

Abstract
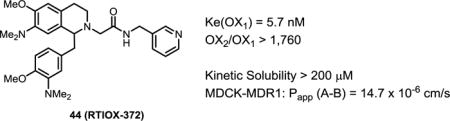
Orexins are hypothalamic neuropeptides playing important roles in many functions including the motivation of addictive behaviors. Blockade of the orexin-1 receptor has been suggested as a potential strategy for the treatment of drug addiction. We have previously reported OX1 receptor antagonists based on the tetrahydroisoquinoline scaffold with excellent OX1 potency and selectivity; however, these compounds had high lipophilicity (clogP > 5) and low to moderate solubility. In an effort to improve their properties, we have designed and synthesized a series of analogs where the 7-position substituents known to favor OX1 potency and selectivity were retained, and groups of different nature were introduced at the 1-position where substitution was generally tolerated as demonstrated in previous studies. Compound 44 with lower lipophilicity (clogP = 3.07) displayed excellent OX1 potency (Ke = 5.7 nM) and selectivity (> 1,760-fold over OX2) in calcium mobilization assays. In preliminary ADME studies, 44 showed excellent kinetic solubility (> 200 μM), good CNS permeability (Papp = 14.7 × 10−6 cm/sec in MDCK assay), and low drug efflux (efflux ratio = 3.3).
Keywords: orexin, antagonist, selective, tetrahydroisoquinoline, ADME
TOC image
Introduction
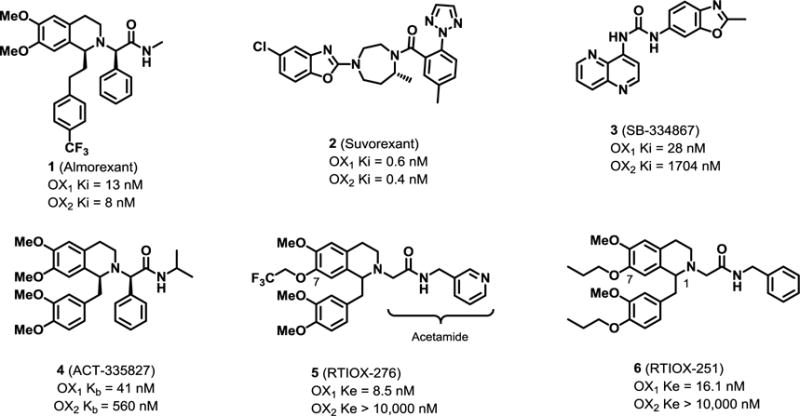
Orexin-A and -B, also known as hypocretin-1 and -2, are two neuropeptides synthesized by neurons in the lateral hypothalamic areas, that activate two G protein-coupled receptors (GPCRs), the orexin-1 (OX1) and -2 (OX2) receptors. Orexins, the orexin receptors and the hypocretin genes were first discovered in 1998 by two research groups,1, 2 and the system has been widely studied since.3, 4 Initially, orexins were named for their appetite-enhancing effects as they were found to play a role in feeding behavior and energy expenditure. The orexin system was later shown to also be a key regulator of arousal and sleep/wake cycles. Hence, the orexin system is a promising target in the development of medications for the treatment of sleep disorders. A number of dual orexin receptor antagonists (DORAs) have been developed for this purpose, such as almorexant (1) and suvorexant (2) (Figure 1).5–9 In particular, 2 developed by Merck & Co became the first orexin antagonist approved by the FDA for the treatment of insomnia in 2014 and is currently marketed under the brand name Belsomra.10
Figure 1.
Representative Orexin antagonists
More recently, the orexins have been implicated in a range of motivation and reward processes, including drug addiction.11–14 Accumulating evidence has confirmed the involvement of the orexin system, particularly the OX1 receptor, in different stages of the addiction process to several drugs of abuse.15, 16 In conditioned place preference (CPP) experiments, the widely used animal model measuring drug rewarding effects, the orexin system showed a role in acquisition, expression and reinstatement of morphine CPP.16, 17 Blockade of the OX1 receptor by systemically or intra-VTA (ventral tegmental area) administrated SB334867 (3, Fig. 1), an OX1 antagonist, attenuated motivated drug seeking behaviors of cocaine.18, 19 Administration of 3 also attenuated cue- and stress-induced reinstatement of extinguished cocaine seeking in animals.19 In addition, ethanol and nicotine self-administration and reinstatement can also be decreased by administration of 3.20–22
Compound 3 was the first reported OX1 selective antagonist and is still widely used in the orexin research community.23 However, 3 suffers from several limitations, including moderate selectivity over OX2 (~50×), off-target interactions with several receptors (serotonin, adenosine), and instability of the hydrochloride salt.24, 25 Several other OX1 selective antagonists have been since described (e.g. ACT-335827, 4, Fig. 1), but most of them still have certain OX2 activities.26–29 While blockade of both orexin receptors has been shown to be effective in some addiction models in a limited number of studies, selective OX1 antagonists provide the benefit of minimized sedation arising from OX2 antagonism.30, 31 In addition, the majority of in vivo studies so far for OX1-specific pathways were done with 3. OX1 antagonists with improved selectivity will help to further elucidate and validate the in vivo role and biology of the OX1 receptors.
Our group has previously reported a series of OX1 selective antagonists based on the tetrahydroisoquinoline (THIQ) scaffold.32–35 In these studies, we explored the structure-activity relationships (SARs) at several locations, and have identified structural features that enhanced the potency and selectivity of OX1 over OX2. As a result, several highly potent and selective OX1 antagonists have been identified, including RTIOX-276 (5) and RTIOX-251 (6). These compounds attenuated the development of conditioned place preference for cocaine,32 or blocked development of locomotor sensitization to cocaine in rats.34 In addition, at doses that attenuated the motivation for cocaine, 5 reduced spontaneous dopamine transient amplitude and cue-evoked dopamine release, and also attenuated cocaine-induced dopamine uptake inhibition at the level of dopamine terminals, confirming its in vivo efficacy.36
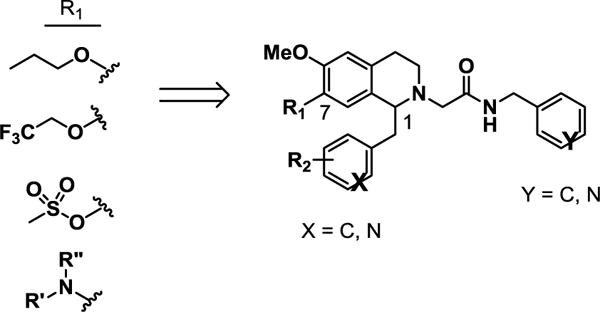
These THIQ-based OX1 antagonists, despite the excellent potency and selectivity, have high lipophilicity (clogP > 5). The logP value of a compound is a well-established measure of a compound’s lipophilicity; high lipophilicity, or low hydrophilicity, causes lower solubility and poor absorption, thus affecting amount of drug reaching its site of action. In general, substituents with increased polarity and/or ionizability (e.g. protonable amino groups) at physiological pH lead to lower lipophilcity.37 Therefore, we aimed to introduce substituents at several positions of the THIQ scaffold that would retain OX1 potency while leading to improved pharmacokinetic properties. Since the 7-position has been demonstrated as a key factor in determining the OX1 potency and selectivity,32 and substituents such as propoxy, trifluoroethoxy, methylsulfonate and dimethylamino groups led to excellent OX1 potency and selectivity, these favorable 7-position substituents were retained (R1, Fig. 2). In addition, the 1-position has been shown to tolerate a variety of substituents, and therefore groups of different nature were introduced at the 1-position (R2, Fig. 2). Among these substitutents at the 1- and 7-positions that have been shown to lead to good OX1 potency and selectivity, several groups were either polar (e.g. trifluoroethoxy, methylsulfonate) or ionizable (e.g. dimethylamino), and are expected to have reduced lipophilicity and increased solubitility. In addition, replacing the benzyl group at the acetamide position with groups such as pyridyl may also reduce lipophilicity. We herein describe the design, synthesis, and evaluation of the OX1 potency and selectivity, as well as preliminary ADME properties, including aqueous solubility and CNS permeability, for these new analogs.
Figure 2.
Structural modifications at the 7-, 1-, and acetamide positions
Results and Discussion
Chemistry
The general approach to this series of THIQ derivatives is shown in Scheme 1, starting from phenylacetic acids 8a-f. Most of these phenylacetic acids were commercially available, except for 8f which was synthesized by O-alkylation of 4-hydroxy-3-methoxyphenylacetic acid methyl ester (7) and hydrolysis under basic conditions. Acids 8a-f were first coupled with 4-hydroxy-3-methoxyphenylethylamine to give the amides 9a-f, which were then cyclized via a Bischler-Napieralski reaction using phosphorus oxychloride in toluene to form the dihydroisoquinolines, followed by reduction to tetrahydroisoquinolines 10a-f using sodium borohydride. N-Alkylation was achieved using N-benzyl bromoacetamide in the presence of base to give intermediates 11a-f. Finally, the phenol was alkylated using 1-iodopropane in the presence of potassium carbonate to afford the target compounds 12a-e,g. The 3-dimethylamino derivative 12f was obtained from the 3-nitro analog 12e via reduction to the aniline using Raney nickel, then reductive amination with formaldehyde and sodium triacetoxyborohydride. An alternate approach was attempted for both 12d and 12g, where O-alkylation was performed at the amide stage (9) prior to Bischler-Napieralski reaction. While this approach generally gave better yields in the following steps, it limited diversification in the later synthesis and was thus not further pursued.
Scheme 1.
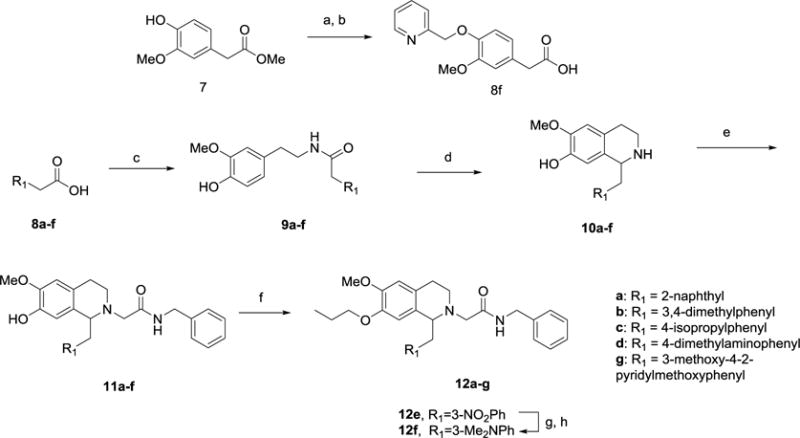
Synthesis of 7-propoxy-6-methoxy THIQ derivatives 12a-g.a
aReagents and Conditions: (a) 2-(bromomethyl)pyridine hydrobromide, K2CO3, Bu4NI, DMF, rt, 16 h; (b) 2N NaOH (aq), EtOH, rt, 16 h; (c) 4-Hydroxy-3-methoxyphenylethylamine, HBTU, iPr2EtN, DMF, rt, 16 h; (d) (i) POCl3, toluene, 90 °C, 2 h; (ii) NaBH4, MeOH, rt, 16 h; (e) BrCH2CONHCH2Ph, iPr2EtN, Bu4NI, DMF, rt, 16 h; (f) 1-iodopropane, K2CO3, DMF, rt, 16 h; (g) Raney Ni, NH2NH2.H2O, EtOH, 50 °C, 1 h; (h) HCHO, NaBH(OAc)3, 1,2-DCE, rt, 16 h.
7-Trifluoroethoxy derivatives 15a-b were made via a similar approach (Scheme 2). The tetrahydroisoquinolines 10a-b were N-alkylated with ethyl bromoacetate to give ester 13a-b. Ester hydrolysis gave the acid, followed by amide coupling with 3-aminomethylpyridine to give 14a-b. O-Alkylation using 1-iodo-2,2,2-trifluoroethane and cesium carbonate gave the desired analogs 15a-b. 1-Pyridylmethyl derivatives 19a-c were synthesized in analogous fashion using the general approach outlined above, starting from the appropriate 2-, 3- or 4-pyridylacetic acid (16a-c) (Scheme 2).
Scheme 2.
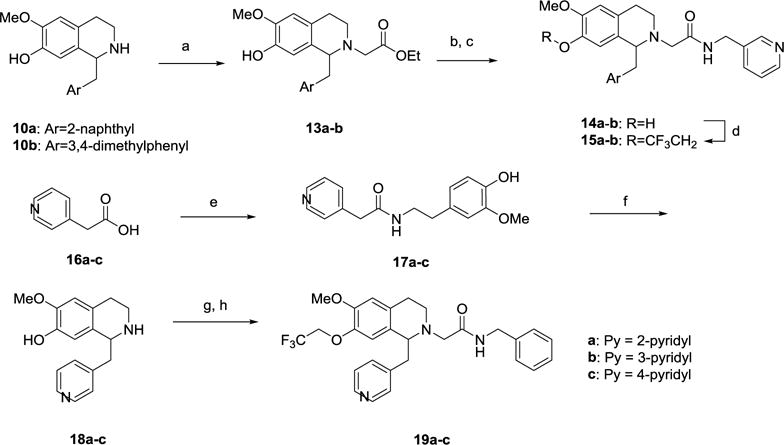
Synthesis of 7-trifluoroethoxy substituted THIQ derivatives 19a-c.a
aReagents and Conditions: (a) BrCH2CO2Et, iPr2EtN, Bu4NI, DMF, rt, 16 h; (b) 2N NaOH (aq), EtOH, rt, 16 h; (c) 3-aminomethylpyridine, HATU, iPr2EtN, DMF, rt, 16 h; (d) CF3CH2I, Cs2CO3, DMF, 100 °C, 2 h; (e) 4-Hydroxy-3-methoxyphenylethylamine, HBTU, iPr2EtN, DMF or DCC, HOBt, Et3N, THF, rt, 16 h; (f) (i) POCl3, CH3CN, reflux, 1 h; (ii) NaBH4, MeOH, 0 °C to rt, 16 h; (g) BrCH2CONHCH2Ph, iPr2EtN, Bu4NI, DMF, rt, 16 h; (h) CF3CH2I, Cs2CO3, DMF, 100 °C, 2 h.
The 7-methylsulfonate derivatives 25a-b (Scheme 3) were prepared from 4-hydroxy-3-methoxyphenylacetic acid 20. Protection of the phenolic hydroxyl group as its benzyl ether gave intermediate 21. Acid 21 was then coupled with 4-hydroxy-3-methoxyphenethylamine to give amide 22, followed by sulfonylation of the phenol group with methanesulfonyl chloride to afford the sulfonate 23. After cyclization to the tetrahydroisoquinoline 24 and N-alkylation as described above, the benzyl group of 25a was removed under acidic conditions and the resulting hydroxyl group was converted to the butyl ether upon treatment with 1-bromobutane to give 25b.
Scheme 3.
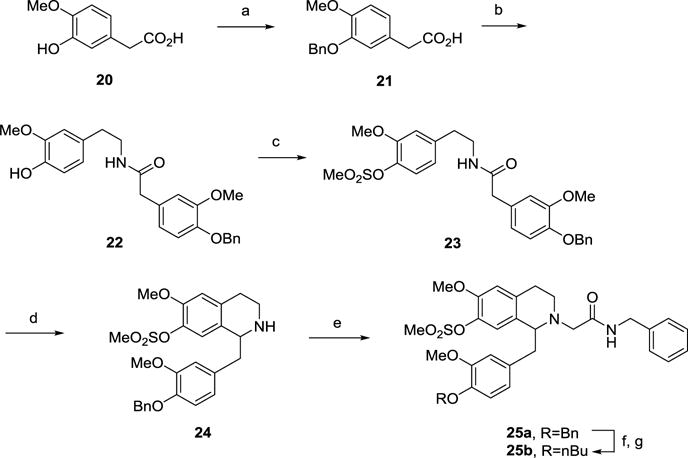
Synthesis of 7-sulfonate substituted THIQ derivatives 25a-b.a
aReagents and Conditions: (a) Na, BnBr, MeOH, reflux, 5 h; (b) 4-Hydroxy-3-methoxyphenylethylamine, HBTU, iPr2EtN, DMF, rt, 16 h; (c) MeSO2Cl, Et3N, CH2Cl2, 0 °C, 4 h; (d) (i) POCl3, toluene, 90 °C, 2 h; (ii) NaBH4, MeOH, rt, 16 h; (e) BrCH2CONHCH2Ph, iPr2EtN, Bu4NI, DMF, rt, 16 h; (f) conc. HCl, MeOH, reflux, 16 h; (g) 1-bromobutane, K2CO3, Bu4NI, DMF, 50 °C, 16 h.
