwa S, Kitagawa K, Hattori T, Isobe T, Otani S, et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. Embo J. 2005;24:160–169. doi: 10.1038/sj.emboj.7600486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang B, Liu K, Lin HY, Bellam N, Ling S, Lin WC. 14-3-3Tau regulates ubiquitin-independent proteasomal degradation of p21, a novel mechanism of p21 downregulation in breast cancer. Mol Cell Biol. 2010;30:1508–1527. doi: 10.1128/MCB.01335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem. 2004;279:16000–16006. doi: 10.1074/jbc.M312264200. [DOI] [PubMed] [Google Scholar]
- 68.Evans SC, Viswanathan M, Grier JD, Narayana M, El-Naggar AK, Lozano G. An alternatively spliced HDM2 product increases p53 activity by inhibiting HDM2. Oncogene. 2001;20:4041–4049. doi: 10.1038/sj.onc.1204533. [DOI] [PubMed] [Google Scholar]
- 69.Perry ME, Mendrysa SM, Saucedo LJ, Tannous P, Holubar M. p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53. J Biol Chem. 2000;275:5733–5738. doi: 10.1074/jbc.275.8.5733. [DOI] [PubMed] [Google Scholar]
- 70.Calabro V, Mansueto G, Parisi T, Vivo M, Calogero RA, La Mantia G. The human MDM2 oncoprotein increases the transcriptional activity and the protein level of the p53 homolog p63. J Biol Chem. 2002;277:2674–2681. doi: 10.1074/jbc.M107173200. [DOI] [PubMed] [Google Scholar]
- 71.Kadakia M, Slader C, Berberich SJ. Regulation of p63 function by Mdm2 and MdmX. DNA Cell Biol. 2001;20:321–330. doi: 10.1089/10445490152122433. [DOI] [PubMed] [Google Scholar]
- 72.Little NA, Jochemsen AG. Hdmx and Mdm2 can repress transcription activation by p53 but not by p63. Oncogene. 2001;20:4576–4580. doi: 10.1038/sj.onc.1204615. [DOI] [PubMed] [Google Scholar]
- 73.Ongkeko WM, Wang XQ, Siu WY, Lau AW, Yamashita K, Harris AL, et al. MDM2 and MDMX bind and stabilize the p53-related protein p73. Curr Biol. 1999;9:829–832. doi: 10.1016/s0960-9822(99)80367-4. [DOI] [PubMed] [Google Scholar]
- 74.Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci USA. 2003;100:8247–8252. doi: 10.1073/pnas.1431613100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol. 2003;23:5113–5121. doi: 10.1128/MCB.23.15.5113-5121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coutts AS, Boulahbel H, Graham A, La Thangue NB. Mdm2 targets the p53 transcription cofactor JMY for degradation. EMBO Rep. 2007;8:84–90. doi: 10.1038/sj.embor.7400855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, et al. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol Cell. 2007;25:739–750. doi: 10.1016/j.molcel.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 78.Shmueli A, Oren M. Mdm2: p53’s Lifesaver? Mol Cell. 2007;25:794–796. doi: 10.1016/j.molcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 79.Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, et al. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. Embo J. 2001;20:6499–6508. doi: 10.1093/emboj/20.22.6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Obeyesekere MN, Tecarro E, Lozano G. Model predictions of MDM2 mediated cell regulation. Cell Cycle. 2004;3:655–661. [PubMed] [Google Scholar]
- 81.Garcia P, Cales C. Endoreplication in megakaryoblastic cell lines is accompanied by sustained expression of G1/S cyclins and downregulation of cdc25C. Oncogene. 1996;13:695–703. [PubMed] [Google Scholar]
- 82.Varmeh-Ziaie S, Manfredi JJ. The dual-specificity phosphatase Cdc25B, but not the closely related Cdc25C, is capable of inhibiting cellular proliferation in a manner dependent upon its catalytic activity. J Biol Chem. 2007;282:24633–24641. doi: 10.1074/jbc.M703105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.