

Abstract
Cardiovascular Disease (CVD) is the most common cause of death in industrialized countries, and myocardial infarction (MI) is a major CVD with significant morbidity and mortality. Following MI, the left ventricle (LV) undergoes a wound healing response to ischemia that results in extracellular matrix (ECM) scar formation to replace necrotic myocytes. While ECM accumulation following MI is termed cardiac fibrosis, this is a generic term that does not differentiate between ECM accumulation that occurs in the infarct region to form a scar that is structurally necessary to preserve left ventricle (LV) wall integrity and ECM accumulation that increases LV wall stiffness to exacerbate dilation and stimulate the progression to heart failure. This review focuses on post-MI LV ECM remodeling, targeting the discussion on ECM biomarkers that could be useful for predicting MI outcomes.
Keywords: extracellular matrix, myocardial infarction, biomarkers, collagen, matrix metalloproteinases
Introduction
Cardiovascular disease (CVD) is the most common cause of death in industrialized countries [1], and myocardial infarction (MI) contributes significantly to CVD mortality [2]. The prevalence of CVD is estimated to increase by 10% over the next 20 years, and by 2030 cause 23.6 million deaths each year worldwide [3]. In addition, public health costs for MI are estimated to increase three-fold over the next 2 decades [4].
Following MI, the left ventricle (LV) undergoes a series of molecular, cellular, and extracellular matrix (ECM) changes that progress over time to alter LV geometry and impair LV physiology [5]. The ECM is a dynamic and complex structure, which serves to preserve LV physiology under homeostasis and regulate the development of the infarct scar in response to MI. The balance between ECM synthesis and degradation defines net ECM accumulation (synthesis minus degradation), with ECM degradation prominent in the early MI response and ECM synthesis prominent in the later post-MI period [6,7]. Evaluating ECM flux over the MI time continuum may provide early diagnostic or prognostic indicators of LV remodeling and allow clinicians to stratify patients based on risk or treatment strategy. Biomarkers are currently used for the timely diagnosis of MI; however, their use to date has shown limited use in heart failure due to lack of specificity and selectivity [8]. For example, while infarct size and the extent of post-MI LV dilation tracks with adverse remodeling and progression to heart failure, there is a lot of individual variation in response that may be due to differences in intensity of the inflammatory response[9]. The development of ECM remodeling biomarkers that detect structural changes during MI would provide information for the time period that occurs after MI and before heart failure development. This review will focus on ECM remodeling in the post-MI LV and concentrates the discussion on ECM changes that could serve as biomarkers to predict MI outcomes.
Cell structural and physiological changes induced by MI
MI is defined as the occurrence of cardiomyocyte cell death due to prolonged ischemia that most frequently occurs due to coronary artery occlusion. MI generates an intense wound repair process, which includes one type of LV remodeling characterized by robust inflammation and scar formation [10]. Disruption of the ECM network due to degradation by matrix metalloproteinases (MMPs) interrupts the structural integrity and induces physiological impairment characterized by reductions in both systolic (due to myocyte loss) and diastolic (due to ECM) function [11]. LV remodeling encapsulates effects in the infarct region, which are structurally important for limiting infarct expansion induced LV dilation, and effects in the remote region, which increase LV wall stiffness [6,12]. A better understanding of the molecular and cellular LV remodeling events is important for maximizing long-term MI survival by limiting progression to heart failure. Figure 1 depicts the molecular and cellular time continuum of the different MI response phases: inflammation, wound healing, and collagen deposition.
Figure 1.
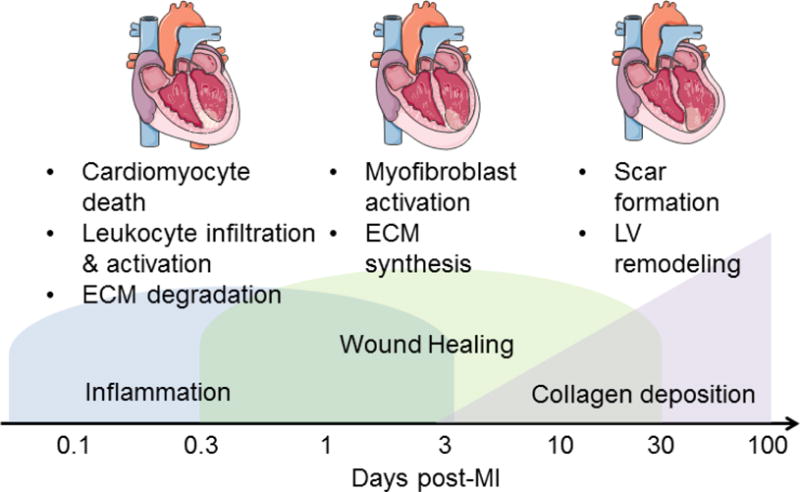
Cellular and molecular events during the three phases 1) Inflammation, 2) Wound Healing and 3) Collagen deposition post-MI.
The fate of the myocardium following MI depends on the balance of several competing events, including pro- vs. anti-inflammatory processes and ECM degradation vs. synthesis pathways. Excessive ECM accumulation can increase myocardial stiffness [13–17] and impair electrical activity [18]. Understanding which factors contribute to this balance will provide mechanistic insight into how the progression to heart failure is stimulated and identify new targets to examine.
Myocyte cell physiology is immediately impaired after the onset of ischemia, and by 30 minutes of ischemia, cardiomyocytes undergo irreversible cell death that stimulates an acute inflammatory response by upregulating the complement pathway [19–21]. Neutrophils and macrophages infiltrate into the infarct region and release inflammatory mediators, including matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinase (TIMP), to stimulate ECM degradation and necrotic cell removal [22]. While macrophage influx occurs first, neutrophil influx rapidly exceeds that of the macrophage and by post-MI day 1,, the neutrophil is the predominant leukocyte component in the infarct region [23]. Neutrophil numbers peak at day 1 post-MI and return to baseline by days 5-7 post-MI [24]. Macrophage numbers (Mac-3+ cells) peak at day 3 post-MI and begin to decline after day 7 [25]. ECM is cleaved by proteases to release necrotic myocytes; and these ECM peptide fragments generated by proteolytic processing stimulate fibroblasts into an activated form (Figure 2). Cardiac fibroblasts are the major ECM producers in the LV, and cardiac fibroblast numbers and polarization begin to increase by day 3 post-MI [26]. The collagen-rich reparative scar begins to form around day 5 post-MI to replace the extensive loss of cardiomyocytes in the infarcted area [27].
Figure 2.
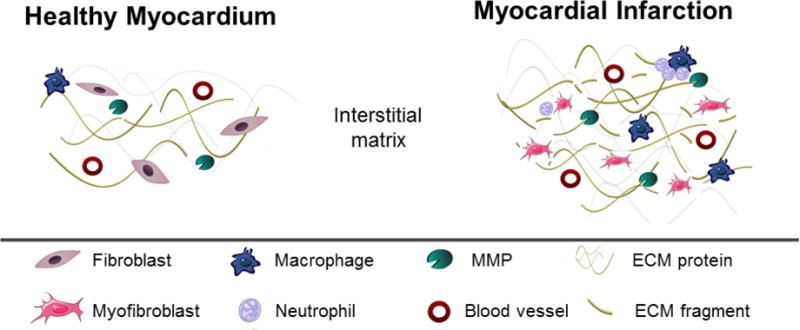
Illustration of the changes occurring in the ECM remodeling during development of MI.
Cardiac relevant ECM proteins induced by MI
The myocardial ECM is comprised of hundreds of components that include collagens, fibronectin, glycoproteins, glycosaminoglycans (GAGs), proteoglycans, and MMPs/TIMPs (Table 1). Some of these proteins can have matricellular properties, hence having a regulatory role rather than a structural one [7,28,29]. The cardiac ECM can be conceptually divided into proteins that reside in the interstitial matrix (e.g., collagen I, collagen III, and fibronectin) and proteins that reside in the basement membrane (collagen types IV, V, VII, X, and XIV and laminin) [30–32]. The basement membrane is the highly organized sheet-like ECM layer found on the surface of the cell sarcolemma and plays a key role in sarcomere formation via interactions with integrins [33].
Table 1.
Major myocardial extracellular matrix proteins and their functions
| Protein | Function | Localization | Change during MI | Reference |
|---|---|---|---|---|
| Collagen I | Tensile strength and structural support | Interstitial matrix | ↑ | [30,31] |
| Collagen III | Tissue elasticity | Interstitial matrix | ↑ | [30,31,34] |
| Collagen IV | Maintain basement membrane architecture | Basement membrane | ↑ | [49] |
| Collagen VI | Organization of fibrillar collagens/Anchoring to basement membrane | Interstitial matrix/basement membrane | ↑ | [158] |
| Elastin | Tissue elasticity and recoil capability | Interstitial matrix | ↑ | [52] |
| Fibronectin | Infiltration of connective tissue and inflammatory cells | Interstitial matrix | ↑ | [56] |
| Galectin-3 | Apposition of ECM proteins and increased collagen turnover. | Interstitial matrix | ↑ | [65] |
| Laminin | Maintain basement membrane architecture, cell migration, and angiogenesis | Basement membrane | ↑ | [46] |
| Osteopontin | Mediate adhesion, migration, growth, and differentiation | Interstitial matrix | ↑ | [159,160] |
| Periostin | Collagen fibrillogenesis and overall organization of ECM | Interstitial matrix | ↑ | [161] |
| Decorin | Bind to collagen type I and III, and affect formation of collagen fibrils | Interstitial matrix | ↑ | [63] |
| Versican | Mediate inflammatory cell-cell and cell-matrix interactions | Interstitial matrix | ↑ | [67] |
Fibrillar collagens
Historically, the cardiac ECM has been reported to be mainly composed of collagen; in the mammalian heart, approximately 85% of fibrillar collagen is type I, whereas type III comprises about 11% of total collagen [30,31,34]. In addition to comprising the bulk of the interstitial matrix, collagens (such as collagen XIII) may also play important structural roles in intercalated discs that form between cardiac myocytes [35]. Collagens are secreted as procollagens into the extracellular space, with the carboxy- and end-terminal peptides being proteolytically cleaved to form a mature collagen fibril [36]. Mature collagen is rendered insoluble and resistant to degradation via cross-linking by the enzymes lysyl oxidase [36]. Myocardial collagen content shows a biphasic nature following MI, with collagen degradation dominating early and synthesis dominating later as collagen is organized into scar tissue. Early post-MI, during the inflammatory phase, collagen is degraded by MMPs released by infiltrating neutrophils and monocytes, allowing these inflammatory cells to effectively phagocytose necrotic cells and creating a favorable environment for angiogenesis [37]. Proteolytically cleaved ECM fragments (matricryptins or matrikines) have important post-MI biological roles, including effects on cell proliferation, migration, differentiation, and inflammation [6]. Following the inflammatory phase, collagen deposition becomes favored over degradation. In rats with MI, collagen III gene expression is induced 2 days post-MI and continues to increase by 21 days post-MI, whereas collagen I gene expression is increased 4 days post-MI, peaks at 7 days post-MI, and remains at that level by 21 days post-MI [38]. The increase in collagen content remains throughout the life of the infarct scar and plays a crucial role in the maintenance or deterioration of cardiac physiology and the progression to heart failure (HF).