The 7-dimethylamino-6-methoxytetrahydroisoquinoline derivatives required synthesis of the phenethylamine 28, which was prepared from 3-methoxyphenylacetonitrile 26 (Scheme 4), by nitration using tetrabutylammonium nitrate in the presence of trifluoroacetic anhydride and 18-crown-6 to give 27, followed by reduction of the nitrile using borane-THF complex at 70 °C. The amine 28 was then coupled with 3,4-dimethoxyphenylacetic acid to give amide 29. The presence of a strong electron withdrawing group such as a nitro group on the phenethylamine portion retards the reactivity of the aromatic system and makes the Bischler-Napieralski cyclization sluggish, so the amide 29 was reduced using Raney nickel to the aniline 30 then protected as the methyl carbamate using methyl chloroformate and diisopropylethylamine to give 31. Bischler-Napieralski cyclization and reduction to the tetrahydroisoquinoline followed by N-alkylation with the acetamide component gave the acetamide 32 and removal of the carbamate using 2N sodium hydroxide solution in methanol provided amine 33. This amine could then be derivatized via sulfonylation, reductive amination or acylation to give a series of derivatives 34a-e.
Scheme 4.
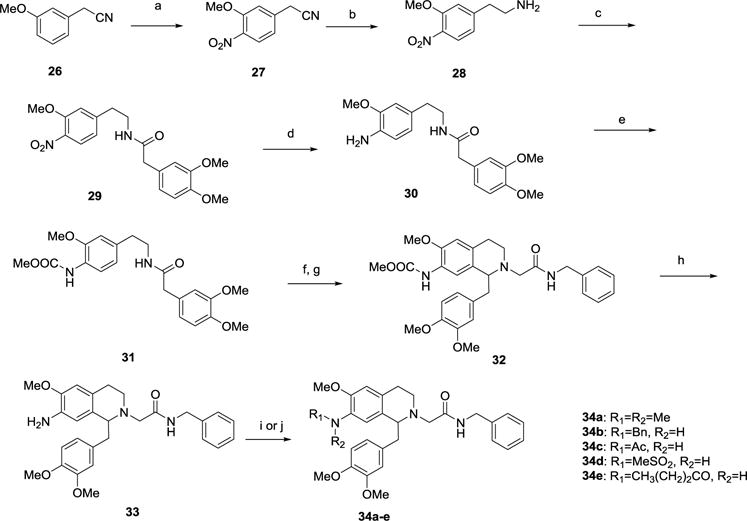
Synthesis of 7-dimethylamino substituted THIQ derivatives 34a-e.a
aReagents and Conditions: (a) Bu4NNO3, (CF3CO)2O, 18-crown-6, CH2Cl2, 0 °C, 30 min; (b) BH3-THF, THF, 70 °C, 2 h; (c) 3,4-dimethoxyphenylacetic acid, HBTU, iPr2EtN, DMF, rt, 16 h; (d) Raney Ni, NH2NH2-H2O, EtOH, 50 °C, 1 h; (e) MeOCOCl, iPr2EtN, CH2Cl2, rt, 16 h; (f) (i) POCl3, toluene, 90 °C, 2 h; (ii) NaBH4, MeOH, rt, 16 h; (g) BrCH2CONHCH2Ph, iPr2EtN, Bu4NI, DMF, rt, 16 h; (h) 2N NaOH, MeOH, 50 °C, 48 h; (i) 34a: HCHO, NaBH(OAc)3, 1,2-DCE, rt, 16 h; 34b: PhCHO, NaHCO3, MeOH, 40 °C, 1h, then NaBH4, rt, 16 h; (j) 34c: Ac2O, iPr2EtN, CH2Cl2, rt, 16 h; 34d: MeSO2Cl, Et3N, CH2Cl2, 0 °C to rt, 16 h; then 3N NaOH, 80 °C, 16 h; 34e: CH3(CH2)2CO2H, BOP, iPr2EtN, CH2Cl2, rt, 16 h.
The bis-dimethylamino derivatives 39 and 42 were made in analogous fashion to the other 7-amino derivatives (Scheme 5). For 39, the phenethylamine 28 was coupled to 4-hydroxy-3-nitrophenylacetic acid, then the phenol was converted to the methyl ether using iodomethane to afford 35. Reduction of both nitro groups with Raney nickel was followed by formation of the bis- methyl carbamate using methyl chloroformate to form 36. Cyclization and then reduction gave the tetrahyroisoquinoline 37 then N-alkylation as before gave 38. Both methyl carbamates were removed using sodium hydroxide and reductive amination provided the bis-dimethylamino product 39. For 42, amine 28 was first coupled with 3-nitrophenylacetic acid to afford amide 40 and both nitro groups were reduced with Raney nickel. Reductive amination gave the bis-dimethylamino amide 41, which was then subjected to Bischler-Napieralski conditions, reduced and N-alkylated to afford the final product 42. Finally, the 1-(3-pyridylmethyl)benzyl analog 44 was prepared from intermediate 37, which underwent alkylation using ethyl bromoacetate, ester hydrolysis, and coupling with 3-aminomethylpyridine to give 43. The final sequence of carbamate removal and reductive amination with formaldehyde afforded the desired 44.
Scheme 5.
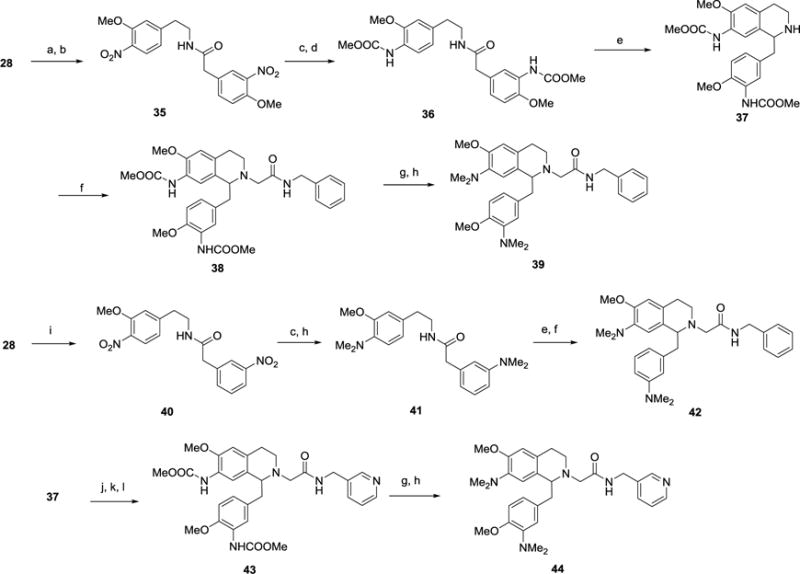
Synthesis of 7-dimethylamino substituted THIQ derivatives 39, 42 and 44.a
aReagents and Conditions: (a) 4-hydroxy-3-nitrophenylacetic acid, HBTU, iPr2EtN, DMF, rt, 16 h; (b) Me-I, K2CO3, DMF, 0-60 °C, 2 h; (c) Raney Ni, NH2NH2.H2O, EtOH, 50 °C, 90 min; (d) MeOCOCl, iPr2EtN, CH2Cl2, rt, 16 h; (e) (i) POCl3, toluene, 90 °C, 2 h; (ii) NaBH4, MeOH, rt, 16 h; (f) BrCH2CONHCH2Ph, iPr2EtN, Bu4NI, DMF, rt, 16 h; (g) 2N NaOH (aq), MeOH, 50 °C, 16 hr; (h) HCHO, NaBH(OAc)3, 1,2-DCE, rt, 16 h; (i) 3-nitrophenylacetic acid, HBTU, iPr2EtN, DMF, rt, 16 h; (j) BrCH2CO2Et, iPr2EtN, Bu4NI, DMF, rt, 16 h; (k) 2N NaOH, EtOH, rt, 16 h; (l) 3-aminomethylpyridine, HATU, iPr2EtN, DMF, rt, 16 h.
Pharmacological Evaluation
Activity of the target compounds at the OX1 and OX2 receptors was evaluated in a calcium mobilization based functional assay. The apparent dissociation constant Ke was calculated from compound-mediated inhibition of orexin A activity as previously described.32, 34, 35, 38 Briefly, EC50 curves of the agonist, orexin A, were obtained alone and together with the test compound, respectively, and the right-shift of the agonist curve was measured. Ke values were then calculated using the equation Ke = [L]/ ((EC50+/EC50−)-1), where [L] is the test compound concentration, EC50+ is the EC50 of orexin A in the presence of test compound, and EC50− is the EC50 of orexin A alone. In these assays, the EC50 for orexin A at OX1 and OX2 is 0.13 ± 0.02 nM and 4.2 ± 0.2 nM, respectively. All the compounds that had OX1 Ke values < 1 μM were also tested for agonist activity at 10 μM final concentration at the OX1 receptor; none of them showed any agonist activity.
We previously reported SAR studies at three different locations of the THIQ core, namely the 7-position, the 1-position and the acetamide, and had identified structural features that led to enhanced potency and OX1 selectivity at these positions.32, 34, 35 At the 7-position, it was discovered that a modest increase in the chain length, from methoxy to propoxy or trifluoroethoxy groups, led to increased OX1 potency and selectivity.32 Similarly, methylsulfonate and dimethylamino groups also led to excellent potency and selectivity. At the 1-position of the THIQ core, a series of aromatic systems provided similar or slightly improved potency than the initial 3,4-dimethoxybenzyl group.34 For example, we found that pyridylmethyl groups were as potent as the benzyl group.32 In this study, we examined the effects of combining these favorable structural modifications on the OX1 potency and selectivity. Given the tolerance for substitution at the 1-position, aromatic groups of different nature were introduced at this position in an effort to improve the PK properties of these compounds.
We first examined a series of 7-propoxy analogs with different substituents at the 1-benzyl position. At the 1-position, several groups that have previously been demonstrated to be tolerated were introduced. As expected, the naphthyl (12a), 3,4-dimethylphenyl (12b), and 4-isopropylphenyl (12c) all showed good OX1 potency, consistent with our previous findings.34 The introduction of a nitrogen atom into the structure (12d, f) did not significantly change the potency, but reduced the lipophilicity of these compounds from 12c (clogP 7.4 vs. 6.17). The 3-dimethylamino analog 12f was the most potent of the series with a Ke value of 21 nM. When a pyridinylmethyl group was introduced at the 4-position (12g), the clogP was further lowered (5.96), the potency remained good (Ke = 96.4 nM), and the selectivity was high (>100-fold). Overall, these results show that substitution at the 1-benzyl position results in compounds with good selectivity for the OX1 receptor over the OX2 receptor, although clogP remained high.
Previously reported compound 5, with a trifluoroethoxy substitution at the 7-position, is a highly potent and selective OX1 antagonist.32 Both the naphthyl analog 15a and 3,4-dimethyl analog 15b showed excellent potency, comparable to the propoxy series (12a-b). The presence of the trifluoroethyl led to lower clogP than the propyl (12b: 6.98 vs. 15b: 5.39). To further reduce clogP, we synthesized 3 different pyridinyl derivatives (19a-c) at the 1-position. Although they have lower clogP (4.47), all three compounds showed significantly lower potency at the OX1 receptor and also displayed insurmountable antagonism in the curve shift assays. As previously reported by us,35 increasing the receptor-test compound incubation period can help to elucidate whether this type of antagonism is due to an allosteric mechanism or an orthosteric antagonist with a slow dissociation rate. For derivatives 19a-c, we found that increasing the incubation time led to activity profiles consistent with competitive antagonism.
Another group that provided satisfactory results in previous SAR studies was the 7-methylsulfonate. Again, these methylsulfonate analogs (25a, b) showed excellent OX1 potency and selectivity. However, both compounds had clogP in the 5-6 range.
All these structural alterations discussed thus far have resulted in potent and selective OX1 antagonists that continue to have high lipophilicity (clogP > 5). Thus, a series of 7-amino compounds were prepared and their potency against OX1 and OX2 receptors was assayed (Table 2). It was expected that the introduction of the amino groups not only would reduce clogP or lipophilicity, but would also facilitate the formation of more soluble ammonium salts.
Table 2.
7-Amino tetrahydroisoquinoline derivatives incorporating modifications at the 1-benzyl position.
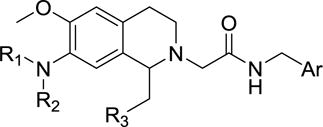
| ||||||||
|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | R3 | Ar | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 | clogP |
| 33 | H | H |
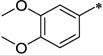
|
Ph | 494 ± 32 | >10,000 | >20 | 3.87 |
| 34a | Me | Me |
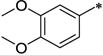
|
Ph | 31.2 ± 3.3 | >10,000 | >321 | 4.92 |
| 34b | Bn | H |
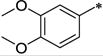
|
Ph | 352 ± 34 | a | >28 | 6.13 |
| 34c | Ac | H |
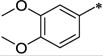
|
Ph | 116 ± 44 | >10,000 | >86 | 3.76 |
| 34d | MeSO2 | H |
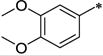
|
Ph | 128 ± 13 | >10,000 | >78 | 3.63 |
| 34e | CH3(CH2)2CO | H |
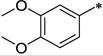
|
Ph | 689 ± 73 | >10,000 | >14.5 | 4.82 |
| 38 | MeOCO | H |
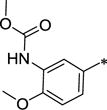
|
Ph | >10,000 | 592 ± 170 | 0.1 | 4.39 |
| 39 | Me | Me |
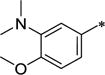
|
Ph | 7.60 ± 2.8 | 857 ± 220 | 113 | 5.11 |
| 42 | Me | Me |
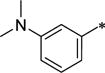
|
Ph | 148 ± 36 | 1,360 ± 84 | 9.1 | 5.30 |
| 44 | Me | Me |
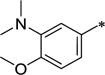
|
3-Py | 5.7 ± 1.0 | >10,000 | >1,760 | 3.07 |
< 35% inhibition at 10 μM final concentration;
Values are the mean of at least three independent experiments in duplicate;
Values are the mean of at least two independent experiments in duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
As can be seen from Table 2, all the compounds in the 7-amino series were generally tolerated for OX1 activity. The unsubstituted aniline 33 was active with a Ke of 494 nM, but showed little selectivity. Dimethylation of the amino group (34a) significantly improved the OX1 potency (494 nM vs. 31.2 nM). The benzylamino analog (34b) was less potent, showing a Ke value of 352 nM, suggesting larger groups may not be well tolerated. When the amino group was acylated (34c, 116 nM) or mesylated (34d, 128 nM), the resulting compounds had good potency and selectivity, as well as lower clogP (< 4). When the acyl group was elongated, the butyric analog (34e) was less potent (Ke = 689 nM), consistent with the limited tolerance for bulk at this position. The methylcarbamate (38) was inactive, and showed increased OX2 activity as well. The fact that 34a (-NMe2) was more potent than all the NH containing alalogs suggests that the presence of an hydrogen bond donor at this position may be unfavorable for OX1 activity. Alternatively, the NH group may be forming an intramolecular hydrogen bond with the methoxy group at the 6-position, thus limiting the rotational freedom and ability of R1 group to engage in favorable interations with the receptor.
Given the encouraging results with the 3-dimethylamino analog (34a), we synthesized several additional analogs with amino groups also on the 1-benzyl position. While the 3-dimethylamino analog (42) had a Ke of 148 nM, the corresponding 3-dimethylamino-4-methoxy derivatve (39) was more potent (Ke = 7.6 nM). Finally, when the benzylacetamide was replaced with the 3-pyridyl group, a potent and more selective OX1 antagonist (44) was obtained. Importantly, the clogP value of this compound was 3.07 and PSA was 70, implying that this modification could enhance solubility.
