

Abstract
Skeletal muscle is a highly plastic tissue in the human body that undergoes extensive adaptation in response to environmental cues, such as physical activity, metabolic perturbation, and disease conditions. The endoplasmic reticulum (ER) plays a pivotal role in protein folding and calcium homeostasis in many mammalian cell types including skeletal muscle. However, overload of misfolded or unfolded proteins in the ER lumen cause stress, which results in the activation of a signaling network called the unfolded protein response (UPR). The UPR is initiated by three ER transmembrane sensors: protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6). The UPR restores ER homeostasis through modulating the rate of protein synthesis and augmenting the gene expression of many ER chaperones and regulatory proteins. However, chronic heightened ER stress can also lead to many pathological consequences including cell death. Accumulating evidence suggests that ER stress-induced UPR pathways play pivotal roles in the regulation of skeletal muscle mass and metabolic function in multiple conditions. They have also been found to be activated in skeletal muscle under catabolic states, degenerative muscle disorders, and various types of myopathies. In this article, we have discussed the recent advancements towards understanding the role and mechanisms through which ER stress and individual arms of the UPR regulate skeletal muscle physiology and pathology.
Keywords: ER Stress, UPR, Myogenesis, Skeletal muscle atrophy, Metabolism, Myopathies
Graphical Abstract
ER stress-induced UPR pathways play important roles in the regulation of tissue homeostasis. Accumulating evidence suggests that UPR pathways modulate skeletal muscle mass and metabolic function in diverse conditions. Moreover, chronic activation of ER stress contributes to myopathy. In this review, we discuss the role and mechanisms by which ER stress and UPR pathways regulate skeletal muscle health and disease.
Introduction
Skeletal muscle is a highly dynamic tissue that represents approximately 40% of the total body weight. The muscle tissue plays diverse roles, such as posture maintenance, breathing, locomotion, whole-body metabolism, and it is a reservoir of amino acids that can support protein synthesis or energy production in vital tissues during starvation or extreme energy shortfalls. Skeletal muscle is also a secretory organ that produces and releases several growth factors, cytokines, and peptides, referred to as myokines [1]. While the number of skeletal muscle fibers remain constant after achieving adulthood, skeletal muscle mass is influenced by various factors such as diet, genetics, hormones, growth factors, and mechanical stimuli [2, 3]. Loss of skeletal muscle mass leads to debilitating consequences causing permanent disability and mortality in many conditions, including: cancer, chronic heart failure, burn injury, chronic kidney disease, type 2 diabetes (T2D), aging, immobilization, spinal cord injury, inflammatory myopathies and various genetic muscle disorders such as muscular dystrophy [4].
The endoplasmic reticulum (ER) is a network of branching ER tubules and flattened sacs that extends from the nuclear membrane throughout the cytosol. One of the major functions of the ER is to orchestrate the synthesis, folding, and structural maturation of cellular proteins. Most proteins that are translated on ER membrane-associated ribosomes either get secreted or become resident in the ER, Golgi apparatus, lysosomes, and plasma membrane [5–8]. The ER also contains high levels of Ca2+ that are supported by the active transport function of the sarco/ER calcium ATPase. Indeed, several ER chaperone proteins and enzymes require high levels of Ca2+ in the ER for proper protein folding and maturation [9]. The protein folding capacity of ER is disrupted in multiple physiological or pathological insults including glucose deprivation, environmental toxins, viral infection, alterations in Ca2+ levels, hypoxia, inflammatory challenges, oxidative stress, and limited or disproportionate availability of nutrients [5]. To alleviate stress and restore ER function, cells initiate a signaling cascade commonly referred to as the unfolded protein response (UPR). The UPR is mediated by three ER transmembrane sensors: protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6) [7, 8, 10].
In the unstressed ER lumen, an ER chaperone protein, GRP78 (BiP), binds to PERK, IRE1α and ATF6 to inhibit their oligomerization and phosphorylation (Figure 1). During ER stress, GRP78 disassociates from these receptors and binds to misfolded proteins. Following ER stress, PERK undergoes oligomerization and auto-phosphorylation leading to its activation. Activated PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α) on serine 51 that integrates general translation repression while selectively increasing the translation of activating transcription factor-4 (ATF4) and expression of stress-responsive genes such as C/EBP homologous protein (CHOP), and BiP [7, 11, 12]. The PERK/eIF2α signaling axis also inhibits the UPR through inducing gene expression of growth arrest and DNA damage-inducible protein, GADD34 that promotes dephosphorylation of eIF2α leading to recovery from translational inhibition [13, 14]. IRE1α is also activated through homodimerization and auto-phosphorylation during ER stress. IRE1α has both kinase and endonuclease activity. Through its endonuclease activity, IRE1α promotes splicing of a 26-base intron from X-box binding protein 1 (XBP1) mRNA [15]. Spliced XBP1 (sXBP1) acts as a transcription factor to increase gene expression of a number of ER chaperones involved in restoring protein folding or ER-associated protein degradation [16]. However, prolonged ER stress increases IRE1α kinase activity, which leads to activation of c-Jun N-terminal kinase (JNK) and nuclear factor-kappa B (NF-κB) pathways [7]. Finally, in response to ER stress, ATF6 moves from the ER to the Golgi apparatus to be cleaved by site 1 protease (S1P) and S2P [17]. The cleaved N-terminal fragment of ATF6 is then transported to the nucleus where it cooperates with sXBP1 to increase gene expression of proteins that function to alleviate ER stress [7, 11, 12, 15].
FIGURE 1. Activation and mode of action of the unfolded protein response (UPR) in physiological processes.

In absence of stress, endoplasmic reticulum (ER) chaperone protein, BiP/GRP78, binds to the UPR sensors: protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), inositol-requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6) to keep them inactive. Upon ER stress, BiP disassociates from these sensors and preferentially binds to the misfolded proteins in the ER lumen. Upon release from BiP, PERK undergoes dimerization and autophosphorylation leading to a cascade of signals including phosphorylation of eIF2α, inhibition of protein synthesis, and selective upregulation of translation of ATF4, a potent transcription factor. The PERK arm of the UPR also regulates mitochondrial biogenesis, antioxidant response, and gene expression of C/EBP homologous protein (CHOP), growth arrest and DNA damage-inducible protein 34 (GADD34), fibroblast growth factor 21 (FGF21), and enzymes involved in amino acid metabolism. IRE1α also becomes activated by dimerization and autophosphorylation during ER stress. Through its endonuclease activity, IRE1α processes the mRNA encoding unspliced XBP1 (uXBP1) to produce an active transcription factor, spliced XBP1 (sXBP1). The sXBP1 controls the transcription of genes encoding ER chaperones, ER-associated protein degradation (ERAD), and protein quality control and phospholipid synthesis. Through interacting with adaptor proteins, such as TNF receptor-associated protein 2 (TRAF2), IRE1α activates c-Jun N-terminal kinase (JNK) and potentially nuclear factor-kappa B (NF-κB) pathways to regulate cell fate. Upon ER stress, ATF6 is transported to the Golgi apparatus, where it is processed by site 1 protease (S1P) and S2P, releasing its cytosolic domain fragment which translocate to the nucleus to induce gene expression of components of ERAD as well as XBP1. Studies using genetic mouse models have revealed that individual arm of the UPR may regulate common or distinct physiological response as depicted here.
Genetic studies have provided strong evidence that the UPR plays a critical role in cell survival and homeostasis of distinct organs especially the liver, pancreas, and antibody-secreting B cells [5, 7]. For example, while Perk1-deficient mice are normal at birth, they die during the post-natal period due to β-cell degeneration the pancreas resulting in diabetes mellitus, reduced secretion of digestive enzymes, and impairment in bone formation [18–21]. Similarly, homozygous Eif2αS51A mutant mice die within 24h after birth by severe hypoglycemia, potentially due to impairments in glycogen synthesis and gluconeogenesis [22]. Both Ire1−/− and Xbp1−/− mice are embryonically lethal due to liver failure, defects in the placenta, and anemia [23, 24]. Single knockout of ATF6α or ATF6β does not cause any developmental defects. However, double deficiency of ATF6α and ATF6β is embryonically lethal [25]. Moreover, ATF6α−/− produce lethality when challenged with ER stress agents, possibly due to liver dysfunction [26]. Studies using tissues-specific knockout mice and cultured cells showed that in addition to restoring ER function, the UPR regulates autophagy, mitochondrial biogenesis and function, and the expression of various anti-oxidant molecules to protect mammalian cells under stressed conditions [27, 28]. Recent studies have also provided evidence that the components of the UPR have fundamental roles in other physiological processes beyond controlling protein folding. For example, agonists of Toll-like receptors (TLRs) activate the IRE1α-XBP1 axis to induce the expression of proinflammatory cytokines, such as interleukin-6, to regulate innate immune responses in macrophages [29]. The phosphorylation of IRE1α is also regulated in response to fluctuations in glucose levels, which may be a mechanism to control insulin levels [6]. The UPR also plays a pivotal role in the survival, self-renewal, differentiation and dedifferentiation of cells in various organs [7, 30, 31]. Although the main function of the UPR is to promote cell survival, chronic unmitigated ER stress can also lead to cell death [7]. Moreover, it is now increasingly clear that each arm may have a distinct role in the regulation of cell fate in naïve and diseased states [7, 8, 10].
Although skeletal muscle contains an extensive network of ER, also known as sarcoplasmic reticulum, the role of ER stress and the UPR in the regulation of skeletal muscle physiology and diseases remains less understood. Recent studies using pharmacological interventions and genetic mouse models suggest that the activation of the UPR can produce both beneficial and deleterious consequences in skeletal muscle [32, 33]. In the following sections, we provide a succinct review of available literature about the role and mechanisms of action of the UPR in skeletal muscle adaptation and myopathies.
Skeletal Muscle Remodeling
Skeletal muscle is a dynamic tissue of the human body that responds adaptively to both intrinsic and extrinsic stimuli. The remodeling responses involve activation of multiple intracellular signaling pathways and genetic reprogramming, resulting in amendments of skeletal muscle mass, contractile properties, and metabolic states. Some cascade of events that result in muscle remodeling are also thought to be initiated by muscle damage. For example, repeated bouts of resistance exercise training lead to muscle injury that ultimately results in compensatory growth (hypertrophy) of skeletal muscle. Several studies have suggested that ER stress and the UPR pathways may regulate not only skeletal muscle adaptation in response to physiological stimuli, but also their formation during embryonic development and regeneration of myofibers in adults [6, 32, 34]. In the following sections, we describe the role of ER stress and the UPR in various aspects of skeletal muscle remodeling and metabolic function (Figure 2).
