

Abstract
Conformational change and modification of proteins are involved in many cellular functions. However, they can also have adverse effects that are implicated in numerous diseases. How structural change promotes disease is generally not well understood. This perspective illustrates how mass spectrometry (MS), followed by toxicological and epidemiological validation, can discover disease-relevant structural changes and therapeutic strategies. We (with our collaborators) set out to characterize the structural and toxic consequences of disease-associated mutations and post-translational modifications (PTMs) of the cytosolic antioxidant protein Cu/Zn-Superoxide dismutase (SOD1). Previous genetic studies discovered > 180 different mutations in the SOD1 gene that caused familial (inherited) amyotrophic lateral sclerosis (fALS). Using HDX-MS, we determined that diverse disease-associated SOD1 mutations cause a common structural defect – perturbation of the SOD1 electrostatic loop. X-ray crystallographic studies had demonstrated that this leads to protein aggregation through a specific interaction between the electrostatic loop and an exposed beta-barrel edge strand. Using epidemiology methods, we then determined that decreased SOD1 stability and increased protein aggregation are powerful risk factors for fALS progression, with a combined hazard ratio > 300 (for comparison, a lifetime of smoking is associated with a hazard ratio of ∼15 for lung cancer). The resulting structural model of fALS etiology supported the hypothesis that some sporadic ALS (sALS, ∼80% of ALS is not associated with a gene defect) could be caused by post-translational protein modification of wild-type SOD1. We developed immunocapture antibodies and high sensitivity top-down MS methods, and characterized PTMs of wild-type SOD1 using human tissue samples. Using global-HDX, X-ray crystallography, and neurotoxicology we then characterized toxic and protective subsets of SOD1 PTMs. To cap this perspective, we present proof-of-concept that post-translational modification can cause disease. We show that numerous mutations (N→D; Q→E), which result in the same chemical structure as the PTM deamidation, cause multiple diseases.
Introduction
Most human proteins are post-translationally modified, often in more than one way, from simple N-terminal acetylation to complex phosphorylation, lipidation, and glycosylation patterns.[1] These processes, among others, result in multiple natural proteoforms of each protein.[2] Conversely, aberrant protein modifications can result from changes in post-translational processing and protein catabolism, xenobiotics (e.g. nutrients, pesticides, pharmaceuticals), and genetic mutation. Changes in protein amino acid composition can lead to changes in their secondary, tertiary, and quaternary structures, including aggregation. For example, modifications as small as a replacing a hydrogen atom with a methyl group (G93A) lead to exposure of a toxic epitope within the Cu/Zn-Superoxide dismutase (SOD1) protein,[3] and a highly penetrant (all carriers develop the disease), rapidly progressing (3.1 years mean survival) [4] form of amyotrophic lateral sclerosis (ALS). Modifications can result in loss-of-function (e.g. the metabolic diseases including Myotonia congenital [5] and the lysosomal storage diseases (LSDs) [6]) or a gain-of-function, which can either augment physiological processes or be cytotoxic.[7, 8]
The consequence of protein modifications depends upon evolutionary context: mutations in the gene encoding hemoglobin protect carriers from malaria while causing sickle-cell anemia. Likewise, the myriad of mutations that cause late-onset diseases were not manifest until human survival increased significantly (more than doubled [9]) during the 19th and 20th centuries. A testament to the disease-relevance of aberrantly modified proteins is that they are histopathological hallmarks of the “proteinopathies.” These include some of the most prevalent – often late onset – diseases. Modified proteins define, to name a few, the Tauopathies (Alzheimer's, Pick's complex, chronic traumatic encephalopathy, etc. [10]), Synucleinopathies (Parkinson's, dementia with Lewy bodies, multiple system atrophy [11]), and secondary Ubiquitinopathies (lysosomal storage disorders, amyotrophic lateral sclerosis, etc. [12-14]).
Amyotrophic lateral sclerosis (ALS), as well as Parkinson's disease (PD) and Alzheimer's disease (AD), are syndromic diseases. Unknown causes, as well as a variety of mutations, somehow converge upon the death of disease-specific populations of selectively vulnerable neurons (e.g. the death of brain and spinal cord motor neurons in ALS). The molecular mechanisms of cell death, including whether common pathways are involved (e.g. proteostasis or RNA processing), is unknown and research is fundamentally limited by the following problems. The majority of disease is idiopathic/sporadic (i.e., no discernable genetic basis) and is not strongly associated with environment (e.g. behavior or xenobiotics). Thus, idiopathic disease cannot be recapitulated in laboratory models, and since biopsy is not an option, studies must involve post-mortem human tissue samples. Unfortunately, most changes in observed end-stage neurodegenerative disease tissues are epiphenomena (e.g. most cells of interest are already dead and inflammation dominates). A minority of disease cases are familial and further subdivided into a myriad of mutations in multiple genes. These can be used to create suitable transgenic laboratory models of disease, with the major caveat of potentially not representing the disease as a whole, and the major benefits of permitting longitudinal disease studies and facilitating drug development.
Research must strike a balance between human tissue samples, which best represent disease variation but underrepresent causative mechanisms, and animal models, which do the opposite. In either case, we use mass spectrometry (MS) to compare disease and control preparations, and validate the MS findings using secondary methods (e.g. toxicology, phenotype, epidemiology, familial diseases models, etc.). We primarily studied SOD1-mediated ALS, which is an adult onset, rapidly progressive (generally ∼2-5 years survival) motor neuron disease that affects ∼ 20,000 Americans at any given time, and results in the death of ∼1/1000 humans (for review see Taylor et al. [15]). Voluntary muscle control is lost as motor neurons in the brain and spinal cord degenerate by a yet-uncharacterized mechanism. The first genetic cause of ALS was found in the gene encoding SOD1.[16] Since this time, more than 180 mutations in the SOD1 gene have been linked to familial ALS (fALS) (http://alsod.iop.kcl.ac.uk/). Like most dominantly inherited diseases, SOD1 fALS results from a toxic gain-of-function rather than the loss of SOD1 enzymatic activity. This new toxic function does not appear to be related to SOD1's canonical function (dismutase activity). One of the leading hypotheses is that the toxic gain-of-function is a modified conformation that leads to SOD1 dimer dissociation [3, 17, 18] followed by aggregation.[3, 19, 20] In the first part of this perspective we demonstrate that diverse SOD1 mutations result in a common structural consequence that can lead to toxic protein aggregation.
Recent evidence, including our collaborative work, extended this association, linking modified wild type SOD1 (SOD1WT) to sporadic ALS (sALS) [16, 21] (for dissenting opinions see [22, 23]). This perspective outlines our hypothesis-driven approach to characterizing the structural and toxic consequences of mutations in SOD1 that cause a subset of fALS, and studies of modifications of the same protein in sporadic ALS. SOD1 mutations are found in only 2-7 % of ALS cases (e.g. ∼ 6 % of US cases), (http://alsod.iop.kcl.ac.uk/)[24] and there are other prevalent mutations that were not studied (e.g. C9orf72, TDP-43, etc.).[16, 25, 26] We concentrated on SOD1-ALS mutations because they cause – are not just associated with – ALS, and because they have diverse, well-characterized severity. For example, carriers of a single allele of either the A4V or H46R SOD1 mutation that live a normal lifespan unfortunately develop ALS. Whereas A4V patients survive an average of 1.2 years after the onset, H46R patients survive an average of 18 years.[4] This allows the structural changes we observe in fALS variants to be rank-ordered in terms of disease severity.
One hypothesis for how modified SOD1WT can lead to sALS asserts that modifications that affect surface charge such as phosphorylation, deamidation, and oxidation, can lead to toxic conformations,[3] instability, and misfolding.[27, 28] Indeed, certain mutations and PTMs induce similar – even identical – structural change. For example, mutations are often used as a mock-PTM (e.g., converting serine or threonine to aspartic acid to imitate permanent phosphorylation). Due to structural similarities between their mutationally- and post-translationally-modified proteoforms, certain proteins (including all of the histopathological hallmarks discussed above) have been considered as possible links between familial and sporadic forms of disease. These include Amyloid-beta [29] and Tau [30] (AD), alpha-synuclein [31] and Parkin [32] (PD), and TDP-43 [33] and SOD1 [21,t145t143 34-36] (ALS). Although there is considerable support for the hypothesis that PTMs can be responsible for sporadic disease,[21, 32, 37, 38] this has never been proven. In the second part of this perspective we present evidence of both toxic and protective (oxidation [3, 28] and cysteinylation,[39] respectively) PTMs in human tissue, and how these informed our current therapeutic strategies. We punctuate this perspective with proof that PTMs can cause disease, if occurring on a large enough scale, by illustrating a variety of diseases (see Table 1 at the end of Results & Discussion) that are caused by mutations that result in a structure that is equivalent to that of post-translational deamidation.