Physiochemical Properties and Preliminary ADME Studies
Compound 44, in general, appeared to have favorable physicochemical properties which would suggest good solubility and blood-brain barrier (BBB) permeability required for successful CNS agents (Table 3, calculated in ACD Labs).39, 40 The lower clogP values suggested 44 (3.07) might have improved solubility over 5 (4.21). When the kinetic solubility was assessed at pH 7.4, 5 showed a solubility of 77.8 ± 4.1 μM (mean ± %CV). Compound 44 was more soluble in aqueous solutions, displaying a kinetic solubility greater than 200 μM.
Table 3.
Physicochemical and Preliminary ADME Properties of Compounds 6 and 44
| Desired value | 5 | 44 | |
|---|---|---|---|
| MW | < 500 | 559.6 | 517.7 |
| ClogP | 1 - 4 | 4.21 | 3.07 |
| PSA | < 70 | 82.1 | 70.2 |
| pKa | < 8 | 14.1, 5.4 | 14.2, 6.3 |
| HBD | < 3 | 1 | 1 |
| HBA | < 7 | 7 | 7 |
| Solubility (μM) | > 60 | 77.8 ± 4.1 | > 200 |
| Papp (10−6 cm/sec) A-to-B |
> 2 | 12.2 | 14.7 |
| Papp (10−6 cm/sec) B-to-A |
36.2 | 47.8 | |
| Efflux ratio | < 2.5 | 3.0 | 3.3 |
HBD: H-bond donor; HBA: H-bond acceptor.
We also determined the CNS permeability of 5 and 44. CNS permeability is an important property for CNS targeting agents, and is generally determined by two factors: the ability to passively permeate through the BBB and efflux by the transport proteins such as the P-glycoprotein.37 The apparent permeability of the compounds through MDCK-MDR1 monolayers were determined in both the apical-to-basolateral (A-to-B) and basolateral-to-apical (B-to-A) directions. Both compounds showed good apparent permeability (Papp > 10 × 10−6 cm/sec). In general, efflux ratios > 2.5 indicate drug efflux occuring.37 Compounds 5 and 44 had efflux ratios of 3.0 and 3.3, respectively, suggesting efflux is present for both compounds, albeit relatively weak. Since these MDR1-transfected cells also express endogenous transporters,41 the observed drug efflux may indicate these compound are P-gp substrates, but the effects could also be mediated by the endogenous transportors.
Conclusions
The orexin system has been implicated in several important functions, including addiction. With the growing interest in the role of the orexin system on the reward pathway and the potential for orexin-1 antagonists as therapeutics in the treatment of drug addiction, there is a need for the development of orexin-1 selective antagonists. Our group has previously reported a series of tetrahydroisoquinoline-based antagonists. While some of the compounds showed high potency and selectivity, they had high lipophilicity and moderate or low solubility. In this study, we aimed to improve the ADME properties of the compounds by introducing structural elements that may lead to more optimal properties. In particular, groups such as propoxy, trifluoroethoxy, methylsulfonate and dimethylamino at the 7-position that had been previously demonstrated to be critical for OX1 potency and selectivity have been retained, whereas groups of different nature were introduced at the 1-position which has been shown to tolerate a variety of substituents. These structural alterations resulted in several compounds with reduced lipophilicity as evidenced by the lower clogP and excellent OX1 potency and selectivity, such as 44 (clogP = 3.07), which had a dimethylamino groups at the 7-position and on the 1-benzyl group, respectively.
Similar to the previously reported 5 which showed efficacy in several in vivo models, 44 displayed high OX1 potency (Ke 5.7 nM vs. 8.5 nM) and excellent selectivity (both >1,000-fold over OX2). These values also represent significant improvement from the commonly used OX1 antagonist SB334867 (IC50 = 40 nM, 50× over OX2). Moreover, 44 showed greater kinetic solubility (>200 μM) than 5 (77.8 μM) and good BBB permeability. Finally, 44 possessed little Pgp activity with an efflux ratio of 3.3. Given its high OX1 potency and selectivity, as well as good ADME properties, 44 may serve as an improved molecular tool in the elucidation and validation of the in vivo roles of the OX1 receptor in many orexin related disorders.
Experimental Procedures
General
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all were purchased from commercial vendors and used as received. Flash column chromatography was done on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used were hexane, ethyl acetate (EtOAc), dichloromethane, methanol and chloroform:methanol:ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds was established by a combination of high pressure liquid chromatography (HPLC), thin layer chromatography (TLC), mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis. 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in Hz. TLC was done on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. Low resolution mass spectra were obtained using a Waters Alliance HT/Micromass ZQ system (ESI). All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column with gradient elution using the mobile phases (A) H2O containing 0.1% CF3COOH and (B) MeCN, with a flow rate of 1.0 mL/min. For ADME analysis, samples were analyzed by LC-MS/MS using a CTC PAL autosampler, Thermo Accela UPLC and a Thermo Quantum Ultra triple quadrupole mass spectrometer. The [M+H]+ adducts of the test compounds and internal standard were monitored using positive mode electrospray ionization in multiple reaction monitoring (MRM) mode. The analytes were injected onto a C18 column and chromatographed using a reverse phase gradient with 0.1% formic acid in water and 0.1% formic acid in 20/80 isopropyl alcohol/acetonitrile mobile phases. Peak integrations were performed using Thermo Xcalibur (v 2.4) software.
Methyl (4-hydroxy-3-methoxyphenyl)acetate (7)
To a solution of 4-hydroxy-3-methoxyphenylacetic acid (2.50 g, 13.72 mmol) in anhydrous methanol (60 mL) was added concentrated sulfuric acid (2.02 g, 1.1 mL, 20.59 mmol) and the reaction heated to reflux for 3 hr. The reaction was cooled, the solvent was removed under reduced pressure and NaHCO3 solution was added. The aqueous solution was extracted three times with dichloromethane and the combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give the methyl ester as a yellow oil (2.69 g, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.84 - 6.90 (m, 1H), 6.80 (d, J = 1.70 Hz, 1H), 6.73 - 6.78 (m, 1H), 5.57 (s, 1H), 3.89 (s, 3H), 3.69 (s, 3H), 3.55 (s, 2H).
2-[3-Methoxy-4-(pyridin-2-ylmethoxy)phenyl]acetic acid (8f)
Phenol 7 (1.0 g, 5.10 mmol), 2-(bromomethyl)-pyridine hydrobromide (1.93 g, 7.65 mmol), potassium carbonate (2.82 g, 20.4 mmol) and tetrabutylammonium iodide (0.38 g, 1.02 mmol) were combined in anhydrous dimethylformamide (50 mL) and heated to 50 °C overnight. The reaction was cooled, diluted with EtOAc, washed twice with water and with brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-60% EtOAc in hexane) to give the desired ether as an orange oil (0.49 g, 34%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.58 (d, J = 4.62 Hz, 1H), 7.69 (dt, J = 1.65, 7.70 Hz, 1H), 7.51 - 7.60 (m, 1H), 7.16 - 7.25 (m, 1H), 6.82 - 6.88 (m, 1H), 6.81 (s, 1H), 6.71 - 6.78 (m, 1H), 5.27 (s, 2H), 3.91 (s, 3H), 3.69 (s, 3H), 3.56 (s, 2H).
To the solution of ester (0.49 g, 1.71 mmol) in methanol (15 mL) was added 2N sodium hydroxide solution (3.4 mL, 6.82 mmol) and the reaction stirred at RT overnight. The pH was adjusted to between 8 and 9 with 2N HCl then all the solvents were removed under reduced pressure. The crude was dissolved in methanol as far as possible, the solids were removed by filtration then the solvent was removed under reduced pressure to give the acid as a yellow solid (0.47 g, 100%). 1H NMR (300 MHz, METHANOL-d4) δ 8.47 - 8.54 (m, 1H), 7.84 (dt, J = 1.70, 7.72 Hz, 1H), 7.62 (d, J = 7.91 Hz, 1H), 7.30 - 7.39 (m, 1H), 7.01 (d, J = 1.88 Hz, 1H), 6.83 - 6.90 (m, 1H), 6.74 - 6.83 (m, 1H), 5.15 (s, 2H), 3.86 (s, 3H), 3.41 (s, 2H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-(naphthalen-2-yl)acetamide (9a)
To 2-naphthylacetic acid (8a) (0.74 g, 3.97 mmol), 4-hydroxy-3-methoxyphenethylamine hydrochloride (0.81 g, 3.97 mmol) and HATU (1.66 g, 4.37 mmol) in anhydrous dimethylformamide (20 mL) was added diisopropylethylamine (2.06 g, 2.8 mL, 15.90 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with 2N aqueous HCl solution, NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure to give the amide as a white solid (1.33 g, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.74 - 7.87 (m, 3H), 7.62 (s, 1H), 7.47 - 7.54 (m, 2H), 7.24 - 7.29 (m, 2H), 6.61 (d, J = 7.91 Hz, 1H), 6.52 (d, J = 1.88 Hz, 1H), 6.40 (dd, J = 1.88, 7.91 Hz, 1H), 5.38 (br. s., 1H), 3.71 (s, 3H), 3.70 (s, 2H), 3.43 (q, J = 6.78 Hz, 2H), 2.63 (t, J = 6.88 Hz, 2H).
2-(3,4-Dimethylphenyl)-N-[2-(4-hydroxy-3-methoxyphenyl)ethyl]acetamide (9b)
This was made by the same method as 9a, using 3,4-dimethylphenylacetic acid (8b). Colorless film. Yield 100%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.25 - 7.29 (m, 1H), 7.06 (d, J = 7.54 Hz, 1H), 6.83 - 6.96 (m, 2H), 6.72 - 6.79 (m, 1H), 6.59 (d, J = 1.51 Hz, 1H), 6.48 (d, J = 7.91 Hz, 1H), 5.39 (br. s., 1H), 3.83 (s, 3H), 3.46 (s, 2H), 3.42 (q, J = 6.78 Hz, 2H), 2.65 (t, J = 6.88 Hz, 2H), 2.25 (s, 3H), 2.22 (s, 3H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-[4-(propan-2-yl)phenyl]acetamide (9c)
This was made by the same method as 9a using 4-isopropylphenylacetic acid (8c). Colorless film. Yield 71%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.14 - 7.21 (m, 2H), 7.05 - 7.12 (m, 2H), 6.76 (d, J = 8.10 Hz, 1H), 6.61 (d, J = 1.51 Hz, 1H), 6.48 (dd, J = 1.60, 8.01 Hz, 1H), 5.53 (s, 1H), 5.40 (br. s., 1H), 3.84 (s, 3H), 3.50 (s, 2H), 3.43 (q, J = 6.78 Hz, 2H), 2.84 - 2.95 (m, 1H), 2.66 (t, J = 6.97 Hz, 2H), 1.25 (d, J = 6.97 Hz, 6H).
2-[4-(Dimethylamino)phenyl]-N-[2-(4-hydroxy-3-methoxyphenyl)ethyl]acetamide (9d)
This was made by the same method as 9a using 4-dimethylaminophenylacetic acid (8d). White sticky solid. Yield 99%. 1H NMR (300 MHz, DMSO-d6) δ 8.70 (s, 1H), 7.89 (t, J = 5.42 Hz, 1H), 7.03 (d, J = 8.57 Hz, 2H), 6.72 (d, J = 1.60 Hz, 1H), 6.61 - 6.68 (m, 3H), 6.53 (dd, J = 1.65, 7.96 Hz, 1H), 3.71 (s, 3H), 3.15 - 3.27 (m, 4H), 2.85 (s, 6H), 2.57 (t, J = 7.30 Hz, 2H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-(3-nitrophenyl)acetamide (9e)
This was made by the same method as 9a using 3-nitrophenylacetic acid (8e). White sticky solid. Yield 100%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.13 (d, J = 8.10 Hz, 1H), 8.08 (s, 1H), 7.44 - 7.59 (m, 2H), 6.79 (d, J = 8.10 Hz, 1H), 6.62 (d, J = 1.51 Hz, 1H), 6.56 (dd, J = 1.70, 8.10 Hz, 1H), 5.57 (s, 1H), 5.51 (br. s., 1H), 3.84 (s, 3H), 3.58 (s, 2H), 3.50 (q, J = 6.66 Hz, 2H), 2.72 (t, J = 6.88 Hz, 2H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-[3-methoxy-4-(pyridin-2-ylmethoxy)phenyl]acetamide (9f)
This was made by the same method as 9a using acid 8f. Yellow sticky solid. Yield 69%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.63 (d, J = 4.14 Hz, 1H), 7.71 - 7.81 (m, 1H), 7.59 (d, J = 7.82 Hz, 1H), 7.27 - 7.32 (m, 1H), 6.67 - 6.81 (m, 3H), 6.58 (dd, J = 1.98, 8.10 Hz, 1H), 6.55 (d, J = 1.88 Hz, 1H), 6.37 (dd, J = 1.88, 8.01 Hz, 1H), 5.32 (br. s., 1H), 5.23 (s, 2H), 3.85 (s, 3H), 3.80 (s, 3H), 3.46 (s, 2H), 3.37 - 3.44 (m, 2H), 2.64 (t, J = 6.64 Hz, 2H).
6-Methoxy-1-(naphthalen-2-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-7-ol (10a)
Amide 9a (1.37 g, 4.09 mmol) was suspended in anhydrous toluene (20 mL) and phosphorus oxychloride (3.13 g, 1.9 mL, 20.42 mmol) was added slowly. The mixture was heated to 90 °C for 2 hr, then cooled to RT and quenched cautiously with water. The reaction was heated for 15 min until the oil formed went into solution. The reaction was again cooled to RT, the toluene layer was removed and the aqueous layer was adjusted to pH 8-9 using 2N aqueous sodium hydroxide solution. It was then extracted 3 times with DCM, the combined extracts were dried over MgSO4 and the solvents were removed under reduced pressure to give the crude dihydroisoquinoline as an orange viscous oil, which was used in the next step without further purification.
The crude material was dissolved in methanol (40 mL) and cooled in an ice bath. Sodium borohydride (0.78 g, 20.4 mmol) was added portionwise then the reaction was allowed to warm slowly to RT overnight. The reaction was quenched with water then the methanol was removed under reduced pressure. The reaction mixture was diluted with water and then extracted 3 times with DCM. The combined extracts were dried over MgSO4 and the solvents were removed under reduced pressure to give the product as a frothy yellow solid (0.41 g, 31%).
N-Benzyl-2-[7-hydroxy-6-methoxy-1-(naphthalen-2-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (11a)
Amine 10a (32 mg, 0.100 mmol), N-benzyl bromoacetamide (34 mg, 0.150 mmol) and tetrabutylammonium iodide (7 mg, 0.020 mmol) were combined in anhydrous DMF (1 mL), then diisopropylethylamine (32 mg, 44 mL, 0.250 mmol) was added. The reaction was stirred at RT overnight. The reaction was diluted with EtOAc and washed with NaHCO3 solution and brine, then dried over MgSO4 and the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0-75% EtOAc/hexane) to give the desired product as a yellow viscous oil (20 mg, 43%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (ddd, J = 3.49, 6.08, 11.73 Hz, 2H), 7.65 - 7.71 (m, 2H), 7.44 - 7.51 (m, 2H), 7.35 (dd, J = 1.70, 8.29 Hz, 1H), 7.08 - 7.16 (m, 3H), 6.75 (s, 1H), 6.55 - 6.65 (m, 4H), 5.52 (s, 1H), 4.13 (dd, J = 8.48, 14.69 Hz, 1H), 3.89 (s, 3H), 3.69 - 3.76 (m, 1H), 3.44 - 3.58 (m, 1H), 3.03 - 3.27 (m, 4H), 2.75 - 2.99 (m, 3H), 2.43 - 2.53 (m, 1H). MS (ESI) m/z 467 (M+H).
N-Benzyl-2-{1-[(3,4-dimethylphenyl)methyl]-7-hydroxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yacetamide (11b)
This was made using the methods outlined for 10a and 11a, starting from 9b to give 11b as an off-white sticky solid in 4% overall yield. MS (ESI) m/z 445 (M+H).
N-Benzyl-2-(7-hydroxy-6-methoxy-1-{[4-(propan-2-yl)phenyl]methyl}-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (11c)
This was made using the methods outlined for 10a and 11a, starting from 9c to give 11c as an off-white sticky solid in 12% overall yield. MS (ESI) m/z 459 (M+H).
N-Benzyl-2-{7-hydroxy-6-methoxy-1-[(3-nitrophenyl)methyl]-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (11e)
This was made using the methods outlined for 10a and 11a, starting from 9e to give 11e as an off-white sticky solid in 8% overall yield. MS (ESI) m/z 462 (M+H).