FIGURE 2. Involvement of ER stress and the UPR in skeletal muscle remodeling.

Many physiological stimuli and disease states lead to the activation of ER stress-induced UPR pathways. Although genetic evidence is still sparse, the UPR can improve or deteriorate skeletal muscle mass and function. Adaptive UPR generally improves skeletal muscle formation and metabolic function and promotes maintenance of skeletal muscle mass during exercise and other perturbation. Adaptive UPR also inhibits ER stress through improving protein folding and restoring Ca2+ homeostasis in ER. Chronic activation of UPR can lead to skeletal muscle wasting through repressing the rate of protein synthesis, activation of proteolytic pathways, inflammation, and development of insulin resistance.
Skeletal Myogenesis
The process of skeletal muscle formation during embryonic development or postnatal life is called myogenesis. During embryonic development, the multi-potential mesodermal cells differentiate into myogenic lineages giving rise to myoblasts. Myoblasts then exit from the cell cycle, differentiate, and fuse with each other to generate multinucleated myotubes, which then mature as myofibers [35]. Myogenesis is a highly regulated process that involves the sequential expression of a number of transcription factors including paired box (Pax) 3 and Pax7 followed by the expression of myogenic regulatory factors (MRFs) such as Myf5, MyoD, myogenin, and MRF4. While myofibers are terminally differentiated cells, skeletal muscle retain regenerative capacity attributed to the presence of muscle stem cells, also called satellite cells. In undamaged tissue, satellite cells reside between the sarcolemma and the basal lamina in a relatively quiescent state. Upon muscle injury, these cells become activated, undergo several rounds of cell division, and eventually differentiate into myoblasts/myocytes, which then fuse with injured myofibers [36, 37]. A vast majority of activated satellite cells differentiate into the myogenic lineage and participate in the repair of damaged myofibers. However, a small fraction of them undergoes self-renewal and returns to a quiescent state to participate in future rounds of muscle injury and repair. It has been found that Pax7 transcription factor is essential not only for the self-renewal of satellite cells, but also for the maintenance of their myogenic potential [35, 38]. Several steps of satellite cell homeostasis and myogenesis can be studied in vitro using isolated myofibers, primary myoblasts from animals, or using established cell lines such as mouse C2C12 or rat L6 myoblasts [35, 38].
Accumulating evidence suggests that ER stress-induced UPR pathways may regulate various aspects of myogenesis (Table 1). Skeletal muscle formation involves selective elimination of differentiation-incompetent myoblasts through apoptosis [39, 40]. One of the consequences of increased ER stress is cell death. Prolonged ER stress activates caspase-12, which leads to the downstream activation of caspase-9 and caspase-3 and eventually results in apoptosis. A few studies have suggested that ER stress is induced during myogenesis. Specifically, levels of ER stress-related proteins such as ATF6, CHOP and GRP74 and the activity of caspase-12 are increased in myoblasts undergoing apoptosis during myogenic differentiation [41]. Heightened ER stress seems to be essential for proper progression of myogenesis because inhibition of ATF6 or caspase-12 reduced the formation of multinucleated myotubes. An increase in caspase-12 activity is also observed during embryonic development of skeletal muscle suggesting that the ATF6 arm of the UPR is required for the removal of a subpopulation of myoblasts that may not be able to sustain cellular stresses [41]. The role of ER stress in myogenesis is also supported by the findings that forced activation of ER stress improves myotube formation [42]. ER stressors, such as tunicamycin and thapsigargin, increase cell death in C2C12 myoblast cultures after induction of differentiation. However, the surviving myoblasts more efficiently differentiate into functional myotubes further suggesting that ER stress is a mechanism to remove differentiation-incompetent myoblasts during myogenesis [42].
TABLE 1.
Role and mechanisms of action of different components of the UPR during myogenesis.
| UPR component | Role in myogenesis |
|---|---|
| PERK | Promotes quiescence and self-renewal of satellite cells [46] Improves survival and differentiation of satellite cells during regenerative myogenesis through inhibiting precocious activation of p38 MAPK [47] Improves survival of cultured myoblasts during myogenesis [47] Improves myogenesis in response to low-intensity extracorporeal shock wave therapy [48] |
| eIF2α | Promotes satellite cell quiescence through repression of general translation [46] Improves survival and differentiation of cultured myoblasts [43] |
| CHOP | Inhibits premature myogenic differentiation through repressing MyoD levels [43] |
| XBP1 | Regulates gene expression of ER chaperones and the components of DNA damage and repair pathways [45] Inhibits myotube formation through upregulation of Mist1 [45] No role in satellite cell-mediated skeletal muscle regeneration [47] |
| ATF6α | Activates caspase-12 to eliminate differentiation-incompetent myoblasts during myogenic differentiation [41, 42] |
The ER stress-induced UPR pathways may also play a role in fine-tuning myogenic differentiation. It has been reported that the phosphorylation of PERK and eIF2α and levels of CHOP are transiently increased in a subset of myoblasts after incubation in differentiation medium. CHOP inhibits myogenic differentiation through repressing the expression of transcription factor MyoD, which could be a mechanism to prevent premature differentiation of myoblasts [43]. IRE1α has an endonuclease activity that mediates the unconventional splicing of XBP1 mRNA [7]. Spliced XBP1 (sXBP1) is a powerful transcription factor that induces UPR target genes, especially those encoding chaperones and the components of ER associated protein degradation (ERAD) [8]. In myogenic cells, the expression of Xbp1 is regulated by MyoD and myogenin [44]. Intriguingly, genome-wide gene expression analysis has shown that XBP1 controls the expression of a large subset of genes in myogenic cells including those involved in maintenance of ER function, growth, and DNA damage and repair pathways [45]. Forced expression of sXBP1 inhibits myogenic differentiation leading to the formation of smaller myotubes in C2C12 cultures after induction of differentiation. In this regard, it was found that XBP1 up-regulates transcription factor, Mist1, a negative regulator of MyoD activity [45].
The role of individual arms of the UPR in the regulation of satellite cell homeostasis and function was recently investigated [46, 47]. Using reporter mouse models, Zismanov et al showed that PERK and eIF2α are constitutively phosphorylated in quiescent satellite cells. Activation of the PERK/eIF2α arm of the UPR may be a mechanism for maintaining satellite cells in a quiescent state through repression of general translation [46]. The study also showed that targeted inducible expression of a phosphorylation resistant mutant of eIF2α (i.e. eIF2αS51A) or deletion of PERK leads to the activation of satellite cells. Remarkably, eIF2αS51A-expressing satellite cells are capable of undergoing differentiation and progress through myogenic lineage. Similarly, it was reported that blocking eIF2α dephosphorylation using Sal003 promotes self-renewal of satellite cells on myofiber explants [46]. Our group used a slightly different approach to investigate the role of PERK in satellite cell homeostasis. We generated mice in which PERK was ablated in satellite cells of adult mice using a tamoxifen-inducible Pax7-Cre line. Surprisingly, we could not find any significant difference in the number of quiescent satellite cells in skeletal muscle of control and satellite cell-specific PERK-knockout (i.e. scPERK-KO) mice. We also found that PERK is required for the survival of satellite cells and regeneration of myofibers upon injury [47]. Our cell culture studies also showed that deletion of PERK causes satellite cell death during myogenesis, which could be attributed to precocious activation of p38 MAPK. Pharmacological inhibition of p38 MAPK prevents cell death and improves myotube formation in PERK−/− satellite cell cultures and augments the regeneration of myofibers in scPERK-KO mice [47]. Consistent with our findings, there is another study which demonstrated that overexpression of eIF2αS51A causes cell death and diminishes the differentiation of cultured myoblasts [43]. More recently, it was shown that low-intensity extracorporeal shock wave therapy improves myogenesis through activating PERK/eIF2α arm of the UPR [48]. Although the exact reason to why two independent studies reached two different conclusions about the role of the PERK/eIF2α pathway in satellite cell homeostasis and regenerative myogenesis remain unknown, some of these discrepancies could be attributed to the different experimental approaches and mouse models utilized in these studies [46, 47]. While we focused our experiments on PERK [47], Zismanov et al used approaches in which the phosphorylation of eIF2α was manipulated [46]. It is now known that eIF2α can also be phosphorylated by three other kinases: double stranded RNA-activated protein kinase R (PKR), heme-regulated inhibitor eIF2α kinase (HRI), and general control nonderepressible-2 (GCN2) under stress conditions [49]. Moreover, PERK is reported to regulate cell survival through mechanisms independent of eIF2α phosphorylation [27, 50–52]. Interestingly, while PERK was shown to play a pivotal role, satellite cell-specific ablation of XBP1 does not affect satellite cell homeostasis or the regeneration of skeletal muscle [47]. Altogether, these studies suggest that the UPR plays an important role in satellite cell homeostasis and myogenesis both in vitro and in vivo (Table 1). More investigations are needed to understand how the components of the UPR interact with other signaling modules to regulate skeletal muscle formation. Furthermore, the role of IRE1α and ATF6α in satellite cell function and regenerative myogenesis need to be investigated using genetic mouse models.
Adaptation to exercise
Regular exercise has a profound benefit on overall health, including the prevention of obesity, cardiovascular disease, and diabetes. Exercise is a potent stimulus to preserve skeletal muscle mass, aerobic fitness, and strength [53]. These adaptations can ameliorate metabolic dysfunction and prevent chronic disease. Aerobic (endurance) and resistance (strength-based) activities represent two extremes of the exercise continuum and produce distinct metabolic and molecular responses. Aerobic exercise imposes a high-frequency (repetition), low-power output (load) demand on muscle contraction. In contrast, resistance exercise imposes a low-frequency, high-resistance demand on musculature [53]. The sarcoplasmic reticulum is the major source of Ca2+ that is required for the initiation of muscle contraction in response to exercise perturbations. Therefore, fluctuations in Ca2+ levels in ER during exercise can lead to the activation of the UPR. Indeed, accumulating evidence suggests that exercise activates all three arms of the UPR in skeletal muscle [54–56]. The markers of ER stress, such as: BiP, GRP94, GADD34, ATF4, CHOP, and sXBP1, are increased in skeletal muscle after a single bout of exhaustive treadmill running [55]. Moreover, the levels of phosphorylated IRE1α, PERK, and eIF2α have been found to be elevated in the skeletal muscle of mice after a downhill treadmill running [57]. The activation of the UPR appears to be an adaptive mechanism because the mice that were previously trained to the exercise paradigm showed upregulation of only BiP and GRP94 in skeletal muscle during successive exercise training [55]. A rapid increase in the markers of ER stress was also observed when rats were subjected to chronic contractile activity and this increase was attenuated with repeated bouts [58]. Similar to rodent models, elevated levels of ER stress markers, such as BiP and sXBP1, have also been observed in musculature of humans after a bout of running [59]. However, the exact role of these molecules in myofiber hypertrophy remain unknown.