Table 1.
Disease-associated mutations that cause protein deamidation.
| Disease | Protein | Mutation |
|---|---|---|
| ALS | SOD1 | Asn86Asp |
| Alzheimer | Presenilin | Asn135Asp[1] |
| Autosomal Recessive Polycystic Kidney Disease | PKHD1 | Asn3175Asp[2] |
| Neuronal Ceroid Lipofuscinosis | PPT1CNL8 | Gln177Glu[3] Gln256Glu[4] |
| Fanconi Anemia | FANCA | Gln1128Glu[5] Gln1235Glu[6] |
| Waardenburg II Deafness | MITF | Asn278D[7] |
| Prostate Cancer | Androgen receptor | Asn222Asp; Asn756Asp[8] |
Crook, R., et al., Early-onset Alzheimer's disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann Neurol, 1997. 42(1): p. 124-8.
Furu, L., et al., Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol, 2003. 14(8): p. 2004-14.
Waliany, S., et al., Identification of three novel mutations of the palmitoyl-protein thioesterase-1 (PPT1) gene in children with neuronal ceroid-lipofuscinosis. Hum Mutat, 2000. 15(2): p. 206-7.
Zelnik, N., et al., A novel mutation of the CLN8 gene: is there a Mediterranean phenotype? Pediatr Neurol, 2007. 36(6): p. 411-3.
Levran, O., et al., Sequence variation in the Fanconi anemia gene FAA. Proc Natl Acad Sci U S A, 1997. 94(24): p. 13051-6.
Levran, O., et al., Spectrum of sequence variations in the FANCA gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat, 2005. 25(2): p. 142-9.
Tassabehji, M., et al., The mutational spectrum in Waardenburg syndrome. Hum Mol Genet, 1995. 4(11): p. 2131-7.
Gottlieb, B., et al., The androgen receptor gene mutations database. Nucleic Acids Res, 1997. 25(1): p. 158-62.
Results & Discussion
To understand most dominantly inherited diseases, and probably most neurodegenerative diseases, we must avoid the common misconception that disease results from the loss of a particular protein's canonical (normal) function. Instead, disease symptoms commonly result from a novel modification-induced function. We hypothesized this toxic-gain-of-function involved a change in SOD1 conformation and employed top-down MS to characterize the structural consequences of ALS-associated SOD1 modifications. Numerous 3D structures (NMR and X-ray), neurotoxicology assays, and epidemiology studies allowed our results to be placed into context, resulting in a structure-based model for SOD1-mediated ALS onset and progression. This knowledge was then translated into therapeutic strategies. This work was highly collaborative (see acknowledgments).
Top-down and ultrahigh resolution mass spectrometry methods to enrich the detection of disease-relevant modifications
Popular MS sample preparation techniques were designed for large-scale protein identification rather than discovering disease-relevant protein modifications. Most proteomics studies involve disulfide reduction, cysteine alkylation, and endoproteinase digestion.[40, 41] Reduction and alkylation result in loss of important prevalent in vivo cysteine modifications (e.g. disulfides, sulfenic acid, nitrosylation, and Sthiolation).[42] This problem greatly affects SOD1 – a disulfide-containing metalloprotein with variable S-thiolation – and all of these PTMs could be involved in ALS etiology. The endoproteinase digestion step results in: 1) loss of metal cofactors (∼30% of proteins are metalloproteins); 2) loss of correlation between PTMs (e.g., phosphorylation of one residue promotes methylation of a second residue); 3) loss of information concerning the relative abundance of PTMs;[43] and 4) scrambling of the sites of disulfide bonds and S-thiolation.[44] All of these problems apply to SOD1 and potentially ALS. The high sensitivity of mass spectrometers, which is generally an advantage, also increases the probability of detecting low-abundance modifications, including artifacts of sample preparation and proteoforms with concentrations that are too low to have a significant physiological effect.
Top-down MS techniques can overcome all of the problems described above: eliminating artifacts of reduction and alkylation, maintaining labile PTMs and their location, and providing information on their relative abundance and functional relationship.[45] In a series of manuscripts, we developed top-down techniques for PTM characterization. The most important of these include the following: high sensitivity immunoaffinity techniques;[20, 39] the debut of the Big Mascot (Mascot TD) search engine, which remains one of the few search engines capable of assigning internal fragment ions (which are much more abundant in top-down studies);[46] a comprehensive model of the dissociation pathways of intact protein collisionally activated dissociation;[47] and methods for detecting S-thiolation, a prevalent PTM that is either missed or location-scrambled in bottom-up studies.[42] Other groups have employed top-down MS to demonstrate the importance of characterizing PTMs to identify disease biomarkers, such as with chronic heart failure.[48] To better define the chemical composition of PTMs and to enable quantitative metabolic labeling experiments in future studies, we developed ultrahigh-resolution MS methods. In this series of manuscripts we presented fast and accurate algorithms and programs for modeling spectra with resolved isotopic fine structure.[49-52] Isotopic fine structure can be used, among other things, to determine the elemental composition of PTMs and to discern between isobaric modifications, including the S1 and O2 modifications we observe on the Cys111 residue of SOD1.
HDX-MS demonstrates that diverse SOD1 mutations result in a common structural perturbation
Given that hundreds of known fALS-related SOD1 mutations cause a similar disease phenotype, ALS, we hypothesized that SOD1 variants shared a common toxic conformation. Whereas biochemical studies[53] and solution x-ray scattering[54] showed that the properties of fALS-linked SOD1 variants differed significantly from SOD1WT, X-ray crystallographic studies of fALS variants showed little structural variation from SOD1WT.[55-58] It is well known that the crystallization process can favor stable conformations, which for fALS variants would lead to the underrepresentation of disease-relevant conformations in X-ray crystallography studies. To better understand the solution structures of fALS variants, we analyzed fourteen biochemically diverse ALS variants using hydrogen-deuterium exchange mass spectrometry (HDX-MS), described in Molnar 2009.[59] HDX-MS allows for protein secondary and tertiary structure to be examined in higher resolution than offered by circular dichroism or charge-state distribution MS, respectively. For a review from a leading HDX-MS group, see Wales et al.[60] We determined that all fALS variants share one thing in common – a perturbed electrostatic loop (Loop VII, residues 121-142, Figure 1). Several variants also had perturbations near the dimer interface and zinc binding loop. Note that the metal content of wild type SOD1 and each variant was determined by inductively coupled plasma-MS (ICP-MS), and while SOD1WT was found to be completely metallated, the variants were metal deficient to varying degrees as detailed in our previous work.[59]
Figure 1.
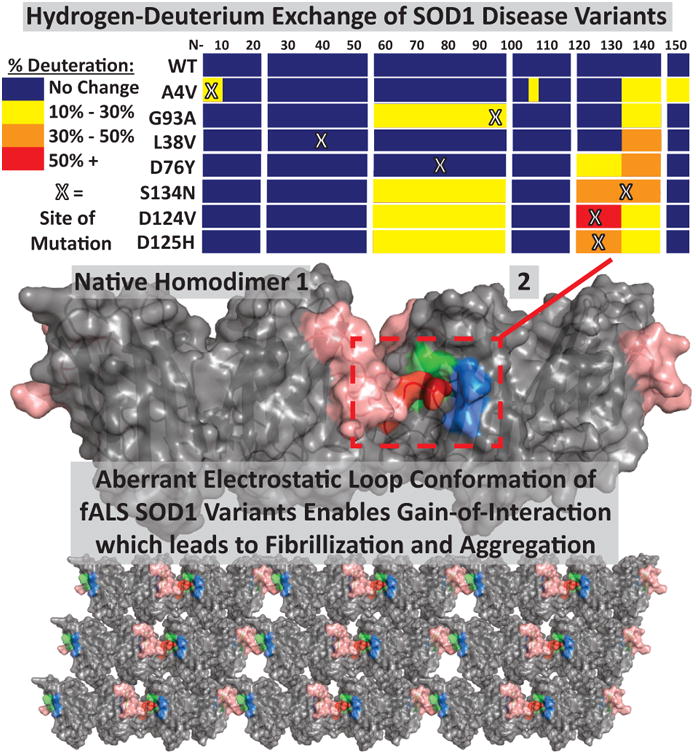
Hypothesized model for SOD1-mediated fALS disease progression. (Top) Mutations at various locations in SOD1, as demonstrated by HDX-MS in Molnar et al. 2009, lead to perturbation of the electrostatic loop (residues 121-142). (Middle) This perturbation exposes a toxic epitope, which enables a gain-of-interaction, possibly between a region of the electrostatic loop (red, rest of loop is pink) of one SOD1 homodimer and the exposed beta strand edges of beta strands V and VI (green and blue) of another SOD1 dimer, as demonstrated by the Hart group (Elam, J.S., et al. 2003, adapted from pdb entry 1OZU). (Bottom) This leads to aberrant interactions of SOD1 dimers, resulting in greater unnatural quaternary structure, fibrillization, and aggregation.
