

Abstract
Septins self-assemble into heteromeric rods and filaments to act as scaffolds and modulate membrane properties. How cells tune the biophysical properties of septin filaments to control filament flexibility and length, and in turn the size, shape, and position of higher-order septin structures, is not well understood. We examined how rod composition and nucleotide availability influence physical properties of septins such as annealing, fragmentation, bundling, and bending. We found that septin complexes have symmetric termini, even when both Shs1 and Cdc11 are coexpressed. The relative proportion of Cdc11/Shs1 septin complexes controls the biophysical properties of filaments and influences the rate of annealing, fragmentation, and filament flexibility. Additionally, the presence and apparent exchange of guanine nucleotide also alters filament length and bundling. An Shs1 mutant that is predicted to alter nucleotide hydrolysis has altered filament length and dynamics in cells and impacts cell morphogenesis. These data show that modulating filament properties through rod composition and nucleotide binding can control formation of septin assemblies that have distinct physical properties and functions.
INTRODUCTION
Septins are filament-forming proteins that belong to a family of P-loop GTPases and are conserved from yeast to humans. A key feature of the septin cytoskeleton is that filaments form from multimeric complexes that show plasticity in composition depending on cell type or developmental stage. In yeast, four septins form an octameric complex where the terminal position can be occupied by two different septins: Cdc11 and Shs1 (Garcia et al., 2011). Multiple distinct septin complexes also coexist in a mammalian cell with different subunit combinations and splice variants possible (Sellin et al., 2011). What is the functional consequence of this compositional heterogeneity on septin form and function?
In principal, varying the mixtures of heteromeric complexes could change the material properties of filaments, the subcellular localization, and/or interaction partners. Mixing different proportions of Shs1 and Cdc11 complexes can give rise to different ultrastructures in vitro (Garcia et al., 2011). Different septin heteromers also generate diverse higher-order structures in Aspergillus nidulans (Hernández-Rodríguez et al., 2014). Similarly, SEPT9 presence in complexes promotes different higher-order structures that function at different times and places in mammalian cells (Kim et al., 2011). Despite clear evidence of functional differences, little is known about how septin complex composition gives rise to these differences.
Recent evidence shows that the septin subunit order and oligomer composition is controlled by GTP binding and hydrolysis rates (Weems and McMurray, 2017). Other studies suggest GTP binding/hydrolysis may also be important for the formation of higher-order septin structures both in vitro and in vivo (Versele et al., 2004; Huang et al., 2006; Nagaraj et al., 2008; Weems et al., 2014; Akhmetova et al., 2015). However, there is little evidence of nucleotide hydrolysis by the septin complex over one yeast cell cycle, raising the issue of whether GTP plays a role beyond a structural one in controlling filament dynamics (Vrabioiu et al., 2004). We examine how septin complex composition and nucleotide state affects filament properties to gain insight into how cells may produce structurally and functionally distinct higher-order septin structures.
RESULTS AND DISCUSSION
Analysis of septin complex formation with coexpressed terminal subunits
In Saccharomyces cerevisiae, two septin subunits, Cdc11 and Shs1, can occupy the terminal position in a heterooctamer. The relative abundance of complexes with different terminal septins changes the size and shape of septin ultrastructures in vitro (Garcia et al., 2011). The overall Cdc11:Shs1 ratio is ∼1:1 in yeast, although the composition and relative abundance of septin complexes has not been directly measured (Chong et al., 2015). In fact, there are many possible combinations of complexes (Figure 1A).
FIGURE 1:

Symmetric rod composition in vitro and in vivo. (A) Schematic depiction of possible rod combinations divided into two symmetry-based classes. (B) Rod length distributions from negative stain TEM images (n ≥ 400 rods) of septins in 300 mM salt. (C) Average intensity distribution for single particles over time using complexes formed when Cdc11 and Shs1 are coexpressed recombinantly. (D) Two-step photobleaching profiles of Cdc11-SNAP and Shs1-GFP particles. (E) Frequency of homotypic and heterotypic particles among purified septin complexes. (F) Average intensity distributions for particles pulled down by α-GFP from yeast extract containing Cdc11-GFP and Shs1-mCherry. (G) A two-step photobleaching profile of particles in F. (H) Frequency of green (Cdc11-GFP), red (Shs1-mCherry), and yellow (Cdc11-GFP + Shs1-mCherry) particles for extract containing Cdc11-GFP and Shs1-mCherry compared with His-GFP extract as a negative control. In C and F, X’s represent particles containing signal in both channels.
We first investigated what complexes are built when Cdc11 and Shs1 are coexpressed. To measure the size distribution of complexes, we coexpressed four (Cdc10, Cdc3, Cdc12, and Cdc11) or five (Cdc10, Cdc3, Cdc12, Cdc11, and Shs1) septins, purified from Escherichia coli and visualized via negative stain transmission electron microscopy (TEM). Longer complexes were observed when both Cdc11 and Shs1 were expressed as opposed to Cdc11 only (p < 0.005, Kolmogorov–Smirnov [KS] test). However, the mode for both distributions remained at 32 nm, the length of an octamer (Figure 1B). Thus, although longer complexes can form, the majority of rods are octameric when Shs1 and Cdc11 are coexpressed.