The role of the UPR in skeletal muscle adaptation to exercise has been validated by a study from Bruce Spiegelman’s group [55]. Peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) is a transcriptional coactivator that plays a key role in mitochondrial biogenesis and metabolic function [60, 61]. Levels of PGC-1α are induced in skeletal muscle following exercise training [62, 63] and transgenic mice overexpressing PGC-1α in the skeletal muscle perform better during voluntary and forced exercise training [64]. Interestingly, PGC-1α interacts with cleaved ATF6α to induce an adaptive UPR in skeletal muscle after exercise [55]. ATF6α-null mice fail to efficiently recover from muscle damage that occurs after acute exercise. While ATF6α participates in the adaptive response, other arms of the UPR may differentially regulate skeletal muscle adaptation and recovery following exercise training. The expression of CHOP is induced through the activation of the PERK/eIF2α arm of the UPR [5]. Interestingly, ablation of CHOP improved exercise tolerance in skeletal muscle specific PGC-1α knockout mice [55] suggesting that the PERK-eIF2α arm of the UPR may negatively regulate skeletal muscle homeostasis following exercise training.
Resistance exercise improves skeletal muscle mass through augmenting the rate of protein synthesis and organelle biogenesis. Synergistic ablation is a model used to study skeletal muscle hypertrophy, similar to that seen from resistance exercise. In this model, the gastrocnemius and soleus muscles are partially ablated leaving the plantaris muscle as the lone hind limb extensor [65–67]. Since the rate of protein synthesis is increased during skeletal muscle hypertrophy, it can be envisioned that the activation of ER stress and the UPR could very well be a mechanism to improve protein folding capacity. Although the phosphorylation of PERK and eIF2α was not studied, a significant increase in the mRNA levels of CHOP has been observed in plantaris muscle of rats from 3 to 12 days post ablation surgery which may be a mechanism to reduce the rate of protein synthesis in an attempt to halt the excessive muscle growth [68]. Protein levels of BiP, PERK, and IRE1α are also increased in skeletal muscle of humans after a unilateral single bout of resistance exercise using the knee extensors [69].
Altogether, these studies indicate that both endurance and resistance exercise cause induction of ER stress and activation of the UPR pathways. In most of the cases, the expression of ER stress markers is diminished during successive exercise suggesting that the UPR becomes activated to novel or challenging exercise stimuli and may be a mechanism for skeletal muscle adaptation. Currently, genetic studies, especially about the role of the eIF2α and IRE1α arms of the UPR, in exercise-induced skeletal muscle adaptation are lacking. Future studies should also focus on investigating the molecular mechanisms by which ER stress-induced UPR pathways promote adaptive or maladaptive response following exercise training.
Atrophy
Skeletal muscle mass is regulated by a dynamic control of the processes of protein synthesis and degradation in combination with systems in place to manage protein folding, trafficking and clearance [2, 70]. In many catabolic states, the rate of protein degradation exceeds that of protein synthesis, leading to overall loss of skeletal muscle mass also known as skeletal muscle atrophy or wasting. The PI3K/Akt/mTOR cascade is a major intracellular pathway that increases the rate of protein synthesis in skeletal muscle [2, 71]. This pathway also inhibits the rate of protein degradation in skeletal muscle through distinct mechanisms [2, 70, 72]. Several proteolytic pathways such as the ubiquitin-proteasome system (UPS), autophagy, caspases, and calpains contribute to protein degradation in skeletal muscle under catabolic states [73]. Among them, the activity of the UPS has been found to be consistently up-regulated in atrophying skeletal muscle [74, 75]. The UPS mediates protein degradation in skeletal muscle through upregulation of several E3 ubiquitin ligases, such as muscle RING-finger protein-1 (MuRF1), muscle atrophy F-box (MAFbx)/Atrogin-1, neural precursor cell-expressed developmentally downregulated gene 4.1 (Nedd4.1), TNF-associated factor 6 (TRAF6), and muscle ubiquitin ligase of SCF complex in atrophy-1 (MUSA1) [70, 76, 77]. MuRF1, MAFbx, Nedd4.1, and MUSA1 label the target proteins for degradation by the 26S proteasome, whereas TRAF6 induces proteolysis through modulating the activity of catabolic signaling pathways [76, 78–80]. Autophagy is another proteolytic system that gets activated and mediates protein degradation in skeletal muscle in catabolic conditions [81]. While a basal level of autophagy is required for the clearance of defunct organelles [82, 83], excessive activation of autophagy mediates skeletal muscle wasting in multiple conditions, including fasting, denervation, and cancer cachexia [70, 84–86]. There is also evidence that the activation of caspases and calpains can cause muscle protein degradation directly or through functioning upstream of the UPS and autophagy [70, 81, 87, 88]. It is also evidenced that a number of signaling pathways such as p38 MAPK, AMP-activated protein kinase (AMPK), and nuclear factor-kappa B (NF-κB) modulates the activity of various proteolytic systems in skeletal muscle under catabolic states [70, 89]. Moreover, recent studies also suggest that mitochondria play a pivotal role in the maintenance of skeletal muscle mass and metabolic function [90]. Perturbations in mitochondrial dynamics and function are responsible for skeletal muscle wasting in multiple pathophysiological conditions [91, 92].
Although ER stress and activation of the UPR have been observed in skeletal muscle in multiple atrophying conditions, their exact role in the regulation of skeletal muscle mass remains poorly understood. ER stress has been implicated in diaphragm contractile dysfunction in a mouse model of sepsis [93]. A previous study found no changes in the levels of GRP78, calreticulin, CHOP, vinculin, the type I D-myo-inositol 1,4,5-trisphosphate receptor, protein kinase R, and eIF2α in skeletal muscle upon hind limb unloading, a model of disuse atrophy [94]. By contrast, BiP and CHOP are increased 14 days after reloading of hind limb and this effect was more pronounced in skeletal muscle of aged mice [95]. ER stress has also been found to be elevated in the skeletal muscle of the elderly, which could be due to reduced expression of ER chaperones [34, 96–98]. While anabolic resistance is one of the important reasons for age-associated skeletal muscle atrophy, a recent study showed that ER stress may not be the cause for anabolic resistance in skeletal muscle during aging [96].
The integrated stress response (ISR) is a complex signaling network that is activated in a wide variety of physiological and pathological conditions including: hypoxia, amino acid deprivation, glucose deprivation, and viral infection [99]. ER stress caused by accumulation of unfolded proteins also activate the ISR in mammalian cells. Activation of ISR results in the phosphorylation of eIF2α on serine 51 residue leading to a reduction in global protein synthesis [99]. Increased ammonia uptake by skeletal muscle leads to skeletal muscle atrophy and poor clinical outcomes in cirrhosis [100, 101]. Hyperammonemia causes muscle atrophy through inhibiting mTORC1 signaling and diminishing the rate of protein synthesis. Interestingly, while hyperammonemia increases the phosphorylation of eIF2α, it does not involve the activation of PERK [102]. Rather, during hyperammonemia, the phosphorylation of eIF2α in skeletal muscle is mediated by amino acid deficiency sensor, GCN2 [102]. These findings suggest that in some catabolic conditions, such as aging and cirrhosis, inhibition in the rate of protein synthesis in skeletal muscle can occur through the activation of ISR, but not UPR.
Through generation of skeletal muscle-specific Fv2E-PERK transgenic (Tg) mice, two recent studies showed that inducible activation of PERK leads to skeletal muscle wasting in adult mice in a dose-dependent manner [54, 103]. Interestingly, while Fv2E-PERK Tg mice undergo atrophy, constitutive activation of PERK in skeletal muscle also leads to increased gene expression of several enzymes involved in amino acid metabolism and upregulation of antioxidant molecule, glutathione. Moreover, forced activation of PERK dramatically increased the expression and secretion of fibroblast growth factor 21 (FGF21), an anti-obesity myokine/hepatokine that stimulates energy expenditure in brown adipose tissue [54]. These findings suggest that while overexpression of PERK causes skeletal muscle atrophy, it also improves metabolic adaptation of the whole body. However, the effects of targeted deletion of PERK in the regulation of skeletal muscle mass and function has not been yet investigated using genetic mouse models.
The role of CHOP, a downstream target of the PERK/eIF2α pathway, has been evaluated by a genetic approach in the denervation-induced muscle atrophy. ER stress markers, including CHOP, are increased in skeletal muscle upon sciatic nerve transection. Similarly, markers of ER stress are also elevated in skeletal muscle of AR113Q mice, a model of spinal and bulbar muscular atrophy (SBMA) [104]. Interestingly, ablation of CHOP accelerates denervation-induced skeletal muscle wasting in normal mice as well as in the AR113Q mice potentially due to the increased activation of autophagy. Inhibition of autophagy through suppression of Beclin-1 attenuates denervation-induced muscle atrophy and increases the lifespan of AR113Q mice [104]. The role ER stress role has also been investigated in superoxide dismutase 1 (SOD1) mutant mice, a model of amyotrophic lateral sclerosis [105, 106]. Interestingly, it was found that induction of autophagy following Xbp1 deletion ameliorated pathology of SOD1 mice due to clearance of mutant SOD1 aggregates [106]. These finding emphasize that depending on the catabolic stimuli or pathological condition, different arms of the UPR may differentially regulate skeletal muscle mass.