N-Benzyl-2-[6-methoxy-1-(naphthalen-2-ylmethyl)-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (12a)
To phenol 11a (20 mg, 0.043 mmol) and potassium carbonate (18 mg, 0.129 mmol) in dimethylformamide (0.5 mL) was added 1-iodopropane (11 mg, 6 μL, 0.064 mmol) then the reaction was stirred at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired product as a yellow solid (17 mg, 77%). m.p. 46-49 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.68 - 7.83 (m, 3H), 7.65 (s, 1H), 7.43 - 7.52 (m, 2H), 7.34 (dd, J = 1.51, 8.29 Hz, 1H), 7.15 (dd, J = 1.70, 4.90 Hz, 3H), 6.64 - 6.80 (m, 3H), 6.60 (s, 1H), 6.48 (s, 1H), 4.19 (dd, J = 8.19, 14.79 Hz, 1H), 3.85 (s, 3H), 3.68 - 3.83 (m, 3H), 3.44 - 3.59 (m, 1H), 3.05 - 3.32 (m, 4H), 2.83 - 3.05 (m, 3H), 2.44 - 2.57 (m, 1H), 1.72 - 1.88 (m, 2H), 0.98 (t, J = 7.44 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C33H37N2O3 [M + H]+ 509.2799, m/z found 509.2817.
N-Benzyl-2-{1-[(3,4-dimethylphenyl)methyl]-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (12b). Prepared as 12a
White solid. Yield 70%. m.p. 105-106 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.19 - 7.37 (m, 4H), 6.86 - 7.08 (m, 5H), 6.59 (s, 1H), 6.48 (s, 1H), 4.44 (dd, J = 8.19, 14.98 Hz, 1H), 3.89 (t, J = 6.88 Hz, 2H), 3.82 - 3.86 (m, 3H), 3.37 - 3.68 (m, 3H), 3.06 - 3.33 (m, 2H), 2.78 - 3.01 (m, 4H), 2.43 - 2.55 (m, 1H), 2.13 (s, 6H), 1.76 - 1.93 (m, 2H), 1.04 (t, J = 7.44 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C31H39N2O3 [M + H]+ 487.2955, m/z found 487.2964.
N-Benzyl-2-(6-methoxy-1-{[4-(propan-2-yl)phenyl]methyl}-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (12c)
Prepared as 12a. Pale yellow solid. Yield 59%. m.p. 80-81 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.20 - 7.36 (m, 3H), 7.03 - 7.14 (m, 7H), 6.58 (s, 1H), 6.41 (s, 1H), 4.37 (dd, J = 7.63, 15.16 Hz, 1H), 3.84 (s, 3H), 3.79 - 3.89 (m, 1H), 3.60 - 3.77 (m, 2H), 3.38 - 3.52 (m, 1H), 3.09 - 3.34 (m, 2H), 2.74 - 3.02 (m, 5H), 2.42 - 2.58 (m, 1H), 1.75 - 1.90 (m, 3H), 1.18 (dd, J = 4.90, 6.78 Hz, 6H), 1.03 (t, J = 7.44 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C32H41N2O3 [M + H]+ 501.3112, m/z found 501.3121.
N-Benzyl-2-(1-{[4-(dimethylamino)phenyl]methyl}-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (12d)
To a solution of amide 9d (3.26 g, 9.93 mmol), potassium carbonate (3.43 g, 24.82 mmol) and tetrabutylammonium iodide (0.73 g, 1.99 mmol) in anhydrous DMF (50 mL) was added 1-bromopropane (1.83 g, 1.35 mL, 14.89 mmol) and the reaction stirred at rt under N2 overnight. An additional aliquot of 1-bromopropane (0.95 g, 0.70 mL, 7.71 mmol) was added and stirring continued for an additional 24 hr. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, then dried over MgSO4 and the solvent was removed under reduced pressure to give the desired intermediate as a white solid (3.17 g, 86%).1H NMR (300 MHz, DMSO-d6) δ 7.89 (t, J = 5.32 Hz, 1H), 7.02 (d, J = 8.57 Hz, 2H), 6.82 (d, J = 8.19 Hz, 1H), 6.76 (s, 1H), 6.59 - 6.68 (m, 3H), 3.85 (t, J = 6.59 Hz, 2H), 3.71 (s, 3H), 3.18 - 3.28 (m, 4H), 2.85 (s, 3H), 2.85 (s, 3H), 2.61 (t, J = 7.16 Hz, 2H), 1.70 (sxt, J = 7.06 Hz, 2H), 0.96 (t, J = 7.35 Hz, 3H).
This intermediate was used in the preparation of 12d using the methods outlined for 10a and 11a as a white solid in 30% overall yield. m.p. 135-138 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.33 (m, 3H), 7.02 - 7.13 (m, 4H), 6.92 - 7.01 (m, 1H), 6.54 - 6.65 (m, 3H), 6.49 (s, 1H), 4.46 (dd, J = 8.10, 14.98 Hz, 1H), 3.90 (t, J = 6.88 Hz, 2H), 3.84 (s, 3H), 3.50 - 3.65 (m, 2H), 3.39 - 3.50 (m, 1H), 3.08 - 3.32 (m, 2H), 2.75 - 2.99 (m, 10H), 2.39 - 2.54 (m, 1H), 1.76 - 1.94 (m, 2H), 1.04 (t, J = 7.44 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C31H40N3O3 [M + H]+ 502.3064, m/z found 502.3074.
N-Benzyl-2-{6-methoxy-1-[(3-nitrophenyl)methyl]-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (12e)
This was prepared using the method for 12a starting from 11e. Colorless film. Yield 68%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.02 (s, 1H), 7.87 (dd, J = 1.22, 8.19 Hz, 1H), 7.43 (d, J = 7.54 Hz, 1H), 7.17 - 7.34 (m, 4H), 6.97 - 7.07 (m, 2H), 6.73 (br. s., 1H), 6.60 (s, 1H), 6.44 (s, 1H), 4.33 (dd, J = 7.06, 14.98 Hz, 1H), 3.85 (s, 3H), 3.81 - 3.95 (m, 2H), 3.71 (dd, J = 5.37, 9.14 Hz, 1H), 3.11 - 3.47 (m, 4H), 2.84 - 3.10 (m, 4H), 2.43 - 2.57 (m, 1H), 1.76 - 1.93 (m, 2H), 0.97 - 1.09 (m, 3H). MS (ESI) m/z 504 (M+H).
N-Benzyl-2-(1-{[3-(dimethylamino)phenyl]methyl}-6-methoxy-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (12f)
To the nitro derivative 12e (15 mg, 0.03 mmol) in ethanol (0.5 mL) was added hydrazine monohydrate (15 mg, 14 μL, 0.298 mmol) and the reaction warmed to 50 °C. Raney nickel (2800 type, as a slurry in water, 5 mg) was added and the reaction stirred at 50 °C for 1 hr, filtered through Celite and the solvent was removed under reduced pressure to give the amine as a colorless film (7 mg, 50%).
To the amine (7 mg, 0.015 mmol) in 1,2-dichloroethane (0.5 mL) was added formaldehyde (37% solution in water, 6 mg, 6 μL, 0.074 mmol) then sodium triacetoxyborohydride (16 mg, 0.074 mmol) and the reaction stirred at RT overnight. The solvent was removed under reduced pressure and redissolved in EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired dimethylamine as a white solid (5 mg, 71%). mp 48-51 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.33 (m, 3H), 6.99 - 7.14 (m, 4H), 6.48 - 6.63 (m, 5H), 4.45 (dd, J = 8.19, 14.98 Hz, 1H), 3.90 (t, J = 6.88 Hz, 2H), 3.85 (s, 3H), 3.54 - 3.70 (m, 2H), 3.38 - 3.53 (m, 1H), 3.08 - 3.32 (m, 2H), 2.79 - 2.99 (m, 10H), 2.41 - 2.53 (m, 1H), 1.78 - 1.92 (m, 2H), 1.04 (t, J = 7.35 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C31H40N3O3 [M + H]+ 502.3064, m/z found 502.3087.
N-Benzyl-2-(6-methoxy-1-{[3-methoxy-4-(pyridin-2-ylmethoxy)phenyl]methyl}-7-propoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)acetamide (12g)
To a mixture of phenol 9f (0.66 g, 1.56 mmol), potassium carbonate (0.65 g, 4.69 mmol) and tetrabutylammonium iodide (0.12 g, 0.31 mmol) in dimethylformamide (10 mL) was added 1-bromopropane (0.29 g, 0.21 mL, 2.34 mmol) and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-20% CMA-80 in EtOAc) to give the desired propoxy derivative as an off-white solid (0.38 g, 53%). m.p. 117-119 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.59 (td, J = 0.84, 4.83 Hz, 1H), 7.71 (dt, J = 1.13, 7.68 Hz, 1H), 7.56 (d, J = 7.82 Hz, 1H), 7.17 - 7.26 (m, 1H), 6.83 (d, J = 8.01 Hz, 1H), 6.69 - 6.76 (m, 2H), 6.60 - 6.68 (m, 2H), 6.50 (dd, J = 1.51, 8.10 Hz, 1H), 5.43 (br. s., 1H), 5.27 (s, 2H), 3.94 (t, J = 6.78 Hz, 2H), 3.86 (d, J = 0.47 Hz, 3H), 3.81 (d, J = 0.47 Hz, 3H), 3.37 - 3.50 (m, 4H), 2.67 (t, J = 6.88 Hz, 2H), 1.84 (sxt, J = 7.18 Hz, 2H), 1.02 (t, J = 7.44 Hz, 3H).
This intermediate was used in the preparation of 12g using the methods outlined for 10a and 11a as a yellow sticky solid in 58% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.58 (d, J = 4.80 Hz, 1H), 7.60 - 7.71 (m, 1H), 7.49 - 7.56 (m, 1H), 7.16 - 7.33 (m, 4H), 7.06 - 7.13 (m, 2H), 6.99 (dd, J = 5.18, 7.72 Hz, 1H), 6.62 - 6.75 (m, 3H), 6.58 (s, 1H), 6.47 (s, 1H), 5.14 (s, 2H), 4.45 (dd, J = 7.91, 14.98 Hz, 1H), 3.89 (t, J = 6.83 Hz, 2H), 3.84 (s, 3H), 3.84 (s, 3H), 3.58 - 3.69 (m, 2H), 3.09 - 3.50 (m, 3H), 2.78 - 2.99 (m, 4H), 2.41 - 2.54 (m, 1H), 1.76 - 1.92 (m, 2H), 1.04 (t, J = 7.44 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C36H42N3O5 [M + H]+ 596.3119, m/z found 596.3130.
Ethyl 2-[7-hydroxy-6-methoxy-1-(naphthalen-2-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetate (13a)
Tetrahydroisoquinoline 10a (0.37 g, 1.158 mmol), ethyl bromoacetate (0.29 g, 0.19 mL, 1.738 mmol) and tetrabutylammonium iodide (86 mg, 0.232 mmol) were combined in anhydrous DMF (12 mL). Diisopropylethylamine (0.37 g, 0.50 mL, 2.896 mmol) was added and the reaction was stirred at RT overnight. The reaction was diluted with water and extracted twice with EtOAc. The combined extracts were washed twice with brine, dried over MgSO4 and then the solvent was removed under reduced pressure. The crude product was purified by chromatography on silica (0-40% EtOAc/hexane) to give the desired acetate as a yellow oil (0.21 g, 44%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.70 - 7.82 (m, 3H), 7.60 (s, 1H), 7.37 - 7.47 (m, 2H), 7.33 (dd, J = 1.60, 8.38 Hz, 1H), 6.53 (s, 1H), 6.44 (s, 1H), 5.38 (s, 1H), 3.96 - 4.14 (m, 3H), 3.85 (s, 3H), 3.32 - 3.48 (m, 2H), 3.19 - 3.31 (m, 2H), 3.04 - 3.13 (m, 1H), 2.97 (td, J = 4.76, 12.72 Hz, 1H), 2.69 - 2.84 (m, 1H), 2.43 - 2.54 (m, 1H), 1.12 (t, J = 7.06 Hz, 3H). MS (ESI) m/z 406 (M+H).
Ethyl 2-{1-[(3,4-dimethylphenyl)methyl]-7-hydroxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetate (13b)
This was made by the same method as 13a using 10b. Yellow oil. Yield 24%. 1H NMR (300 MHz, CHLOROFORM-d) δ 6.99 - 7.03 (m, 1H), 6.94 (s, 1H), 6.85 - 6.91 (m, 1H), 6.54 (s, 1H), 6.35 (s, 1H), 5.38 (br. s., 1H), 4.11 (quin, J = 7.06 Hz, 2H), 3.92 (t, J = 6.31 Hz, 1H), 3.85 (s, 3H), 3.22 - 3.46 (m, 3H), 2.91 - 3.10 (m, 2H), 2.74 - 2.86 (m, 2H), 2.47 - 2.57 (m, 1H), 2.22 (s, 6H), 1.20 (t, J = 7.16 Hz, 3H). MS (ESI) m/z 384 (M+H).
2-[6-Methoxy-1-(naphthalen-2-ylmethyl)-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl]-N-(pyridin-3-ylmethyl)acetamide (14a)
2N Sodium hydroxide solution (1.02 mL, 2.04 mmol) was added to a solution of ester 13a (207 mg, 0.51 mmol) in ethanol (5 mL) and stirred at RT overnight. The pH was adjusted to 7-8 using 2N HCl, then all solvents were removed under reduced pressure. The residue was dissolved as far as possible in methanol, filtered and the solvent was removed under reduced pressure to give the crude acid as a white solid which was used without further purification.
The crude acid was combined with 3-pyridylmethylamine (54 mg, 0.50 mmol) and HATU (230 mg, 0.60 mmol) in anhydrous DMF (5 mL), then diisopropylethylamine (195 mg, 0.26 mmL, 1.51 mmol) was added. The reaction was stirred at RT overnight. The reaction was diluted with EtOAc, then washed with aqueous NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0-30% CMA-80/EtOAc) to give the desired amide as a white solid (106 mg, 45% over 2 steps). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.34 - 8.43 (m, 1H), 7.63 - 7.85 (m, 5H), 7.44 - 7.54 (m, 2H), 7.32 - 7.44 (m, 1H), 6.99 - 7.07 (m, 1H), 6.89 - 6.99 (m, 1H), 6.79 (s, 1H), 6.60 (s, 1H), 6.46 - 6.55 (m, 1H), 3.99 (dd, J = 8.38, 15.16 Hz, 1H), 3.89 (s, 3H), 3.79 - 3.87 (m, 1H), 3.61 - 3.73 (m, 2H), 3.02 - 3.28 (m, 4H), 2.83 - 3.01 (m, 2H), 2.67 (dd, J = 4.71, 15.07 Hz, 1H), 2.43 - 2.55 (m, 1H).
2-{1-[(3,4-Dimethylphenyl)methyl]-7-hydroxy-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (14b)
This was prepared by the same method as 14a from 13b. White solid. Yield 27% over 2 steps. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.45 - 8.51 (m, 1H), 8.24 (s, 1H), 7.35 (d, J = 8.10 Hz, 1H), 7.21 (dd, J = 4.99, 7.63 Hz, 1H), 6.95 - 7.06 (m, 3H), 6.76 - 6.85 (m, 1H), 6.72 (s, 1H), 6.58 (s, 1H), 4.33 (dd, J = 8.19, 15.35 Hz, 1H), 3.88 (s, 3H), 3.38 - 3.59 (m, 3H), 3.04 - 3.29 (m, 2H), 2.82 - 2.99 (m, 4H), 2.41 - 2.52 (m, 1H), 2.13 (s, 6H).