Expression of collagen cross-linking enzymes is also elevated during the maturation phase to enhance overall LV stiffness. Lysyl oxidase is ~5-fold elevated at day 3 post-MI, remains elevated by day 7, and then decreases [39]. Mice lacking matrix metalloproteinase-9 (MMP-9) are protected from MI-induced LV dilation, in part due to increased lysyl oxidase expression and collagen cross-linking [40]. In a rat model of cardiac volume overload-induced heart failure, inhibition of lysyl oxidase with beta-aminoproprionitrile also offers cardioprotection [41,42]. Advanced glycation end-product (AGE) are another source of collagen cross-linking that have been associated with poorer outcome post-MI [43]. Unlike cross-links produced by LOX, AGEs are formed spontaneously. In mice, methylglyoxal-AGEs are produced in the post-MI LV, suggesting a role for accumulation of collagen and functional loss of the heart [43]. These results indicate that the effect of LV stiffness on dilation is U-shaped rather than linear, with too much or too little having detrimental effects.
Basement Membrane Proteins
The basement membrane is a dense network of ECM proteins that surrounds cardiomyocytes, and includes laminins, collagen type IV, and a number of proteoglycans [44,45]. Laminin α1 is involved in basement membrane assembly and architecture, and plays an important role in cell migration through interactions with integrins α3β1 and α6β1 [46]. Fragmentation of the cardiac laminin network occurs after 1 hour of occlusion and continues for up to 7 days following reperfusion [47]. Studies have shown that basement membrane thickness is significantly increased around intramyocardial capillaries in patients with acute MI, which may contribute to impairment of oxygen diffusion and hypoxic stress [48]. Autoantibodies against collagen type IV are increased in post-MI patients, which may be causative or contributory to impaired basement membrane structural integrity and consequently endothelial cell dysfunction [49]. Laminin-derived peptides stimulate wound healing by facilitating angiogenesis [50]. Peptides derived from the degradation of collagen IV and perlecan play critical roles in angiogenesis, vascular structural integrity, and cell-cell interactions following myocardial ischemic injury [51].
Elastin
Elastin provides elasticity to tissues such as blood vessels and skin, and its expression is relatively low in the adult heart [33]. Elastin expression decreases in the post-MI scar, leading to decreased elasticity and resiliency to recoil, and contributing to stiffening of the scar tissue [52–55]. Implantation of COS-7 cells overexpressing elastin into the infarcted myocardium 6 days post-MI partially attenuated the decreases in fractional shortening (58% decrease in control rats versus 29% decrease in treated rats) and decreased infarct expansion by nearly 2-fold at 8-weeks post-MI, suggesting that elastin may be a therapeutic target for improving infarct strength and elasticity [42]. Like collagen, elastin can be cross-linked by lysyl oxidase, altering its tensile strength [33].
Fibronectin
Fibronectin is a glycoprotein that provides a scaffold for the infiltration of connective tissue and inflammatory cells [56]. Fibronectin is expressed in the developing heart and is a minor component of the adult uninjured LV [57]. Fibronectin is induced in the adult heart in response to MI, produced mainly by fibroblasts and endothelial cells. The alternatively spliced extra domain A (EDA) of fibronectin acts as a pro-inflammatory agent [58]. Fibronectin increases more than 10-fold in the infarcted area at day 2 post-MI and is believed to contribute to an increase in mechanical strength of the infarcted wall.
Proteoglycans
Proteoglycans are proteins covalently bound to long polysaccharide chains called glycosaminoglycans, including chondroitin/dermatan sulfate, keratin sulfate, and heparan sulfate [43,44]. Proteoglycans have been implicated in regulating the extent and organization of the collagen matrix [59]. Hyaluronan is a unique glycosaminoglycan in that it is not bound to a core protein, nor is it sulfated. Hyaluronan is increased post-MI in the LV infarct, and the generation of hyaluronan fragments promote inflammation, such that clearance of hyaluronan fragments is necessary for resolution of post-MI inflammation [60,61]. Administration of IL-10, an anti-inflammatory cytokine, may improve functional outcomes post-MI by downregulating hyaluronidase-3 and thuspreventing hyaluronan fragmentation and improving inflammation resolution [62].
Decorin binds and regulates assembly of collagen I and III fibrils and binds the pro-fibrotic transforming growth factor-beta (TGF-β) to inhibit its activity [48]. In vitro, collagen fibrils form more slowly and are of smaller diameter in the presence of decorin than those formed spontaneously, linking decorin with maintaining spatial order of collagen fibrils [63–65]. In rats, decorin increases by over 30% at week 5 post-MI and doubles by week 13 [63]. Mice lacking decorin show disorganized collagen fibril formation (lower packing density and increased heterogeneity in fibril size), more dilation (25% increase in LV circumference 2 weeks post-MI), and impaired physiology (ejection fraction reduced ~2-fold at 8 weeks post-MI) [66].
Versican, the major hyalectan (hyaluronan-binding proteoglycan) expressed in the LV, mediates inflammatory cell-cell and cell-matrix interactions in the infarcted heart and is primarily expressed by infiltrating monocytes [67]. Versican is induced 6 hours following MI and peaks at 2 days, then declines gradually. Versican is also necessary for normal cardiac and vascular development [68].
Matricellular Proteins
Matricellular proteins such as osteopontin, galectin-3, periostin, secreted protein acidic and rich in cysteine (SPARC), thrombospondin, and tenascins are secreted by myocardial resident and infiltrating cells and play non-structural roles in the cardiac ECM [69]. Matricellular proteins mediate processes such as cell adhesion, migration, growth, and differentiation.
Osteopontin is present at very low levels under basal conditions [70]. After MI, osteopontin expression markedly increases by 40-fold in the infarct region [70,71]. Osteopontin expression coincides with the development of pressure overload-induced heart failure, and may play a contributory role through its stimulatory effects on inducible nitric oxide synthase expression [71]. However, osteopontin may be cardioprotective post-MI, as osteopontin null mice show 1.6-fold increased normalized LV volume without concomitant increases in infarct collagen content 14 days following MI, suggesting that osteopontin is essential for collagen deposition and healing post-MI [72]. Osteopontin is cleaved at multiple sites by MMPs, including MMP-9, which may decrease its cardioprotective actions [72].
Galectin-3, a lectin expressed by inflammatory cells, particularly macrophages, plays critical roles in stimulating both inflammation and fibrosis, mainly by activating TGF-β, IL-1, and IL-2 signaling [44]. In mice, increased galectin-3 expression can be detected as early as 30 minutes post-MI, and is increased 3-fold by 24 hours [73]. At 7 days post-MI, mice lacking galectin-3 have more dilation (~10% increase in end-diastolic diameter), and decreased LV physiology (25% decreased ejection fraction and fractional shortening) [74].
Periostin, a collagen chaperone, is essentially undetectable in the adult myocardium except in valvular tissue, and is highly expressed in the infarct region 3 days post-MI mainly in cardiac fibroblasts [75]. Periostin appears to play dichotomous roles post-MI, being critical for normal healing and scar repair in the short term, but also contributing to cardiac fibrosis and LV stiffening in the long term. Mice lacking periostin have over 2-fold increased rupture rates 7 days post-MI and display 2-fold lower collagen content and 3-fold decreased cross-linked collagen [61]. In a porcine model, delivery of a periostin-derived peptide 2 days post-MI improves ejection fraction by about 25% at 3 months, but also leads to increases in myocardial fibrosis (6-fold increases in collagen content) in the remote region at one and 12 weeks post-treatment, limiting its clinical application as a viable therapeutic strategy [76]. ECM peptides have been well-studied in wound healing therapeutics, and there are a number of reasons for using a peptide rather than a whole molecule [77,78]. Peptides are easier to deliver, and cleaved protein fragments often have different functions than the whole molecule.
SPARC is a matricellular protein expressed by cardiac fibroblasts and endothelial cells that serves as an essential chaperone for procollagen processing and assembly of collagen fibrils [81]. SPARC expression is increased ~2-fold at 3 days post-MI, and mice lacking SPARC show less LV dilation (25% lower end-diastolic volume) and 33% increased ejection fraction following MI [81]. Cartilage oligomeric matrix protein (COMP) is a matricellular calcium-binding protein belonging to the thrombospondin family, which participates in cellular responses to growth factors and cytokines [79]. High levels of COMP have shown to be associated with lower risk of incident MI, suggesting a beneficial role in individuals without incident cardiovascular disease [80]. In this study, intact COMP was measured by a commercially available ELISA. As COMP is known to be degraded by ADAMTS7 [81] an assay measuring ADAMTS7-generated cleavage fragments of COMP may reveal a relationship between ADAMTS7 and COMP that has not yet been explored in the MI setting.