While the role of individual arms of the UPR remains poorly understood, physiological levels of ER stress may be beneficial for the maintenance of skeletal muscle mass in naïve condition and during cancer growth. 4-phenylbutyrate (4-PBA) is a chemical chaperone and a FDA approved drug that inhibits ER stress both in vivo and in vitro [107, 108]. Intriguingly, we found that chronic administration of 4-PBA leads to significant loss of skeletal muscle mass and strength in naïve mice [109]. Markers of ER stress are highly elevated in the skeletal muscle of two widely studied mouse models of cancer cachexia: Lewis lung carcinoma (LLC)-bearing mice and ApcMin/+ mice [109]. The activation of ER stress during cancer cachexia is attributed to the factors produced by tumor cells evidenced by the findings that treatment of cultured myotubes with LLC conditioned medium increases the phosphorylation of eIF2α, splicing of XBP1, and gene expression of several ER stress markers. Interestingly, pan-inhibition of ER stress using 4-PBA leads to accelerated muscle loss in LLC-bearing mice [109]. Moreover, 4-PBA induces rapid atrophy in cultured myotubes, which is further increased in the presence of LLC conditioned medium. Our study also showed that 4-PBA inhibits the rate of protein synthesis and Akt/mTOR pathway and augments the activation of proteolytic systems during cancer cachexia [109]. It has been reported that ATF4, which is activated by PERK/eIF2α arm of the UPR, is involved in skeletal muscle wasting during starvation [79, 110]. Even though muscle wasting is increased, chronic treatment of mice with 4-PBA reduces the levels of ATF4 in LLC-bearing mice suggesting that ATF4 may not have any significant role during cancer cachexia. [109].
While the studies in our laboratory provided evidence that pan-inhibition of ER stress causes muscle wasting in naïve mice and during cancer cachexia, other groups have recently reported that treatment with 4-PBA can ameliorate skeletal muscle wasting in a few conditions especially those involving release of excessive Ca2+ ions from the sarcoplasmic reticulum. Severe thermal injury is followed by a profound hypermetabolic response that leads to whole body catabolism, increased resting energy expenditure, and multi-organ dysfunction. Skeletal muscle wasting is one of the important consequences of severe burn injury. A recent study showed that an experimental burn injury in rats leads to the activation of GRP78, CHOP, p-eIF2α, XBP1 and ATF6 and disruption of Ca2+ homeostasis in skeletal muscle. Interestingly, treatment with 4-PBA ameliorates structural damage of myofibrils, activation of calpains, and gene expression of MAFbx in skeletal muscle after burn injury [111]. Moreover, 4-PBA also improves skeletal muscle mass and strength in mice with an I4895T mutation in the type 1 ryanodine receptor/Ca2+ release channel [112]. Different outcomes of 4-PBA in different models of skeletal muscle wasting could be attributed to the dose and duration of treatment, age and strain of the animals, and underlying etiology. Although 4-PBA is one of the commonly used pharmacological inhibitors of ER stress, it is possible that its prolonged use can also influence some other pathways. Therefore, contribution of ER stress and the role of individual arms of the UPR in the regulation of skeletal muscle mass needs to be investigated in depth using genetic mouse models.
One of the most important consequences of ER stress is the induction of autophagy. In this regard, protein kinase C theta (PKCθ) functions as an ER stress sensor and is required for ER stress-dependent activation of autophagy in skeletal muscle [113]. Genetic ablation of PKCθ prevents the activation of autophagy and skeletal muscle wasting in response to starvation or immobilization [113]. TRAF6 is an E3 ubiquitin ligase and an adaptor protein that mediates the activation of NF-κB, p38 MAPK, and PI3K/Akt pathways in response to certain cytokines and microbial products. Skeletal-muscle specific inhibition of TRAF6 attenuates skeletal muscle wasting in distinct catabolic conditions including starvation through inhibition of autophagy and the UPS [79, 80]. Although the exact mechanisms remain unknown, TRAF6 is also involved in the activation of the UPR because targeted ablation of TRAF6 reduces the levels of ATF4, CHOP, GRP94, and GADD34 in skeletal muscle of fasted mice [79]. These findings suggest that activation of the UPR may involve signaling cross-talk with other pathways involved in the regulation of skeletal muscle mass. Collectively, it is now evidenced that ER stress and the UPR are activated in skeletal muscle under multiple catabolic states and depending on the stimulus, they may attenuate or accelerate the loss of skeletal muscle mass.
Insulin resistance
Insulin resistance is defined as an impairment in the ability of insulin to appropriately regulate glucose and lipid metabolism. Insulin resistance is the major cause for obesity, type 2 diabetes (T2D), and other disorders of metabolism. While the mechanisms of insulin resistance are still less understood, a strong association exists between insulin resistance and excess lipid accumulation in nonadipose tissues, particularly skeletal muscle and liver [114]. The stress-activated kinase, JNK, mediates insulin resistance through inhibitory phosphorylation of insulin receptor substrate [115]. One of the consequences of the ER stress is the activation of JNK that is mediated by the IRE1α arm of the UPR. A seminal study has previously showed that ER stress is highly activated in mice fed with a high-fat diet (HFD) as well as in a genetic model of murine obesity (ob/ob) and plays a key role in the development of insulin resistance in liver [116]. Forced activation of ER stress or reduction in the compensatory capacity through down-regulation of XBP1 results in the suppression of insulin signaling in intact cells through IRE1α-dependent activation of JNK [116]. Furthermore, inhibition of ER stress using 4-PBA has been found to improve insulin receptor signaling in obese and diabetic mice [107].
Skeletal muscle is the major site of glucose disposal in the body. Recent studies have suggested that ER stress may play a role in the development of insulin resistance in skeletal muscle [117, 118]. For example, mice fed with a HFD show increased levels of BiP, IRE1α, and phospho-PERK and higher gene expression of XBP1, ATF4, and CHOP in skeletal muscle. Moreover, ER stressors tunicamycin, thapsigargin, or palmitic acid lead to the inhibition of mTORC1 activity in cultured C2C12 myotubes [117].
A putative kinase tribbles homolog 3 (TRB3) is expressed in multiple tissues and is involved in the regulation of metabolism [119]. TRB3 inhibits insulin signaling in the liver through inhibiting Akt activation [120]. Levels of TRB3 have been found to be increased in muscle biopsies from patients with T2D [121]. The increased levels of TRB3 contribute to insulin resistance, which is supported by the findings that forced expression of TRB3 in C2C12 myotubes inhibits insulin-stimulated glucose uptake and phosphorylation of Akt [122]. Moreover, ER stressors tunicamycin and thapsigargin increase the levels of TRB3 in skeletal muscle. Finally, down-regulation of TRB3 restores insulin signaling and glucose uptake in skeletal muscle of mice fed with a HFD [123].
AMPK is a potent regulator of skeletal muscle metabolism and gene expression. AMPK activity is increased during adaptation of skeletal muscle to exercise training. One of the mechanisms by which AMPK improves glucose utilization is through increasing the expression of glucose transport protein 4 (GLUT4). Upon activation, GLUT4 is translocated to the plasma membrane, where it increases glucose uptake in skeletal muscle, independent of insulin [124]. Mice lacking AMPK2α display reduced glucose utilization in skeletal muscle [125]. It has been reported that ER stress inhibits AMPK activity through activation of extracellular-regulated kinase 1/2 (ERK1/2) in cultured L6 myotubes [126]. Moreover, the anti-cancer drug Bortezomib inhibits ER stress and improves insulin resistance in skeletal muscle both in vivo and in vitro. Importantly, the beneficial effects of Bortezomib on insulin signaling were diminished after knockdown of AMPK in cultured myotubes [127]. Collectively, these reports suggest that ER stress-mediated inhibition of AMPK activity is one of the important mechanisms for insulin resistance and reduced glucose utilization in skeletal muscle.
Chronic low-grade inflammation, glucose intolerance, and insulin resistance are common features of obesity [128]. There appears to be a link between the inflammatory response and ER stress in response to excessive caloric uptake. ER stress and expression of TLRs and inflammatory cytokines are increased in liver, skeletal muscle, and adipose tissue of mice fed with a HFD. Mice lacking TLR4 show reduced ER stress in metabolic tissues including skeletal muscle and are protected from body weight gain and glucose intolerance following a HFD [129]. Peroxisome proliferator-activated receptors (PPARs) are transcription factors of nuclear hormone receptor superfamily that have been shown to reduce inflammation [130]. GW501516, an agonist of PPARβ/δ, prevents palmitate, thapsigargin, or tunicamycin-induced ER stress in C2C12 myotubes. Furthermore, the activation of PPARβ/δ inhibits ER stress-associated inflammation and insulin resistance. Myotubes treated with compound C, an AMPK inhibitor, failed to demonstrate the beneficial effect of PPARβ/δ, further suggesting an essential role of AMPK in PPARβ/δ prevention of inflammation and insulin resistance [131]. Finally, PPARα agonist fenofibrate also reduces ER stress in skeletal muscle and improves insulin sensitivity in HFD-fed mice [132].
FGF21 is an important myokine that protects the body from obesity and insulin resistance [133]. Mice lacking FGF21 become obese and develop glucose intolerance [134]. Interestingly, lack tuberous sclerosis complex 1 (TSC1) in mice results in a constitutively active mammalian target of rapamycin complex 1 (mTORC1) and increased levels of FGF21 in plasma. Moreover, these mice have increased insulin sensitivity and fatty acid oxidation. Furthermore, activation of mTORC1 triggers the activation of ER stress, specifically the PERK-eIF2α-ATF4 arm of the UPR, which leads to the secretion of FGF21 [135]. As discussed above, the Fv2E-PERK Tg mice show increased levels of FGF21 in skeletal muscle and plasma. This study also showed that FGF21 reduces fat accumulation by stimulating energy consumption in brown adipose tissue, but not in skeletal muscle [54]. Collectively, ER stress and UPR pathways play important roles in the regulation of insulin signaling in various tissues including skeletal muscle. More investigation using knock-in and knockout approaches are needed to understand the role of individual arms of the UPR in the regulation of metabolic function and insulin resistance in skeletal muscle.
Myopathies
Myopathy refers to a systemic clinical disease in which the myofiber’s structure and metabolism are disrupted leading to varying degrees of weakness and dysfunction, especially in proximal muscles. A number of factors, including genetic mutations, drugs, toxins, inflammation, infection, and endocrine and electrolyte imbalances, can cause myopathy. Inherited myopathies (e.g. congenital, mitochondrial and metabolic myopathies and muscular dystrophy) have an early age of onset with a relatively longer duration and more severe progression, whereas acquired myopathies (e.g. inflammatory, toxin- or drug-related myopathies, and myopathies associated with systemic conditions) are relatively less severe and generally present at a later age. Emerging evidence suggests that ER stress and the UPR are also activated in various types of myopathies (Figure 3). In the following sections, we provide a succinct review of published reports about the potential role of ER stress in skeletal muscle diseases.