A structure-based mechanism of neurodegeneration: The survival of SOD1-mediated ALS patients is decreased by protein instability and protein aggregation
Before we demonstrated that fALS variants shared a common structural defect, perturbation of the electrostatic loop, the only universally shared characteristic was the formation of intracellular aggregates. Many researchers considered fALS SOD1 aggregates to be epiphenomena, perhaps even the result of a protective sequestering process. Likewise, some researchers asserted that fALS SOD1 variants were less stable than SOD1WT,[61] while other researchers found no correlation with stability and disease – noting that certain mutations had WT-like stability and activity.[62] Other neurodegenerative diseases were plagued by a similar lack of consensus as to whether aggregation was toxic, an epiphenomenon, or even protective. To address the importance of protein stability and aggregation in ALS, we assembled the largest fALS patient epidemiology database, the largest data set of SOD1 variant's stabilities, and the largest data set of protein aggregation rates. Using the theory introduced by Chris Dobson, we generated a model that could predict the rate of protein aggregation based upon the physicochemical changes induced by a protein modification (Δ charge, Δ hydrophobicity, Δ entropy).
In the first study of its kind, we used well-established epidemiology hazard/risk-analysis methods to model how protein physicochemical changes affect disease progression.[4] We found that decreased protein stability (hazard ratio of 24) and increased aggregation propensity (hazard ratio of 13) are risk factors for SOD1-fALS. Taken alone, the hazard-ratio of loss-of-stability (or aggregation propensity) with respect to SOD-fALS progression is similar to that of smoking with respect to lung cancer (hazard ratio ∼15).[63] Simply put, smokers have a 15-fold increased risk of dying from lung cancer, and the rate of fALS progression (severity) increases in proportion to both protein instability and aggregation propensity. Taken together, loss-of-stability and aggregation propensity have a hazard ratio of 333, indicating their effects are synergistic and potent. A model combining loss-of-stability and aggregation could account for more than two-thirds of the large variability in SOD1-fALS patient survival times. Given that SOD1 mutations are highly penetrant (i.e. generally accepted as causing fALS) and the physicochemical changes are irreducible (i.e. not prone to spurious associations), this model accounts for most of what causes fALS progression. Combined with the relatively high cellular concentrations of SOD1 in motor neurons, this model also provides a potential mechanism for the selective vulnerability of motor neurons in ALS. Although the use of epidemiology methods to qualify protein structural change was unprecedented and initially controversial, this work has since been confirmed in numerous ALS-related studies[64-66] and extended to animal models of AD.[67]
Combined with our structural characterization of fALS SOD1 variants and the X-ray crystallographic studies of the Hart group,[68] a mechanism of fALS etiology emerged. Diverse mutations in the SOD1 gene result in perturbation of the electrostatic loop and loss of stability, which promotes a novel interaction with an exposed beta-barrel. This novel interaction, as well as the dissociation of the SOD1 dimer [69, 70] provide templates that nucleate SOD1 aggregation. Following nucleation, the rate of SOD1 aggregate propagation (and patient's disease progression) changes quantifiably based upon the physicochemical properties of the variant (specifically aggregation increases in proportion to losses in charge and conformational entropy and gain of hydrophobicity). (Figure 1) Additionally, the Hart group and collaborators have shown that numerous individual peptides isolated from SOD1 form fibril-like aggregates, even in their fALS-mutated form.[71]
Can PTMs of SOD1 lead to sporadic ALS?
In the 1950s Denham Harman proposed the free radical theory of aging, where deleterious oxidative modifications of DNA, the equivalent of somatic mutations, increased with age.[72] This theory was extended to other biomolecules, including lipids and proteins, and gave rise to the theory that aging resulted from a viscous cycle involving increasing oxidative damage to proteins that resulted in the inhibition of proteolytic enzymes. These theories, combined with the observation that just about any structural modification of SOD1 causes ALS (180/184 known coding mutations), apparently through similar mechanisms, led to the hypothesis that PTMs of SOD1 can cause sporadic (no SOD1 mutation) ALS. The first step in addressing this hypothesis was to characterize whether SOD1 becomes modified in vivo during ALS. There are no animal models of sALS and tissue biopsies of brain and spinal cord of ALS patients are not performed. Our studies therefore involved post-mortem brain and spinal cord samples, but were augmented by (living) human blood and mice overexpressing SOD1. It should be noted that end-stage neurodegenerative disease tissue is highly inflammatory, and consequently there are bound to be numerous modifications that are the result of inflammation rather than primary causes. Parsing the toxic modifications from the epiphenomena would require additional neurotoxicology studies.
The discovery of in vivo SOD1 PTMs including oxidation and S-thiolation
We began by developing a quick and comprehensive isolation for SOD1 from human samples. Using SOD1 purified from human erythrocytes, and then modified using SOD1's enzymatic end-product, peroxide, until a subpopulation of the SOD1 contained oxidative PTMs, we raised polyclonal antibodies to SOD1.[20] SOD1 specific antibodies were affinity-purified and could deplete all SOD1 from tissue samples (as confirmed by western blot using commercial SOD1 antibodies). Using SOD1 purified by our antibody affinity methods, we identified oxidative modifications in a fALS SOD1G93A mouse models, in human erythrocytes, and ALS tissues. Top-down MS detected the relative abundance of the modified forms; including oxidation; metallation and disulfide bond status; relative tertiary structure through charge state distribution; and localization of the oxidative modifications (confirmed by endoproteinase digestion). The most abundant modification was cysteinylation of Cys111, followed by oxidative modifications of residues Trp32 and Cys111. These studies showed that Trp32 and Cys111 were both modified by oxygen (1-3), and the predominant Cys111 modification was to cysteine sulfonic acid.
The identification of toxic and protective SOD1 PTMS
Toxic oxidative modifications of Trp32
Research had suggested that the SOD1 mutations associated with fALS promoted higher levels of PTMs,[53, 73, 74] SOD1WT could also be modified, and that oxidative modifications of SOD1 induce aggregation as shown in vitro.[35, 75, 76] We investigated whether the oxidative modifications to Trp32 that we observed in humans promoted aggregation and toxicity. A Trp32Phe mutation was introduced to minimize SOD1 oxidation at residue 32. Using a well-established primary cell culture model of toxicity, the survival and aggregation of neurons injected with the fALS SOD1G93A were compared to those injected with SOD1G93A/W32F (and to SOD1W32F and SOD1WT controls). The Trp32Phe mutation was able to fully “rescue” G93A mutations, with the SOD1G93A/W32F double mutant having its toxicity reduced to that of normal SOD1WT while also producing fewer aggregates. (Figure 2) Recent studies have shown that SOD1 aggregation can occur through Trp32 oxidation that leads to di-tryptophan covalent cross-links between SOD1 monomers.[77]
Figure 2.
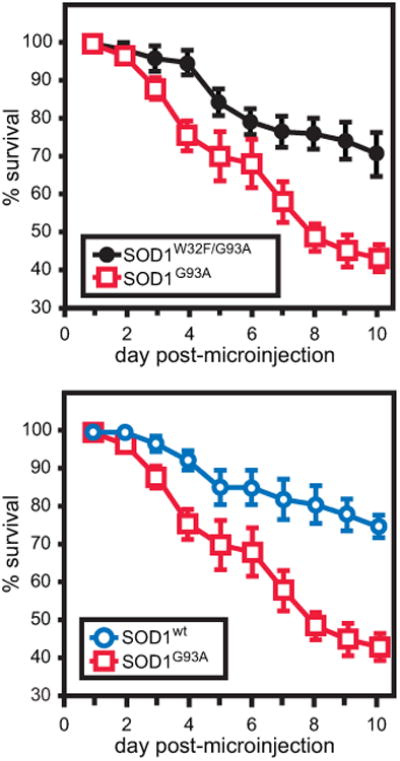
Over the course of 10 days, motor neurons expressing wild-type, G93A, and W32F/G93A SOD1 were monitored for viability. (Top) SOD1G93A proved to be significantly more toxic to motor neurons than wild-type SOD1. (Bottom) Mutating SOD1G93A's Trp32 to Phe resulted in a rescue of the pathogenic variant's toxicity to motor neurons, demonstrating similar viability to wild-type SOD1. (This research was originally published in Taylor, D.M., et al. Tryptophan 32 Potentiates Aggregation and Cytotoxicity of a Copper/Zinc Superoxide Dismutase Mutant Associated with Familial Amyotrophic Lateral Sclerosis. Journal of Biological Chemistry. 2007; 282(22):16329-16335. © the American Society for Biochemistry and Molecular Biology.)