2-[6-Methoxy-1-(naphthalen-2-ylmethyl)-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl]-N-(pyridin-3-ylmethyl)acetamide (15a)
In a sealed tube, tetrahydroisoquinoline 14a (30 mg, 0.064 mmol) was combined with cesium carbonate (42 mg, 0.128 mmol) in anhydrous DMF (0.75 mL), then 2,2,2-trifluoroiodoethane (20 mg, 10 μL, 0.096 mmol) was added. The vessel was sealed and heated to 100 °C for 2 hr. The reaction was cooled and diluted with water. The mixture was extracted twice with EtOAc and the combined extracts were washed with brine, dried over MgSO4 and the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (10-30% CMA-80/EtOAc) to give the desired product as a yellow sticky solid (16 mg, 46%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.40 (dd, J = 1.70, 4.71 Hz, 1H), 7.66 - 7.86 (m, 5H), 7.44 - 7.54 (m, 2H), 7.38 (dd, J = 1.51, 8.29 Hz, 1H), 7.00 - 7.08 (m, 1H), 6.90 - 7.00 (m, 1H), 6.78 (s, 1H), 6.67 (s, 1H), 6.52 (d, J = 7.35 Hz, 1H), 4.26 - 4.42 (m, 2H), 4.01 (dd, J = 8.38, 14.98 Hz, 1H), 3.82 - 3.91 (m, 3H), 3.48 - 3.77 (m, 2H), 3.04 - 3.28 (m, 4H), 2.85 - 3.02 (m, 2H), 2.70 - 2.81 (m, 1H), 2.49 - 2.63 (m, 1H). 19F NMR (282 MHz, CHLOROFORM-d) δ -74.1 (t, J = 8.8 Hz). HRMS (ESI, CH3OH) m/z calcd for C31H31F3N3O3 [M + H]+ 550.2312, m/z found 550.2339.
2-{1-[(3,4-Dimethylphenyl)methyl]-6-methoxy-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-(pyridin-3-ylmethyl)acetamide (15b)
This was prepared by the same method as 15a using 14b. White solid. Yield 56%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.49 (d, J = 4.71 Hz, 1H), 8.26 (s, 1H), 7.37 (d, J = 7.91 Hz, 1H), 7.15 - 7.30 (m, 1H), 6.93 - 7.04 (m, 3H), 6.75 - 6.87 (m, 1H), 6.69 (s, 1H), 6.64 (s, 1H), 4.26 - 4.44 (m, 3H), 3.86 (s, 3H), 3.42 - 3.65 (m, 3H), 3.04 - 3.30 (m, 2H), 2.76 - 3.00 (m, 4H), 2.47 - 2.59 (m, 1H), 2.14 (s, 6H). HRMS (ESI, CH3OH) m/z calcd for C29H33F3N3O3 [M + H]+ 528.2469, m/z found 528.2490.
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-(pyridin-2-yl)acetamide (17a)
2-Pyridylacetic acid hydrochloride (16a) (1.0 g, 5.76 mmol), DCC (1.43 g, 6.91 mmol) and HOBt monohydrate (0.93 g, 6.91 mmol) were combined in anhydrous THF (40 mL) and stirred at RT under N2 for 2 hr. A solution of 4-hydroxy-3-methoxyphenthylamine hydrochloride (1.29 g, 6.34 mmol) and triethylamine (2.91 g, 4.0 mL, 28.80 mmol) in THF (20 mL) was added and the resulting mixture stirred at RT overnight. The reaction was diluted with EtOAc and filtered, then the filtrate was washed with brine and dried over MgSO4, then the solvent was removed under reduced pressure. The crude material was purified by chromatography on silica (0-10% MeOH/DCM) to give the desired amide as an yellow oil (0.57 g, 35%).1H NMR (300 MHz, CHLOROFORM-d) δ 8.40 - 8.45 (m, 1H), 7.65 (dt, J = 1.79, 7.68 Hz, 1H), 7.29 - 7.38 (m, 1H), 7.15 - 7.26 (m, 2H), 6.73 - 6.82 (m, 1H), 6.54 - 6.70 (m, 2H), 5.78 (br. s., 1H), 3.82 (s, 3H), 3.69 (s, 2H), 3.43 - 3.54 (m, 2H), 2.71 (t, J = 6.97 Hz, 2H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-(pyridin-3-yl)acetamide (17b)
4-Hydroxy-3-methoxyphenethylamine hydrochloride (1.17 g, 5.76 mmol), 3-pyridylacetic acid hydrochloride (16b) (1.0 g, 5.76 mmol) and HBTU (2.40 g, 6.34 mmol) were combined in anhydrous DMF (30 mL). Diisopropylethylamine (2.98 g, 4.0 mL, 23.04 mmol) was added and the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, the solution was washed with aqueous NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-10% MeOH/DCM) to give the desired amide as a yellow oil (0.52 g, 32%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.52 (dd, J = 1.51, 4.71 Hz, 1H), 8.44 (d, J = 1.70 Hz, 1H), 7.53 - 7.60 (m, 1H), 7.22 - 7.30 (m, 2H), 6.80 (d, J = 7.91 Hz, 1H), 6.61 (d, J = 1.70 Hz, 1H), 6.55 (dd, J = 1.88, 7.91 Hz, 1H), 5.51 (br. s., 1H), 3.84 (s, 3H), 3.43 - 3.54 (m, 4H), 2.70 (t, J = 6.97 Hz, 2H).
N-[2-(4-Hydroxy-3-methoxyphenyl)ethyl]-2-(pyridin-4-yl)acetamide (17c)
This was prepared by the method used for 17b using 4-pyridylacetic acid hydrochloride (16c). Yellow oil.Yield 35%.1H NMR (300 MHz, CHLOROFORM-d) δ 8.51 - 8.57 (m, 2H), 7.13 (d, J = 5.84 Hz, 2H), 6.82 (d, J = 8.10 Hz, 1H), 6.61 (d, J = 1.70 Hz, 1H), 6.55 (dd, J = 1.60, 8.01 Hz, 1H), 5.59 (br. s., 1H), 5.37 (br. s., 1H), 3.83 (s, 3H), 3.44 - 3.54 (m, 4H), 2.71 (t, J = 6.88 Hz, 2H).
6-Methoxy-1-(pyridin-2-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-7-ol (18a)
Amide 17a (0.56 g, 1.96 mmol) was dissolved in anhydrous acetonitrile (10 mL) with warming. Phosphorus oxychloride (1.92 g, 1.2 mL, 12.52 mmol) was added and the reaction heated at reflux for 1 hr. The reaction was cooled and all solvents removed under reduced pressure. The crude was suspended in water and then heated until fully dissolved. The solution was cooled and the pH was adjusted to 8 with conc. ammonium hydroxide solution, then extracted 3 times with DCM. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give an orange viscous oil.
The crude dihydroisoquinoline was dissolved in anhydrous methanol (10 mL) and cooled in an ice bath under N2. Sodium borohydride (0.15 g, 3.91 mmol) was added portionwise, the mixture was stirred in ice for 30 min then at RT overnight. The reaction was quenched with water, the methanol was removed under reduced pressure, then the mixture was diluted further with water and extracted 3 times with DCM. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-10% MeOH/DCM) to give the desired tetrahydroisoquinoline as an orange solid (0.36 g, 68%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.57 (dd, J = 0.85, 4.80 Hz, 1H), 7.61 (dt, J = 1.79, 7.68 Hz, 1H), 7.11 - 7.21 (m, 2H), 6.73 (s, 1H), 6.55 (s, 1H), 4.39 (dd, J = 3.20, 9.98 Hz, 1H), 3.82 (s, 3H), 3.25 - 3.33 (m, 1H), 3.21 (dd, J = 5.75, 11.96 Hz, 1H), 3.06 (dd, J = 10.08, 14.03 Hz, 1H), 2.95 (td, J = 5.91, 12.10 Hz, 1H), 2.67 - 2.77 (m, 2H).
6-Methoxy-1-(pyridin-3-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-7-ol (18b)
This was prepared using the method for 18a using 17b. Orange oil.Yield 50%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.44 - 8.52 (m, 2H), 7.57 (d, J = 7.72 Hz, 1H), 7.20 - 7.29 (m, 1H), 6.75 (s, 1H), 6.56 (s, 1H), 4.11 (dd, J = 3.49, 9.51 Hz, 1H), 3.81 (s, 3H), 3.09 - 3.22 (m, 2H), 2.83 - 2.97 (m, 2H), 2.60 - 2.79 (m, 2H).
6-Methoxy-1-(pyridin-4-ylmethyl)-1,2,3,4-tetrahydroisoquinolin-7-ol (18c)
This was prepared using the method for 18a using 17c. Orange oil. Yield 27%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.50 - 8.55 (m, 2H), 7.19 (d, J = 6.03 Hz, 2H), 6.78 (s, 1H), 6.58 (s, 1H), 4.16 (dd, J = 3.49, 9.70 Hz, 1H), 3.86 (s, 3H), 3.12 - 3.23 (m, 2H), 2.60 - 2.98 (m, 4H).
N-Benzyl-2-[6-methoxy-1-(pyridin-2-ylmethyl)-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (19a)
This was prepared by the methods used for 11a and 16a, starting from 18a. Orange sticky solid. Yield 20% over 2 steps. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.19 - 8.30 (m, 1H), 7.53 - 7.69 (m, 1H), 7.23 - 7.45 (m, 4H), 7.13 - 7.22 (m, 2H), 6.88 - 7.01 (m, 2H), 6.57 - 6.76 (m, 2H), 4.41 - 4.53 (m, 1H), 4.23 - 4.39 (m, 2H), 4.02 - 4.17 (m, 2H), 3.80 - 3.88 (m, 3H), 3.15 - 3.40 (m, 3H), 3.11 (d, J = 6.97 Hz, 2H), 2.73 - 2.91 (m, 2H), 2.35 - 2.48 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C27H29F3N3O3 [M + H]+ 500.2156, m/z found 500.2173.
N-Benzyl-2-[6-methoxy-1-(pyridin-3-ylmethyl)-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (19b)
This was prepared by the methods used for 11a and 16a, starting from 18b. Yellow solid. Yield 9% over 2 steps. m.p. 49-50 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.43 (br. s., 1H), 8.37 (d, J = 3.58 Hz, 1H), 7.36 - 7.46 (m, 1H), 7.23 - 7.36 (m, 3H), 7.11 (d, J = 6.78 Hz, 2H), 7.01 (dd, J = 4.52, 7.72 Hz, 1H), 6.66 - 6.75 (m, 1H), 6.64 (s, 1H), 6.61 (s, 1H), 4.25 - 4.46 (m, 3H), 3.86 (s, 3H), 3.81 - 3.93 (m, 1H), 3.60 - 3.73 (m, 1H), 3.07 - 3.45 (m, 3H), 2.81 - 3.03 (m, 4H), 2.46 - 2.61 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C27H29F3N3O3 [M + H]+ 500.2156, m/z found 500.2181.
N-Benzyl-2-[6-methoxy-1-(pyridin-4-ylmethyl)-7-(2,2,2-trifluoroethoxy)-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (19c)
This was prepared by the methods used for 11a and 16a, starting from 18c. Yellow solid. Yield 17% over 2 steps. m.p. 56-58 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.40 (d, J = 5.84 Hz, 1H), 7.24 - 7.39 (m, 4H), 7.10 - 7.20 (m, 2H), 7.01 - 7.07 (m, 2H), 6.61 - 6.79 (m, 3H), 4.23 - 4.46 (m, 3H), 3.86 (s, 3H), 3.64 - 3.84 (m, 2H), 3.06 - 3.42 (m, 3H), 2.78 - 3.03 (m, 4H), 2.44 - 2.61 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C27H29F3N3O3 [M + H]+ 500.2156, m/z found 500.2180.
2-[4-(Benzyloxy)-3-methoxyphenyl]acetic acid (21)
Sodium metal (0.76 g, 32.94 mmol) was added portionwise to anhydrous methanol (17 mL) under N2 at RT. When dissolution was complete, acid 20 (3.0 g, 16.47 mmol) was added in one portion and the mixture warmed until all solids had dissolved. Benzyl bromide (5.63 g, 3.9 mL, 32.94 mmol) was added and the reaction heated to reflux for 5 hr then allowed to stand at RT overnight. The reaction was diluted with ether and the solids collected by filtration. These were rinsed with EtOAc, dissolved in 1N HCl as far as possible and the solids collected by filtration to give the benzyl ether as a white solid (3.22 g, 72%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.23 - 7.47 (m, 5H), 6.79 - 6.86 (m, 2H), 6.70 - 6.77 (m, 1H), 5.13 (s, 2H), 3.88 (s, 3H), 3.57 (s, 2H).
2-[4-(Benzyloxy)-3-methoxyphenyl]-N-[2-(4-hydroxy-3-methoxyphenyl)ethyl]acetamide (22)
This was prepared by the method used for 9a, starting from acid 21 and 4-hydroxy-3-methoxyphenethylamine, with HBTU as coupling agent. White sticky solid. Yield 96%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.42 - 7.48 (m, 2H), 7.28 - 7.41 (m, 3H), 6.78 (dd, J = 8.05, 12.10 Hz, 2H), 6.70 (d, J = 1.98 Hz, 1H), 6.57 - 6.64 (m, 2H), 6.48 (dd, J = 1.88, 8.01 Hz, 1H), 5.57 (s, 1H), 5.41 (br. s., 1H), 5.15 (s, 2H), 3.84 (s, 3H), 3.82 (s, 3H), 3.45 (s, 2H), 3.38 - 3.44 (m, 2H), 2.66 (t, J = 6.92 Hz, 2H).
4-(2-{2-[4-(Benzyloxy)-3-methoxyphenyl]acetamido}ethyl)-2-methoxyphenyl methanesulfonate (23)
To a solution of amide 22 (0.67 g, 1.58 mmol) in dichloromethane (2 mL) cooled in an ice bath under N2 was added triethylamine (0.24 g, 0.33 mL, 2.37 mmol) then slowly methanesulfonyl chloride (0.22 g, 0.15 mL, 1.90 mmol). The reaction was stirred in an ice bath for 4 hr, quenched by the addition of water then the layers separated. The aqueous portion was extracted twice with dichloromethane and the combined organic fractions were dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the sulfonate as a white solid (0.51 g, 65%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.41 - 7.48 (m, 2H), 7.28 - 7.41 (m, 3H), 7.13 (d, J = 8.19 Hz, 1H), 6.83 (d, J = 8.10 Hz, 1H), 6.74 (dd, J = 1.84, 4.10 Hz, 2H), 6.56 - 6.64 (m, 2H), 5.53 (t, J = 5.37 Hz, 1H), 5.14 (s, 2H), 3.85 (s, 3H), 3.82 (s, 3H), 3.40 - 3.50 (m, 4H), 3.14 (s, 3H), 2.73 (t, J = 7.02 Hz, 2H).
1-{[4-(Benzyloxy)-3-methoxyphenyl]methyl}-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl methanesulfonate (24)
This was prepared using the method shown for 10a using amide 23 to give 24 as a frothy white solid in 89% yield over 2 steps. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.41 - 7.48 (m, 2H), 7.28 - 7.40 (m, 3H), 7.18 (br. s., 1H), 6.79 - 6.87 (m, 1H), 6.67 - 6.78 (m, 3H), 5.14 (s, 2H), 4.12 (dd, J = 2.45, 9.23 Hz, 1H), 3.86 (s, 6H), 3.17 (s, 3H), 3.11 - 3.26 (m, 1H), 2.67 - 2.97 (m, 5H).
2-[(Benzylcarbamoyl)methyl]-1-{[4-(benzyloxy)-3-methoxyphenyl]methyl}-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl methanesulfonate (25a)
This was prepared using the method shown for 11a starting from 24 to give 25a as a clear film in 65% yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.36 - 7.43 (m, 2H), 7.35 (s, 1H), 7.19 - 7.33 (m, 4H), 7.09 (d, J = 6.59 Hz, 2H), 7.05 (s, 1H), 6.83 - 6.92 (m, 1H), 6.61 - 6.76 (m, 5H), 4.97 (s, 2H), 4.42 (dd, J = 8.10, 14.98 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.54 - 3.73 (m, 2H), 3.32 - 3.48 (m, 1H), 3.17 (s, 3H), 3.06 - 3.32 (m, 2H), 2.80 - 3.02 (m, 4H), 2.48 - 2.59 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C35H39N2O7S [M + H]+ 631.2472, m/z found 631.2475.
2-[(Benzylcarbamoyl)methyl]-1-[(4-butoxy-3-methoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl methanesulfonate (25b)
To a solution of benzyl ether (0.16 g, 0.25 mmol) in methanol (5 mL) was added concentrated HCl (2.5 mL) and the reaction heated to reflux overnight. Methanol was removed under reduced pressure then the acid was carefully poured into NaHCO3 solution and then extracted three times with EtOAc. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the phenol as a colorless film (27 mg, 20%).