FIGURE 3. Role of ER stress in myopathies.
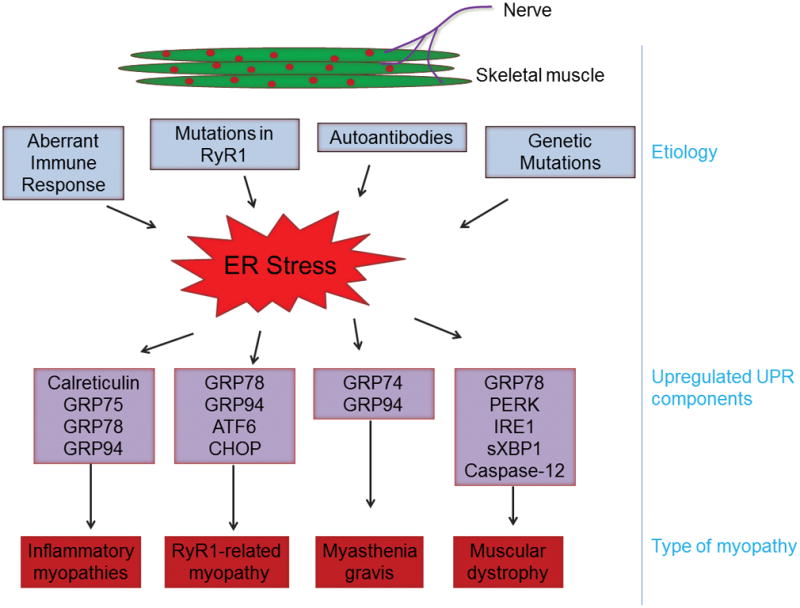
Chronic activation of ER stress occurs in several types of myopathies. The underlying cause and various components of ER stress and the UPR found to be activated in different types of myopathy are summarized here. RyR1, Ryanodine receptor type 1.
Inflammatory myopathies
Dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM) are heterogeneous inflammatory myopathies in which myofibers are damaged as a result of the immune response [136]. IBM is divided into sporadic-IBM (s-IBM) and hereditary-IBM (h-IBM). The s-IBM is the most common muscle disease that starts after age 50 years and leads to severe disability. The h-IBM is characterized by pathologic features that strikingly resemble those of s-IBM except that skeletal muscle do not involve lymphocyte infiltration. Upregulation of class I major histocompatibility complex (MHC) in skeletal muscle fibers is a prominent feature of human inflammatory myopathies. Several studies have shown that the levels of ER stress markers, such as: calreticulin, GRP75, GRP78, and GRP94, are elevated in muscle biopsies from human myositis patients and in mouse models of inflammatory myopathy [137–139]. Moreover, overexpression of mouse class I MHC (H-2K(b)) induces ER stress in cultured skeletal muscle cells [138]. While IBM involves inflammation in skeletal muscle, a few studies have indicated that inflammation alone does not cause myositis [140]. A conditional transgenic line in which class I MHC (H-2K(b)) is overexpressed specifically in skeletal muscle has been proposed as a mouse model of IBM. Interestingly, ER chaperone protein GRP78 is increased in the skeletal muscle of these transgenic mice [138]. In a separate study, mice overexpressing class I MHC (H-2K(b)) were crossed with the immunodeficient Rag2−/− mice to examine whether the upregulation of ER stress and myopathy are due to the inflammatory response. Interestingly, even in the absence of significant inflammatory cell infiltrations, myopathic changes, muscle weakness, and elevated levels of ER stress markers GRP78 and ATF6 were present [141].
Major characteristics of s-IBM are vacuolar muscle fibers, intramuscular fiber inclusions, and various degrees of mononuclear cell inflammation [142, 143]. Like Alzheimer’s disease, s-IBM also involves accumulation of amyloid-β (Aβ) and phosphorylated tau proteins [142, 143]. Interestingly, muscle biopsies from patients with s-IBM indicate that all major ER chaperones are increased and aggregate with the Aβ proteins in skeletal muscle [144] suggesting a potential role of ER chaperone proteins in folding and trafficking of Aβ in skeletal muscle. A more recent study also demonstrated the activation of the UPR in skeletal muscle of s-IBM patients [145]. In addition to IBM, elevated levels of ER stress-specific chaperone protein GRP74 have been observed in serum of patients with DM or PM suggesting that ER stress and activation of the UPR is a common feature in inflammatory myopathies [146]. While the exact role and mechanisms by which ER stress causes inflammatory myopathies remain unknown, it may be related to oxidative stress, mitochondrial dysfunction, and production of inflammatory cytokines [147].
Ryanodine receptor 1-related myopathy
Type 1 ryanodine receptor (RyR1), a major sarcoplasmic reticulum calcium release channel, plays a pivotal role in the excitation-contraction coupling in skeletal muscle. Mutations in RyR1 result in a constant calcium leak at rest, defective excitation-contraction coupling, and increased mitochondrial oxidative stress leading to morbidity and risk for early mortality in patients [148]. The Y524S mutation in RyR1 gene causes malignant hyperthermia, myopathy, increased heat sensitivity and oxidative stress, mitochondrial abnormalities, and a temperature-dependent sarcoplasmic reticulum Ca2+ leak [149]. Moreover mice with an I4895T mutation in RyR1 display muscle weakness and atrophy, which may be attributed to a decrease in the amplitude of the sarcoplasmic reticulum Ca2+ gradients, diminished cytosolic Ca2+ levels, and increases in mitochondria-mediated oxidative stress. These mice also show increases in ER stress and activation of the UPR in skeletal muscle. Interestingly, treatment of I4895T mutation knock-in mice with 4-PBA reduced ER stress and improved skeletal muscle function suggesting that inhibition of ER stress can be a potential therapeutic intervention for RyR1-related myopathies [112].
Myasthenia gravis (MG)
MG is an autoimmune neuromuscular disease that causes weakness in skeletal muscles, especially those that control eye and eyelid movement, facial expression, chewing, talking, and swallowing. The main cause for this disease is autoantibodies that block, alter, or destroy the acetylcholine receptors at the neuromuscular junction leading to impairment in muscle contraction, weakness, and fatigability [150]. It has been proposed that damage to acetylcholine receptor can induce ER stress in skeletal muscle. Indeed, elevated levels of GRP74 have been reported in muscle biopsies from patients with MG [151]. Moreover, higher binding of autoantibodies to ER chaperone GRP94 has been found to be associated with a subset of MG patients [152]. ER stress alone may also contribute to a loss of acetylcholine receptor in skeletal muscle cells. A recent study demonstrated that the ER stressor tunicamycin stimulates the loss of the acetylcholine receptors in C2C12 myotubes through endocytosis and lysosomal degradation. Moreover, knockdown of XBP1 in C2C12 myotubes attenuates the degradation of acetylcholine receptor by endocytosis [153] suggesting that inhibition of ER stress and/or endocytosis can be a potential therapeutic approach for MG.
Muscular Dystrophy
Muscular dystrophy is a group of skeletal muscle diseases that involves progressive degeneration of myofibers, myopathy, and fibrosis. Muscular dystrophy arises due to mutations in individual genes that encode extracellular matrix proteins, transmembrane and membrane-associated proteins, cytoplasmic enzymes, and nuclear matrix proteins [154, 155]. Duchenne muscular dystrophy (DMD) is one of the most prevalent forms of muscular dystrophies that results from total or partial deficiency of dystrophin protein [155]. Pathogenesis of DMD includes an inflammatory response, loss of calcium homeostasis, hypoxia, and oxidative stress, all of which can cause ER stress [32, 154]. Dystrophin-deficient mdx mice are widely used as a model to study the pathogenesis of DMD. ER stress markers, such as: GRP78, PERK, eIF2α, IRE1α, and sXBP1 and caspase-12, are increased in dystrophic muscle of mdx mice as well as in muscle biopsies from patients with DMD suggesting that dystrophin-deficiency disrupts ER homeostasis in skeletal muscle [156, 157]. ER stress specifically activates caspase-12, which leads to the activation of caspase-9 and caspase-3 to induce apoptosis. Interestingly, genetic ablation of caspase-12 significantly reduces myofiber degeneration and improves skeletal muscle strength in mdx mice [157]. Another study recently demonstrated that ER stress inhibitors restore ER-mitochondria links, mitochondrial Ca2+ uptake, and improve contractility of the diaphragm in mdx mice [158] further implying that heightened ER stress and UPR pathways contribute to the dystrophic phenotype.
Myotonic dystrophy (DM1) is an inherited autosomal-dominant muscular dystrophy caused by an abnormal amount of CTG trinucleotide repeats in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) gene. The pathogenesis of DM1 involves conduction defects, myofiber atrophy, and abnormal Ca2+ homeostasis. Ikezoe et al analyzed the markers of ER stress and the UPR in skeletal muscle of patients with DM1. They reported a significant increase in the levels of GRP78 and calnexin, and phosphorylation of PERK and eIF2α in atrophic myofibers of DM1 patients suggesting that enhanced ER stress may contribute to muscle wasting in DM1 patients [159]. There are also reports suggesting that ER stress is increased in dysferlin-related and collagen VI-related myopathies [160–162].
Tibial muscular dystrophy (TMD) is an autosomal dominant late-onset distal myopathy that results from heterogeneous mutations in the last two exons of the Titin gene. TMD is characterized by myofiber atrophy and weakness in the skeletal muscle of the anterior compartment of the lower leg [163]. Gene profiling studies provided evidence that ER stress could be one of the potential reasons for muscle weakness in TMD patients [164]. Altogether, available literature suggests that ER stress and the UPR could be important players in the pathogenesis of various types of muscular dystrophy.