Toxic oxidative modifications of Cys111
In similar studies, the Borchelt, Carri, and Araki groups demonstrated that C111S mutations, which prevented the oxidation we observed in Cys111, could rescue the toxicity of a variety of SOD1 mutants.[78-80] A collaborative study with the Bosco and Brown groups employed an important structural tool for SOD1-mediated ALS, the C4F6 conformation-specific anti-SOD1 antibody. This monoclonal antibody was raised against disease-variant SOD1G93A in mice,[81] and does not interact with unmodified SOD1WT. This tool serves as an important complement to MS and X-ray crystallography. In this study, top-down MS/MS using electron capture dissociation (ECD) determined the oxidative site of modification on SOD1 and enabled the production of SOD1 modified (by SOD1s end product, peroxide) exclusively at Cys111. Recombinant SOD1WT with Cys111 oxidized to sulfonic acid was shown to be toxic – indeed as toxic as fALS variants – through blocking of anterograde Fast Axonal Transport (FAT) in a squid axoplasm assay. SOD1WT extracted from C4F6-immunoreactive sALS tissue was also shown to inhibit FAT, while recombinant SOD1WT and SOD1 extracted from control tissue did not. Also of note was that inclusion of the C4F6 antibody in FAT-inhibition assays prevents fALS-associated SOD1 variants from inhibiting FAT, indicating that the conformational epitope that causes this inhibition is masked when the antibody is bound.[3]
Identification of the epitope recognized by conformation specific antibodies: SOD1's electrostatic loop and zinc binding region mediate the exposure of a hidden toxic epitope
With misfolded SOD1 established as a possible link between certain cases of fALS and sALS, and potential oxidative mechanisms and methods to protect against this having been identified, we felt it was important to determine the epitope of toxic-SOD1 antibody C4F6.[3, 81] Epitope mapping can be a difficult process, most often completed through cross-linking MS with various chemical functionalities and spacer arm lengths, resulting in a rough idea of the binding site, not necessarily an exact picture. Site-directed mutagenesis was again a useful tool helping elucidate which residues in the identified loops (IV and VII, the electrostatic loop and zinc binding region, respectively) were important for antibody recognition.[82] This revealed that residues Asp92 and Asp96 are crucial for C4F6 binding, whether as part of the epitope or in exposing the epitope. An important note is that removal of loops IV and VII still results in a folded protein that interacts with C4F6 as well as the original antigen. This further confirms that the destabilization of SOD1 through mutations or PTMs results in instability of the electrostatic loop and zinc binding regions in some forms of SOD1-mediated ALS, exposing a previously concealed toxic epitope and releasing coordinated copper and zinc. Simple de-metalation of SOD1WT also exposes this epitope to some degree, indicating that any metal-deficient disease-variant or PTM-product SOD1 could potentially be neurotoxic.
Protective S-thiolation of Cys111
SOD1's Cys111 can be oxidized,[83, 84] and modified by copper,[85] glutathione,[28, 86] and cysteine.[85, 87] We isolated SOD1 from human nervous tissue and identified both oxidation and cysteinylation of Cys111.[39, 88] Cysteinylation had not been previously identified on SOD1, and rarely is identified as standard bottom-up proteomic workflows eliminate the possibility of detecting thiol-modifications due to reduction and alkylation steps and the general nature of disulfide scrambling during MS analysis. Quantitative accuracy allowed for the determination of relative amounts of each species, and showed that unmodified SOD1 was not present in the oxidatively-stressed sample. (Figure 3) MS, with the help of X-ray crystallography, showed that cysteinylation was only possible at a ratio of one modification per dimer. However, while one monomer's Cys111 was blocked from oxidation, it did not necessarily block oxidation of the adjacent cysteine in the monomer-pair.
Figure 3.
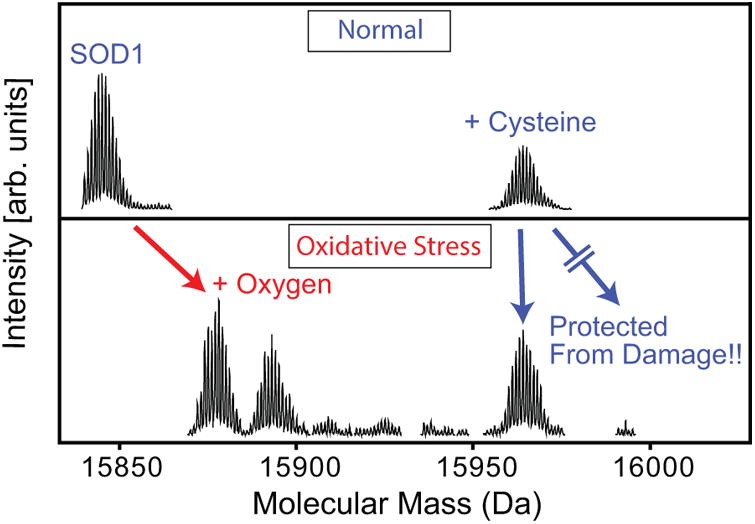
Cysteinylation protects SOD1's Cys111 from oxidative damage. Mass spectrometry showed that SOD1wt underwent oxidation at Cys111 (MS/MS not shown), resulting in a mix of SOD1-Cys111 Sulfinic and Sulfonic acid (2 and 3 oxygens on Cysteine, respectively) with no unmodified SOD1wt remaining. SOD1wt that had been cysteinylated at Cys111 was protected from oxidative damage, showing very little oxidative products and remaining cysteinylated. (Reprinted with permission from Auclair, J.R., et al., Post Translational Modification by Cysteine Protects Cu/Zn-Superoxide Dismutase from Oxidative Damage. Biochemistry, 2013. 52(36):6137-6144. © 2013 American Chemical Society.)
Further thermodynamic experiments with the Cys111-cysteinylated form of SOD1 revealed that oxidation of SOD1 decreased the protein's melting temperature by 23°C, whereas cysteinylation only decreased the melting temperature by 5°C.[88] This suggests that modification by cysteine is slightly destabilizing, but prevents the much more destabilizing modification of oxidation. The cellular concentration of cystine, the dimeric oxidized form of cysteine, has been shown to increase (along with oxygen) in cells with age and oxidative stress.[89] This supports our hypothesis that SOD1 may become cysteinylated in vivo in a protective manner as a method to compensate for increased oxidative stress loads, protecting proteins from aberrant cysteine oxidation. In this regard, the therapeutic approach of cross-linking the cysteine dyad pairs on non-covalently bound monomer pairs also serves to protect Cys111 from oxidation.
Mass spectrometry-informed therapeutic strategies
Stabilizing the SOD1 dimer using cross-linkers
We hypothesized that covalently cross-linking the SOD1 dimer using Cys111 residues at the dimer interface [17] would simultaneously prevent the toxicity associated with oxidative modification of Cys111, while kinetically stabilizing the SOD1 dimer. Our proof-of-concept study demonstrated this using dithiobismaleimidoethane (DTME) and bismaleimidoethane (BMOE), which are not practical as therapeutics due to their lack of specificity for this target. This idea has since evolved to the use of cyclic disulfides, as they specifically target proximal cysteine dyads and do not permanently fix themselves to lone cysteines.[90] The modification sites and products of protein cross-linking reactions were confirmed using top-down and bottom-up MS. Because the process of electrospray ionization disrupts the natural noncovalent dimer of SOD1, a cross-linking assay where two monomers are tethered together in solution is easy to monitor, as a shift in mass is observed from monomer mass to the mass of the dimer plus the mass of what remains attached to the protein of the cross-linker (Figure 4). This assay was also used to show that pathogenic variants of SOD1, like the G93A variant, can be stabilized with cross-linking.
Figure 4.