MMP roles in MI
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases responsible for ECM degradation and proteolytic activation of ECM proteins, inflammatory mediators, matricellular proteins, and other MMPs within the infarct region (Table 2) [82,83]Increased MMP activity in the vasculature can also lead to collagen degradation in fibro-atheromas, potentially resulting in plaque rupture and subsequent MI [82]. MMPs, therefore, are involved in all stages from atherosclerosis development to MI, post-MI wound healing, and progression to HF. MMP activity is dependent on the concentration of active enzyme and on the presence of a family of naturally occurring tissue inhibitors of metalloproteinases (TIMPs) [84]. Following MI, MMPs facilitate ECM degradation and recruit inflammatory cells to remove the necrotic cardiomyocytes. Initially the upregulation of pro-inflammatory cytokines results in robust MMP activation. After long-term stimulation of pro-inflammatory cytokines, TIMP level increase leading to decreased MMP/TIMP ratio and increases in fibrillar collagen deposition [85].
Table 2.
MMPs and TIMPs involved in MI remodeling
| MMP/TIMP | Expression post-MI | Biological roles | Reference |
|---|---|---|---|
| MMP-1 | ↑ | Degrade collagen type I and III MMP-1:TIMP-1 ratio is correlated with ejection fraction | [86] |
| MMP-2 | ↑ | Increased LV rupture and delay post-MI remodeling | [91–93] |
| MMP-3 | ↑ | Increased risk of long-term MI and independent predictor of LV systolic dysfunction | [162] |
| MMP-7 | ↑ | Deletion attenuates LV dilation post-MI | [21] |
| MMP-8 | ↑ | Early increase of MMP-8 is correlated with poor prognosis and promote infarct rupture in humans by degradation of collagens | [163] |
| MMP-9 | ↑ | MMP-9 deletion attenuated LV dilation post-MI | [95,96] |
| MMP-12 | ↑ | MMP-12 inhibition exacerbates cardiac dysfunction | [61] |
| MMP-28 | ↑ | MMP-28 deletion aggravated MI-induced LV dysfunction Increased LV rupture | [5] |
| TIMP-1 | ↑ | Associate with markers of HF severity, systemic inflammation and LV remodeling MMP-1:TIMP-1 ratio is correlated with ejection fraction | [107,109] |
| TIMP-2 | ↑ | Correlate with ECM deposition | [91] |
| TIMP-3 | ↓ | Cardioprotective and reduced post-MI | [114,116] |
| TIMP-4 | ↓ | Deletion attenuates increased LV rupture and mortality | [114] |
Of the 25 MMPs described to date, human MMP-1 has the highest affinity for fibrillar collagen and preferentially degrades collagen type I and collagen type III. Increases in MMP-1 synthesis have been reported in the infarcted myocardium in pigs and humans [86]. Inactivation of MMP-1 synthesis in the infarcted myocardium results in collagen accumulation, which in turn leads to a stiff and non-compliant LV and gradual cardiac dysfunction [87]. The interaction between MMP-1 and TIMP-1 is of critical relevance in the maintenance of the post-MI integrity of the cardiac collagen network. The MMP-1:TIMP-1 ratio has been inversely correlated with ejection fraction in patients with either systolic or diastolic heart failure [88]. Using an MMP to TIMP ratio is artificial, however, because there are more than one MMP and more than one TIMP present in the post-MI myocardium.
MMP-2 and -9 are the most widely studied MMPs, because they have historically been the easiest to measure using gel zymograms [89,90]. In a rat MI model, MMP-2 and -9 expression increases within 24 hours post-MI. At day 14, MMP-2 expression peaks and returns to baseline after 10 weeks, whereas MMP-9 expression continues to be elevated until 16 weeks post-MI [91,92]. There are species differences in MMP and TIMP expression, as MMP-9 is returning to baseline values by day 7 post-MI in the mouse. Excessive MMP activity post-MI leads to ECM degradation, which increases LV compliance and promotes cardiac rupture [55]. As such, MMP-2 deletion improves survival rates after MI by limiting cardiac rupture [93]. Infiltrating macrophages are a major source of MMP-9 [94], and MMP-9 deletion attenuates LV dilation post-MI [95,96]. This is mainly due to MMP-9 regulation of inflammation through its proteolytic activity of both ECM and cytokine substrates. MMP-9 mediates the post-MI degradation of CD36 to decrease macrophage phagocytosis of apoptotic neutrophils; MMP-9 deletion increased phagocytosis and neutrophil apoptosis to improve post-MI LV dilation [97]. MMP-9 promotes a pro-inflammatory macrophage phenotype, as MMP-9 deletion increases pro-reparative M2 markers in macrophages following MI [94]. Although a number of in vitro MMP-9 substrates have been identified, including collagens (type I, IV, V, VII, X and XIV), fibronectin, elastin, IL-8, Cxcl4, and IL-1β, the mechanisms whereby MMP-9 modulates LV remodeling post-MI have not been completely elucidated [98,99].
MMP-3 is a stromelysin sub-type, along with MMP-10 and MMP-11, and is actively involved in pro-MMP proteolysis [100]. Unlike MMP- 9, the active form of MMP-3 is down-regulated by day 3 in the infarct region [101]. Elevated levels of MMP-3 associate with increased MI risk, most likely due to MMP-3 acting as an upstream regulatory step in the activation of many MMPs [102]. MMP-7 is responsible for degrading a number of ECM and matricellular proteins, as well as non-ECM proteins such as connexin-43, contributing to the development of MI-related arrhythmias [21]. MMP-7 protein levels are up-regulated 3-fold in both remote and infarct regions at 7 days post-MI [103]. Similar to MMP-9, MMP-7 deletion has been shown to be protective post-MI albeit through different mechanisms [104].
MMP-8 and -13 are two of the major collagenases in the heart [105]. In rats, MMP-8 levels increase after 2 weeks and remain high until 16 weeks post-MI [91]. In cryoinfarcted mice, MMP-8 levels are increased 3 days post-MI, but return to normal by 2 weeks, suggesting potentially different roles for this MMP in rats and mice [105]. MMP-13 shows an identical post-MI pattern of expression to MMP-8, thus mediating collagen degradation early post-MI [105]. In patients, elevated levels of plasma MMP-8 early post-MI is correlated with poor prognosis whereas higher levels more than 20 months after MI is associated with relative preservation of LV systolic function, indicating a biphasic profile [106]. MMP-12 (macrophage metalloelastase) increases 3-fold in the LV infarct region at day 1 post-MI and remains upregulated until day 7. MMP-12 inhibition exacerbates cardiac dysfunction by prolonging neutrophil mediated inflammation [61]. Contrastingly, MMP-28 decreases post-MI as the source moves from the highly abundant myocyte to macrophage. MMP-28 deletion aggravated MI-induced LV dysfunction and rupture as a result of defective inflammatory response and scar formation by suppressing M2 macrophage activation[5].
TIMP-1 (29 kDa), a glycoprotein member of the TIMP family, co-localizes with MMP-1 in normal myocardium and is expressed by cardiac fibroblasts and myocytes [107–110]. TIMP-1 expression increases early, peaks at day 1 and remains elevated through day 21 post-MI [91,111,112]. Circulating TIMP-1 levels closely associate with markers of HF severity, systemic inflammation, and LV remodeling [113]. In addition, high levels of TIMP-1 are associated with diastolic dysfunction [114]. TIMP-1 null mice have a greater degree of LV dilation post-MI [114,115]. TIMP-2 levels increase by 2 weeks post-MI, correlating with deposition of ECM by infiltrating myofibroblasts, and return to baseline after 8 weeks during the late remodeling phase [91]. In contrast, TIMP-3 and -4 are significantly reduced post-MI [91,112]. TIMP-3 has cardioprotective functions, as TIMP-3 deletion leads to spontaneous dilated cardiomyopathy, and delivery of recombinant TIMP-3 post-MI improves LV ejection fraction, decreases LV dilation, and attenuates infarct expansion [116].
Biomarkers of MI: current state of the field
The use of biomarkers for early diagnosis of acute MI has been extremely valuable over the past 40 years [117]. The most widely established diagnostic biomarker of MI is cardiac troponin (cTn). The available assays target the C, I, or T subunits of troponin, which control the calcium mediated interaction of actin and myosin, leading to contraction and relaxation of the striated muscle [118]. Troponin I (cTnI) and troponin T (cTnT) are measured when they are released from cardiac myocytes upon myocardial damage. This biomarker is highly specific to cardiac tissue, since cTnI and cTnT have additional residues at the N- terminal end compared to the skeletal isoform of troponin [119]. Assays for troponins do present some disadvantages, including: high variability across commercial source; there could be misleading cTn levels in patients who receive reperfusion therapy; and there is no correlation between cTn and histopathology [10]. Nevertheless, cTn has since the year 2000 been the preferred biomarker for MI diagnosis [10].
C-reactive protein (CRP), myoglobin, and creatinine kinase (CK-MB) have been proposed as alternatives to cTn. CRP is a nonspecific acute-phase reactant protein produced in the liver used as marker of acute inflammation. Its use in MI is limited, since levels vary according to ethnicity, gender, genetics, and body weight. CRP is also non-specific for MI over other inflammatory conditions and has actually been described as a good indicator for non-cardiac pathologies [120,121]. CK-MB is an enzyme primarily located in the cardiac muscle. The MB isoenzyme is released upon myocardial injury [122]. During acute MI, the level of CK-MB doubles in the first 6 hours, and peaks within 12-24 hours [123,124]. CK-MB is a good indicator of infarct size and predicts risk of re-infarction. This marker elevates later than cTn and has in several cases been shown to be less sensitive and specific than cTn. Myoglobin is a small protein localized in the cytoplasm of cardiac and skeletal muscle cells. Due to its small size, myoglobin is rapidly released into the circulation upon injury and is generally one of the earliest to appear in the circulation. Elevated levels can be observed 0.2-2 hours after acute MI, which is considered rapid; due to its presence in skeletal muscle, however, this marker lacks tissue specificity [125,126].