Concluding Remarks
ER stress and the UPR were initially viewed as a pathological events that occurred downstream of primary etiology. However, studies using genetically modified animals and small molecules that selectively target specific mediators have revealed that the UPR can have distinct outcomes in different diseases and when different physiological components are manipulated. Moreover, the UPR can have opposite effects depending on the disease stage. Initially, the UPR may work as a pro-survival factor to sustain proteostasis, but its chronic activation can lead to cell damage during late symptomatic phases. Like many other organs, ER stress and the UPR are activated in skeletal muscle in multiple conditions. However, due to the complexity of multiple UPR pathways, signaling cross-talk, and several downstream targets, their exact role in skeletal muscle remains less understood. Available evidence suggests that the UPR promotes skeletal muscle formation and adaptation in response to exercise. Low levels of ER stress may also be required for maintaining the pool of satellite cells in adult skeletal muscle. While there are several studies suggesting that ER stress and the UPR pathways are perturbed in skeletal muscle under catabolic states, it remains unknown how these pathways regulate skeletal muscle mass. Consistent with the notion that chronic unresolved stress can lead to insulin resistance, inflammation, and cell death, the markers of ER stress have been found to be elevated in models of metabolic disorders and in a number of myopathies. Indeed, most of the published reports indicate that ER stress contributes to skeletal muscle wasting in disease conditions. Altogether, it is increasingly evident that the modulation of ER stress and the UPR can have therapeutic importance for skeletal muscle diseases. A number of ER stress activators and inhibitors have now been developed and can potentially be used for treatment of skeletal muscle diseases as well.
There are also several outstanding questions about the role of ER stress and the UPR in the regulation of skeletal muscle mass. For example, it is of critical importance to examine the role of individual arms of the UPR in skeletal muscle physiology and various disease conditions using genetic mouse models. Similarly, the role of the downstream effectors of the UPR in skeletal muscle needs to be investigated using molecular and genetic approaches. It is also increasingly evidenced that some of the components of UPR can be activated independent of ER stress and modulate the activity of other signaling pathways. Therefore, signaling cross-talk of UPR with other intracellular pathways that are involved in regulation of skeletal muscle mass, needs to be further investigated. Finally, the role of UPR in metabolic perturbations in skeletal muscle including oxidative phosphorylation should be investigated using molecular, genetic, and proteomics approaches. Nevertheless, available literature provides compelling evidence that ER stress and the UPR pathways regulate skeletal muscle health and disease. With the availability of genetic mouse models and specific inhibitors and activators of various components of the UPR, there is a renewed interest towards understanding the role and mechanisms of action of the UPR in skeletal muscle biology.
Acknowledgments
FUNDING SOURCE: This work was supported by the U.S. National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases Grants AR059810 and AR068313 and National Institute on Aging Grant AG029623 (to A.K.).
Authors thank members of Ashok Kumar’s laboratory for their critically reading the manuscript. We appreciate Dr. Guangyan Xiong for help with figures. We apologize to those investigators whose work has not been cited due to space constraints or oversight.
ABBREVIATIONS
- 4-PBA
4-phenylbutyrate
- AMPK
5′ AMP-activated protein kinase
- ATF
Activating transcription factor
- BiP
immunoglobulin heavy chain binding protein
- CHOP
C/EBP homologous protein
- DMD
Duchenne muscular dystrophy
- eIF2α
Eukaryotic translation initiation factor 2α
- ER
Endoplasmic reticulum
- FGF21
Fibroblast growth factor 21
- GADD34
Growth arrest and DNA damage-inducible protein
- GRP
Glucose-regulating protein
- HFD
High-fat diet
- IBM
inclusion body myositis
- IRE1
Inositol-requiring protein 1
- ISR
integrated stress response
- JNK
c-Jun N-terminal kinase
- LLC
Lewis lung carcinoma
- mTOR
mammalian target of rapamycin
- MAPK
Mitogen-activated protein kinase
- NF-κB
Nuclear factor-kappa B
- PERK
protein kinase R (PKR)-like endoplasmic reticulum kinase
- PI3K
Phosphoinositide 3-kinase
- RyR1
Ryanodine receptor type 1
- sXBP1
Spliced X-box-binding protein 1
- TRAF6
Tumor necrosis factor receptor associated factor 6
- TRB3
tribbles homolog 3
- UPR
Unfolding protein response
- UPS
Ubiquitin-proteasome system
- XBP1
X-box-binding protein
Footnotes
Conflict of interest statement: The authors report no conflicts of interest.
AUTHOR CONTRIBUTIONS: Both authors contributed equally to the conceptualization and writing of the manuscript.
References
- 1.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–65. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 2.Egerman MA, Glass DJ. Signaling pathways controlling skeletal muscle mass. Crit Rev Biochem Mol Biol. 2014;49:59–68. doi: 10.3109/10409238.2013.857291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glass DJ. Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol. 2005;37:1974–84. doi: 10.1016/j.biocel.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 4.Shin JY, Mendez-Lopez I, Wang Y, Hays AP, Tanji K, Lefkowitch JH, Schulze PC, Worman HJ, Dauer WT. Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev Cell. 2013;26:591–603. doi: 10.1016/j.devcel.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17:829–38. doi: 10.1038/ncb3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 8.Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer. 2014;14:581–97. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 9.Michalak M, Robert Parker JM, Opas M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 2002;32:269–78. doi: 10.1016/s0143416002001884. [DOI] [PubMed] [Google Scholar]
- 10.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ. 2006;13:374–84. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 11.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 12.Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–65. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- 13.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 14.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flamment M, Hajduch E, Ferre P, Foufelle F. New insights into ER stress-induced insulin resistance. Trends Endocrinol Metab. 2012;23:381–90. doi: 10.1016/j.tem.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–24. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–74. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–7. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 21.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 22.Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–76. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 23.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–7. [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. 2005;115:268–81. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–76. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto K, Takahara K, Oyadomari S, Okada T, Sato T, Harada A, Mori K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–86. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mollereau B, Manie S, Napoletano F. Getting the better of ER stress. J Cell Commun Signal. 2014;8:311–21. doi: 10.1007/s12079-014-0251-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–8. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, Marciniak SJ, Goodall JC, Green AR, Wouters BG, Wienholds E, Dick JE. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510:268–72. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 31.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–9. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 32.Bohnert KR, McMillan JD, Kumar A. Emerging roles of ER stress and unfolded protein response pathways in skeletal muscle health and disease. J Cell Physiol. 2018;233:67–78. doi: 10.1002/jcp.25852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rayavarapu S, Coley W, Nagaraju K. Endoplasmic reticulum stress in skeletal muscle homeostasis and disease. Curr Rheumatol Rep. 2012;14:238–43. doi: 10.1007/s11926-012-0247-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deldicque L. Endoplasmic reticulum stress in human skeletal muscle: any contribution to sarcopenia? Front Physiol. 2013;4:236. doi: 10.3389/fphys.2013.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentzinger CF, Wang YX, Rudnicki MA. Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Relaix F, Zammit PS. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–56. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- 37.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiological reviews. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F. The formation of skeletal muscle: from somite to limb. J Anat. 2003;202:59–68. doi: 10.1046/j.1469-7580.2003.00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olson EN. Interplay between proliferation and differentiation within the myogenic lineage. Dev Biol. 1992;154:261–72. doi: 10.1016/0012-1606(92)90066-p. [DOI] [PubMed] [Google Scholar]
- 40.Fidzianska A, Goebel HH. Human ontogenesis. 3. Cell death in fetal muscle. Acta Neuropathol. 1991;81:572–7. doi: 10.1007/BF00310140. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–60. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakanishi K, Dohmae N, Morishima N. Endoplasmic reticulum stress increases myofiber formation in vitro. FASEB J. 2007;21:2994–3003. doi: 10.1096/fj.06-6408com. [DOI] [PubMed] [Google Scholar]