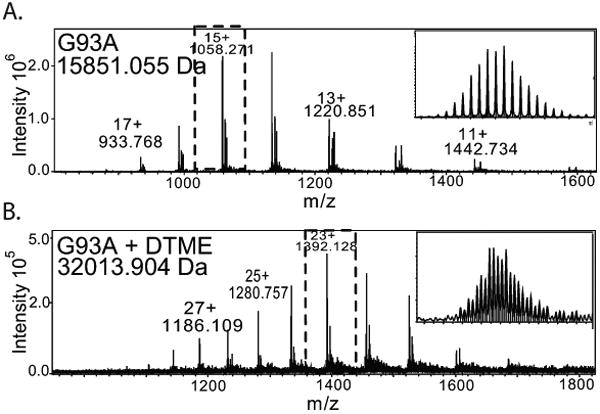
Covalent dimerization of pathogenic G93A variant of SOD1 with DTME – A therapeutic stabilization strategy. (A) Mass spectrum of SOD1G93A. (B) Mass spectrum of DTME-cross-linked SOD1G93A stabilizes the non-covalent dimer with a covalent cross-link that is not lost upon ionization. (Figure adapted from Auclair, J.R., et al. Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A, 2010. 107(50):21394-9.)
Studies by other groups indicate that increased SOD1 dimer dissociation occurs in several fALS variants. A large scale thermodynamic analysis of SOD1 dimer and monomer stability by the Dokholyan group showed that 70 out of 75 pathogenic SOD1 variants demonstrate decreased dimer stability and/or increased dimer dissociation.[70] Our HDX studies of demetallated fALS variants (which are predominantly monomeric) are consistent with these observations. However, we did not detect perturbation at the dimer interface in some of the metallated variants, indicating that they are not the major species. Given that all of the fALS variants destabilize SOD1 (either its metallated or apo form) and that the rate-limiting step of SOD1 aggregation is dimer dissociation, the balance of the evidence supports that stabilizing the SOD1 dimer is one viable therapeutic approach (Figure 5).
Figure 5.
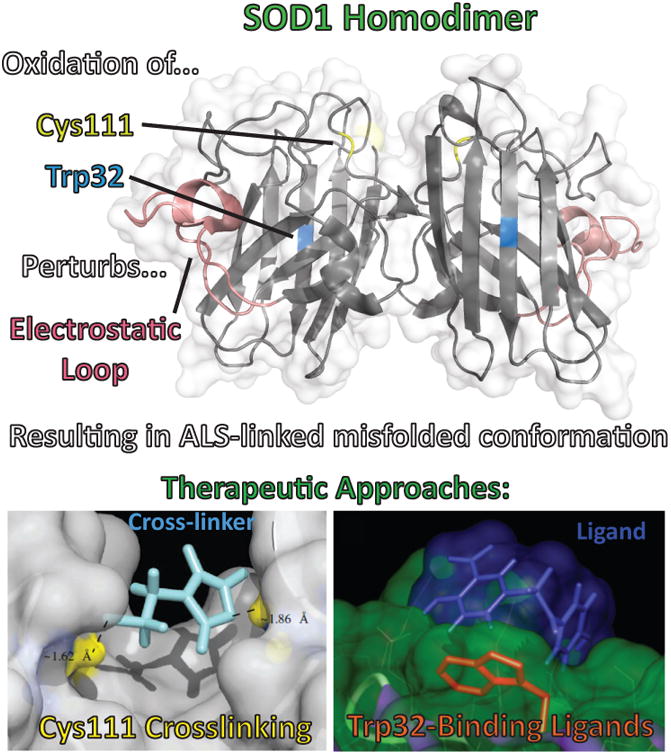
(Top) Oxidation of SOD1's Cys111 and Trp32 perturb the structure of the protein in a similar way as any of the pathogenic disease variants, leading to ALS-linked misfolded conformations of the protein. (Bottom) Therapeutic approaches that have been proposed to prevent these oxidation events are (Left) cross-linking proximal Cys111's on SOD1 monomer pairs to block the cysteines from oxidation and maintain the homodimer and (Right) blocking oxidation of Trp32 with a ligand, which also serves to stabilize the protein structure.
Trp32 binding pharmacological chaperones
Because SOD1's Trp32 was shown to be oxidized in vivo and had high-potential to be involved in protein-protein interactions, we hypothesized that blocking this residue with small-molecule binding could replicate the effects we observe when mutating this residue to the less oxidation-prone phenylalanine. Mimicking this rescue phenotype would allow for increased protein stability and may prevent SOD1-mediated disease onset and progression. Our group began further examination of this binding site and its potential as a therapeutic route to combat SOD1-mediated ALS through a combination of in silico docking, protein stability assays, and competitive MS binding assays. [91] The docking was examined using GLIDE,[92] searching for compounds that have high theoretical propensity of binding to this site (Figure 5). Three compounds previously shown to bind to Trp32 and three new compounds from the in silico docking were chosen, and were incubated with unstable fALS-variants of the protein. Binding affinity and protein stability before and after compound binding were evaluated with microscale thermophoresis and differential scanning fluorimetry. These compounds were also evaluated by MS through a competitive binding assay under light electrospray conditions. We found that, of the compounds that bound, they each offered only slight increases in protein stability, their greater contribution likely being protection from oxidation. In fact, Trp32 and a small ligand binding site in loop II of the protein are the only native surface sites that have been confirmed as drug-targetable.[93] Previously, X-ray crystallography had determined that 5-fluorouridine (5-FUrd) binds to Trp32 through aromatic stacking, offering increased stability to the protein upon binding.[54]
“Deamidating” mutations provide proof of concept that PTMs can cause disease
One of the most commonly studied instances of a PTM causing disease, and one with the most obvious potential connection between familial and sporadic disease onset, is found with deamidation. Numerous familial diseases, including the neurodegenerative disorders AD and ALS, provide genetic proof of the concept that deamidation can cause disease. For example, the fALS SOD1 mutation, Asn86Asp (asparagine to aspartic acid mutation at residue 86) results in a protein that is chemically identical to SOD1WT that underwent post-translational asparagine deamidation at asparagine 86. A cursory review of the mutations that cause various diseases (Table 1) reveals that deamidation mutations (Asn→Asp or Gln→Glu) occur in AD;[94] Autosomal Recessive Polycystic Kidney Disease;[95] Neuronal Ceroid Lipofuscinosis;[96, 97] Fanconi Anemia;[98] Waardenburg II Deafness;[99] and Prostate Cancer.[100] If these protein variants lead to disease, deamidation at the same residue as indicated here could lead to the same disease progression if it occurs on a large scale.
There have also been documented cases of misdiagnoses when symptoms manifest in similar ways, as has been the case with ALS and (the deamidation-dependent) Celiac disease. In Celiac disease, transglutaminase-mediated deamidation of a wheat gluten-derived peptide renders the peptide antigenic,[101-104] thereby eliciting an autoimmune disorder. Celiac disease is usually associated with neurological symptoms,[105, 106] including pure motor variants resembling ALS.[106, 107] These and more recent studies provide evidence of overlap in Celiac disease and ALS symptoms to the extent that misdiagnosis of Celiac disease for ALS has occurred.[108] Transglutaminase 6 antibodies have been found in ALS patient serum.[109] Additionally, cerebrospinal fluid of ALS patients has shown elevated levels of transglutaminase,[110] and other findings show that transglutaminase is involved in aberrant misfolded SOD1 assembly leading to neuroinflammation and ALS disease progression in mice.[111] This implies that sporadic ALS etiology may have substantial overlap with Celiac disease.
Conclusion & Perspective
Mass spectrometry has proven an indispensable tool in aiding with localization of modification sites, elucidation of the structural dynamics of protein variants, and revelation of the location of pathogenic epitopes in the protein. As modern medicine allows for further increases in life expectancy, neurological disorders have become more prominent and are accounting for more and more deaths each year.[86] In fact, AD is practically inevitable if one lives long enough, increasing exponentially with age and reaching ∼50% prevalence by age 95.[112] There are comprehensive reviews available about the current state of knowledge on several diseases discussed here, such as ALS,[15] AD,[113] PD,[114] and prion diseases.[115] We note that RNA metabolism appears to be critical to additional (non-SOD1) genetic variants of ALS, and was not addressed by our studies. Another theme common to most, if not all, genetic variants of ALS and indeed neurodegeneration in general, is that modified proteins inhibit proteolysis, promoting a vicious cycle that ends in the accumulation and aggregation of misfolded proteins. We have shown that fALS SOD1 mutations selectively inhibit proteasome activity in affected motor neurons and tissues.[116-119]
Using a variety of toxicology assays, we have determined that oxidative and mutation-based modifications of SOD1 have similar toxicities. Using traditional HDX-MS we have determined that 14 of the most prevalent fALS mutations result in perturbation of the electrostatic loop, but we have not yet performed these studies with all 180+ fALS variants or with oxidative modifications. Instead, evidence for perturbation of the electrostatic loop of oxygen-modified SOD1 comes from conformation specific antibodies, and evidence for destabilization from reduction of their unfolding temperatures and from increased rates of global HDX. We have no direct evidence that electrostatic loop perturbation is toxic, per se, only x-ray crystallographic studies showing how electrostatic loop perturbation can lead to aggregation. We also do not understand the allostery of SOD1 - in particular, how distant structural perturbations affect the electrostatic loop or how electrostatic loop perturbation affects dimer dissociation.