To determine whether octamers are symmetric or asymmetric (Figure 1A, class 1 vs. class 2), we analyzed single septin complexes via total internal reflection fluorescence (TIRF) microscopy. Purified complexes containing Cdc11-SNAP and Shs1-GFP were added at very low concentrations (pM) to detect single particles. Signal intensity analysis revealed two distinct complex populations containing either Shs1-GFP or Cdc11-SNAP but none with both signals (Figure 1C). Two-step bleaching profiles confirmed that each complex contained two Cdc11-SNAP or two Shs1-GFP molecules (Figure 1D). A very small population of particles contained both Shs1-GFP and Cdc11-SNAP signal that was difficult to distinguish from noise (Figure 1, C and E). These data suggest that when Cdc11 and Shs1 are coexpressed, septin rods form two distinct, symmetric complexes with either Cdc11 or Shs1 present.
To determine the composition of native yeast complexes, we used single-molecule pull down (SiMPull) on extracts from cells expressing both Cdc11-GFP and Shs1-mCherry (Jain et al., 2009). Anti-GFP antibodies were immobilized on a coverslip and captured puncta were analyzed for mCherry signal. Shs1-mCherry signals were not detected at the same positions as Cdc11-GFP signals, suggesting that two distinct populations exist in yeast cytoplasm containing either Cdc11 or Shs1 (Figure 1F). Bleach step profiles revealed two Cdc11-GFP molecules per punctum consistent with rods containing Cdc11 on both ends (Figure 1G). Puncta detectable in the red channel generally did not colocalize with Cdc11 signal and were indistinguishable from noise when compared with extract containing unlabeled septins and monomeric eGFP (Figure 1H). It is possible that the extremely low abundance puncta with both colors may represent a composition present in vivo, but it would be a rare, minority population. Therefore, yeast septins likely form exclusively symmetric complexes both in vitro and in vivo even in the presence of alternate subunits.
Symmetric rod formation suggests a mechanism to coordinate the complex composition across tens of nanometers. How heteromeric complexes of these dimensions assemble so that the terminal positions “match” is challenging to envision. One explanation would be that subunits could be locally translated with cotranslational complex assembly. Evidence supports coordinated septin translation in Ustilago maydis, where septin mRNA is transported and translated on endosomes (Baumann et al., 2014; Zander et al., 2016) and is supported by heterokaryon experiments in yeast (Weems et al., 2014). Alternatively, allostery or some type of assembly selection may bias complex composition. Cdc3/Cdc12 nucleotide state may bias its incorporation with Cdc10 core dimers to promote symmetry, or the Cdc12 nucleotide state conformation can propagate through the oligomer to coordinate the termini states prior to Cdc11 or Shs1 binding.
Shs1 changes the biophysical properties of septin filaments
What is the consequence of having two pools of septin complexes with different termini? Genetic and FRET-based assays have shown that Shs1 complexes do not polymerize with other Shs1 complexes (Finnigan et al., 2015; Booth and Thorner, 2016). Additionally, changing the ratio of Cdc11:Shs1 complexes changes higher-order structure size in vitro (Garcia et al., 2011). We investigated how the ratio of Cdc11- to Shs1-containing rods affects biophysical properties of septin filaments such as filament length, flexibility, annealing and fragmentation events, and bundling.
On supported lipid bilayers (SLBs), filament length was inversely related to Shs1-complex abundance (Figure 2A and Supplemental Videos 1 and 2). High Shs1 concentrations led to more dynamic filaments with increased annealing and fragmentation event frequencies (Figure 2, B and C). The septin filament persistence length also decreased (i.e., increased flexibility) with increasing proportions of Shs1 rods (Figure 2E). The flexural rigidity differences may reflect affinity differences between Shs1-Cdc11 and Cdc11-Cdc11 interactions (Booth et al., 2015). Previous studies have shown that Shs1-containing complexes can associate laterally, whereas Cdc11-Cdc11 interactions are end on (Garcia et al., 2011; Bridges et al., 2014). In our experiments, bundling/unbundling events increased with Shs1 proportion (Figure 2D). The orientation or strength of Shs1-Cdc11 interactions may promote filament flexibility compared with end-on Cdc11-Cdc11 interactions (Kaplan et al., 2015).
FIGURE 2:

Ratio of Cdc11 and Shs1 complexes modulates septin filament properties. (A) Septin filament length, (B) fragmentation/length, (C) annealing events/length, and (D) bundling and unbundling per unit length with varying Cdc11:Shs1 ratios. (A) n > 250 filaments and (B–D) n ≥ 49 filaments for each ratio; error bars represent SE. *p < 0.05, **p < 0.005, KS test, arrowheads represent measured events. (E) Filament persistence length decreases from a median of 6.4 μm for Cdc11 only to 2.4 μm for 1:1 Cdc11:Shs1 ratio filaments (n ≥ 15 filaments). (F) Septin adsorption on different bead sizes at different Cdc11:Shs1 ratios using a total of 100 nM protein (n ≥ 50, p < 0.005, Dunn test). (G) Fold difference heat map of septin adsorption to different bead sizes with different Cdc11:Shs1 ratios, normalized to the lowest septin adsorption. (H) Shs1 complexes show curvature preference when recruited via NTA-Ni lipids using the His-tag (p < 0.005, Dunn test).
Video S1.