General perspectives on ideal biomarker(s) of MI remodeling: opportunities for identifying targets
The ideal biomarker of MI response should be as non-invasive as possible and should monitor disease progression and predict long-term prognosis following MI. The biomarker assay should be easy to perform, reproducible, accurate, fast, and inexpensive. While cardiac-specificity is important to ensure no cross reactivity to other biological processes, it is not essential if MI is the only inflammatory pathology present [118,127]. Such non-invasive biomarkers may provide patients and their physicians with a better and timely assessment of diagnosis as well as surveillance of progression and response to treatment. The ECM proteome, which is strongly regulated during MI, may be a large source of potential new targets.
Neo-epitope biomarkers of ECM peptides: an opportunity for post-MI monitoring
A finely regulated balance between ECM degradation and synthesis is required for maintenance of tissue homeostasis. In pathological conditions, such as cardiac fibrosis, this balance is disrupted. An excessive production and accumulation of ECM proteins combined with dysregulated turnover contributes to post-MI cardiac remodeling and its progression to HF. The neo-epitope technology takes into consideration the action of proteases such as MMPs, which are active post-MI and work on ECM proteins at specific sites, to identify specific markers [128]. Immunoassays detecting neo-epitope peptides generated after protease cleavage and released into circulation would provide information on the dynamic turnover of the ECM and distinguish between responses that favor ECM degradation over formation (Figure 3). Targeting either protease-mediated fragmentation products (indicative of protein degradation) or the pro-peptide that is cleaved off the molecule during its maturation (indicative of protein formation) will provide information on ECM turnover. ECM degradation fragments not only reflect active connective tissue remodeling, the fragments can also acquire bioactive properties and be key mechanistic players in LV remodeling [78].
Figure 3. Neo-epitope markers detecting ECM remodeling.
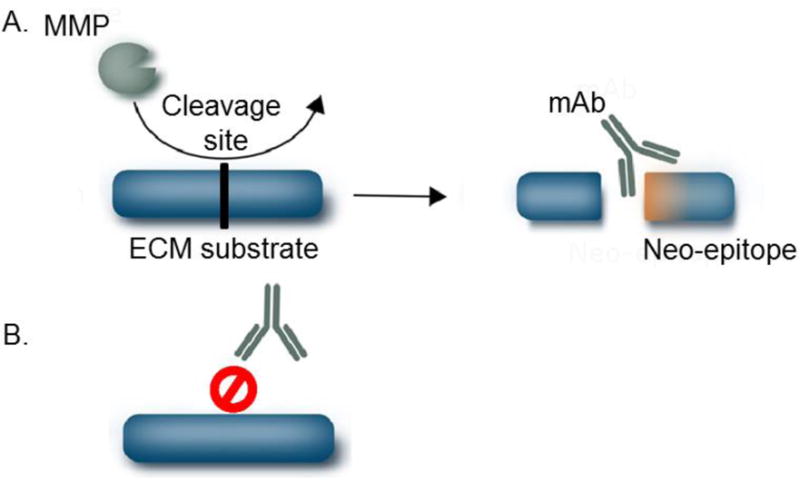
A) Formation of detectable neo-epitopes generated by cleavage of ECM proteins by specific proteases. B) In the absence of proteolytic cleavage, the ECM protein is not recognized by the antibody.
Soluble ST2, which is a receptor for cardiac fibroblast-derived interleukin-33, is elevated in the circulation of patients with heart failure with preserved ejection fraction and is strongly associated with myocardial fibrosis [36]. Both cardiac fibroblasts and cardiac myocytes are sources of IL-33 [36].
Neo-epitope biomarkers have already proven useful as serological and urinary markers of ECM remodeling in several organ injury models, including liver, lung [129–133], and kidney [134–136] and could potentially be applied to myocardial fibrosis following MI, Elevated circulating carboxyterminal telopeptide of collagen type I (CITP), a biomarker of collagen degradation, is elevated 2 days post-MI, peaks at 3 days, and remains at peak levels for 14 days [27], Elevated CITP is associated with poor long-term clinical outcomes including mortality, cardiac arrest, heart failure, and recurrent MI for up to 1 year post-MI [27], Other circulating peptides released during collagen synthesis, such as aminoterminal propeptide of type I procollagen (PINP) and type III procollagen (PIIINP), TIMP-1, and type I collagen telopeptide (ICTP), have prognostic value following acute MI in human patients, and in combination with brain natriuretic peptide (BNP) levels may help predict development of a primary composite event following MI [137]. Poor outcome events such as death, development of HF, and recurrent MI significantly increase in patients presenting with elevated serum collagen matricryptins (matrikines) [27–31].
Matricellular proteins such as SPARC, thrombospondin, osteopontin, and galectin-3 are also elevated in the circulation following myocardial injury [36]. Galectin-3 is up-regulated in animal models of HF and can be detected before the development of clinical HF [143–145]. Serum levels of galectin-3 increase in patients following acute MI, and progressive increases in circulating galectin-3 correlate with declining ejection fraction 24 weeks post-MI in patients [146]. Furthermore, patients with higher baseline levels of galectin-3 have a higher prevalence of MI [147]. Circulating MMP-9 is a potential prognostic biomarker for risk of cardiovascular disease and mortality, and reflects LV dilation and dysfunction following MI [148]. In addition to collagen degradation, circulating MMP-9 levels can also reflect inflammation status [148].
The use of mass spectrometry based proteomics has gained increasing interest, due to its ability to determine changes in the ECM of the heart and other tissues [6,149]. Proteomics can be used to identify new biomarker targets in a variety of sample types and is used in biomarker assays [150,151]. Several excellent recent reviews have highlighted how proteomics has contributed to our understanding of ECM remodeling [28,152–157]. A major current limitation of ECM biomarkers for MI wound healing responses is that other organs may also contribute to the pool of neo-epitope peptides released in serum, since the same ECM proteins are present in more than one organ. Whether there is a signature biomarker panel that reflects MI response remains to be determined.
Final considerations
The development of non-invasive ECM specific biomarkers could predict imbalanced tissue turnover, and thereby post-MI outcomes. Such biomarkers could facilitate clinical studies to both identify patients at risk of progressing to HF after MI and monitoring response to treatment strategies.
Highlights.
This review focuses on post-MI LV ECM remodeling, targeting the discussion on ECM biomarkers that could be useful for predicting MI outcomes.
Acknowledgments
We would like to thank past and present members of the Lindsey lab for their contributions to this knowledge. We acknowledge grant support from The Danish Research Foundation and the National Institute of Health HL075360, HL129823, HL051971, GM104357, GM114833, and GM115428, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 and IK2BX003922. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Danish Research Foundation, the National Institutes of Health, or the Veterans Administration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heidenreich Pa, Trogdon JG, Khavjou Oa, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson Sa, Nichol G, Orenstein D, Wilson PWF, Woo YJ. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, De Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB. Heart disease and stroke statistics-2015 update : A report from the American Heart Association. 2015 doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 3.Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:e442. doi: 10.1371/journal.pmed.0030442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PWF, Woo YJ. Forecasting the future of cardiovascular disease in the United States: A policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, Jin YF, Han HC, Manicone AM, Lindsey ML. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ Res. 2013;112:675–88. doi: 10.1161/CIRCRESAHA.111.300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spinale FG, Frangogiannis NG, Hinz B, Holmes JW, Kassiri Z, Lindsey ML. Crossing into the Next Frontier of Cardiac Extracellular Matrix Research. Circ Res. 2016;119:1040–1045. doi: 10.1161/CIRCRESAHA.116.309916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin YF, Lange RA, Tokmina-Roszyk D, Fields GB, De Castro Brás LE. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J Am Coll Cardiol. 2015;66:1364–1374. doi: 10.1016/j.jacc.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iqbal N, Wentworth B, Choudhary R, Landa ADLP, Kipper B, Fard A, Maisel AS. Cardiac biomarkers: new tools for heart failure management. Cardiovasc Diagn Ther. 2012;2:147–64. doi: 10.3978/j.issn.2223-3652.2012.06.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westman PC, Lipinski MJ, Luger D, Waksman R, Bonow RO, Wu E, Epstein SE. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J Am Coll Cardiol. 2016;67:2050–60. doi: 10.1016/j.jacc.2016.01.073. [DOI] [PubMed] [Google Scholar]
- 10.Joint T, Society E, College A. Myocardial infarction redefined — A consensus document of The Joint European Society of Cardiology / American College of Cardiology Committee for the Redefinition of Myocardial Infarction. The Joint European Society of Cardiology / American College of Card. 2000;4636:1502–1513. doi: 10.1053/euhj.2000.2305. [DOI] [PubMed] [Google Scholar]
- 11.van den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–7. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 12.Díez J, Querejeta R, López B, González A, Larman M, Martínez Ubago JL. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–7. doi: 10.1161/01.cir.0000017264.66561.3d. http://www.ncbi.nlm.nih.gov/pubmed/12034658. [DOI] [PubMed] [Google Scholar]