This perspective concerns oxidative modifications of proteins. Metals, as discussed above, are also important structural determinants of many proteins, including SOD1.[120, 121] For example, loss of copper and zinc result in toxic proteoforms of SOD1 (Reviewed [122]). Restoring these metals has resulted in the most effective treatment to date in animal models of ALS, CuATSM.[123, 124] Our model for SOD1-mediated ALS indicates that the toxicity of mutations in the SOD1 gene result from changes to SOD1 protein structure. The correlation of changes in SOD1 physicochemical properties to both fALS progression and in vitro toxicity models supports this. Whether changes in protein structure are responsible for all ALS or only a subset of the disease is unknown. On the one hand, additional prevalent genetic risk factors for fALS (FUS, TDP-43, Ubiquilin 2, C9orf72, etc.) generally result in protein aggregation. On the other hand, these same risk factors also effect RNA function. The disease-relevance of changes to protein and RNA function remains to be determined.
With few selective pressures against the plethora of different (late onset) fALS SOD1 mutations existing until lifespan rapidly increased in the 20th century, it appears that SOD1 evolved into a space of narrow protein stability. Even the slightest perturbations to SOD1 structure lead to disease. This is consistent with the concept of an entatic state – in which a protein is held in a highly strained and delicate state to promote catalysis. Our studies indicate that fALS mutations exert their toxic effects through a common, aggregation prone, structural intermediate. In human tissue, we identified oxidative PTMs that also destabilize SOD1 and are toxic. Our current therapeutic approach involves preventing these PTMs and kinetically stabilizing SOD1. We have not demonstrated that oxidative modifications of SOD1WT occur on a large enough scale to match the pathogenicity of fALS-linked SOD1 variants. Whether oxidative PTMs occur selectively in the highly metabolic motor neuron remains to be determined. Others have proposed that SOD1-linked sALS can progress through a small population of aberrant modifications that lead to protein-templating to convert SOD1WT to a toxic conformation.[125]
Further creative uses of bio-chemical and -physical experiments coupled with mass spectrometry show great promise in advancing our understanding of disease at the molecular level.
Acknowledgments
These studies began during Agar's post-doctoral fellowship at the Montreal Neurological Institute in Heather Durham's neurotoxicology laboratory, and involving numerous clinical, biochemical, and structural collaborators, including the laboratories of Ashutosh Tiwari, Larry Hayward, Yoshi Hamuro, Bernard Gibbs, Greg Petsko, Dagmar Ringe, Bob Brown Jr., Jared Auclair, Nathalie Agar, and Daryl Bosco, and our ongoing collaboration with Bruker Daltonics. We'd also like to thank all former Agar lab members for their contributions to the research presented.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angewandte Chemie International Edition. 2005;44(45):7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–7. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, et al. Protein Aggregation and Protein Instability Govern Familial Amyotrophic Lateral Sclerosis Patient Survival. PLOS Biology. 2008;6(7):e170. doi: 10.1371/journal.pbio.0060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ségalat L. Loss-of-function genetic diseases and the concept of pharmaceutical targets. Orphanet Journal of Rare Diseases. 2007;2:30–30. doi: 10.1186/1750-1172-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidransky E, Ginns EI. Genetic basis of Gaucher disease. J Pediatr. 1995;127(3):510. doi: 10.1016/s0022-3476(95)70104-4. [DOI] [PubMed] [Google Scholar]
- 7.Mair B, et al. Gain- and Loss-of-Function Mutations in the Breast Cancer Gene GATA3 Result in Differential Drug Sensitivity. PLOS Genetics. 2016;12(9):e1006279. doi: 10.1371/journal.pgen.1006279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2(2):a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Human Mortality Database. University of California, Berkeley (USA), and Max Planck Institute for Demographic Research (Germany). Available at www.mortality.org http://www.mortality.org/or www.humanmortality.de (data downloaded on March 2, 2017)
- 10.Dickson DW. Neuropathology of Non-Alzheimer Degenerative Disorders. International Journal of Clinical and Experimental Pathology. 2010;3(1):1–23. [PMC free article] [PubMed] [Google Scholar]
- 11.McCann H, et al. alpha-Synucleinopathy phenotypes. Parkinsonism Relat Disord. 2014;20 Suppl 1:S62–7. doi: 10.1016/S1353-8020(13)70017-8. [DOI] [PubMed] [Google Scholar]
- 12.Cullen V, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol. 2011;69(6):940–53. doi: 10.1002/ana.22400. [DOI] [PubMed] [Google Scholar]
- 13.Dickson DW. Chapter 7 Ubiquitinopathies. Blue Books of Neurology. 2007;30:165–185. [Google Scholar]
- 14.Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2006;1762(11 – 12):1038–1050. doi: 10.1016/j.bbadis.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 15.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 17.Auclair JR, et al. Strategies for stabilizing superoxide dismutase (SOD1), the protein destabilized in the most common form of familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2010;107(50):21394–9. doi: 10.1073/pnas.1015463107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molnar KS, et al. A Common Property of Amyotrophic Lateral Sclerosis-associated Variants: DESTABILIZATION OF THE COPPER/ZINC SUPEROXIDE DISMUTASE ELECTROSTATIC LOOP. Journal of Biological Chemistry. 2009;284(45):30965–30973. doi: 10.1074/jbc.M109.023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw BF, et al. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J Biol Chem. 2006;281(26):18167–76. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- 20.Taylor DM, et al. Tryptophan 32 Potentiates Aggregation and Cytotoxicity of a Copper/Zinc Superoxide Dismutase Mutant Associated with Familial Amyotrophic Lateral Sclerosis. Journal of Biological Chemistry. 2007;282(22):16329–16335. doi: 10.1074/jbc.M610119200. [DOI] [PubMed] [Google Scholar]
- 21.Bredesen DE, et al. Do posttranslational modifications of CuZnSOD lead to sporadic amyotrophic lateral sclerosis? Ann Neurol. 1997;42(2):135–7. doi: 10.1002/ana.410420202. [DOI] [PubMed] [Google Scholar]
- 22.Ayers JI, et al. Distinct conformers of transmissible misfolded SOD1 distinguish human SOD1-FALS from other forms of familial and sporadic ALS. Acta Neuropathol. 2016;132(6):827–840. doi: 10.1007/s00401-016-1623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Da Cruz S, et al. Misfolded SOD1 is not a primary component of sporadic ALS. Acta Neuropathol. 2017 doi: 10.1007/s00401-017-1688-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abel O, et al. Development of a Smartphone App for a Genetics Website: The Amyotrophic Lateral Sclerosis Online Genetics Database (ALSoD) JMIR mHealth and uHealth. 2013;1(2):e18. doi: 10.2196/mhealth.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 26.Siddique T, Deng HX. Genetics of amyotrophic lateral sclerosis. Hum Mol Genet. 1996;5 Spec No:1465–70. doi: 10.1093/hmg/5.supplement_1.1465. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, et al. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J Am Chem Soc. 2013;135(42):15897–908. doi: 10.1021/ja407801x. [DOI] [PubMed] [Google Scholar]
- 28.Wilcox KC, et al. Modifications of superoxide dismutase (SOD1) in human erythrocytes: a possible role in amyotrophic lateral sclerosis. J Biol Chem. 2009;284(20):13940–7. doi: 10.1074/jbc.M809687200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu T, et al. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer's disease. J Neurosci Res. 2002;70(3):451–61. doi: 10.1002/jnr.10350. [DOI] [PubMed] [Google Scholar]
- 30.Yan SD, et al. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(16):7787–7791. doi: 10.1073/pnas.91.16.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giasson BI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–9. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 32.LaVoie MJ, et al. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11(11):1214–21. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- 33.Mackenzie IR, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61(5):427–34. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 34.Kabashi E, et al. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann Neurol. 2007;62(6):553–9. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- 35.Rakhit R, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277(49):47551–6. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- 36.Shibata N, et al. Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett. 1994;179(1-2):149–52. doi: 10.1016/0304-3940(94)90956-3. [DOI] [PubMed] [Google Scholar]
- 37.Ihara Y, et al. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer's disease. J Biochem. 1986;99(6):1807–10. doi: 10.1093/oxfordjournals.jbchem.a135662. [DOI] [PubMed] [Google Scholar]