Filaments formed on a SLB by addition of 5 nM total protein containing Cdc11 only complexes, corresponding to Figure 2. Time lapse was acquired via TIRF microscopy with a 100 ms exposure and a 250 ms interval.
Video S2.
Filaments formed on a SLB by mixing Cdc11 and Shs1 complexes in a 1:1 ratio to a final protein concentration of 5 nM, corresponding to Figure 2. Video was acquired with a 100 ms exposure and 250 ms time interval.
Next we found that Shs1:Cdc11 ratios do not alter the micron-scale curvature preference of septins (Figure 2F) (Bridges et al., 2016). Notably, however, increasing Shs1 abundance increased total septin adsorption on beads of all sizes tested (Figure 2, F and G). This increase may arise due to differential filament packing on a curved surface, facilitated by Shs1. Shs1 complexes alone showed minimal binding at all curvatures (Figure 2, F and G), consistent with previous work showing polymerization is required for stable association to curved surfaces (Finnigan et al., 2015; Booth and Thorner, 2016; Bridges et al., 2016). Shs1 complexes did display a curvature preference when recruited artificially via the His-tag and Ni2+-NTA modified lipids (Figure 2H). Thus, Shs1 influences the flexibility, assembly, disassembly, and bundling of filaments while not changing micron-scale curvature preference.
These results suggest that varying septin complex proportions is a mechanism to tune septin biophysical properties. While Shs1 and Cdc11 are expressed approximately stoichiometrically in yeast cells, locally controlling Cdc11 and Shs1 complex ratios within the septin structure can be used to generate filaments with distinct properties and diversity in structures.
Magnesium and guanine nucleotides promote septin filament bundles
A second prominent feature of septins that could influence filament properties is the presence and state of the guanine nucleotide. Septin complexes purified from E. coli, yeast, or mammalian cells contain a mixture of guanosine triphosphate (GTP) and guanosine diphosphate (GDP) (Field, 1996; Vrabioiu et al., 2004). Recent work illuminated a key role of nucleotide state for directing the assembly order of yeast septin complexes (Weems and McMurray, 2017). It is unclear whether nucleotide exchange/hydrolysis influences filament properties that arise after complex formation. We assessed polymerization of Cdc11-containing complexes in the presence of nucleotide and magnesium to stabilize bound nucleotide (Sirajuddin et al., 2009). Strikingly, Mg2+ alone causes septin filaments to bundle at physiological ion concentrations (1–3 mM) in both the presence and absence of an anionic lipid (75% PC, 25% PI) SLB (Figure 3, A and B). Intensity analysis of filaments formed in the presence of Mg2+ on SLBs shows that they are twice as bright as single filaments suggesting filament pairing. We suspect that, similarly to F-actin, Mg2+ promotes bundling by shielding negative charge and reducing repulsion between actin filaments (Tang and Janmey, 1996). Alternatively, Mg2+ binding may displace bound GTP to a more surface exposed position and provide a new interface for filament bundling, similarly to FtsZ (Marrington et al., 2004).
FIGURE 3:

Mg2+ and GTP/GDP promote septin filament bundling. (A) Mg2+ causes septin filament bundling on SLBs and PEG. The 5 nM (on SLB) and 100 nM (on PEG) septins were allowed to polymerize with and without Mg2+. (B) Filaments formed in the presence of Mg2+ on SLBs were 2× brighter than filaments formed in the absence of Mg2+. (C) Septins show bundling with GTP and GDP on SLBs. (D) Septins form long stable filaments in the presence of GTP and GDP on PEG-coated coverslips. Insets show enhance contrasted filaments.
To disentangle the bundling effect of Mg2+ from a nucleotide state, we modified nucleotide exchange protocols for septins (Sheffield et al., 2003; Huang et al., 2006). Septins were incubated with excess nucleotide in the presence of 1 mM Mg2+ before dilution to 20 μM Mg2+ and low salt to promote polymerization. Under these conditions, we observed normal, flexible filaments without additional nucleotide but substantial bundling in the presence of GTP or GDP. Notably, ATP did not promote bundling (Figure 3C). No differences were observed between GTP and GDP, suggesting nucleotide binding, rather than hydrolysis, promotes lateral interactions. As no bundles were detected without pretreatment to promote the apparent exchange, increased bundling may be due to incorporation or possibly exchange of previously bound nucleotide. Bundling was nonuniform throughout the membrane, possibly due to slow nucleotide turnover and Mg2+ requirement for GTP binding. These data suggest that GTP/GDP can produce stiff filaments with increased lateral associations (Supplemental Video 3).
Video S3.
Filaments formed in absence or presence of nucleotide using 5 nM protein and 5 μM nucleotide, corresponding to Figure 3.
Guanine-nucleotide induced bundles were longer, thicker, and sequestered more protein when septins were polymerized in solution and visualized on polyethylene glycol (PEG)-treated glass instead of assembling on a SLB (Figure 3D). This raises the possibility of a common interface between membrane binding and filament-filament interactions, indicating possible competition between these two types of interactions. Some cells normally have septin bundles, including nuclear-associated septins in mammalian cells, assemblies in yeast mating projections, and bars that specify septation in filamentous fungi (Ford and Pringle, 1991; Alvarez-Tabarés and Pérez-Martín, 2010). Previous work has also implicated the G-domain in septin filament lateral interactions (Bertin et al., 2010). It is difficult to know whether these bundles seen in vitro mimic those found in cells, and how nucleotide state, interacting proteins, and membrane binding affects septin bundling in vivo requires further study.