- 13.McLenachan JM, Dargie HJ. Ventricular arrhythmias in hypertensive left ventricular hypertrophy. Relationship to coronary artery disease, left ventricular dysfunction, and myocardial fibrosis. Am J Hypertens. 1990;3:735–40. doi: 10.1093/ajh/3.10.735. http://www.ncbi.nlm.nih.gov/pubmed/2145865. [DOI] [PubMed] [Google Scholar]
- 14.Díez J. Mechanisms of cardiac fibrosis in hypertension. J Clin Hypertens (Greenwich) 2007;9:546–50. doi: 10.1111/j.1524-6175.2007.06626.x. http://www.ncbi.nlm.nih.gov/pubmed/17617765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan D, et al. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:1–13. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JAC. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. doi: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janicki JS, Brower GL. The role of myocardial fibrillar collagen in ventricular remodeling and function. J Card Fail. 2002;8:S319–25. doi: 10.1054/jcaf.2002.129260. [DOI] [PubMed] [Google Scholar]
- 19.Frangogiannis NG. The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest. 2017;127:1600–1612. doi: 10.1172/JCI87491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeLeon-Pennell KY, Meschiari CA, Jung M, Lindsey ML. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog Mol Biol Transl Sci. 2017;147:75–100. doi: 10.1016/bs.pmbts.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol. 2016;91:114–22. doi: 10.1016/j.yjmcc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Y, De Castro Brás LE, Toba H, Iyer RP, Hall ME, Winniford MD, Lange RA, Tyagi SC, Lindsey ML. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch Eur J Physiol. 2014;466:1113–1127. doi: 10.1007/s00424-014-1463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frodermann V, Nahrendorf M. Neutrophil–macrophage cross-talk in acute myocardial infarction. Eur Heart J. 2016;38:ehw085. doi: 10.1093/eurheartj/ehw085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 2013;28:391–403. doi: 10.1152/physiol.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindsey ML. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. AJP Hear Circ Physiol. 2005;290:H232–H239. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- 26.Ma Y, Iyer RP, Jung M, Czubryt MP, Lindsey ML. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol Sci. 2017;38:448–458. doi: 10.1016/j.tips.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Y, Halade GV, Lindsey ML. Extracellular matrix and fibroblast communication following myocardial infarction. J Cardiovasc Transl Res. 2012;5:848–857. doi: 10.1161/CIRCULATIONAHA.111.087940.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barallobre-Barreiro J, Lynch M, Yin X, Mayr M. Systems biology-opportunities and challenges: the application of proteomics to study the cardiovascular extracellular matrix. Cardiovasc Res. 2016;112:626–636. doi: 10.1093/cvr/cvw206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldsmith EC, Bradshaw AD, Zile MR, Spinale FG. Myocardial fibroblast-matrix interactions and potential therapeutic targets. J Mol Cell Cardiol. 2014;70:92–9. doi: 10.1016/j.yjmcc.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix. J Pathol. 2003;200:423–8. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 31.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–87. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 32.Paulssons M, Saladin K. Mouse heart laminin. Purification of the native protein and structural comparison with Engelbreth-Holm-Swarm tumor laminin. J Biol Chem. 1989;264:18726–18732. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=2478549. [PubMed] [Google Scholar]
- 33.Yang H, Borg TK, Liu H, Gao BZ. Interactive relationship between basement-membrane development and sarcomerogenesis in single cardiomyocytes. Exp Cell Res. 2015;330:222–32. doi: 10.1016/j.yexcr.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwak HB. Aging, exercise, and extracellular matrix in the heart. J Exerc Rehabil. 2013;9:338–47. doi: 10.12965/jer.130049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hägg P, Väisänen T, Tuomisto A, Rehn M, Tu H, Huhtala P, Eskelinen S, Pihlajaniemi T. Type XIII collagen: a novel cell adhesion component present in a range of cell-matrix adhesions and in the intercalated discs between cardiac muscle cells. Matrix Biol. 2001;19:727–42. doi: 10.1016/s0945-053x(00)00119-0. http://www.ncbi.nlm.nih.gov/pubmed/11223332. [DOI] [PubMed] [Google Scholar]
- 36.Zile MR, Baicu CF. Biomarkers of diastolic dysfunction and myocardial fibrosis: application to heart failure with a preserved ejection fraction. J Cardiovasc Transl Res. 2013;6:501–15. doi: 10.1007/s12265-013-9472-1. [DOI] [PubMed] [Google Scholar]
- 37.Jourdan-LeSaux C, Zhang J, Lindsey ML. Extracellular matrix roles during cardiac repair. Life Sci. 2010;87:391–400. doi: 10.1016/j.lfs.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol. 1995;147:325–38. http://www.ncbi.nlm.nih.gov/pubmed/7639329. [PMC free article] [PubMed] [Google Scholar]
- 39.González-Santamaría J, Villalba M, Busnadiego O, López-Olañeta MM, Sandoval P, Snabel J, López-Cabrera M, Erler JT, Hanemaaijer R, Lara-Pezzi E, Rodríguez-Pascual F. Matrix cross-linking lysyl oxidases are induced in response to myocardial infarction and promote cardiac dysfunction. Cardiovasc Res. 2016;109:67–78. doi: 10.1093/cvr/cvv214. [DOI] [PubMed] [Google Scholar]
- 40.Voorhees AP, DeLeon-Pennell KY, Ma Y, Halade GV, Yabluchanskiy A, Iyer RP, Flynn E, Cates CA, Lindsey ML, Han HC. Building a better infarct: Modulation of collagen cross-linking to increase infarct stiffness and reduce left ventricular dilation post-myocardial infarction. J Mol Cell Cardiol. 2015;85:229–39. doi: 10.1016/j.yjmcc.2015.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El Hajj EC, El Hajj MC, Ninh VK, Bradley JM, Claudino MA, Gardner JD. Detrimental role of lysyl oxidase in cardiac remodeling. J Mol Cell Cardiol. 2017;109:17–26. doi: 10.1016/j.yjmcc.2017.06.013. [DOI] [PubMed] [Google Scholar]
- 42.El Hajj EC, El Hajj MC, Ninh VK, Gardner JD. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med (Maywood) 2016;241:539–49. doi: 10.1177/1535370215616511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blackburn NJR, Vulesevic B, McNeill B, Cimenci CE, Ahmadi A, Gonzalez-Gomez M, Ostojic A, Zhong Z, Brownlee M, Beisswenger PJ, Milne RW, Suuronen EJ. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res Cardiol. 2017;112:57. doi: 10.1007/s00395-017-0646-x. [DOI] [PubMed] [Google Scholar]
- 44.Yang H, Borg TK, Wang Z, Ma Z, Gao BZ. Role of the basement membrane in regulation of cardiac electrical properties. Ann Biomed Eng. 2014;42:1148–57. doi: 10.1007/s10439-014-0992-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11. doi: 10.1016/j.matbio.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tzu J, Marinkovich MP. Bridging structure with function: structural, regulatory, and developmental role of laminins. Int J Biochem Cell Biol. 2008;40:199–214. doi: 10.1016/j.biocel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324:475–88. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 48.Begieneman MPV, van de Goot FRW, Krijnen PAJ, Fritz J, Paulus WJ, Spreeuwenberg MD, van Hinsbergh VWM, Niessen HWM. The basement membrane of intramyocardial capillaries is thickened in patients with acute myocardial infarction. J Vasc Res. 2010;47:54–60. doi: 10.1159/000231721. [DOI] [PubMed] [Google Scholar]
- 49.McLeod O, Dunér P, Samnegård A, Tornvall P, Nilsson J, Hamsten A, Bengtsson E. Autoantibodies against basement membrane collagen type IV are associated with myocardial infarction. IJC Hear Vasc. 2015;6:42–47. doi: 10.1016/j.ijcha.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malinda KM, Wysocki AB, Koblinski JE, Kleinman HK, Ponce ML. Angiogenic lamininderived peptides stimulate wound healing. Int J Biochem Cell Biol. 2008;40:2771–80. doi: 10.1016/j.biocel.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lauten A, Gerhard-Garcia A, Suhr F, Fischer JH, Figulla HR, Bloch W. Impact of ischemia-reperfusion on extracellular matrix processing and structure of the basement membrane of the heart. PLoS One. 2014;9:e92833. doi: 10.1371/journal.pone.0092833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kielty CM, Sherratt MJ, Shuttleworth CA. Elastic fibres. J Cell Sci. 2002;115:2817–28. doi: 10.1242/jcs.115.14.2817. http://www.ncbi.nlm.nih.gov/pubmed/12082143. [DOI] [PubMed] [Google Scholar]
- 53.Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. 2002;415:168–71. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 54.Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng C-F, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J, Jr, Honjo T, Chien KR. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature. 2002;415:171–5. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno T, Mickle DAG, Kiani CG, Li RK. Overexpression of elastin fragments in infarcted myocardium attenuates scar expansion and heart dysfunction. Am J Physiol Heart Circ Physiol. 2005;288:H2819–27. doi: 10.1152/ajpheart.00862.2004. [DOI] [PubMed] [Google Scholar]
- 56.Casscells W, Kimura H, Sanchez JA, Yu ZX, Ferrans VJ. Immunohistochemical study of fibronectin in experimental myocardial infarction. Am J Pathol. 1990;137:801–10. http://www.ncbi.nlm.nih.gov/pubmed/2221013. [PMC free article] [PubMed] [Google Scholar]