The phenol (40 mg, 0.074 mmol), potassium carbonate (26 mg, 0.185 mmol) and tetrabutylammonium iodide (6 mg, 0.015 mmol) were combined in dry dimethylformamide (1 mL) and 1-bromobutane (15 mg, 12 μL, 0.111 mmol) was added. The reaction was heated to 50 °C overnight, then diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-80% EtOAc in hexane) to give the desired ether as a clear film (39 mg, 91%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.04 - 7.14 (m, 3H), 6.82 - 6.93 (m, 1H), 6.60 - 6.76 (m, 4H), 4.47 (dd, J = 8.15, 14.93 Hz, 1H), 3.88 (s, 3H), 3.79 (s, 3H), 3.84 (td, J = 3.33, 6.81 Hz, 2H), 3.53 - 3.74 (m, 2H), 3.34 - 3.48 (m, 1H), 3.18 (s, 3H), 3.07 - 3.32 (m, 2H), 2.81 - 3.02 (m, 4H), 2.45 - 2.61 (m, 1H), 1.72 - 1.87 (m, 2H), 1.40 - 1.55 (m, 2H), 0.97 (t, J = 7.35 Hz, 3H). m/z 597 (M+H). HRMS (ESI, CH3OH) m/z calcd for C32H41N2O7S [M + H]+ 597.2629, m/z found 597.2643.
(3-Methoxy-4-nitrophenyl)acetonitrile (27)
To a solution of tetrabutylammonium nitrate (7.86 g, 25.82 mmol) and 18-crown-6 (0.11 g, 0.41 mmol) in dry dichloromethane (150 mL) cooled in an ice bath under N2 was added dropwise trifluoroacetic anhydride (13.13 g, 8.8 mL, 62.5 mmol). The solution was stirred in ice for 30 min, then transferred to an addition funnel and slowly added to a solution of 3-methoxyphenylacetonitrile (26) (4.0 g, 3.8 mL, 27.18 mmol) in dichloromethane (300 mL). The resulting mixture was stirred at RT overnight and washed with NaHCO3 solution. The aqueous layer was extracted once with CH2Cl2, then the combined organic fractions were dried over MgSO4 and solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-40% EtOAc in hexane) to give the desired 4-nitro derivative as a viscous orange oil (2.71 g, 56%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.89 (d, J = 8.29 Hz, 1H), 7.05 - 7.12 (m, 1H), 7.00 (td, J = 0.80, 8.29 Hz, 1H), 4.01 (s, 3H), 3.84 (s, 2H).
2-(3-Methoxy-4-nitrophenyl)ethanamine (28)
To a solution of nitrile 27 (2.7 g, 14.05 mmol) in anhydrous tetrahydrofuran (75 mL) cooled in an ice bath under N2 was slowly added a solution of borane-THF complex (1M solution in THF, 56 mL). Upon completion of addition, the reaction was warmed to RT then heated to 70 °C for 2 hr. The reaction was cooled then 2N HCl (7 mL) was carefully added to quench the reaction, then the solution made basic with 2N NaOH solution. It was extracted twice with EtOAc, then the combined extracts were dried over MgSO4 and the solvent removed under reduced pressure to give the phenylethanamine (2.7 g, 97%) as a dark orange oil. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.83 (d, J = 8.19 Hz, 1H), 6.83 - 6.97 (m, 2H), 3.96 (s, 3H), 3.02 (t, J = 6.78 Hz, 2H), 2.81 (t, J = 6.97 Hz, 2H).
2-(3,4-Dimethoxyphenyl)-N-[2-(3-methoxy-4-nitrophenyl)ethyl]acetamide (29)
3,4-Dimethoxyphenylacetic acid (0.53 g, 2.70 mmol) and amine 28 (0.53 g, 2.70 mmol) were combined in anhydrous DMF (15 mL), then BOP (1.20 g, 2.70 mmol) was added, followed by diisopropylethylamine (0.87 g, 1.2 mL, 6.75 mmol), then the reaction stirred at RT under N2 overnight. The reaction was diluted with EtOAc, then washed with 2N HCl, NaHCO3 solution and brine, dried over MgSO4 then the solvent was removed under reduced pressure to give the desired amide as an orange oil (1.01 g, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.75 (d, J = 8.19 Hz, 1H), 6.64 - 6.85 (m, 5H), 5.50 (br. s., 1H), 3.90 (s, 3H), 3.88 (s, 3H), 3.85 (s, 3H), 3.44 - 3.57 (m, 4H), 2.82 (t, J = 6.97 Hz, 2H)
N-[2-(4-Amino-3-methoxyphenyl)ethyl]-2-(3,4-dimethoxyphenyl)acetamide (30)
To a solution of amide 29 (1.02 g, 2.72 mmol) in ethanol was added hydrazine monohydrate (1.36 g, 1.32 mL, 27.2 mmol) then Raney nickel (as a slurry in water, 0.20 g) was added. The reaction was heated at 50 °C for 1 hr, cooled to RT then filtered through Celite, rinsing with ethanol. The solvent was removed under reduced pressure to give the aniline as an orange oil (0.94 g, 100%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.75 - 6.84 (m, 2H), 6.60 - 6.74 (m, 3H), 6.56 (d, J = 7.72 Hz, 1H), 6.51 (d, J = 1.51 Hz, 1H), 6.40 (dd, J = 1.65, 7.77 Hz, 1H), 5.42 (br. s., 1H), 3.89 (s, 3H), 3.83 (s, 3H), 3.79 (s, 3H), 3.47 (s, 2H), 3.42 (q, J = 6.72 Hz, 2H), 2.63 (t, J = 6.83 Hz, 2H).
Methyl N-(4-{2-[2-(3,4-dimethoxyphenyl)acetamido]ethyl}-2-methoxyphenyl)carbamate (31)
To a solution of aniline 30 (0.94 g, 2.72 mmol) in DCM (15 mL) cooled in ice under N2 was added diisopropylethylamine (0.88 g, 1.18 mL, 6.80 mmol) then methyl chloroformate (0.51 g, 0.42 mL, 5.44 mmol). The reaction was allowed to warm slowly to RT overnight. The reaction was quenched with methanol, then the solvents were removed under reduced pressure. The crude was purified by chromatography on silica (0-3% MeOH/DCM) to give the carbamate as a yellow oil (0.44 g, 40%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.93 (d, J = 7.35 Hz, 1H), 7.16 (br. s., 1H), 6.77 - 6.84 (m, 1H), 6.57 - 6.72 (m, 4H), 5.45 (br. s., 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.76 - 3.79 (m, 3H), 3.40 - 3.51 (m, 4H), 2.70 (t, J = 6.88 Hz, 2H).
Methyl N-{2-[(benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl}carbamate (32)
This was prepared using the methods used to make 10a and 11a sequentially, starting from 31. Yellow oil. Yield 55% over 3 steps. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.92 (br. s., 1H), 7.17 - 7.33 (m, 4H), 7.06 (d, J = 6.88 Hz, 2H), 6.78 - 6.86 (m, 1H), 6.71 - 6.77 (m, 2H), 6.56 - 6.63 (m, 2H), 4.47 (dd, J = 8.38, 15.07 Hz, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.71 (s, 3H), 3.67 (dd, J = 4.19, 11.07 Hz, 1H), 3.53 (dd, J = 4.66, 15.12 Hz, 1H), 3.38 - 3.48 (m, 1H), 3.05 - 3.32 (m, 2H), 2.78 - 3.03 (m, 4H), 2.47 (dd, J = 4.62, 16.20 Hz, 1H).
2-{7-Amino-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}-N-benzylacetamide (33)
To a solution of carbamate 32 (0.36 g, 0.67 mmol) in methanol (30 mL) was added sodium hydroxide solution (2N, 3.35 mL, 6.70 mmol) and the reaction heated at 50 °C for 48 hr. The reaction was cooled and the methanol removed under reduced pressure. It was further diluted with water and extracted 3 times with EtOAc. The combined extracts were dried over MgSO4 and the solvent removed under reduced pressure. The crude material was purified by chromatography on silica (0-100% EtOAc/hexane) to give the free aniline as a white solid (0.19 g, 59%). m.p. 65-67 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 - 7.34 (m, 4H), 7.08 (d, J = 6.97 Hz, 2H), 6.93 (dd, J = 4.85, 7.30 Hz, 1H), 6.56 - 6.77 (m, 4H), 6.50 (s, 1H), 6.44 (s, 1H), 4.47 (dd, J = 8.24, 15.12 Hz, 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.73 (s, 3H), 3.52 - 3.63 (m, 2H), 3.33 - 3.49 (m, 1H), 3.09 - 3.32 (m, 2H), 2.78 - 2.97 (m, 4H), 2.37 - 2.50 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C28H34N3O4 [M + H]+ 476.2544, m/z found 476.2546.
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-(dimethylamino)-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (34a)
To amine 33 (7 mg, 0.015 mmol) in 1,2-dichloroethane (0.5 mL) was added formaldehyde (37% solution in water, 6 mg, 6 μL, 0.074 mmol) then sodium triacetoxyborohydride (16 mg, 0.074 mmol) and the reaction stirred at RT overnight. The solvent was removed under reduced pressure and redissolved in EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-50% EtOAc in hexane) to give the desired dimethylamine as a white solid (5 mg, 71%). m.p. 128-130 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.07 - 7.15 (m, 2H), 7.00 (dd, J = 5.13, 7.30 Hz, 1H), 6.60 - 6.74 (m, 3H), 6.56 (s, 1H), 6.51 (s, 1H), 4.50 (dd, J = 8.01, 14.98 Hz, 1H), 3.87 (s, 3H), 3.81 (s, 3H), 3.75 (s, 3H), 3.66 (dd, J = 4.99, 15.07 Hz, 2H), 3.34 - 3.48 (m, 1H), 3.13 - 3.34 (m, 2H), 2.79 - 2.99 (m, 4H), 2.73 (s, 6H), 2.42 - 2.57 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C30H38N3O4 [M + H]+ 504.2857, m/z found 504.2867.
N-Benzyl-2-[7-(benzylamino)-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (34b)
Aniline 33 (30 mg, 0.063 mmol) was combined with sodium bicarbonate (2 mg, 0.025 mmol) and benzaldehyde (7 mg, 7 μL, 0.069 mmol) in anhydrous methanol (0.5 mL) and heated to 40 °C for 1 hour. The reaction was then cooled in an ice bath and sodium borohydride (2 mg, 0.041 mmol) was added. The reaction was allowed to warm to RT slowly overnight. The solvent was removed under reduced pressure and the crude purified by chromatography on silica (0-100% EtOAc/hexane) to give the benzylamine as a clear film (3 mg, 8%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.32 - 7.44 (m, 4H), 7.18 - 7.31 (m, 5H), 7.08 (d, J = 6.59 Hz, 2H), 6.92 - 7.00 (m, 1H), 6.57 - 6.73 (m, 3H), 6.48 (s, 1H), 6.21 (s, 1H), 4.48 (dd, J = 8.05, 14.93 Hz, 1H), 4.30 (s, 2H), 3.83 (s, 3H), 3.79 (s, 3H), 3.73 (s, 3H), 3.50 - 3.63 (m, 2H), 3.33 - 3.47 (m, 1H), 3.10 - 3.32 (m, 2H), 2.70 - 2.98 (m, 4H), 2.38 - 2.49 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C35H40N3O4 [M + H]+ 566.3013, m/z found 566.3031.
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-acetamido-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (34c)
To a solution of aniline 33 (30 mg, 0.063 mmol) and diisopropylethylamine (20 mg, 27 μL, 0.158 mmol) in anhydrous DCM (0.5 mL) was added acetic anhydride (13 mg, 12 μL, 0.126 mmol) and the reaction stirred at RT overnight. The reaction mixture was applied directly to chromatography on silica (0-100% EtOAc/hexane) to give the desired amide as a white solid (24 mg, 73%). m.p. 81-84 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.23 (s, 1H), 7.75 (s, 1H), 7.17 - 7.34 (m, 3H), 7.06 (d, J = 6.97 Hz, 2H), 6.69 - 6.88 (m, 3H), 6.55 - 6.65 (m, 2H), 4.47 (dd, J = 8.38, 15.07 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.71 (s, 3H), 3.62 - 3.70 (m, 1H), 3.37 - 3.58 (m, 2H), 3.05 - 3.31 (m, 2H), 2.78 - 3.03 (m, 4H), 2.48 (dd, J = 4.52, 16.29 Hz, 1H), 2.22 (s, 3H). HRMS (ESI, CH3OH) m/z calcd for C30H36N3O5 [M + H]+ 518.2649, m/z found 518.2658.
N-Benzyl-2-{1-[(3,4-dimethoxyphenyl)methyl]-7-methanesulfonamido-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl}acetamide (34d)
To a solution of aniline 33 (50 mg, 0.105 mmol) in DCM (0.5 mL) cooled in ice was added triethylamine (43 mg, 59 μL, 0.420 mmol) then methanesulfonyl chloride (36 mg, 24 μL, 0.315 mmol). The reaction was allowed to warm slowly to RT overnight. The crude reaction was directly applied to chromatography on silica (0-100% EtOAc/hexane) to give a mixture of mono- and bis-sulfonamides (mono 17 mg, 29%; bis 23 mg, 35%). The bis-sulfonamide was suspended in aqueous sodium hydroxide (3N, 1 mL) and heated to 80 °C overnight. The reaction was cooled and the pH adjusted to 7 with 2N HCl, forming a precipitate. The mixture was extracted 3 times with EtOAc, the combined extracts were then dried over MgSO4 and the solvent was removed under reduced pressure to give the mono-sulfonamide as a clear oil (17 mg, 85%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.35 (m, 3H), 7.07 (d, J = 7.44 Hz, 2H), 6.77 - 6.85 (m, 1H), 6.58 - 6.76 (m, 5H), 4.49 (dd, J = 8.24, 15.02 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.75 - 3.90 (m, 1H), 3.73 (s, 3H), 3.62 - 3.71 (m, 1H), 3.37 - 3.60 (m, 2H), 3.07 - 3.34 (m, 2H), 2.94 (s, 3H), 2.79 - 3.04 (m, 4H), 2.47 - 2.59 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C29H36N3O6S [M + H]+ 554.2319, m/z found 554.2341.
N-{2-[(Benzylcarbamoyl)methyl]-1-[(3,4-dimethoxyphenyl)methyl]-6-methoxy-1,2,3,4-tetrahydroisoquinolin-7-yl}butanamide (34e)
To a solution of aniline 33 (30 mg, 0.063 mmol), butyric acid (6 mg, 6 μL, 0.063 mmol) and BOP (33 mg, 0.076 mmol) in anhydrous DCM (0.5 mL) was added diisopropylethylamine (24 mg, 33 μL, 0.189 mmol) and the reaction was stirred at RT overnight. The solution was applied directly to chromatography on silica (0-100% EtOAc/hexane) to give the desired amide as a yellow solid (28 mg, 82%). m.p. 101-103 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.28 (s, 1H), 7.75 (s, 1H), 7.17 - 7.33 (m, 3H), 7.06 (d, J = 6.88 Hz, 2H), 6.70 - 6.86 (m, 3H), 6.55 - 6.63 (m, 2H), 4.46 (dd, J = 8.38, 15.07 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.71 (s, 3H), 3.62 - 3.75 (m, 1H), 3.38 - 3.58 (m, 2H), 3.03 - 3.31 (m, 2H), 2.77 - 3.03 (m, 4H), 2.48 (dd, J = 4.52, 16.48 Hz, 1H), 2.39 (t, J = 7.39 Hz, 2H), 1.71 - 1.86 (m, 2H), 1.03 (t, J = 7.35 Hz, 3H). HRMS (ESI, CH3OH) m/z calcd for C32H40N3O5 [M + H]+ 546.2962, m/z found 546.2974.