Nucleotide pocket mutants show increased lateral interactions
We further examined the role of nucleotide binding in septin filament assembly by generating several mutants predicted to alter nucleotide state. A predicted hydrolysis-defective Cdc12 mutant (T75A) and a putative gain-of-hydrolysis Cdc11 mutant (D65T) were generated based on structural studies and sequence homology (Sirajuddin et al., 2009). The mutants showed no complex formation defects (Figure 4A). Two other nucleotide pocket mutants (Cdc11 R35E and Cdc12 G247E) showed complex formation defects and did not form filaments (unpublished data). Both the Cdc11 D65T and Cdc12 T75A complexes formed small, bundled filaments in addition to the unbundled filaments typical of yeast septins on SLBs. The frequency of bundles was higher in Cdc11 D65T compared with Cdc12 T75A or when the two mutations were combined (Figure 4B). As for nucleotide-exchanged conditions, bundling was substantially enhanced when septins were polymerized in solution (Figure 4D). The majority of Cdc11 D65T filaments had four- to fivefold greater intensity (or “bundling factor”) compared with the wild type (WT). A large population of Cdc12 T75A and the double mutant displayed a bundling factor of 1–2× compared with the WT (Figure 4E). Consistent with increased lateral interactions, filament flexibility decreased, with Cdc11 D65T displaying the highest rigidity corresponding to the most bundled filaments (Figure 4F). Remarkably, the double mutant protein showed a less severe phenotype than Cdc11 D65T alone, possibly due to increased compatibility of the mutant G-domain conformations (Weems et al., 2014). It is possible that these conformational changes are a result of the residue mutations instead of nucleotide state changes. These data suggest that changing the state of the bound nucleotide by different septin subunits may cause bundling through conformational changes.
FIGURE 4:

Nucleotide binding pocket mutants show an increase in bundling. (A) Coomassie-stained SDS gel showing normal complex formation by the mutants. (B) The 5 nM septins polymerized on an SLB. Mutants form bundled filaments. (C) The 100 nM septins polymerized on a PEG surface. Mutants display increased lateral interactions in absence of a membrane. Insets show enhance contrasted filament. (D) Intensity distribution of filaments formed on PEG surface. (E) Bundling factor of mutants polymerized on PEG compared with the WT. Percentage frequency captures filaments displaying at least a bundling of factor x. (F) Persistence length of WT and nucleotide mutant filaments (n ≥ 20 filaments, **p < 0.005, KS test).
Changing Shs1 proportion in branch collars results in polarity defects
A prediction of these in vitro experiments is that cells may be able to control features of higher-order assembly based on the relative composition of complexes of different identities and/or local nucleotide exchange. Ashbya gossypii forms spatially distinct septin structures that form at sites of different cellular curvatures (DeMay et al., 2009; Bridges et al., 2016) and, therefore, serves as a backdrop to examine how cells locally exploit different ratios of distinct complexes. Indeed, unlike in S. cerevisiae, Shs1 is essential for spore production and/or viability, suggesting that modulating this terminal position is critical in these more morphologically complex cells (unpublished data). Ashbya gossypii cells predominantly contain two types of higher-order septin structures: interregion rings and branch-associated collars. We found that although Cdc11 was generally more abundant than Shs1 in higher-order structures, the ratio of Shs1/Cdc11 appeared the same in both types of assemblies (Figure 5A).
FIGURE 5:

Analysis of Shs1:Cdc11 ratios and function of nucleotide state in A. gossypii. (A) Cdc11 and Shs1 intensities in different higher-order septin structures. (B) Sequence alignment showing conserved serine residue in different fungi (arrow). (C) Western blot of A. gossypii extract showing total Shs1 and Cdc11 levels. (D) GFP fluorescence intensity in interregion rings (n ≥ 35) and branch collars (n ≥ 80) for Shs11-GFP and shs1 S35V-GFP. (E) shs1 S35V-GFP has slower dynamics but higher mobile fraction than Shs1-GFP at branch collars (n ≥ 8 rings). (F) Representative images of Shs1-GFP and shs1 S35V-GFP. (G) Interbranch distance in shs1-GFP and shs1 S35V-GFP cells (n = 108 branches). *p < 0.05, **p < 0.005, KS test.
To examine the effect of nucleotide, we mutated a serine residue in Shs1 (S35V), predicted to be important for GTP binding, and conserved in other fungi but not in S. cerevisiae (Figure 5B). The conservation suggests an important nucleotide-related function that may have been lost and not needed in budding yeast septin assembly. The mutant expression level was comparable to WT Shs1 and did not alter Cdc11 abundance (Figure 5C). Interestingly, we detected no inter-region ring intensity differences between Shs1-GFP and shs1 S35V-GFP but observed a significant decrease in shs1 S35V-GFP incorporation at branch collars compared with Shs1-GFP (Figure 5D, p < 0.005 KS test). FRAP experiments demonstrated that Shs1-GFP S35V recovered more slowly with a higher mobile fraction than Shs1-GFP (Figure 5E), suggesting that oligomeric interactions and/or curved membrane affinity is perturbed. We also observed branch formation defects, revealed by increased interbranch distance (Figure 5, F and G). Although the Cdc11:Shs1 ratio is comparable in different septin structures in WT cells, the nucleotide mutant selectively compromised Shs1 incorporation into branch collars and altered cell morphology.