- 38.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83(11):4044–8. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Auclair JR, et al. Post-Translational Modification by Cysteine Protects Cu/Zn-Superoxide Dismutase from Oxidative Damage. Biochemistry. 2013;52(36):6137–6144. doi: 10.1021/bi4006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shevchenko A, et al. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68(5):850–8. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 41.Kuljanin M, et al. Comparison of sample preparation techniques for large-scale proteomics. PROTEOMICS. 2017;17(1-2):1600337–n/a. doi: 10.1002/pmic.201600337. [DOI] [PubMed] [Google Scholar]
- 42.Auclair JR, et al. Artifacts to avoid while taking advantage of top-down mass spectrometry based detection of protein S-thiolation. Proteomics. 2014;14(10):1152–7. doi: 10.1002/pmic.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, et al. Protein Analysis by Shotgun/Bottom-up Proteomics. Chemical reviews. 2013;113(4):2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sung WC, et al. Evaluation of disulfide scrambling during the enzymatic digestion of bevacizumab at various pH values using mass spectrometry. Biochim Biophys Acta. 2016;1864(9):1188–94. doi: 10.1016/j.bbapap.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 45.Catherman AD, Skinner OS, Kelleher NL. Top Down Proteomics: Facts and Perspectives. Biochemical and biophysical research communications. 2014;445(4):683–693. doi: 10.1016/j.bbrc.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karabacak NM, et al. Sensitive and Specific Identification of Wild Type and Variant Proteins from 8 to 669 kDa Using Top-down Mass Spectrometry. Molecular & Cellular Proteomics: MCP. 2009;8(4):846–856. doi: 10.1074/mcp.M800099-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cobb JS, Easterling ML, Agar JN. Structural characterization of intact proteins is enhanced by prevalent fragmentation pathways rarely observed for peptides. Journal of the American Society for Mass Spectrometry. 2010;21(6):949–959. doi: 10.1016/j.jasms.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, et al. Top-Down Quantitative Proteomics Identified Phosphorylation of Cardiac Troponin I as a Candidate Biomarker for Chronic Heart Failure. Journal of Proteome Research. 2011;10(9):4054–4065. doi: 10.1021/pr200258m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, et al. Memory-efficient calculation of the isotopic mass states of a molecule. Rapid Communications in Mass Spectrometry. 2010;24(18):2689–2696. doi: 10.1002/rcm.4666. [DOI] [PubMed] [Google Scholar]
- 50.Li L, et al. A Hierarchical Algorithm for Calculating the Isotopic Fine Structures of Molecules. Journal of the American Society for Mass Spectrometry. 2008;19(12):1867–1874. doi: 10.1016/j.jasms.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 51.Liu Q, Easterling ML, Agar JN. Resolving Isotopic Fine Structure to Detect and Quantify Natural Abundance- and Hydrogen/Deuterium Exchange-Derived Isotopomers. Analytical Chemistry. 2014;86(1):820–825. doi: 10.1021/ac403365g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salisbury JP, Liu Q, Agar JN. QUDeX-MS: hydrogen/deuterium exchange calculation for mass spectra with resolved isotopic fine structure. BMC Bioinformatics. 2014;15(1):403. doi: 10.1186/s12859-014-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tiwari A, Xu Z, Hayward LJ. Aberrantly Increased Hydrophobicity Shared by Mutants of Cu, Zn-Superoxide Dismutase in Familial Amyotrophic Lateral Sclerosis. Journal of Biological Chemistry. 2005;280(33):29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- 54.Wright GS, et al. Ligand binding and aggregation of pathogenic SOD1. Nat Commun. 2013;4:1758. doi: 10.1038/ncomms2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao X, et al. Structures of the G85R Variant of SOD1 in Familial Amyotrophic Lateral Sclerosis. The Journal of Biological Chemistry. 2008;283(23):16169–16177. doi: 10.1074/jbc.M801522200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galaleldeen A, et al. Structural and Biophysical Properties of Metal-Free Pathogenic SOD1 Mutants A4V and G93A. Archives of biochemistry and biophysics. 2009;492(1-2):40–47. doi: 10.1016/j.abb.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rakhit R, Chakrabartty A. Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2006;1762:11–12. 1025–1037. doi: 10.1016/j.bbadis.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Schmidlin T, Kennedy BK, Daggett V. Structural Changes to Monomeric CuZn Superoxide Dismutase Caused by the Familial Amyotrophic Lateral Sclerosis-Associated Mutation A4V. Biophysical Journal. 2009;97(6):1709–1718. doi: 10.1016/j.bpj.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Molnar KS, et al. A common property of amyotrophic lateral sclerosis-associated variants: destabilization of the copper/zinc superoxide dismutase electrostatic loop. J Biol Chem. 2009;284 doi: 10.1074/jbc.M109.023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrometry Reviews. 2006;25(1):158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 61.Lindberg MJ, et al. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(28):9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–93. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 63.Jha P, et al. 21st-Century Hazards of Smoking and Benefits of Cessation in the United States. New England Journal of Medicine. 2013;368(4):341–350. doi: 10.1056/NEJMsa1211128. [DOI] [PubMed] [Google Scholar]
- 64.Sekhar A, et al. Probing the free energy landscapes of ALS disease mutants of SOD1 by NMR spectroscopy. Proceedings of the National Academy of Sciences. 2016;113(45):E6939–E6945. doi: 10.1073/pnas.1611418113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma Q, et al. The Contrasting Effect of Macromolecular Crowding on Amyloid Fibril Formation. PLOS ONE. 2012;7(4):e36288. doi: 10.1371/journal.pone.0036288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prudencio M, et al. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet. 2009;18(17):3217–26. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Speretta E, et al. Expression in Drosophila of Tandem Amyloid β Peptides Provides Insights into Links between Aggregation and Neurotoxicity. Journal of Biological Chemistry. 2012;287(24):20748–20754. doi: 10.1074/jbc.M112.350124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Elam JS, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10(6):461–7. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- 69.Ray SS, et al. An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43(17):4899–905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 70.Khare SD, Caplow M, Dokholyan NV. FALS mutations in Cu, Zn superoxide dismutase destabilize the dimer and increase dimer dissociation propensity: a large-scale thermodynamic analysis. Amyloid. 2006;13(4):226–35. doi: 10.1080/13506120600960486. [DOI] [PubMed] [Google Scholar]
- 71.Ivanova MI, et al. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111(1):197–201. 72. doi: 10.1073/pnas.1320786110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harman D. Aging: A Theory Based on Free Radical and Radiation Chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 73.Andrus PK, et al. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71(5):2041–8. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- 74.Poon HF, et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice--a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39(4):453–62. doi: 10.1016/j.freeradbiomed.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 75.Davies KJ. Protein damage and degradation by oxygen radicals. I. general aspects. J Biol Chem. 1987;262(20):9895–901. [PubMed] [Google Scholar]
- 76.Zhang H, et al. Mass spectral evidence for carbonate-anion-radical-induced posttranslational modification of tryptophan to kynurenine in human Cu, Zn superoxide dismutase. Free Radic Biol Med. 2004;37(12):2018–26. doi: 10.1016/j.freeradbiomed.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 77.Coelho FR, et al. Oxidation of the tryptophan 32 residue of human superoxide dismutase 1 caused by its bicarbonate-dependent peroxidase activity triggers the non-amyloid aggregation of the enzyme. J Biol Chem. 2014;289(44):30690–701. doi: 10.1074/jbc.M114.586370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cozzolino M, et al. Cysteine 111 Affects Aggregation and Cytotoxicity of Mutant Cu, Znsuperoxide Dismutase Associated with Familial Amyotrophic Lateral Sclerosis. The Journal of biological chemistry. 2008;283(2):866–874. doi: 10.1074/jbc.M705657200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagano S, et al. A cysteine residue affects the conformational state and neuronal toxicity of mutant SOD1 in mice: relevance to the pathogenesis of ALS. Hum Mol Genet. 2015;24(12):3427–39. doi: 10.1093/hmg/ddv093. [DOI] [PubMed] [Google Scholar]