- 57.Konstandin MH, Toko H, Gastelum GM, Quijada P, De La Torre A, Quintana M, Collins B, Din S, Avitabile D, Völkers M, Gude N, Fässler R, Sussman MA. Fibronectin is essential for reparative cardiac progenitor cell response after myocardial infarction. Circ Res. 2013;113:115–25. doi: 10.1161/CIRCRESAHA.113.301152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res. 2011;108:582–92. doi: 10.1161/CIRCRESAHA.110.224428. [DOI] [PubMed] [Google Scholar]
- 59.Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339:237–46. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- 60.Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res. 2014;114:872–88. doi: 10.1161/CIRCRESAHA.114.302533. [DOI] [PubMed] [Google Scholar]
- 61.Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, Lindsey ML. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. 2015;185:198–208. doi: 10.1016/j.ijcard.2015.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33. doi: 10.1007/s00395-017-0622-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimmerman SD, Thomas DP, Velleman SG, Li X, Hansen TR, McCormick RJ. Time course of collagen and decorin changes in rat cardiac and skeletal muscle post-MI. Am J Physiol Heart Circ Physiol. 2001;281:H1816–22. doi: 10.1152/ajpheart.2001.281.4.H1816. http://www.ncbi.nlm.nih.gov/pubmed/11557576. [DOI] [PubMed] [Google Scholar]
- 64.Vogel KG, Trotter JA. The effect of proteoglycans on the morphology of collagen fibrils formed in vitro. Coll Relat Res. 1987;7:105–14. doi: 10.1016/s0174-173x(87)80002-x. http://www.ncbi.nlm.nih.gov/pubmed/3621881. [DOI] [PubMed] [Google Scholar]
- 65.Vogel KG, Paulsson M, Heinegård D. Specific inhibition of type I and type II collagen fibrillogenesis by the small proteoglycan of tendon. Biochem J. 1984;223:587–97. doi: 10.1042/bj2230587. http://www.ncbi.nlm.nih.gov/pubmed/6439184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weis SM, Zimmerman SD, Shah M, Covell JW, Omens JH, Ross J, Dalton N, Jones Y, Reed CC, Iozzo RV, McCulloch AD. A role for decorin in the remodeling of myocardial infarction. Matrix Biol. 2005;24:313–24. doi: 10.1016/j.matbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 67.Toeda K, Nakamura K, Hirohata S, Hatipoglu OF, Demircan K, Yamawaki H, Ogawa H, Kusachi S, Shiratori Y, Ninomiya Y. Versican is induced in infiltrating monocytes in myocardial infarction. Mol Cell Biochem. 2005;280:47–56. doi: 10.1007/s11010-005-8051-4. [DOI] [PubMed] [Google Scholar]
- 68.Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol. 2010;29:304–16. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rienks M, Papageorgiou AP, Frangogiannis NG, Heymans S. Myocardial extracellular matrix: an ever-changing and diverse entity. Circ Res. 2014;114:872–88. doi: 10.1161/CIRCRESAHA.114.302533. [DOI] [PubMed] [Google Scholar]
- 70.Trueblood Na, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88:1080–1087. doi: 10.1161/hh1001.090842. [DOI] [PubMed] [Google Scholar]
- 71.Singh K, Sirokman G, Communal C, Robinson KG, Conrad CH, Brooks WW, Bing OH, Colucci WS. Myocardial osteopontin expression coincides with the development of heart failure. Hypertension. 1999;33:663–670. doi: 10.1161/01.HYP.33.2.663. [DOI] [PubMed] [Google Scholar]
- 72.Lindsey ML, Zouein FA, Tian Y, Iyer RP, De Castro Brás LE. Osteopontin is proteolytically processed by matrix metalloproteinase 9 1 HHS Public Access. Can J Physiol Pharmacol. 2015;93:879–886. doi: 10.1139/cjpp-2015-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hashmi S, Al-Salam S. Galectin-3 is expressed in the myocardium very early post-myocardial infarction. Cardiovasc Pathol. 2015;24:213–23. doi: 10.1016/j.carpath.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 74.González GE, Cassaglia P, Noli Truant S, Fernández MM, Wilensky L, Volberg V, Malchiodi EL, Morales C, Gelpi RJ. Galectin-3 is essential for early wound healing and ventricular remodeling after myocardial infarction in mice. Int J Cardiol. 2014;176:1423–5. doi: 10.1016/j.ijcard.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 75.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ladage D, Yaniz-Galende E, Rapti K, Ishikawa K, Tilemann L, Shapiro S, Takewa Y, Muller-Ehmsen J, Schwarz M, Garcia MJ, Sanz J, Hajjar RJ, Kawase Y. Stimulating myocardial regeneration with periostin Peptide in large mammals improves function post-myocardial infarction but increases myocardial fibrosis. PLoS One. 2013;8:e59656. doi: 10.1371/journal.pone.0059656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Genovese F, Karsdal MA. Protein degradation fragments as diagnostic and prognostic biomarkers of connective tissue diseases: understanding the extracellular matrix message and implication for current and future serological biomarkers. Expert Rev Proteomics. 2016;13:213–225. doi: 10.1586/14789450.2016.1134327. [DOI] [PubMed] [Google Scholar]
- 79.Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. http://www.ncbi.nlm.nih.gov/pubmed/11102749. [DOI] [PubMed] [Google Scholar]
- 80.Ueland T, Laugsand LE, Vatten LJ, Janszky I, Platou C, Michelsen AE, Damås JK, Aukrust P, Åsvold BO. Extracellular matrix markers and risk of myocardial infarction: The HUNT Study in Norway. Eur J Prev Cardiol. 2017;24:1161–1167. doi: 10.1177/2047487317703826. [DOI] [PubMed] [Google Scholar]
- 81.Liu CJ, Kong W, Ilalov K, Yu S, Xu K, Prazak L, Fajardo M, Sehgal B, Di Cesare PE. ADAMTS-7: a metalloproteinase that directly binds to and degrades cartilage oligomeric matrix protein. FASEB J. 2006;20:988–90. doi: 10.1096/fj.05-3877fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newby AC. Metalloproteinases promote plaque rupture and myocardial infarction: A persuasive concept waiting for clinical translation. Matrix Biol. 2015;44–46:157–66. doi: 10.1016/j.matbio.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 83.Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015;44–46:113–21. doi: 10.1016/j.matbio.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 84.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–39. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 85.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. 2002;91:988–98. doi: 10.1161/01.res.0000043825.01705.1b. http://www.ncbi.nlm.nih.gov/pubmed/12456484. [DOI] [PubMed] [Google Scholar]
- 86.Spinale FG, Coker ML, Bond BR, Zellner JL. Myocardial matrix degradation and metalloproteinase activation in the failing heart: a potential therapeutic target. Cardiovasc Res. 2000;46:225–38. doi: 10.1016/s0008-6363(99)00431-9. http://www.ncbi.nlm.nih.gov/pubmed/10773226. [DOI] [PubMed] [Google Scholar]
- 87.Foronjy RF, Sun J, Lemaitre V, D’Armiento JM. Transgenic expression of matrix metalloproteinase-1 inhibits myocardial fibrosis and prevents the transition to heart failure in a pressure overload mouse model. Hypertens Res. 2008;31:725–35. doi: 10.1291/hypres.31.725. [DOI] [PubMed] [Google Scholar]
- 88.López B, González A, Querejeta R, Larman M, Díez J. Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol. 2006;48:89–96. doi: 10.1016/j.jacc.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 89.Iyer RP, Patterson NL, Fields GB, Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. AJP Hear Circ Physiol. 2012;303:H919–H930. doi: 10.1152/ajpheart.00577.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iyer RP, De Castro Brás LE, Jin YF, Lindsey ML. Translating Koch’s postulates to identify matrix metalloproteinase roles in postmyocardial infarction remodeling: Cardiac metalloproteinase actions (carMA) postulates. Circ Res. 2014;114:860–871. doi: 10.1161/CIRCRESAHA.114.301673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peterson JT, Li H, Dillon L, Bryant JW. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res. 2000;46:307–15. doi: 10.1016/s0008-6363(00)00029-8. http://www.ncbi.nlm.nih.gov/pubmed/10773235. [DOI] [PubMed] [Google Scholar]
- 92.Deten A, Volz HC, Holzl A, Briest W, Zimmer HG. Effect of propranolol on cardiac cytokine expression after myocardial infarction in rats. Mol Cell Biochem. 2003;251:127–37. http://www.ncbi.nlm.nih.gov/pubmed/14575314. [PubMed] [Google Scholar]
- 93.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest. 2005;115:599–609. doi: 10.1172/JCI22304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yabluchanskiy A, Ma Y, DeLeon-Pennell KY, Altara R, Halade GV, Voorhees AP, Nguyen NT, Jin YF, Winniford MD, Hall ME, Han HC, Lindsey ML. Myocardial Infarction Superimposed on Aging: MMP-9 Deletion Promotes M2 Macrophage Polarization. J Gerontol A Biol Sci Med Sci. 2016;71:475–83. doi: 10.1093/gerona/glv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–42. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 97.DeLeon-Pennell KY, Tian Y, Zhang B, Cates CA, Iyer RP, Cannon P, Shah P, Aiyetan P, Halade GV, Ma Y, Flynn E, Zhang Z, Jin YF, Zhang H, Lindsey ML. CD36 Is a Matrix Metalloproteinase-9 Substrate That Stimulates Neutrophil Apoptosis and Removal During Cardiac Remodeling. Circ Cardiovasc Genet. 2016;9:14–25. doi: 10.1161/CIRCGENETICS.115.001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, Weintraub ST, Lindsey ML. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10:2214–23. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zitka O, Kukacka J, Krizkova S, Huska D, Adam V, Masarik M, Prusa R, Kizek R. Matrix metalloproteinases. Curr Med Chem. 2010;17:3751–68. doi: 10.2174/092986710793213724. http://www.ncbi.nlm.nih.gov/pubmed/20846107. [DOI] [PubMed] [Google Scholar]