We show that septin biophysical properties can be modulated by varying the incorporation of different types of complexes and the available nucleotide. Changing the Cdc11:Shs1 complex ratio can control filament length, flexibility, and dynamics and that nucleotide can influence filament bundling capacity. This work indicates multiple factors contribute to septin filament biophysical properties, and these help to explain the diverse septin structures found inside cells.
MATERIALS AND METHODS
Protein expression and purification
BL21 (DE3) E. coli cells were transformed with duet septin expression plasmids immediately prior to production. Protein expression was induced by adding 1 mM isopropyl β-d-1-thiogalactopyranoside at OD600 0.6–0.7 at 22°C for 24 h with shaking. Cells were then harvested by centrifugation and pellets were either stored at -80°C until lysis or lysed immediately. Cells were incubated with lysis buffer (50 mM KH2PO4, pH 8.0, 1 M KCl, 1 mM MgCl2, 1% Tween-20, 10% glycerol, 1× protease inhibitor [Roche], and 20 mM imidazole) and 1 mg/ml lysozyme for 30 min on ice. Cells were then sonicated twice for 20 s each, and the lysate was collected by spinning at 4°C for 30 min at 15,000 rpm using a SS-34 rotor in a Sorvall RC-6 centrifuge. The clarified lysate was filtered through a 0.45-μm filter before loading onto an equilibrated Ni2+-NTA agarose (Thermo Scientific) column. Bound protein was washed 3 times with wash buffer (50 mM KH2PO4, pH 8.0, 1 M KCl, and 20 mM imidazole) before elution by high imidazole buffer (50 mM KH2PO4, pH 8.0, 1 M KCl, and 500 mM imidazole). Protein was then dialyzed into septin storage buffer (50 mM Tris, pH 8.0, 300 mM KCl, and 1 mM β-mercaptoethanol) by using a 100,000 MW cutoff dialysis cassette (Thermo Fisher Scientific). Where applicable, the 6XHIS tag on Cdc12 was subsequently cleaved using ProTEV Plus Protease (Promega), and the protein was run over the Ni2+-NTA column to remove the cleaved 6XHIS tag. Protein was again dialyzed into septin storage buffer. Purity was analyzed by SDS gel electrophoresis, and the total protein concentration was determined using Bradford reagent.
Transmission electron microscopy
After purification, septin complexes were diluted to a concentration of 5 μg/ml in septin storage buffer. The diluted samples were adsorbed onto Formvar carbon coated grids (Ted Pella) after glow discharge. The grids were then washed twice with water and stained with 2% uranyl acetate.
Electron micrographs of the prepared samples were taken using an electron microscope (JEOL 1230 TEM) operated at 120 kV. Micrographs were acquired at 150,000× magnification using a charge-coupled device (CCD) camera (SC1000; Gatan Orius). Length measurements were performed using Fiji (Schindelin et al., 2012).
Single-molecule analysis using recombinant protein
Recombinantly expressed proteins were diluted to pM concentrations before being immobilized on a clean glass surface. Glass coverslips were prepared by sequential sonication steps of 10 mins each in 3 N KOH, 100% ethanol, and double-distilled water (ddH2O). The coverslips were washed with ddH2O five times between each step. Coverslips were then dried and plasma cleaned for 15 min. Plastic chambers were glued onto coverslips. Septins were diluted in buffer containing 50 mM Tris, pH 8, 300 mM KCl, and 1 mM β-mercaptoethanol. The chamber was washed once with buffer containing 50 mM Tris, pH 8, 300 mM KCl, 1 mg/ml BSA, and 1 mM β-mercaptoethanol before adding septins to a final pM concentration in the chamber. The protein was allowed to settle for 5 min before imaging.
Single-molecule pull down on yeast extract
Single-molecule pull down (SiMPull) was performed as previously described (Jain et al., 2009). Yeast extracts were prepared by growing 1 l of S. cerevisiae cells to an OD of 0.6–0.7 in yeast peptone dextrose (YPD) medium with selection. Cells were then spun down at 5000 × g for 15 min, and the pellets were resuspended in 1 ml of YPD. The yeast cells were then flash frozen in liquid nitrogen and ground using a coffee grinder. The ground paste was suspended in lysis buffer containing 50 mM HEPES, pH 7.6, 1 mM KCl, 1 mM EGTA (ethylene glycol tetraacetic acid), 5% glycerol, 0.01% Tween, and 1× protease inhibitor mix. Bradford reagent was used to determine protein concentration of the lysates. A goat α-rabbit biotin conjugated (Invitrogen) secondary antibody was immobilized on biotin PEG-coated coverslips. Rabbit α-GFP (Invitrogen) was used to recruit Cdc11-GFP from the extracts. Single molecules were imaged using time-lapse acquisition with an exposure time and frame rate of 200 ms.