- 43.Alter J, Bengal E. Stress-induced C/EBP homology protein (CHOP) represses MyoD transcription to delay myoblast differentiation. PLoS One. 2011;6:e29498. doi: 10.1371/journal.pone.0029498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blais A, Tsikitis M, Acosta-Alvear D, Sharan R, Kluger Y, Dynlacht BD. An initial blueprint for myogenic differentiation. Genes Dev. 2005;19:553–69. doi: 10.1101/gad.1281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y, Dynlacht BD. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell. 2007;27:53–66. doi: 10.1016/j.molcel.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 46.Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas AE, Crist C. Phosphorylation of eIF2alpha Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell. 2016;18:79–90. doi: 10.1016/j.stem.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 47.Xiong G, Hindi SM, Mann AK, Gallot YS, Bohnert KR, Cavener DR, Whittemore SR, Kumar A. The PERK arm of the unfolded protein response regulates satellite cell-mediated skeletal muscle regeneration. Elife. 2017:6. doi: 10.7554/eLife.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang B, Zhou J, Banie L, Reed-Maldonado AB, Ning H, Lu Z, Ruan Y, Zhou T, Wang HS, Oh BS, Wang G, Qi SL, Lin G, Lue TF. Low-intensity extracorporeal shock wave therapy promotes myogenesis through PERK/ATF4 pathway. Neurourol Urodyn. 2017 doi: 10.1002/nau.23380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab. 2009;20:436–43. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616–29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004;23:169–79. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cavener DR, Gupta S, McGrath BC. PERK in beta cell biology and insulin biogenesis. Trends Endocrinol Metab. 2010;21:714–21. doi: 10.1016/j.tem.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–84. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 54.Miyake M, Nomura A, Ogura A, Takehana K, Kitahara Y, Takahara K, Tsugawa K, Miyamoto C, Miura N, Sato R, Kurahashi K, Harding HP, Oyadomari M, Ron D, Oyadomari S. Skeletal muscle-specific eukaryotic translation initiation factor 2alpha phosphorylation controls amino acid metabolism and fibroblast growth factor 21-mediated non-cell-autonomous energy metabolism. FASEB J. 2016;30:798–812. doi: 10.1096/fj.15-275990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Bostrom P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab. 2011;13:160–9. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mesbah Moosavi ZS, Hood DA. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am J Physiol Cell Physiol. 2017;312:C583–C594. doi: 10.1152/ajpcell.00320.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pereira BC, da Rocha AL, Pinto AP, Pauli JR, de Souza CT, Cintra DE, Ropelle ER, de Freitas EC, Zagatto AM, da Silva AS. Excessive eccentric exercise-induced overtraining model leads to endoplasmic reticulum stress in mice skeletal muscles. Life Sci. 2016;145:144–51. doi: 10.1016/j.lfs.2015.12.037. [DOI] [PubMed] [Google Scholar]
- 58.Memme JM, Oliveira AN, Hood DA. Chronology of UPR activation in skeletal muscle adaptations to chronic contractile activity. Am J Physiol Cell Physiol. 2016;310:C1024–36. doi: 10.1152/ajpcell.00009.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim HJ, Jamart C, Deldicque L, An GL, Lee YH, Kim CK, Raymackers JM, Francaux M. Endoplasmic reticulum stress markers and ubiquitin-proteasome pathway activity in response to a 200-km run. Med Sci Sports Exerc. 2011;43:18–25. doi: 10.1249/MSS.0b013e3181e4c5d1. [DOI] [PubMed] [Google Scholar]
- 60.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem. 2007;282:30014–21. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 61.Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 62.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J. 2002;16:1879–86. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 63.Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino JP, Deriaz O. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes. 2003;52:2874–81. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- 64.Calvo JA, Daniels TG, Wang X, Paul A, Lin J, Spiegelman BM, Stevenson SC, Rangwala SM. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J Appl Physiol (1985) 2008;104:1304–12. doi: 10.1152/japplphysiol.01231.2007. [DOI] [PubMed] [Google Scholar]
- 65.Miyazaki M, McCarthy JJ, Fedele MJ, Esser KA. Early activation of mTORC1 signalling in response to mechanical overload is independent of phosphoinositide 3-kinase/Akt signalling. J Physiol. 2011;589:1831–46. doi: 10.1113/jphysiol.2011.205658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Serrano AL, Baeza-Raja B, Perdiguero E, Jardi M, Munoz-Canoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008;7:33–44. doi: 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 67.Armstrong DD, Esser KA. Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol Cell Physiol. 2005;289:C853–9. doi: 10.1152/ajpcell.00093.2005. [DOI] [PubMed] [Google Scholar]
- 68.Hamilton DL, Philp A, MacKenzie MG, Patton A, Towler MC, Gallagher IJ, Bodine SC, Baar K. Molecular brakes regulating mTORC1 activation in skeletal muscle following synergist ablation. Am J Physiol Endocrinol Metab. 2014;307:E365–73. doi: 10.1152/ajpendo.00674.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ogborn DI, McKay BR, Crane JD, Parise G, Tarnopolsky MA. The unfolded protein response is triggered following a single, unaccustomed resistance-exercise bout. Am J Physiol Regul Integr Comp Physiol. 2014;307:R664–9. doi: 10.1152/ajpregu.00511.2013. [DOI] [PubMed] [Google Scholar]
- 70.Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol. 2001;3:1014–9. doi: 10.1038/ncb1101-1014. [DOI] [PubMed] [Google Scholar]
- 72.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–83. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 73.Tajrishi MM, Zheng TS, Burkly LC, Kumar A. The TWEAK-Fn14 pathway: a potent regulator of skeletal muscle biology in health and disease. Cytokine Growth Factor Rev. 2014;25:215–25. doi: 10.1016/j.cytogfr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–23. doi: 10.1172/JCI200418330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen S, Brault JJ, Gygi SP, Glass DJ, Valenzuela DM, Gartner C, Latres E, Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol. 2009;185:1083–95. doi: 10.1083/jcb.200901052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab. 2014;307:E469–84. doi: 10.1152/ajpendo.00204.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, Stricker S, Goldberg AL, Dupont S, Piccolo S, Amthor H, Sandri M. BMP signaling controls muscle mass. Nat Genet. 2013;45:1309–18. doi: 10.1038/ng.2772. [DOI] [PubMed] [Google Scholar]
- 78.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 79.Paul PK, Bhatnagar S, Mishra V, Srivastava S, Darnay BG, Choi Y, Kumar A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol Cell Biol. 2012;32:1248–59. doi: 10.1128/MCB.06351-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, Kumar A. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191:1395–411. doi: 10.1083/jcb.201006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–6. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 82.Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell reports. 2014;8:1509–21. doi: 10.1016/j.celrep.2014.07.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–15. doi: 10.1016/j.cmet.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 84.Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–70. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- 85.Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, Cavallini G, Bonelli G, Baccino FM, Costelli P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol. 2013;182:1367–78. doi: 10.1016/j.ajpath.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 87.Sandri M. Protein breakdown in cancer cachexia. Semin Cell Dev Biol. 2015 doi: 10.1016/j.semcdb.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 88.Bartoli M, Richard I. Calpains in muscle wasting. Int J Biochem Cell Biol. 2005;37:2115–33. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 89.Li H, Malhotra S, Kumar A. Nuclear factor-kappa B signaling in skeletal muscle atrophy. J Mol Med (Berl) 2008;86:1113–26. doi: 10.1007/s00109-008-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Romanello V, Sandri M. Mitochondrial Quality Control and Muscle Mass Maintenance. Front Physiol. 2015;6:422. doi: 10.3389/fphys.2015.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–85. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiao G, Hao L, Wang M, Zhong B, Yu M, Zhao S, Wang P, Feng R, Tan S, Chen L. Upregulation of endoplasmic reticulum stress is associated with diaphragm contractile dysfunction in a rat model of sepsis. Mol Med Rep. 2017;15:366–374. doi: 10.3892/mmr.2016.6014. [DOI] [PubMed] [Google Scholar]
- 94.Hunter RB, Mitchell-Felton H, Essig DA, Kandarian SC. Expression of endoplasmic reticulum stress proteins during skeletal muscle disuse atrophy. Am J Physiol Cell Physiol. 2001;281:C1285–90. doi: 10.1152/ajpcell.2001.281.4.C1285. [DOI] [PubMed] [Google Scholar]
- 95.Baehr LM, West DW, Marcotte G, Marshall AG, De Sousa LG, Baar K, Bodine SC. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 2016;8:127–46. doi: 10.18632/aging.100879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chalil S, Pierre N, Bakker AD, Manders RJ, Pletsers A, Francaux M, Klein-Nulend J, Jaspers RT, Deldicque L. Aging related ER stress is not responsible for anabolic resistance in mouse skeletal muscle. Biochem Biophys Res Commun. 2015 doi: 10.1016/j.bbrc.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 97.Ogata T, Machida S, Oishi Y, Higuchi M, Muraoka I. Differential cell death regulation between adult-unloaded and aged rat soleus muscle. Mech Ageing Dev. 2009;130:328–36. doi: 10.1016/j.mad.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 98.O’Leary MF, Vainshtein A, Iqbal S, Ostojic O, Hood DA. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304:C422–30. doi: 10.1152/ajpcell.00240.2012. [DOI] [PubMed] [Google Scholar]
- 99.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dasarathy S, Merli M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J Hepatol. 2016;65:1232–1244. doi: 10.1016/j.jhep.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dasarathy S, Hatzoglou M. Hyperammonemia and proteostasis in cirrhosis. Curr Opin Clin Nutr Metab Care. 2017 doi: 10.1097/MCO.0000000000000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davuluri G, Krokowski D, Guan BJ, Kumar A, Thapaliya S, Singh D, Hatzoglou M, Dasarathy S. Metabolic adaptation of skeletal muscle to hyperammonemia drives the beneficial effects of l-leucine in cirrhosis. J Hepatol. 2016;65:929–937. doi: 10.1016/j.jhep.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Miyake M, Kuroda M, Kiyonari H, Takehana K, Hisanaga S, Morimoto M, Zhang J, Oyadomari M, Sakaue H, Oyadomari S. Ligand-induced rapid skeletal muscle atrophy in HSA-Fv2E-PERK transgenic mice. PLoS One. 2017;12:e0179955. doi: 10.1371/journal.pone.0179955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, Robins DM, Lieberman AP. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011;7:e1002321. doi: 10.1371/journal.pgen.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen D, Wang Y, Chin ER. Activation of the endoplasmic reticulum stress response in skeletal muscle of G93A*SOD1 amyotrophic lateral sclerosis mice. Front Cell Neurosci. 2015;9:170. doi: 10.3389/fncel.2015.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, Martinez G, Cuervo AM, Brown RH, Glimcher LH. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest. 2015;125:3303. doi: 10.1172/JCI82799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bohnert KR, Gallot YS, Sato S, Xiong G, Hindi SM, Kumar A. Inhibition of ER stress and unfolding protein response pathways causes skeletal muscle wasting during cancer cachexia. FASEB J. 2016;30:3053–68. doi: 10.1096/fj.201600250RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK, Dierdorff JM, Foster ED, Adams CM. Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem. 2012;287:27290–301. doi: 10.1074/jbc.M112.374777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma L, Chu W, Chai J, Shen C, Li D, Wang X. ER stress and subsequent activated calpain play a pivotal role in skeletal muscle wasting after severe burn injury. PLoS One. 2017;12:e0186128. doi: 10.1371/journal.pone.0186128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee CS, Hanna AD, Wang H, Dagnino-Acosta A, Joshi AD, Knoblauch M, Xia Y, Georgiou DK, Xu J, Long C, Amano H, Reynolds C, Dong K, Martin JC, Lagor WR, Rodney GG, Sahin E, Sewry C, Hamilton SL. A chemical chaperone improves muscle function in mice with a RyR1 mutation. Nat Commun. 2017;8:14659. doi: 10.1038/ncomms14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Madaro L, Marrocco V, Carnio S, Sandri M, Bouche M. Intracellular signaling in ER stress-induced autophagy in skeletal muscle cells. FASEB J. 2013;27:1990–2000. doi: 10.1096/fj.12-215475. [DOI] [PubMed] [Google Scholar]