- 100.Chen Q, Jin M, Yang F, Zhu J, Xiao Q, Zhang L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013;2013 doi: 10.1155/2013/928315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2011;301:H497–505. doi: 10.1152/ajpheart.01070.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cavusoglu E, Marmur JD, Kassotis JT, Yanamadala S, Chopra V, Eng C. Usefulness of Plasma Matrix Metalloproteinase-3 Levels to Predict Myocardial Infarction in Men With and Without Acute Coronary Syndrome. Am J Cardiol. 2016;117:881–6. doi: 10.1016/j.amjcard.2015.12.022. [DOI] [PubMed] [Google Scholar]
- 103.Lindsey ML, Escobar GP, Dobrucki LW, Goshorn DK, Bouges S, Mingoia JT, McClister DM, Su H, Gannon J, MacGillivray C, Lee RT, Sinusas AJ, Spinale FG. Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am J Physiol Heart Circ Physiol. 2006;290:H232–9. doi: 10.1152/ajpheart.00457.2005. [DOI] [PubMed] [Google Scholar]
- 104.Yabluchanskiy A, Li Y, de Castro Brás LE, Hakala K, Weintraub ST, Lindsey ML. Proteomic analysis of the left ventricle post-myocardial infarction to identify in vivo candidate matrix metalloproteinase substrates. Methods Mol Biol. 2013;1066:185–99. doi: 10.1007/978-1-62703-604-7_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Miyasato SK, Loeffler J, Shohet R, Zhang J, Lindsey M, Le Saux CJ. Caveolin-1 modulates TGF-β1 signaling in cardiac remodeling. Matrix Biol. 2011;30:318–29. doi: 10.1016/j.matbio.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Erkol A, Pala S, Oduncu V, Kılıcgedik A, Kızılırmak F, Karabay C, Güler A, Kırma C. Relation of plasma matrix metalloproteinase-8 levels late after myocardial infarction with left ventricular volumes and ejection fraction. Turk Kardiyol Dern Arsivi-Archives Turkish Soc Cardiol. 2013;41:617–624. doi: 10.5543/tkda.2013.68625. [DOI] [PubMed] [Google Scholar]
- 107.Lindsey ML, Goshorn DK, Squires CE, Escobar GP, Hendrick JW, Mingoia JT, Sweterlitsch SE, Spinale FG. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc Res. 2005;66:410–9. doi: 10.1016/j.cardiores.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 108.Romanic AM, Burns-Kurtis CL, Gout B, Berrebi-Bertrand I, Ohlstein EH. Matrix metalloproteinase expression in cardiac myocytes following myocardial infarction in the rabbit. Life Sci. 2001;68:799–814. doi: 10.1016/s0024-3205(00)00982-6. http://www.ncbi.nlm.nih.gov/pubmed/11205871. [DOI] [PubMed] [Google Scholar]
- 109.Tyagi SC, Kumar S, Glover G. Induction of tissue inhibitor and matrix metalloproteinase by serum in human heart-derived fibroblast and endomyocardial endothelial cells. J Cell Biochem. 1995;58:360–71. doi: 10.1002/jcb.240580309. [DOI] [PubMed] [Google Scholar]
- 110.Arpino V, Brock M, Gill SE. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015;44–46:247–54. doi: 10.1016/j.matbio.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 111.Lu L, Zhang JQ, Ramires FJ, Sun Y. Molecular and cellular events at the site of myocardial infarction: from the perspective of rebuilding myocardial tissue. Biochem Biophys Res Commun. 2004;320:907–13. doi: 10.1016/j.bbrc.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 112.Kandalam V, Basu R, Abraham T, Wang X, Awad A, Wang W, Lopaschuk GD, Maeda N, Oudit GY, Kassiri Z. Early activation of matrix metalloproteinases underlies the exacerbated systolic and diastolic dysfunction in mice lacking TIMP3 following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;299:H1012–23. doi: 10.1152/ajpheart.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cavusoglu E, Ruwende C, Chopra V, Yanamadala S, Eng C, Clark LT, Pinsky DJ, Marmur JD. Tissue inhibitor of metalloproteinase-1 (TIMP-1) is an independent predictor of all-cause mortality, cardiac mortality, and myocardial infarction. Am Heart J. 2006;151:1101.e1–8. doi: 10.1016/j.ahj.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 114.Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR. Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease. Circulation. 2006;113:2089–96. doi: 10.1161/CIRCULATIONAHA.105.573865. [DOI] [PubMed] [Google Scholar]
- 115.Creemers EEJM, Davis JN, Parkhurst AM, Leenders P, Dowdy KB, Hapke E, Hauet AM, Escobar PG, Cleutjens JPM, Smits JFM, Daemen MJAP, Zile MR, Spinale FG. Deficiency of TIMP-1 exacerbates LV remodeling after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2003;284:H364–71. doi: 10.1152/ajpheart.00511.2002. [DOI] [PubMed] [Google Scholar]
- 116.Eckhouse SR, Purcell BP, McGarvey JR, Lobb D, Logdon CB, Doviak H, O’Neill JW, Shuman JA, Novack CP, Zellars KN, Pettaway S, Black RA, Khakoo A, Lee T, Mukherjee R, Gorman JH, Gorman RC, Burdick JA, Spinale FG. Local hydrogel release of recombinant TIMP-3 attenuates adverse left ventricular remodeling after experimental myocardial infarction. Sci Transl Med. 2014;6:223ra21. doi: 10.1126/scitranslmed.3007244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meds JR, Zhang A, Meds TF. Biomarkers of myocardial infarction : past, present and future. 2015:23–25. [Google Scholar]
- 118.Ahmad MI, Sharma N. Biomarkers in Acute Myocardial Infarction. 2012;3 doi: 10.4172/2155-9880.1000222. [DOI] [Google Scholar]
- 119.Gerhardt W, Katus H, Ravkilde J, L P, Hnmm C, J PJ, Peheim E, Ljungdahl L. S-Troponin T in Suspected lschemic Myocardial Injury Compared with Mass and Catalytic Concentrations of S-Creatine Kinase lsoenzyme MB. 1991;37:1405–1411. [PubMed] [Google Scholar]
- 120.Sepulveda JL, Mehta JL. C-reactive protein and cardiovascular disease: a critical appraisal. Curr Opin Cardiol. 2005;20:407–16. doi: 10.1097/01.hco.0000175518.57804.94. [DOI] [PubMed] [Google Scholar]
- 121.Dietrich M, Jialal I. The effect of weight loss on a stable biomarker of inflammation, C-reactive protein. Nutr Rev. 2005;63:22–8. doi: 10.1111/j.1753-4887.2005.tb00107.x. http://www.ncbi.nlm.nih.gov/pubmed/15730232. [DOI] [PubMed] [Google Scholar]
- 122.Young GP, Gibler WB, Hedges JR, Hoekstra JW, Slovis C, Aghababian R, Smith M, Rubison M, Ellis J. Serial creatine kinase-MB results are a sensitive indicator of acute myocardial infarction in chest pain patients with nondiagnostic electrocardiograms: the second Emergency Medicine Cardiac Research Group Study. Acad Emerg Med. 1997;4:869–77. doi: 10.1111/j.1553-2712.1997.tb03812.x. http://www.ncbi.nlm.nih.gov/pubmed/9305428. [DOI] [PubMed] [Google Scholar]
- 123.Jurlander B, Clemmensen P, Wagner GS, Grande P. Very early diagnosis and risk stratification of patients admitted with suspected acute myocardial infarction by the combined evaluation of a single serum value of cardiac troponin-T, myoglobin, and creatine kinase MB(mass) Eur Heart J. 2000;21:382–389. doi: 10.1053/euhj.1999.1760. [DOI] [PubMed] [Google Scholar]
- 124.Newby LK, Storrow AB, Gibler WB, Garvey JL, Tucker JF, Kaplan AL, Schreiber DH, Tuttle RH, Mcnulty SE, Ohman EM. Clinical Investigation and Reports Bedside Multimarker Testing for Risk Stratification. Circulation. 2001:1832–1837. doi: 10.1161/01.cir.103.14.1832. [DOI] [PubMed] [Google Scholar]
- 125.Sallach SM, Nowak R, Hudson MP, Tokarski G, Khoury N, Tomlanovich MC, Jacobsen G, de Lemos JA, McCord J. A change in serum myoglobin to detect acute myocardial infarction in patients with normal troponin I levels. Am J Cardiol. 2004;94:864–867. doi: 10.1016/j.amjcard.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 126.Vassiliadis E, Barascuk N, Didangelos A, Karsdal Ma. Novel cardiac-specific biomarkers and the cardiovascular continuum. Biomark Insights. 2012;7:45–57. doi: 10.4137/BMI.S9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Warburton FG, Bernstein A, Wright ANNC. SERUM CREATINE PHOSPHOKINASE ESTIMATIONS IN. 1965 doi: 10.1136/hrt.27.5.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Karsdal Ma, Nielsen MJ, S JM, Henriksen K, Genovese F, Bay-Jensen A-C, Smith V, Adamkewicz JI, Christiansen C, Leeming DJ. Extracellular matrix remodeling: the common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. Assay Drug Dev Technol. 2013;11:70–92. doi: 10.1089/adt.2012.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, Molyneaux PL, McKeever TM, Wells AU, Flynn A, Hubbard RB, Leeming DJ, Marshall RP, Karsdal MA, Lukey PT, Maher TM. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. 2015;3:462–472. doi: 10.1016/S2213-2600(15)00048-X. [DOI] [PubMed] [Google Scholar]
- 130.Sand JMB, Leeming DJ, Byrjalsen I, Bihlet AR, Lange P, Tal-Singer R, Miller BE, Karsdal MA, Vestbo J. High levels of biomarkers of collagen remodeling are associated with increased mortality in COPD ? results from the ECLIPSE study. Respir Res. 2016;17:125. doi: 10.1186/s12931-016-0440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stolz D, Leeming DJ, Kristensen JHE, Karsdal MA, Boersma W, Louis R, Milenkovic B, Kostikas K, Blasi F, Aerts J, Sand JMB, Wouters EFM, Rohde G, Prat C, Torres A, Welte T, Roth M, Papakonstantinou E, Tamm M. Systemic Biomarkers of Collagen and Elastin Turnover Are Associated With Clinically Relevant Outcomes in COPD. Chest. 2017;151:47–59. doi: 10.1016/j.chest.2016.08.1440. [DOI] [PubMed] [Google Scholar]