Supported lipid bilayer preparation
Glass coverslips were cleaned and SLBs were prepared with 75% (mol%) 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC; Avanti Polar Lipids 850375) and 25% (mol%) l-α-phosphatidylinositol (liver, bovine; Avanti Lipids 840042) as described previously (Bridges et al., 2014). Proteins were added to a final concentration of 5 nM total protein in Reaction Buffer A containing 50 mM Tris, pH 8.0, 50 mM KCl, 1 mg/ml fatty acid–free bovine serum albumin (BSA) (Sigma A6003) and 1 mM β-mercaptoethanol
Septin polymerization on PEG surface
To investigate septin polymerization in the absence of a lipid bilayer, purified septins were allowed to polymerize on PEG-passivated coverslips. Plastic chambers were glued onto the coverslip, and septins were added to a final concentration of 100 nM in Reaction Buffer B containing 50 mM Tris, pH 8, 50 mM KCl, 1 mg/ml fatty acid–free BSA, 0.1% methyl cellulose and 1 mM β-mercaptoethanol. Septins were allowed to polymerize for ∼1 h before imaging unless otherwise specified.
Septin polymerization in the presence of Mg2+ and/or nucleotide
To determine the effect of magnesium on septin polymerization, 5 nM total protein was allowed to polymerize in Reaction Buffer A + 1 mM MgCl2 on an SLB. For polymerization on PEG, 100 nM total protein was polymerized in Reaction Buffer B + 3 mM MgCl2.
Nucleotide exchange for septins was done by adopting previously published methods (Sheffield et al., 2003; Huang et al., 2006). Total septin protein (250 nM) was incubated with 250 μM nucleotide and 1 mM MgCl2 in high salt (300 mM KCl) for 3 h at 30°C to allow nucleotide exchange. For SLB experiments, the protein was diluted to a final concentration of 5 nM total protein, 5 μM nucleotide, and 20 μM MgCl2 in Reaction Buffer A and allowed to polymerize for ∼30 min before imaging.
For polymerization on a PEG surface, higher protein concentration was typically required to obtain filaments that can be visualized using TIRF due to three-dimensional instead of two-dimensional diffusion. For this reason, the same exchange protocol was applied but using a higher starting protein concentration. Total protein (400 nM) was incubated with 400 μM nucleotide and 3 mM MgCl2 in high salt for 3 h at 30°C. For polymerization, the protein was diluted to a final concentration of 100 nM, 100 μM nucleotide, and 1 mM MgCl2 in Reaction Buffer B. The protein was allowed to polymerize overnight on PEG before imaging.
Total internal reflection fluorescence microscopy
Septin filaments on supported lipid bilayers, PEG passivated glass, and single molecules were imaged using TIRF microscopy to minimize background. Images were acquired on a Nikon TiE TIRF system using an Apo 100×/1.49 NA oil objective equipped with a solid-state laser system (15 mW; Nikon LUn-4) and a complementary metal-oxide-semiconductor (sCMOS) camera (Photometrics Prime 95B).
Preparing SLB microspheres and curvature analysis
To measure septin adsorption on different curved surfaces, SLB-covered microspheres were prepared as described previously (Bridges et al., 2016). Small unilamellar vesicles (SUVs) were prepared with a lipid composition of 75% DOPC (Avanti Polar Lipids 850375), 25% l-α-phosphatidylinositol (liver, bovine; Avanti Lipids 840042), and >0.1% l-α-phosphatidylethanolamine-N-(lissamine rhodamine B sulfonyl) (Rh-PE; egg-transphosphatidylated, chicken; Avanti Polar Lipids 810146). SUVs were then adsorbed onto silica microspheres (0.96, 3.17, and 5.06 μm; Bangs Laboratories). Plastic chambers were glued onto PEG-passivated coverslips. Septins were then incubated with the lipid-coated bead mixture at a final concentration of 100 nM (unless otherwise stated) for 1 h at room temperature to reach equilibrium in buffer containing 50 mM Tris, pH 8, 100 mM KCl, 1 mg/ml fatty acid–free BSA, 0.1% methyl cellulose, and 1 mM β-mercaptoethanol. Images were acquired using a Nikon TiE widefield with a Plan Apo 100×/1.45 NA oil objective equipped with a sCMOS camera (Andor Zyla 4.2 plus). Septin binding analysis was done in Imaris 8.1.2 (Bitplane) by creating surfaces on the beads and determining the sum intensity for both the lipid mateialsand the septin channel. Septin adsorption was calculated by dividing septin sum intensity by the lipid septin intensity. Subsequent analysis and plots were generated using RStudio, version 1.0.136 (Wilkinson, 2011). Statistical analysis was done by performing a Kruskal-Wallis rank sum test followed by a Bonferonni-adjusted Dunn test as previously described (Bridges et al., 2016).
Cdc11 and Shs1 ratio experiments
Different proportions of purified Cdc11 and Shs1 complexes were diluted in septin storage buffer before addition to the SLB at a final protein concentration of 5 nM. Septin polymerization was followed for a total of 15 min using TIRF (described above). Short videos to determine persistence length were acquired with a time interval of 250-ms and a 100-ms exposure.
For curvature analysis using different Cdc11 and Shs1 proportions, the two complexes were mixed in varying proportion in septin storage buffer to a total protein concentration of 400 nM. For curvature binding the mixtures containing different proportions of Cdc11 and Shs1 were added to the beads to a total protein concentration of 400 nM. The rest of the experiment was performed as described above for curvature analysis.