- 114.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–71. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Solinas G, Becattini B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol Metab. 2017;6:174–184. doi: 10.1016/j.molmet.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 117.Deldicque L, Cani PD, Philp A, Raymackers JM, Meakin PJ, Ashford ML, Delzenne NM, Francaux M, Baar K. The unfolded protein response is activated in skeletal muscle by high-fat feeding: potential role in the downregulation of protein synthesis. Am J Physiol Endocrinol Metab. 2010;299:E695–705. doi: 10.1152/ajpendo.00038.2010. [DOI] [PubMed] [Google Scholar]
- 118.Rieusset J, Chauvin MA, Durand A, Bravard A, Laugerette F, Michalski MC, Vidal H. Reduction of endoplasmic reticulum stress using chemical chaperones or Grp78 overexpression does not protect muscle cells from palmitate-induced insulin resistance. Biochem Biophys Res Commun. 2012;417:439–45. doi: 10.1016/j.bbrc.2011.11.135. [DOI] [PubMed] [Google Scholar]
- 119.Eyers PA, Keeshan K, Kannan N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2016 doi: 10.1016/j.tcb.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300:1574–7. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 121.Liu J, Wu X, Franklin JL, Messina JL, Hill HS, Moellering DR, Walton RG, Martin M, Garvey WT. Mammalian Tribbles homolog 3 impairs insulin action in skeletal muscle: role in glucose-induced insulin resistance. Am J Physiol Endocrinol Metab. 2010;298:E565–76. doi: 10.1152/ajpendo.00467.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–27. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Koh HJ, Toyoda T, Didesch MM, Lee MY, Sleeman MW, Kulkarni RN, Musi N, Hirshman MF, Goodyear LJ. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat Commun. 2013;4:1871. doi: 10.1038/ncomms2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hayashi T, Hirshman MF, Fujii N, Habinowski SA, Witters LA, Goodyear LJ. Metabolic stress and altered glucose transport: activation of AMP-activated protein kinase as a unifying coupling mechanism. Diabetes. 2000;49:527–31. doi: 10.2337/diabetes.49.4.527. [DOI] [PubMed] [Google Scholar]
- 125.Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–8. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hwang SL, Jeong YT, Li X, Kim YD, Lu Y, Chang YC, Lee IK, Chang HW. Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br J Pharmacol. 2013;169:69–81. doi: 10.1111/bph.12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kwak HJ, Choi HE, Jang J, Park SK, Bae YA, Cheon HG. Bortezomib attenuates palmitic acid-induced ER stress, inflammation and insulin resistance in myotubes via AMPK dependent mechanism. Cell Signal. 2016;28:788–97. doi: 10.1016/j.cellsig.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 128.Greenberg AS, Obin MS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. 2006;83:461S–465S. doi: 10.1093/ajcn/83.2.461S. [DOI] [PubMed] [Google Scholar]
- 129.Pierre N, Deldicque L, Barbe C, Naslain D, Cani PD, Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One. 2013;8:e65061. doi: 10.1371/journal.pone.0065061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–59. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 131.Salvado L, Barroso E, Gomez-Foix AM, Palomer X, Michalik L, Wahli W, Vazquez-Carrera M. PPARbeta/delta prevents endoplasmic reticulum stress-associated inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia. 2014;57:2126–35. doi: 10.1007/s00125-014-3331-8. [DOI] [PubMed] [Google Scholar]
- 132.Dai F, Jiang T, Bao YY, Chen GJ, Chen L, Zhang Q, Lu YX. Fenofibrate improves high-fat diet-induced and palmitate-induced endoplasmic reticulum stress and inflammation in skeletal muscle. Life Sci. 2016;157:158–67. doi: 10.1016/j.lfs.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 133.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee MS. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 134.Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–40. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guridi M, Tintignac LA, Lin S, Kupr B, Castets P, Ruegg MA. Activation of mTORC1 in skeletal muscle regulates whole-body metabolism through FGF21. Sci Signal. 2015;8:ra113. doi: 10.1126/scisignal.aab3715. [DOI] [PubMed] [Google Scholar]
- 136.Lundberg IE. Idiopathic inflammatory myopathies: why do the muscles become weak? Curr Opin Rheumatol. 2001;13:457–60. doi: 10.1097/00002281-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 137.Vitadello M, Doria A, Tarricone E, Ghirardello A, Gorza L. Myofiber stress-response in myositis: parallel investigations on patients and experimental animal models of muscle regeneration and systemic inflammation. Arthritis Res Ther. 2010;12:R52. doi: 10.1186/ar2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, Chang J, Dwivedi S, Mitsak M, Chen YW, Plotz P, Rosen A, Hoffman E, Raben N. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–35. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 139.Li CK, Knopp P, Moncrieffe H, Singh B, Shah S, Nagaraju K, Varsani H, Gao B, Wedderburn LR. Overexpression of MHC class I heavy chain protein in young skeletal muscle leads to severe myositis: implications for juvenile myositis. Am J Pathol. 2009;175:1030–40. doi: 10.2353/ajpath.2009.090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.DeVere R, Bradley WG. Polymyositis: its presentation, morbidity and mortality. Brain. 1975;98:637–66. doi: 10.1093/brain/98.4.637. [DOI] [PubMed] [Google Scholar]
- 141.Freret M, Drouot L, Obry A, Ahmed-Lacheheb S, Dauly C, Adriouch S, Cosette P, Authier FJ, Boyer O. Overexpression of MHC class I in muscle of lymphocyte-deficient mice causes a severe myopathy with induction of the unfolded protein response. Am J Pathol. 2013;183:893–904. doi: 10.1016/j.ajpath.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 142.Askanas V, Engel WK. Inclusion-body myositis and myopathies: different etiologies, possibly similar pathogenic mechanisms. Curr Opin Neurol. 2002;15:525–31. doi: 10.1097/00019052-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 143.Askanas V, Engel WK. Inclusion-body myositis: newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1–14. doi: 10.1093/jnen/60.1.1. [DOI] [PubMed] [Google Scholar]
- 144.Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nogalska A, D’Agostino C, Engel WK, Cacciottolo M, Asada S, Mori K, Askanas V. Activation of the Unfolded Protein Response in Sporadic Inclusion-Body Myositis but Not in Hereditary GNE Inclusion-Body Myopathy. J Neuropathol Exp Neurol. 2015;74:538–46. doi: 10.1097/NEN.0000000000000196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xiao F, Tan JZ, Xu XY, Wang XF. Increased levels of HSPA5 in the serum of patients with inflammatory myopathies--preliminary findings. Clin Rheumatol. 2015;34:715–20. doi: 10.1007/s10067-015-2911-4. [DOI] [PubMed] [Google Scholar]
- 147.Lightfoot AP, Nagaraju K, McArdle A, Cooper RG. Understanding the origin of non-immune cell-mediated weakness in the idiopathic inflammatory myopathies - potential role of ER stress pathways. Curr Opin Rheumatol. 2015;27:580–5. doi: 10.1097/BOR.0000000000000212. [DOI] [PubMed] [Google Scholar]
- 148.Witherspoon JW, Meilleur KG. Review of RyR1 pathway and associated pathomechanisms. Acta Neuropathol Commun. 2016;4:121. doi: 10.1186/s40478-016-0392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, Galvan DL, Gilman CP, Baker MR, Shirokova N, Protasi F, Dirksen R, Hamilton SL. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol. 2009;8:475–90. doi: 10.1016/S1474-4422(09)70063-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Iwasa K, Nambu Y, Motozaki Y, Furukawa Y, Yoshikawa H, Yamada M. Increased skeletal muscle expression of the endoplasmic reticulum chaperone GRP78 in patients with myasthenia gravis. J Neuroimmunol. 2014;273:72–6. doi: 10.1016/j.jneuroim.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 152.Suzuki S, Utsugisawa K, Iwasa K, Satoh T, Nagane Y, Yoshikawa H, Kuwana M, Suzuki N. Autoimmunity to endoplasmic reticulum chaperone GRP94 in myasthenia gravis. J Neuroimmunol. 2011;237:87–92. doi: 10.1016/j.jneuroim.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 153.Du A, Huang S, Zhao X, Zhang Y, Zhu L, Ding J, Xu C. Endoplasmic reticulum stress contributes to acetylcholine receptor degradation by promoting endocytosis in skeletal muscle cells. J Neuroimmunol. 2016;290:109–14. doi: 10.1016/j.jneuroim.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 154.Shin J, Tajrishi MM, Ogura Y, Kumar A. Wasting mechanisms in muscular dystrophy. Int J Biochem Cell Biol. 2013;45:2266–79. doi: 10.1016/j.biocel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–95. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 156.Hulmi JJ, Hentila J, DeRuisseau KC, Oliveira BM, Papaioannou KG, Autio R, Kujala UM, Ritvos O, Kainulainen H, Korkmaz A, Atalay M. Effects of muscular dystrophy, exercise and blocking activin receptor IIB ligands on the unfolded protein response and oxidative stress. Free Radic Biol Med. 2016;99:308–322. doi: 10.1016/j.freeradbiomed.2016.08.017. [DOI] [PubMed] [Google Scholar]
- 157.Moorwood C, Barton ER. Caspase-12 ablation preserves muscle function in the mdx mouse. Hum Mol Genet. 2014;23:5325–41. doi: 10.1093/hmg/ddu249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pauly M, Angebault-Prouteau C, Dridi H, Notarnicola C, Scheuermann V, Lacampagne A, Matecki S, Fauconnier J. ER stress disturbs SR/ER-mitochondria Ca2+ transfer: Implications in Duchenne muscular dystrophy. Biochim Biophys Acta. 2017;1863:2229–2239. doi: 10.1016/j.bbadis.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 159.Ikezoe K, Nakamori M, Furuya H, Arahata H, Kanemoto S, Kimura T, Imaizumi K, Takahashi MP, Sakoda S, Fujii N, Kira J. Endoplasmic reticulum stress in myotonic dystrophy type 1 muscle. Acta Neuropathol. 2007;114:527–35. doi: 10.1007/s00401-007-0267-9. [DOI] [PubMed] [Google Scholar]
- 160.Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, Hayashi YK, Momoi T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II) Hum Mol Genet. 2007;16:618–29. doi: 10.1093/hmg/ddm002. [DOI] [PubMed] [Google Scholar]
- 161.Ikezoe K, Furuya H, Ohyagi Y, Osoegawa M, Nishino I, Nonaka I, Kira J. Dysferlin expression in tubular aggregates: their possible relationship to endoplasmic reticulum stress. Acta Neuropathol. 2003;105:603–9. doi: 10.1007/s00401-003-0686-1. [DOI] [PubMed] [Google Scholar]
- 162.De Palma S, Capitanio D, Vasso M, Braghetta P, Scotton C, Bonaldo P, Lochmuller H, Muntoni F, Ferlini A, Gelfi C. Muscle proteomics reveals novel insights into the pathophysiological mechanisms of collagen VI myopathies. J Proteome Res. 2014;13:5022–30. doi: 10.1021/pr500675e. [DOI] [PubMed] [Google Scholar]
- 163.Udd B. Limb-girdle type muscular dystrophy in a large family with distal myopathy: homozygous manifestation of a dominant gene? J Med Genet. 1992;29:383–9. doi: 10.1136/jmg.29.6.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Screen M, Raheem O, Holmlund-Hampf J, Jonson PH, Huovinen S, Hackman P, Udd B. Gene expression profiling in tibial muscular dystrophy reveals unfolded protein response and altered autophagy. PLoS One. 2014;9:e90819. doi: 10.1371/journal.pone.0090819. [DOI] [PMC free article] [PubMed] [Google Scholar]