2-(4-Methoxy-3-nitrophenyl)-N-[2-(3-methoxy-4-nitrophenyl)ethyl]acetamide (35)
To a solution of 4-hydroxy-3-nitrophenylacetic acid (1.97 g, 10 mmol), amine 28 (1.96 g, 10 mmol) and HBTU (4.17 g, 11 mmol) in dimethylformamide (50 mL) was added diisopropylethylamine (3.23 g, 4.4 mL, 25 mmol) and the reaction stirred at RT under N2 overnight. It was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was absorbed onto Celite and purified by chromatography on silica (0-100% EtOAc in hexane) to give the amide (2.35 g, 63%) as a dark orange oil. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.00 (s, 1H), 7.96 (d, J = 1.98 Hz, 1H), 7.78 (d, J = 8.29 Hz, 1H), 7.47 (dd, J = 2.12, 8.62 Hz, 1H), 6.88 (s, 1H), 6.78 (dd, J = 1.37, 8.34 Hz, 1H), 5.81 (br. s., 1H), 3.92 (s, 3H), 3.56 (d, J = 6.78 Hz, 2H), 3.49 (s, 2H), 2.83 - 2.92 (m, 2H).
To the phenol (2.35 g, 6.26 mmol) and potassium carbonate (1.30 g, 9.39 mmol) in dimethylformamide (30 mL) cooled in ice under N2 was added iodomethane (1.07 g, 0.47 mL, 7.51 mmol) and the reaction stirred in ice for 1 hr, then additional aliquots of potassium carbonate (1.30 g) and iodomethane (0.47 mL) were added and the reaction heated at 60 °C for 1 hr. The reaction was cooled and diluted with EtOAc. It was washed with water and brine, dried over MgSO4 and the solvent removed under reduced pressure to give the desired amide (2.38 g, 98%) as a dark orange oil. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.76 (d, J = 8.29 Hz, 1H), 7.72 (d, J = 2.17 Hz, 1H), 7.38 (dd, J = 2.31, 8.71 Hz, 1H), 7.04 (d, J = 8.57 Hz, 1H), 6.86 (s, 1H), 6.77 (dd, J = 1.22, 8.29 Hz, 1H), 5.87 (br. s., 1H), 3.96 (s, 3H), 3.91 (s, 3H), 3.55 (q, J = 6.78 Hz, 2H), 3.48 (s, 2H), 2.87 (t, J = 6.88 Hz, 2H).
N-{2-Methoxy-4-[2-(2-{4-methoxy-3-[(methoxycarbonyl)amino]phenyl}acetamido)ethyl]phenyl}carbamate (36)
To the nitro derivative 35 (2.38 g, 6.11 mmol) in ethanol (60 mL) was added hydrazine monohydrate (3.06 g, 3.0 mL, 61.1 mmol) then Raney nickel (2800 type as a slurry in water, 0.48 g). The reaction was heated to 50 °C for 90 min, cooled then filtered through Celite, rinsing thoroughly with ethanol. The solvent was removed under reduced pressure to give the aniline as a yellow oil which was used in the next step without further purification (1.98 g, 99%). 1H NMR (300 MHz, CHLOROFORM-d) δ 6.62 - 6.76 (m, 2H), 6.56 - 6.62 (m, 1H), 6.51 - 6.56 (m, 1H), 6.40 - 6.51 (m, 2H), 5.52 (br. s., 1H), 3.84 (s, 3H), 3.80 (s, 3H), 3.35 - 3.42 (m, 4H), 2.63 (t, J = 6.88 Hz, 2H).
To a mixture of the aniline (1.98 g, 6.01 mmol) and diisopropylethylamine (3.11 g, 4.2 mL, 24.04 mmol) in dichloromethane (30 mL) cooled in an ice bath under N2 was added slowly methyl chloroformate (1.70 g, 1.4 mL, 18.03 mmol). The reaction was allowed to warm to RT overnight, then quenched with NaHCO3 solution. The layers were separated, the aqueous extracted once with dichloromethane, then the combined extracts were dried over MgSO4 and the solvents removed under reduced pressure. The crude was absorbed onto Celite and purified by chromatography on silica (0-3% methanol in dichloromethane) to give the bis-carbamate as a yellow oil (1.61 g, 60%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.79 - 7.95 (m, 1H), 7.14 (br. s., 1H), 6.78 (s, 2H), 6.50 - 6.62 (m, 2H), 5.42 (br. s., 1H), 3.87 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 3.78 (s, 3H), 3.38 - 3.52 (m, 4H), 2.67 (t, J = 6.83 Hz, 2H).
Methyl N-[6-Methoxy-1-({4-methoxy-3-[(methoxycarbonyl)amino]phenyl}methyl)-1,2,3,4-tetrahydroisoquinolin-7-yl]carbamate (37)
To amide 36 (1.61 g, 3.61 mmol) suspended in anhydrous toluene (20 mL) was added phosphorus oxychloride (2.77 g, 1.7 mL, 18.1 mmol) and the reaction heated to 90 °C for 2 hr. The reaction was cooled and poured carefully into water and stirred vigorously for 15 min. The layers were separated then the aqueous layer was adjusted to pH 8-9 with NaHCO3 solution and extracted three times with dichloromethane. The combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure to give a viscous orange oil.
The crude was dissolved in methanol (40 mL) and cooled in an ice bath under N2. Sodium borohydride (0.69 g, 18.1 mmol) was added portionwise and the reaction allowed to warm slowly to RT overnight. The reaction was quenched with water, the methanol was removed under reduced pressure and the aqueous solution was extracted three times with dichloromethane. The combined extracts were dried over MgSO4 and the solvent removed under reduced pressure to give the desired tetrahydroisoquinoline (1.41 g, 91%) as a frothy solid. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.00 (br. s., 1H), 7.20 (d, J = 15.16 Hz, 1H), 6.87 - 6.96 (m, 1H), 6.77 - 6.85 (m, 1H), 6.57 (s, 1H), 4.14 (dd, J = 3.20, 10.55 Hz, 1H), 3.69 - 3.92 (m, 13H), 3.15 - 3.32 (m, 2H), 2.67 - 2.98 (m, 4H).
Methyl N-{2-[(Benzylcarbamoyl)methyl]-6-methoxy-1-({4-methoxy-3-[(methoxycarbonyl)amino]phenyl}methyl)-1,2,3,4-tetrahydroisoquinolin-7-yl}carbamate (38)
To amine 37 (0.94 g, 2.19 mmol), N-benzyl bromoacetamide (0.75 g, 3.28 mmol) and tetrabutylammonium iodide (0.16 g, 0.44 mmol) in dimethylformamide (22 mL) was added diisopropylethylamine (0.71 g, 0.95 mL, 5.47 mmol) and the reaction stirred under N2 at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution, water and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the desired product (0.74 g, 59%) as a frothy orange solid. m.p. 88-90 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.99 (br. s., 1H), 7.88 (br. s., 1H), 7.12 - 7.32 (m, 5H), 6.92 - 7.06 (m, 3H), 6.81 - 6.91 (m, 1H), 6.50 - 6.67 (m, 2H), 4.27 - 4.46 (m, 1H), 3.58 - 3.91 (m, 14H), 3.41 - 3.55 (m, 1H), 3.19 - 3.35 (m, 1H), 2.76 - 3.13 (m, 5H), 2.38 - 2.55 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C31H37N4O7 [M + H]+ 577.2657, m/z found 577.2668.
N-Benzyl-2-[7-(dimethylamino)-1-{[3-(dimethylamino)-4-methoxyphenyl]methyl}-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (39)
To a solution of carbamate 38 (0.73 g, 1.26 mmol) in methanol (15 mL) was added sodium hydroxide solution (2N, 8 mL, 16 mmol) and the cloudy reaction heated to 50 °C overnight. The reaction was cooled, the methanol was removed under reduced pressure and the reaction was diluted with water. It was extracted three times with EtOAc then the combined extracts were dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the desired amine as a light yellow oil (0.47 g, 81%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 4H), 7.06 - 7.12 (m, 2H), 6.95 - 7.04 (m, 1H), 6.56 (dd, J = 1.74, 6.55 Hz, 2H), 6.50 (s, 1H), 6.45 (s, 1H), 4.48 (dd, J = 8.29, 15.16 Hz, 1H), 3.83 (s, 3H), 3.71 (s, 3H), 3.34 - 3.61 (m, 3H), 3.05 - 3.31 (m, 2H), 2.74 - 2.99 (m, 4H), 2.37 - 2.50 (m, 1H).
To a solution of the intermediate amine (50 mg, 0.11 mmol) and formaldehyde (37% aqueous, 81 μL, 1.09 mmol) in 1,2-dichloroethane (1 mL) was added portionwise sodium triacetoxyborohydride (230 mg, 1.09 mmol) and the reaction stirred at RT overnight. The reaction was diluted with EtOAc, washed with NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-5% methanol in dichloromethane) to give the desired product as a yellow solid (33 mg, 58%). m.p. 127-129 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.18 - 7.34 (m, 3H), 7.03 - 7.15 (m, 3H), 6.70 - 6.79 (m, 2H), 6.58 - 6.64 (m, 1H), 6.55 (s, 1H), 6.50 (s, 1H), 4.48 (dd, J = 7.91, 15.07 Hz, 1H), 3.87 (s, 3H), 3.75 (s, 3H), 3.61 - 3.72 (m, 2H), 3.35 - 3.50 (m, 1H), 3.13 - 3.34 (m, 2H), 2.79 - 3.00 (m, 4H), 2.75 (s, 6H), 2.73 (s, 6H), 2.44 - 2.57 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C31H41N4O3 [M + H]+ 517.3173, m/z found 517.3178.
N-[2-(3-Methoxy-4-nitrophenyl)ethyl]-2-(3-nitrophenyl)acetamide (40)
To a solution of 3-nitrophenylacetic acid (0.25 g, 1.38 mmol), amine 28 (0.27 g, 1.38 mmol) and BOP (0.67 g, 1.52 mmol) in dimethylformamide (7 mL) was added diisopropylethylamine (0.45 g, 0.6 mL, 3.45 mmol) and the reaction stirred at RT under N2 overnight. It was diluted with EtOAc, washed with 2N HCl, NaHCO3 solution and brine, dried over MgSO4 and the solvent was removed under reduced pressure. The crude was purified by chromatography on silica (0-100% EtOAc in hexane) to give the amide as an orange solid (0.28 g, 56%). 1H NMR (300 MHz, CHLOROFORM-d) δ 8.06 - 8.18 (m, 2H), 7.78 (s, 1H), 7.56 - 7.64 (m, 1H), 7.47 - 7.54 (m, 1H), 6.87 (d, J = 1.32 Hz, 1H), 6.78 (dd, J = 1.51, 8.29 Hz, 1H), 5.73 (br. s., 1H), 3.91 (s, 3H), 3.61 (s, 2H), 3.52 - 3.59 (m, 2H), 2.83 - 2.92 (m, 2H).
N-{2-[4-(Dimethylamino)-3-methoxyphenyl]ethyl}-2-[3-(dimethylamino)phenyl]acetamide (41)
To amide 40 (0.28 g, 0.78 mmol) in ethanol (10 mL) was added hydrazine monohydrate (0.39 g, 0.38 mL, 7.79 mmol) then Raney nickel (2800 type as a slurry in water, 56 mg). The reaction was heated to 50 °C for 60 min, cooled then filtered through Celite, rinsing thoroughly with ethanol. The solvent was removed under reduced pressure to give the aniline as a light yellow oil which was used in the next step without further purification (0.21 g, 91%). 1H NMR (300 MHz, CHLOROFORM-d) δ 7.00 - 7.14 (m, 1H), 6.33 - 6.74 (m, 6H), 5.55 (br. s., 1H), 3.79 (s, 3H), 3.31 - 3.48 (m, 4H), 2.55 - 2.70 (m, 2H).
The aniline intermediate was converted to 41, using the method for 39. Orange oil. Yield 68%. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.17 (t, J = 7.86 Hz, 1H), 6.78 (d, J = 7.91 Hz, 1H), 6.47 - 6.68 (m, 5H), 5.58 (br. s., 1H), 3.82 (s, 3H), 3.50 (s, 2H), 3.43 (q, J = 6.72 Hz, 2H), 2.93 (s, 6H), 2.75 (s, 6H), 2.67 (t, J = 6.92 Hz, 2H).
N-Benzyl-2-[7-(dimethylamino)-1-{[3-(dimethylamino)phenyl]methyl}-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]acetamide (42)
This was prepared from 41 using the procedures described for 37 and 38 to give 42 as a white solid in 4% overall yield. 1H NMR (300 MHz, CHLOROFORM-d) δ 7.16 - 7.38 (m, 4H), 7.00 - 7.15 (m, 3H), 6.47 - 6.65 (m, 5H), 4.38 - 4.54 (m, 1H), 3.80 - 3.89 (m, 3H), 3.38 - 3.73 (m, 3H), 3.07 - 3.33 (m, 2H), 2.87 (s, 6H), 2.78 - 3.00 (m, 3H), 2.74 (s, 6H), 2.68 (dd, J = 3.81, 10.60 Hz, 1H), 2.41 - 2.55 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C30H39N4O2 [M + H]+ 487.3068, m/z found 487.3092.
Methyl N-[6-methoxy-1-({4-methoxy-3-[(methoxycarbonyl)amino]phenyl}methyl)-2-{[(pyridin-3-ylmethyl)carbamoyl]methyl}-1,2,3,4-tetrahydroisoquinolin-7-yl]carbamate (43)
This was prepared using the methods for 13a and 14a starting from 37. Yellow oil. Yield 39%. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.36 - 8.55 (m, 2H), 7.91 (br. s., 1H), 7.01 - 7.23 (m, 4H), 6.85 - 6.97 (m, 1H), 6.63 - 6.73 (m, 1H), 6.59 (d, J = 4.52 Hz, 2H), 6.56 (s, 1H), 4.05 - 4.36 (m, 3H), 3.85 (s, 3H), 3.86 (s, 3H), 3.80 (s, 3H), 3.75 (s, 3H), 3.64 - 3.72 (m, 1H), 3.23 - 3.38 (m, 2H), 2.74 - 3.18 (m, 4H), 2.42 - 2.64 (m, 1H).
2-[7-(Dimethylamino)-1-{[3-(dimethylamino)-4-methoxyphenyl]methyl}-6-methoxy-1,2,3,4-tetrahydroisoquinolin-2-yl]-N-(pyridin-3-ylmethyl)acetamide (44)
This was prepared by the method used for 39 using 43. Yellow solid. Yield 35%. m.p. 68-70 °C. 1H NMR (300 MHz, CHLOROFORM-d) δ 8.48 (dd, J = 1.51, 4.71 Hz, 1H), 8.32 (d, J = 1.70 Hz, 1H), 7.40 - 7.47 (m, 1H), 7.21 (dd, J = 4.90, 7.35 Hz, 1H), 6.97 - 7.05 (m, 1H), 6.77 - 6.81 (m, 2H), 6.65 (d, J = 8.67 Hz, 1H), 6.57 (d, J = 1.88 Hz, 2H), 4.39 (dd, J = 7.91, 15.26 Hz, 1H), 3.87 (s, 3H), 3.78 (s, 3H), 3.55 - 3.64 (m, 2H), 3.40 - 3.53 (m, 1H), 3.12 - 3.32 (m, 2H), 2.81 - 2.99 (m, 4H), 2.77 (s, 6H), 2.76 (s, 6H), 2.45 - 2.56 (m, 1H). HRMS (ESI, CH3OH) m/z calcd for C30H40N5O3 [M + H]+ 518.3126, m/z found 518.3141.
Calcium Mobilization Ke Assays
Two individual stable cell lines were created by over-expressing human OX1 and OX2 receptors in CHO-RD-HGA16 (Molecular Devices) cells. The day before the assay, cells were plated into 96-well black wall/clear bottom assay plates at 25,000 cells/well in Ham’s F12 supplemented with 10% fetal bovine serum, 100 units of penicillin and streptomycin, and 100 μg/mL normocin™. The cells were incubated overnight at 37°C, 5% CO2. Prior to the assay, Calcium 5 dye (Molecular Devices) was reconstituted according to the manufacturer instructions. The reconstituted dye was diluted 1:40 in pre-warmed (37°C) assay buffer (1X HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37°C). Growth medium was removed and the cells were gently washed with 100 μL of pre-warmed (37°C) assay buffer. The cells were incubated for 45 minutes at 37°C, 5% CO2 in 200 μL of the diluted Calcium 5 dye. A single concentration of each test compound was prepared at 10× the desired final concentration in 2.5% BSA/8% DMSO/assay buffer. Serial dilutions of orexin A were prepared at 10× the desired final concentration in 0.25% BSA/1% DMSO/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37°C for 15 min. After the dye-loading incubation period, the cells were pre-treated with 25 μL of the test compounds and incubated for 15 min at 37°C. After the pre-treatment incubation period, the plate was read with a FlexStation II (Molecular Devices). Calcium-mediated changes in fluorescence were monitored every 1.52 seconds over a 60 second time period, with the FlexStation II adding 25 μL of the orexin A serial dilutions at the 19 second time point (excitation at 485 nm, detection at 525 nm). Peak kinetic reduction (SoftMax, Molecular Devices) relative fluorescent units (RFU) were plotted against the log of compound concentration. Data were fit to a three-parameter logistic curve to generate EC50 values (GraphPad Prism, GraphPad Software, Inc., San Diego, CA). Apparent Ke values were calculated using the equation Ke = [L]/((EC50+/EC50−) − 1) where [L] is the concentration of test compound, EC50+ is the EC50 of orexin A with test compound, and EC50− is the EC50 of orexin A alone. Ke values were considered valid when the EC50+/EC50− ratio was at least 4.