Fragmentation, annealing, and bundling frequency analysis
For fragmentation, annealing, bundling, and unbundling analysis, isolated filaments were identified and followed for ∼50 frames, and all of these events were recorded. Each interaction was considered authentic and not transient if it persisted for at least two frames. If a filament changed length considerably after an annealing event, then it was given a new identity for tracking purposes. Initial filament lengths were recorded. For each filament tracked the number of events was divided by the starting length of the filament to obtain a measure of annealing, fragmentation, bundling, or unbundling per unit length.
Bundling factor computation
For bundling analysis, “tophat” morphological filtering was applied to the images before thresholding to create filament masks in MATLAB R2016a (MathWorks). Using the masks, each isolated filament was identified, and a frequency distribution of intensity was determined. To account for noise, intensity values that accounted for less than 2% of the filament size were not considered. The 2% threshold was empirically selected as a strong criterion for rejection of noise-based outliers. The intensity ranges were then divided by the average intensity of the WT or untreated filaments to acquire the distribution of the metric we term the “bundling factor.”
Single-particle tracking
Particle centers were segmented within time-lapse data using a convolutional neural network (Newby et al., 2017). Linking particles across consecutive frames and matching particles between channels was formulated as a bipartite graph matching problem, a common optimization problem that selects the best match between two sets of points that minimizes the total cost, in our case distance. The matching problem was solved using the Hungarian-Munkres algorithm with a cutoff distance of five pixels (Newby et al., 2017), that is, particles were required to be closer than five pixels apart in consecutive frames to be a valid match. Maximum particle intensities were plotted for display.
Persistence length analysis
Filament persistence length was determined as described by Bridges et al. (2014), using previously published methodology for actin and microtubules (Gittes, 1993). Fifteen or more filaments were selected with little change in length and tracked for ≥6 frames. Two-dimensional coordinates over the length of the filament were determined using Fiji. MATLAB was then used to determine and report the median persistence length of the filaments. Plots were generated using the RStudio ggplot2 package.
Western blotting
Ashbya gossypii cultures were grown in Ashbya full medium (AFM) containing antibiotics and selection for 15–17 h at 30°C with shaking. Cells were collected by vacuum filtration, suspended in lysis buffer (50 mM HEPES, pH 7.8, 1 M KCl, 1 mM MgCl2, 1 mM EGTA, 0.1% Tween, 2× protease inhibitor, 5% glycerol), and lysed using a bead beater. Lysate was collected by spinning at 13,000 rpm for 10 min at 4°C. Bradford assay was used to determine the lysate protein concentration. Equal amounts of protein were loaded onto a SDS gel for Western blotting. The membranes were probed with rabbit polyclonal α-GFP (Invitrogen) for Shs1-GFP and shs1 S35V-GFP, rabbit polyclonal α-Cdc11 (Santa Cruz) for Cdc11 and rabbit α-PSTAIR (Abcam) as a loading control, all at 1:1000 dilution.
FRAP microscope setup and analysis
Ashbya gossypii was grown as above. Fluorescence recovery after photobleaching (FRAP) experiments were performed on a Nikon Eclipse Ti with a Yokogawa spinning disk using a Plan Apo 100×/1.49 NA oil objective. Photobleaching of septin branch collars was performed using a 405-nm laser (450 mW) in conjunction with a mosaic micromirror array system. Images were acquired using an electron-multiplying-CCD camera (Hamamatsu). Individual branch collars for each strain were bleached and imaged every 30 s for 12 min, and the fluorescence intensity was recorded at each time point.
To measure septin branch collar intensity over time, a region of interest (ROI) was drawn around the ring, and the mean fluorescence was measured. The mean fluorescence of an adjacent area in the cytosol was determined and subtracted from the septin intensity measurements to account for background. ROI position was adjusted to account for branch collars moving due to cell growth. There was minimum photobleaching over the course of the experiment. The mobile fractions were determined by determining the ratio of the percentage of recovery to the percentage of bleach. t1/2 was calculated using the formula
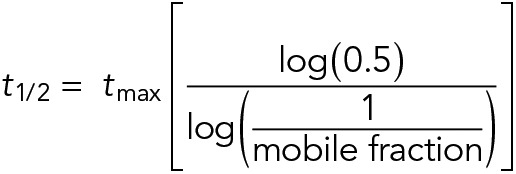 |
where tmax is the time at which recovery plateaus. Plots were generated using the ggplot2 package in RStudio.
Ashbya gossypii growth and imaging
Ashbya gossypii cultures were grown in Ashbya full medium (AFM) containing antibiotics and appropriate selection for 15–17 h at 30°C with shaking. To image GFP-tagged septins, cells were collected by centrifugation at low speed and washed twice with low-fluorescence minimal media and mounted onto 2% agarose gel pads made of low-fluorescence minimal media. Images were acquired on a Zeiss Axioimage-M1 upright light microscope (Carl Zeiss, Jena, Germany) equipped with a Plan-Apochromat 63×/1.4 NA oil objective and an Exfo X-Cite 120 lamp. Z-stacks were acquired using a step size of 0.3 μm and ∼20 slices per image. Image analysis was performed in Fiji. Septin structure intensity was determined by drawing a ROI and taking the sum intensity measurements from five different points along the structure. An adjacent area of the cytosol was measured using the same ROI for five different regions and the mean sum intensity of the cytosol was subtracted from the mean sum intensity of the septin structures to account for background subtraction. Plots were generated using the ggplot2 package in RStudio.