- 80.Roberts BLT, et al. Role of Disulfide Cross-Linking of Mutant SOD1 in the Formation of Inclusion-Body-Like Structures. PLOS ONE. 2012;7(10):e47838. doi: 10.1371/journal.pone.0047838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104(7):2495–500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rotunno MS, et al. Identification of a Misfolded Region in Superoxide Dismutase 1 that is Exposed in Amyotrophic Lateral Sclerosis. Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.M114.581801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fujiwara N, et al. Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem. 2007;282(49):35933–44. doi: 10.1074/jbc.M702941200. [DOI] [PubMed] [Google Scholar]
- 84.Guareschi S, et al. An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proceedings of the National Academy of Sciences. 2012;109(13):5074–5079. doi: 10.1073/pnas.1115402109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu H, et al. Copper(2+) binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant. Biochemistry. 2000;39(28):8125–32. doi: 10.1021/bi000846f. [DOI] [PubMed] [Google Scholar]
- 86.Redler RL, et al. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry. 2011;50(32):7057–66. doi: 10.1021/bi200614y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nakanishi T, et al. Simple and defined method to detect the SOD-1 mutants from patients with familial amyotrophic lateral sclerosis by mass spectrometry. J Neurosci Methods. 1998;81(1-2):41–4. 88. doi: 10.1016/s0165-0270(98)00012-0. [DOI] [PubMed] [Google Scholar]
- 88.Auclair JR, et al. Structural consequences of cysteinylation of Cu/Zn-superoxide dismutase. Biochemistry. 2013;52(36):6145–50. doi: 10.1021/bi400613h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson JM, et al. A rapid LC-FTMS method for analysis of cysteine, cystine and cysteine/cystine steady-stateredox potential in human plasma. Clinica chimica acta; international journal of clinical chemistry. 2008;396(1-2):43–48. 90. doi: 10.1016/j.cca.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Agar JN, Salisbury J. Tethering cysteine residues using cyclic disulfides. 2016 Google Patents. [Google Scholar]
- 91.Isim S. Targeting Trp32 and Cys111 to Stabilize the ALS-Associated Protein Cu/Zn Superoxide Dismutase. Biology. 2014:1–48. Brandeis. [Google Scholar]
- 92.Friesner RA, et al. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. Journal of Medicinal Chemistry. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 93.Antonyuk S, Strange RW, Hasnain SS. Structural Discovery of Small Molecule Binding Sites in Cu–Zn Human Superoxide Dismutase Familial Amyotrophic Lateral Sclerosis Mutants Provides Insights for Lead Optimization. Journal of Medicinal Chemistry. 2010;53(3):1402–1406. doi: 10.1021/jm9017948. [DOI] [PubMed] [Google Scholar]
- 94.Crook R, et al. Early-onset Alzheimer's disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann Neurol. 1997;42(1):124–8. doi: 10.1002/ana.410420121. [DOI] [PubMed] [Google Scholar]
- 95.Furu L, et al. Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol. 2003;14(8):2004–14. doi: 10.1097/01.asn.0000078805.87038.05. [DOI] [PubMed] [Google Scholar]
- 96.Waliany S, et al. Identification of three novel mutations of the palmitoyl-protein thioesterase-1 (PPT1) gene in children with neuronal ceroid-lipofuscinosis. Hum Mutat. 2000;15(2):206–7. doi: 10.1002/(SICI)1098-1004(200002)15:2<206::AID-HUMU14>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 97.Zelnik N, et al. A novel mutation of the CLN8 gene: is there a Mediterranean phenotype? Pediatr Neurol. 2007;36(6):411–3. doi: 10.1016/j.pediatrneurol.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 98.Levran O, et al. Sequence variation in the Fanconi anemia gene FAA. Proc Natl Acad Sci U S A. 1997;94(24):13051–6. doi: 10.1073/pnas.94.24.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tassabehji M, et al. The mutational spectrum in Waardenburg syndrome. Hum Mol Genet. 1995;4(11):2131–7. doi: 10.1093/hmg/4.11.2131. [DOI] [PubMed] [Google Scholar]
- 100.Gottlieb B, et al. The androgen receptor gene mutations database. Nucleic Acids Res. 1997;25(1):158–62. doi: 10.1093/nar/25.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bruce SE, Bjarnason I, Peters TJ. Human jejunal transglutaminase: demonstration of activity, enzyme kinetics and substrate specificity with special relation to gliadin and coeliac disease. Clin Sci (Lond) 1985;68(5):573–9. doi: 10.1042/cs0680573. [DOI] [PubMed] [Google Scholar]
- 102.Kim CY, et al. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A. 2004;101(12):4175–9. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Molberg O, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–7. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 104.van de Wal Y, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161(4):1585–8. [PubMed] [Google Scholar]
- 105.Cooke WT, Smith WT. Neurological disorders associated with adult coeliac disease. Brain. 1966;89(4):683–722. doi: 10.1093/brain/89.4.683. [DOI] [PubMed] [Google Scholar]
- 106.Hadjivassiliou M, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet. 1998;352(9140):1582–5. doi: 10.1016/s0140-6736(98)05342-2. [DOI] [PubMed] [Google Scholar]
- 107.Hadjivassiliou M, et al. Neuropathy associated with gluten sensitivity. J Neurol Neurosurg Psychiatry. 2006;77(11):1262–6. doi: 10.1136/jnnp.2006.093534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turner MR, et al. A case of celiac disease mimicking amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2007;3(10):581–4. doi: 10.1038/ncpneuro0631. [DOI] [PubMed] [Google Scholar]
- 109.Gadoth A, et al. Transglutaminase 6 antibodies in the serum of patients with amyotrophic lateral sclerosis. JAMA Neurology. 2015;72(6):676–681. doi: 10.1001/jamaneurol.2015.48. [DOI] [PubMed] [Google Scholar]
- 110.Fujita K, et al. Transglutaminase activity in serum and cerebrospinal fluid in sporadic amyotrophic lateral sclerosis: a possible use as an indicator of extent of the motor neuron loss. Journal of the neurological sciences. 1998;158(1):53–57. doi: 10.1016/s0022-510x(98)00088-4. [DOI] [PubMed] [Google Scholar]
- 111.Oono M, et al. Transglutaminase 2 accelerates neuroinflammation in amyotrophic lateral sclerosis through interaction with misfolded superoxide dismutase 1. Journal of Neurochemistry. 2014;128(3):403–418. doi: 10.1111/jnc.12441. [DOI] [PubMed] [Google Scholar]
- 112.Prince MJ, et al. World Alzheimer Report 2015 - The Global Impact of Dementia. An analysis of prevalence, incidence, cost and trends. Alzheimer's Disease International 2015 [Google Scholar]
- 113.Reiman EM. Alzheimer's disease: Attack on amyloid-[beta] protein. Nature. 2016;537(7618):36–37. doi: 10.1038/537036a. [DOI] [PubMed] [Google Scholar]
- 114.Klingelhoefer L, Reichmann H. Pathogenesis of Parkinson disease[mdash]the gut-brain axis and environmental factors. Nat Rev Neurol. 2015;11(11):625–636. doi: 10.1038/nrneurol.2015.197. [DOI] [PubMed] [Google Scholar]
- 115.Aguzzi A, Nuvolone M, Zhu C. The immunobiology of prion diseases. Nat Rev Immunol. 2013;13(12):888–902. doi: 10.1038/nri3553. [DOI] [PubMed] [Google Scholar]
- 116.Bendotti C, et al. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol. 2012;97(2):101–26. doi: 10.1016/j.pneurobio.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 117.Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin– proteasome system in Alzheimer's disease. Neurological Research. 2014;36(3):276–282. doi: 10.1179/1743132813Y.0000000288. [DOI] [PubMed] [Google Scholar]
- 118.Kabashi E, et al. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(4):367–71. doi: 10.3109/17482968.2012.686511. [DOI] [PubMed] [Google Scholar]
- 119.McNaught KS, et al. Proteasomal dysfunction in sporadic Parkinson's disease. Neurology. 2006;66(10 Suppl 4):S37–49. doi: 10.1212/wnl.66.10_suppl_4.s37. [DOI] [PubMed] [Google Scholar]
- 120.Trumbull KA, Beckman JS. A Role for Copper in the Toxicity of Zinc-Deficient Superoxide Dismutase to Motor Neurons in Amyotrophic Lateral Sclerosis. Antioxidants & Redox Signaling. 2009;11(7):1627–1639. doi: 10.1089/ars.2009.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Estevez AG, et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286(5449):2498–500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 122.Hilton JB, White AR, Crouch PJ. Metal-deficient SOD1 in amyotrophic lateral sclerosis. Journal of Molecular Medicine (Berlin, Germany) 2015;93(5):481–487. doi: 10.1007/s00109-015-1273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Soon CP, et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286(51):44035–44. doi: 10.1074/jbc.M111.274407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Williams JR, et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol Dis. 2016;89:1–9. doi: 10.1016/j.nbd.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ayers JI, et al. Prion-like propagation of mutant SOD1 misfolding and motor neuron disease spread along neuroanatomical pathways. Acta Neuropathologica. 2016;131(1):103–114. doi: 10.1007/s00401-015-1514-0. [DOI] [PMC free article] [PubMed] [Google Scholar]