Kinetic Solubility
The kinetic solubility of test compounds was measured in commercial PBS, pH 7.4 consisting of potassium phosphate monobasic 1 mM, sodium phosphate dibasic 3 mM and sodium chloride 155 mM.
A 10 μL of test compound stock solution (10 mM DMSO) was combined with 490 μL of PBS buffer. The solution was agitated on a VX-2500 multi-tube vortexer (VWR) for 2 hours at room temperature. Following agitation, the sample was filtrated on a glass-fiber filter (1 μm) and the eluate was diluted 200-fold with a mixture of acetonitrile: water (1:1). On each experimental occasion, nicardipine and imipramine were assessed as reference compounds for low and high solubility, respectively. All samples were assessed in triplicate and analyzed by LC-MS/MS using electrospray ionization against standards prepared in the same matrix.
BBB Permeability
MCK-mdr1 cells at passage 5 were seeded onto permeable polycarbonate supports in 12-well Costar Transwell plates and allowed to grow and differentiate for 3 days. On day 3, culture medium (DMEM supplemented with 10% FBS) was removed from both sides of the transwell inserts and cells were rinsed with warm HBSS. After the rinse step, the chambers were filled with warm transport buffer (HBSS containing 10 mM HEPES, 0.25% BSA, pH 7.4) and the plates were incubated at 37 °C for 30 min prior to TEER (Trans Epithelial Electric Resistance) measurements.
The buffer in the donor chamber (apical side for A-to-B assay, basolateral side for B-to-A assay) was removed and replaced with the working solution (10 μM test article in transport buffer). The plates were then placed at 37 °C under light agitation. At designated time points (30, 60 and 90 min), an aliquot of transport buffer from the receiver chamber was removed and replenished with fresh transport buffer. Samples were quenched with ice-cold ACN containing internal standard and then centrifuged to pellet protein. Resulting supernatants are further diluted with 50/50 ACN/H2O (H2O only for Atenolol) and submitted for LC-MS/MS analysis. Reported apparent permeability (Papp) values were calculated from single determination. Atenolol and propranolol were tested as low and moderate permeability references. Bidirectional transport of digoxin was assessed to demonstrate Pgp activity/expression.
The apparent permeability (Papp, measured in cm/s) of a compound is determined according to the following formula:
where dQ/dt is the net rate of appearance in the receiver compartment, A is the area of the Transwell measured in cm2 (1.12 cm2), Ci is the initial concentration of compound added to the donor chamber and 60 is the conversion factor for minute to second.
Table 1.
The effect of changes at the 7-position on OX antagonism incorporating the most active 1-position analogs
| |||||||
|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | Ar | Ke (OX1, nM)b | Ke (OX2, nM)c | OX2/OX1 | clogP |
| 6 | Pr |
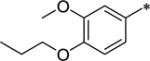
|
Ph | 16.1 ± 3.6 | >10,000 | >621 | 6.86 |
| 12a | Pr |
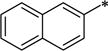
|
Ph | 121 ± 35 | a | >82.6 | 7.29 |
| 12b | Pr |
|
Ph | 28.8 ± 4.4 | a | >347 | 6.98 |
| 12c | Pr |
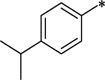
|
Ph | 141 ± 42 | a | >70.9 | 7.40 |
| 12d | Pr |
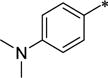
|
Ph | 173 ± 5 | >10,000 | >58 | 6.17 |
| 12f | Pr |
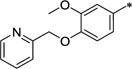
|
Ph | 20.6 ± 2.8 | a | >485 | 6.17 |
| 12g | Pr |
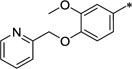
|
Ph | 96.4 ± 34 | >10,000 | >104 | 5.96 |
| 15a | CF3CH2 |
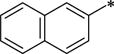
|
3-Py | 29.5 ± 2.2 | 1750 ± 230 | 59 | 5.70 |
| 15b | CF3CH2 |
|
3-Py | 20.4 ± 2.9 | 1360 ± 150 | 67 | 5.39 |
| 19a | CF3CH2 |
|
Ph | 884 ± 70d | a | >11 | 4.47 |
| 19b | CF3CH2 |
|
Ph | >10,000d | a | 0 | 4.47 |
| 19c | CF3CH2 |
|
Ph | 1,860 ± 140d | a | >5.4 | 4.47 |
| 25a | MeSO2 |
|
Ph | 43.0 ± 18 | a | >232 | 5.25 |
| 25b | MeSO2 |
|
Ph | 5.24 ± 1.0 | 1,200 ± 360 | 229 | 5.19 |
< 50% inhibition at 10 μM final concentration;
Values are the mean of at least three independent experiments in duplicate;
Values are the mean of at least two independent experiments in duplicate; for compounds with Ke < 100 nM at OX1 at least three independent experiments in duplicate were performed.
Compound appeared insurmountable during standard assay procedure, so incubation was increased to 1 hour.
Acknowledgments
We are grateful to National Institute on Drug Abuse, National Institutes of Health, U.S. (Grants DA032837 and DA040693 to Y.Z.) for the financial support of this research. We thank Tiffany Langston, Keith Warner, Allyson Smith, and Dr. Elaine Gay for technical assistance and Dr. Thuy Nguyen for the suggestions on manuscript preparation.
ABBREVIATIONS
- BOP
(Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HBTU
O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate
- HPLC
high performance liquid chromatography
- OX1
orexin-1 receptor
- OX2
orexin-2 receptor
- SAR
structure-activity relationship
- TLC
thin layer chromatography
- m.p
melting point
- MDCK
Madin-Darby Canine Kidney
- ADME
absorption, distribution, metabolism, and excretion
Footnotes
Supporting Information. HPLC analysis of target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
ORCID: Yanan Zhang: 0000-0001-7153-4358
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- 2.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsujino N, Sakurai T. Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 2009;61:162–176. doi: 10.1124/pr.109.001321. [DOI] [PubMed] [Google Scholar]
- 4.Sakurai T. The role of orexin in motivated behaviours. Nat Rev Neurosci. 2014;15:719–731. doi: 10.1038/nrn3837. [DOI] [PubMed] [Google Scholar]
- 5.Roecker AJ, Coleman PJ. Orexin receptor antagonists: medicinal chemistry and therapeutic potential. Curr Top Med Chem. 2008;8:977–987. doi: 10.2174/156802608784936746. [DOI] [PubMed] [Google Scholar]
- 6.Boss C, Brisbare-Roch C, Jenck F. Biomedical application of orexin/hypocretin receptor ligands in neuroscience. J Med Chem. 2009;52:891–903. doi: 10.1021/jm801296d. [DOI] [PubMed] [Google Scholar]
- 7.Gatfield J, Brisbare-Roch C, Jenck F, Boss C. Orexin receptor antagonists: a new concept in CNS disorders? ChemMedChem. 2010;5:1197–1214. doi: 10.1002/cmdc.201000132. [DOI] [PubMed] [Google Scholar]
- 8.Kodadek T, Cai D. Chemistry and biology of orexin signaling. Mol Biosyst. 2010;6:1366–1375. doi: 10.1039/c003468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scammell TE, Winrow CJ. Orexin receptors: pharmacology and therapeutic opportunities. Annu Rev Pharmacol Toxicol. 2011;51:243–266. doi: 10.1146/annurev-pharmtox-010510-100528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang LP. Suvorexant: First Global Approval. Drugs. 2014 doi: 10.1007/s40265-014-0294-5. [DOI] [PubMed] [Google Scholar]
- 11.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nambu T, Sakurai T, Mizukami K, Hosoya Y, Yanagisawa M, Goto K. Distribution of orexin neurons in the adult rat brain. Brain Res. 1999;827:243–260. doi: 10.1016/s0006-8993(99)01336-0. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Hu Z, de Lecea L. The hypocretins/orexins: integrators of multiple physiological functions. Br J Pharmacol. 2014;171:332–350. doi: 10.1111/bph.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nature Neurosci. 2014;17:1298–1303. doi: 10.1038/nn.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baimel C, Bartlett SE, Chiou LC, Lawrence AJ, Muschamp JW, Patkar O, Tung LW, Borgland SL. Orexin/hypocretin role in reward: implications for opioid and other addictions. Br J Pharmacol. 2015;172:334–348. doi: 10.1111/bph.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G. Multiple roles for orexin/hypocretin in addiction. Prog Brain Res. 2012;198:79–121. doi: 10.1016/B978-0-444-59489-1.00007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- 18.Boutrel B, Kenny PJ, Specio SE, Martin-Fardon R, Markou A, Koob GF, de Lecea L. Role for hypocretin in mediating stress-induced reinstatement of cocaine-seeking behavior. Proc Natl Acad Sci U S A. 2005;102:19168–19173. doi: 10.1073/pnas.0507480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou L, Ghee SM, Chan C, Lin L, Cameron MD, Kenny PJ, See RE. Orexin-1 receptor mediation of cocaine seeking in male and female rats. J Pharmacol Exp Ther. 2012;340:801–809. doi: 10.1124/jpet.111.187567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lawrence AJ, Cowen MS, Yang HJ, Chen F, Oldfield B. The orexin system regulates alcohol-seeking in rats. Br J Pharmacol. 2006;148:752–759. doi: 10.1038/sj.bjp.0706789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards JK, Simms JA, Steensland P, Taha SA, Borgland SL, Bonci A, Bartlett SE. Inhibition of orexin-1/hypocretin-1 receptors inhibits yohimbine-induced reinstatement of ethanol and sucrose seeking in Long-Evans rats. Psychopharmacology. 2008;199:109–117. doi: 10.1007/s00213-008-1136-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plaza-Zabala A, Flores A, Martin-Garcia E, Saravia R, Maldonado R, Berrendero F. A role for hypocretin/orexin receptor-1 in cue-induced reinstatement of nicotine-seeking behavior. Neuropsychopharmacology. 2013;38:1724–1736. doi: 10.1038/npp.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter RA, Chan WN, Coulton S, Johns A, Hadley MS, Widdowson K, Jerman JC, Brough SJ, Coldwell M, Smart D, Jewitt F, Jeffrey P, Austin N. 1,3-Biarylureas as selective non-peptide antagonists of the orexin-1 receptor. Bioorg Med Chem Lett. 2001;11:1907–1910. doi: 10.1016/s0960-894x(01)00343-2. [DOI] [PubMed] [Google Scholar]
- 24.McElhinny CJ, Jr, Lewin AH, Mascarella SW, Runyon S, Brieaddy L, Carroll FI. Hydrolytic instability of the important orexin 1 receptor antagonist SB-334867: possible confounding effects on in vivo and in vitro studies. Bioorg Med Chem Lett. 2012;22:6661–6664. doi: 10.1016/j.bmcl.2012.08.109. [DOI] [PubMed] [Google Scholar]
- 25.Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ. International Union of Basic and Clinical Pharmacology. LXXXVI Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev. 2012;64:389–420. doi: 10.1124/pr.111.005546. [DOI] [PubMed] [Google Scholar]
- 26.Lebold TP, Bonaventure P, Shireman BT. Selective orexin receptor antagonists. Bioorg Med Chem Lett. 2013;23:4761–4769. doi: 10.1016/j.bmcl.2013.06.057. [DOI] [PubMed] [Google Scholar]
- 27.Gozzi A, Turrini G, Piccoli L, Massagrande M, Amantini D, Antolini M, Martinelli P, Cesari N, Montanari D, Tessari M, Corsi M, Bifone A. Functional magnetic resonance imaging reveals different neural substrates for the effects of orexin-1 and orexin-2 receptor antagonists. PLoS One. 2011;6:e16406. doi: 10.1371/journal.pone.0016406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiner MA, Gatfield J, Brisbare-Roch C, Dietrich H, Treiber A, Jenck F, Boss C. Discovery and Characterization of ACT-335827, an Orally Available, Brain Penetrant Orexin Receptor Type 1 Selective Antagonist. ChemMedChem. 2013;8:898–903. doi: 10.1002/cmdc.201300003. [DOI] [PubMed] [Google Scholar]
- 29.Jiang R, Song X, Bali P, Smith A, Bayona CR, Lin L, Cameron MD, McDonald PH, Kenny PJ, Kamenecka TM. Disubstituted piperidines as potent orexin (hypocretin) receptor antagonists. Bioorg Med Chem Lett. 2012;22:3890–3894. doi: 10.1016/j.bmcl.2012.04.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khoo SY, Brown RM. Orexin/hypocretin based pharmacotherapies for the treatment of addiction: DORA or SORA? CNS drugs. 2014;28:713–730. doi: 10.1007/s40263-014-0179-x. [DOI] [PubMed] [Google Scholar]
- 31.Yeoh JW, Campbell EJ, James MH, Graham BA, Dayas CV. Orexin antagonists for neuropsychiatric disease: progress and potential pitfalls. Front Neurosci. 2014;8:36. doi: 10.3389/fnins.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perrey DA, German NA, Gilmour BP, Li JX, Harris DL, Thomas BF, Zhang Y. Substituted tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor. J Med Chem. 2013;56:6901–6916. doi: 10.1021/jm400720h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrey DA, Gilmour BP, Thomas BF, Zhang Y. Toward the Development of Bivalent Ligand Probes of Cannabinoid CB1 and Orexin OX1 Receptor Heterodimers. ACS Med Chem Lett. 2014;5:634–638. doi: 10.1021/ml4004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perrey DA, German NA, Decker AM, Thorn D, Li JX, Gilmour BP, Thomas BF, Harris DL, Runyon SP, Zhang Y. Effect of 1-substitution on tetrahydroisoquinolines as selective antagonists for the orexin-1 receptor. ACS Chem Neurosci. 2015;6:599–614. doi: 10.1021/cn500330v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrey DA, Decker AM, Li JX, Gilmour BP, Thomas BF, Harris DL, Runyon SP, Zhang Y. The importance of the 6- and 7-positions of tetrahydroisoquinolines as selective antagonists for the orexin 1 receptor. Biorg Med Chem. 2015;23:5709–5724. doi: 10.1016/j.bmc.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy KA, Brodnik ZD, Shaw JK, Perrey DA, Zhang Y, Espana RA. Hypocretin receptor 1 blockade produces bimodal modulation of cocaine-associated mesolimbic dopamine signaling. Psychopharmacology. 2017 doi: 10.1007/s00213-017-4673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di L, Kerns EH. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization. Academic Press; 2010. [Google Scholar]
- 38.Perrey DA, Gilmour BP, Runyon SP, Thomas BF, Zhang Y. Diaryl urea analogues of SB-334867 as orexin-1 receptor antagonists. Bioorg Med Chem Lett. 2011;21:2980–2985. doi: 10.1016/j.bmcl.2011.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rankovic Z. CNS drug design: balancing physicochemical properties for optimal brain exposure. J Med Chem. 2015;58:2584–2608. doi: 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
- 40.Wenlock MC, Austin RP, Barton P, Davis AM, Leeson PD. A comparison of physiochemical property profiles of development and marketed oral drugs. J Med Chem. 2003;46:1250–1256. doi: 10.1021/jm021053p. [DOI] [PubMed] [Google Scholar]
- 41.Kuteykin-Teplyakov K, Luna-Tortos C, Ambroziak K, Loscher W. Differences in the expression of endogenous efflux transporters in MDR1-transfected versus wildtype cell lines affect P-glycoprotein mediated drug transport. Br J Pharmacol. 2010;160:1453–1463. doi: 10.1111/j.1476-5381.2010.00801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]