- 132.Sand JMB, Martinez G, Midjord AK, Karsdal MA, Leeming DJ, Lange P. Characterization of serological neo-epitope biomarkers reflecting collagen remodeling in clinically stable chronic obstructive pulmonary disease. Clin Biochem. 2016;49:1144–1151. doi: 10.1016/j.clinbiochem.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 133.Bihlet AR, Karsdal MA, Sand JMB, Leeming DJ, Roberts M, White W, Bowler R. Biomarkers of extracellular matrix turnover are associated with emphysema and eosinophilic-bronchitis in COPD. Respir Res. 2017;18:22. doi: 10.1186/s12931-017-0509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stribos EGD, Nielsen SH, Brix S, Karsdal MA, Seelen MA, van Goor H, Bakker SJL, Olinga P, Mutsaers HAM, Genovese F. Non-invasive quantification of collagen turnover in renal transplant recipients. PLoS One. 2017;12:e0175898. doi: 10.1371/journal.pone.0175898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fenton A, Jesky MD, Ferro CJ, S?rensen J, Karsdal MA, Cockwell P, Genovese F. Serum endotrophin, a type VI collagen cleavage product, is associated with increased mortality in chronic kidney disease. PLoS One. 2017;12:e0175200. doi: 10.1371/journal.pone.0175200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Genovese F, Boor P, Papasotiriou M, Leeming DJ, Karsdal MA, Floege J. Turnover of type III collagen reflects disease severity and is associated with progression and microinflammation in patients with IgA nephropathy. Nephrol Dial Transplant. 2016;31:472–479. doi: 10.1093/ndt/gfv301. [DOI] [PubMed] [Google Scholar]
- 137.Eschalier R, Fertin M, Fay R, Bauters C, Zannad F, Pinet F, Rossignol P. Extracellular matrix turnover biomarkers predict long-term left ventricular remodeling after myocardial infarction: insights from the REVE-2 study. Circ Heart Fail. 2013;6:1199–205. doi: 10.1161/CIRCHEARTFAILURE.113.000403. [DOI] [PubMed] [Google Scholar]
- 138.Barthélémy O, Beygui F, Vicaut E, Rouanet S, Van Belle E, Baulac C, Degrandsart A, Dallongeville J, Montalescot G, OPERA Investigators Relation of high concentrations of plasma carboxy-terminal telopeptide of collagen type I with outcome in acute myocardial infarction. Am J Cardiol. 2009;104:904–9. doi: 10.1016/j.amjcard.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 139.Barasch E, Gottdiener JS, Aurigemma G, Kitzman DW, Han J, Kop WJ, Tracy RP. Association between elevated fibrosis markers and heart failure in the elderly: the cardiovascular health study. Circ Heart Fail. 2009;2:303–10. doi: 10.1161/CIRCHEARTFAILURE.108.828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lombardi R, Betocchi S, Losi MA, Tocchetti CG, Aversa M, Miranda M, D’Alessandro G, Cacace A, Ciampi Q, Chiariello M. Myocardial collagen turnover in hypertrophic cardiomyopathy. Circulation. 2003;108:1455–60. doi: 10.1161/01.CIR.0000090687.97972.10. [DOI] [PubMed] [Google Scholar]
- 141.Martos R, Baugh J, Ledwidge M, O’Loughlin C, Conlon C, Patle A, Donnelly SC, McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–95. doi: 10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 142.Plaksej R, Kosmala W, Frantz S, Herrmann S, Niemann M, Störk S, Wachter R, Angermann CE, Ertl G, Bijnens B, Weidemann F. Relation of circulating markers of fibrosis and progression of left and right ventricular dysfunction in hypertensive patients with heart failure. J Hypertens. 2009;27:2483–91. doi: 10.1097/HJH.0b013e3283316c4d. [DOI] [PubMed] [Google Scholar]
- 143.Kamal FA, Watanabe K, Ma M, Abe Y, Elbarbary R, Kodama M, Aizawa Y. A novel phenylpyridazinone, T-3999, reduces the progression of autoimmune myocarditis to dilated cardiomyopathy. Heart Vessels. 2011;26:81–90. doi: 10.1007/s00380-010-0018-z. [DOI] [PubMed] [Google Scholar]
- 144.Liu Y-H, D’Ambrosio M, Liao T, Peng H, Rhaleb N-E, Sharma U, André S, Gabius H-J, Carretero OA. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol. 2009;296:H404–12. doi: 10.1152/ajpheart.00747.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Psarras S, Mavroidis M, Sanoudou D, Davos CH, Xanthou G, Varela AE, Panoutsakopoulou V, Capetanaki Y. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur Heart J. 2012;33:1954–63. doi: 10.1093/eurheartj/ehr119. [DOI] [PubMed] [Google Scholar]
- 146.Weir RAP, Petrie CJ, Murphy CA, Clements S, Steedman T, Miller AM, McInnes IB, Squire IB, Ng LL, Dargie HJ, McMurray JJV. Galectin-3 and cardiac function in survivors of acute myocardial infarction. Circ Heart Fail. 2013;6:492–8. doi: 10.1161/CIRCHEARTFAILURE.112.000146. [DOI] [PubMed] [Google Scholar]
- 147.van der Velde AR, Meijers WC, Ho JE, Brouwers FP, Rienstra M, Bakker SJL, Muller Kobold AC, van Veldhuisen DJ, van Gilst WH, van der Harst P, de Boer RA. Serial galectin-3 and future cardiovascular disease in the general population. Heart. 2016;102:1134–41. doi: 10.1136/heartjnl-2015-308975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Halade GV, Jin YF, Lindsey ML. Matrix metalloproteinase (MMP)-9: a proximal biomarker for cardiac remodeling and a distal biomarker for inflammation. Pharmacol Ther. 2013;139:32–40. doi: 10.1016/j.pharmthera.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lindsey ML, Hall ME, Harmancey R, Ma Y. Adapting extracellular matrix proteomics for clinical studies on cardiac remodeling post-myocardial infarction. Clin Proteomics. 2016;13:19. doi: 10.1186/s12014-016-9120-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Quinn KP, Sullivan KE, Liu Z, Ballard Z, Siokatas C, Georgakoudi I, Black LD. Optical metrics of the extracellular matrix predict compositional and mechanical changes after myocardial infarction. Sci Rep. 2016;6:35823. doi: 10.1038/srep35823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin Y-F, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Brás LE. A Novel Collagen Matricryptin Reduces Left Ventricular Dilation Post-Myocardial Infarction by Promoting Scar Formation and Angiogenesis. J Am Coll Cardiol. 2015;66:1364–74. doi: 10.1016/j.jacc.2015.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Iyer RP, Jung M, Lindsey ML. MMP-9 signaling in the left ventricle following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;311:H190–8. doi: 10.1152/ajpheart.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lindsey ML, Iyer RP, Jung M, DeLeon-Pennell KY, Ma Y. Matrix metalloproteinases as input and output signals for post-myocardial infarction remodeling. J Mol Cell Cardiol. 2016;91:134–40. doi: 10.1016/j.yjmcc.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lindsey ML, Mayr M, Gomes AV, Delles C, Arrell DK, Murphy AM, Lange RA, Costello CE, Jin YF, Laskowitz DT, Sam F, Terzic A, Van Eyk J, Srinivas PR, American Heart Association Council on Functional Genomics and Translational Biology, Council on Cardiovascular Disease in the Young, Council on Clinical Cardiology, Council on Cardiovascular and Stroke Nursing, Council on Hypertension, and Stroke Council Transformative Impact of Proteomics on Cardiovascular Health and Disease: A Scientific Statement From the American Heart Association. Circulation. 2015;132:852–72. doi: 10.1161/CIR.0000000000000226. [DOI] [PubMed] [Google Scholar]
- 155.Amar S, Minond D, Fields GB. Clinical Implications of Compounds Designed to Inhibit ECM-Modifying Metalloproteinases. Proteomics. 2017:1600389. doi: 10.1002/pmic.201600389. [DOI] [PubMed] [Google Scholar]
- 156.Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML. The impact of aging on cardiac extracellular matrix. GeroScience. 2017;39:7–18. doi: 10.1007/s11357-017-9959-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Manninen A, Varjosalo M. A proteomics view on integrin-mediated adhesions. Proteomics. 2017;17:1600022. doi: 10.1002/pmic.201600022. [DOI] [PubMed] [Google Scholar]
- 158.Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, Bonaldo P, Chilian WM, Meszaros JG. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ Res. 2012;110:851–6. doi: 10.1161/CIRCRESAHA.111.252734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res. 2001;88:1080–7. doi: 10.1161/hh1001.090842. http://www.ncbi.nlm.nih.gov/pubmed/11375279. [DOI] [PubMed] [Google Scholar]
- 160.Singh K, Sirokman G, Communal C, Robinson KG, Conrad CH, Brooks WW, Bing OH, Colucci WS. Myocardial osteopontin expression coincides with the development of heart failure. Hypertens (Dallas, Tex 1979) 1999;33:663–70. doi: 10.1161/01.hyp.33.2.663. http://www.ncbi.nlm.nih.gov/pubmed/10024324. [DOI] [PubMed] [Google Scholar]
- 161.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–47. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kelly D, Khan S, Cockerill G, Ng LL, Thompson M, Samani NJ, Squire IB. Circulating Stromelysin-1 (MMP-3): A novel predictor of LV dysfunction, remodelling and all-cause mortality after acute myocardial infarction. Eur J Heart Fail. 2008;10:133–139. doi: 10.1016/j.ejheart.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.van den Borne SWM, Cleutjens JPM, Hanemaaijer R, Creemers EE, Smits JFM, Daemen MJAP, Blankesteijn WM. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc Pathol. 2009;18:37–43. doi: 10.1016/j.carpath.2007.12.012. [DOI] [PubMed] [Google Scholar]