Strain construction
The strains, plasmids, and oligonucleotides used in this study are listed in Supplemental Table S1. Custom oligonucleotides were from Integrated DNA Technologies (IDT, Coralville, IA), and Phusion polymerase was used for PCR (NEB, Ipswitch, MA). To generate plasmid pMVB128 HIS-Sccdc12 T75A/ScCDC10 (AGB561), AGO1340 and AGO1341 were used to introduce the point mutation using the Agilent XL QuickChange kit. This created an additional EcoRV site at 349 base pairs. The correct product was verified using digestion with EcoRV and sequencing using AGO1192 and AGO1342. To generate plasmid pMVB133 ScCDC3/Sccdc11 D65T-GFP (AGB562), primers AGO1347 and AGO1348 were used to introduce the point mutation as well as a SalI site at 4004 base pairs using the Agilent XL QuickChange kit. The correct product was verified by digestion with SalI and HindIII as well as sequencing using primers AGO1349 and AGO1350.
To generate plasmid pMVB133 ScCDC3/ScSHS1-GFP (AGB548), assembly cloning was used. ScSHS11 was PCR amplified using genomic DNA and primers AGO1273 and AGO1274 to produce the insert fragment. The 1712–base pair product was gel extracted using the EZNA gel extraction kit (Omega Bio-Tek, Norcross, GA). The plasmid vector was generated by PCR using AGB454 and primers AGO1271 and AGO1272 to generate a 6511–base pair product. The PCR product was purified by gel extraction. The vector and the insert were ligated using NEB HiFi DNA Assembly Master Mix. The resulting plasmid was isolated from transformants using the EZNA Plasmid Mini Kit (Omega Bio-Tek). The correct product was verified by digestion with HindIII and SacI. To generate plasmid pCOLA ScSHS1-GFP (AGB895), assembly cloning was used. The insert was generated by PCR using AGB598 as a template and primers AGO1807 and AGO1808 to get a 944–base pair product and was treated with DpnI to digest the template. The vector was generated through PCR using AGB560 as a template and primers AGO1805 and AGO1806 to produce a 5282–base pair product. Vector was purified by gel extraction. The vector and insert were assembled using NEB HiFi DNA Assembly Master Mix. The resulting plasmid was isolated by mini kit and tested by NruI and PacI double digestion. The correctly digested plasmid was sequence verified using AGO1496 and AGO1320.
To generate AGshs1 S35V-GFP strain (AG372.1), primers AGO537 and AGO538 were used to introduce the S35V point mutation in plasmid AGB88 using the Stratagene QuickChange II XL site-directed mutagenesis kit (AGB211). AGO541 and AGO412 were used to verify the point mutation. The plasmid AGB211 was linearized by cutting with BlpI and the 5263-nucleotide fragment was transformed into AG416. Genomic DNA was isolated using a Qiagen DNeasy Plant Mini kit (Qiagen, Venlo, The Netherlands). Heterokaryons and homokaryons were verified using AGO592 and AGO593.
Sequence alignment
Protein sequences were BLASTED against A. gossypii Shs1 using BLASTp on the Saccharomyces Genome Database. The protein that showed the highest consensus with the BLAST query among the fungal species listed in Figure 5 was selected. Sequence alignment was performed using ClustalW (Thompson et al., 2002; Goujon et al., 2010). JalView (Version 2.10.2b2) was used to display and view the sequence alignment (Waterhouse et al., 2009).
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (MCB MCB-0719126). We thank all Gladfelter lab members, J. Moseley, E. Smith, and H. Higgs for useful discussions; V. Madden for support with TEM work; M. Baig for help with bundling analysis; and S. Dundon for critically reading the manuscript.
Abbreviations used:
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- TIRF
total internal reflection fluorescence
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-10-0608) on January 10, 2018.
REFERENCES
- Akhmetova K, Balasov M, Huijbregts RPH, Chesnokov I. (2015). Functional insight into the role of Orc6 in septin complex filament formation in Drosophila. Mol Biol Cell , 15–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Tabarés I, Pérez-Martín J. (2010). Septins from the phytopathogenic fungus Ustilago maydis are required for proper morphogenesis but dispensable for virulence. PLoS One , e12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S, König J, Koepke J, Feldbrügge M. (2014). Endosomal transport of septin mRNA and protein indicates local translation on endosomes and is required for correct septin filamentation. EMBO Rep , 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, McMurray MA, Thai L, Garcia G, III, Votin V, Grob P, Allyn T, Thorner J, Nogales E. (2010). Phosphatidylinositol-4,5-bisphosphate promotes budding yeast septin filament assembly and organization. J Mol Biol , 711–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth EA, Thorner J. (2016). A FRET-based method for monitoring septin polymerization and binding of septin-associated proteins. Methods Cell Biol , 35–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth EA, Vane EW, Dovala D, Thorner J. (2015). A Förster resonance energy transfer (FRET)-based system provides insight into the ordered assembly of yeast septin hetero-octamers. J Biol Chem , 28388–28401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Jentzsch MS, Oakes PW, Occhipinti P, Gladfelter AS. (2016). Micron-scale plasma membrane curvature is recognized by the septin cytoskeleton. J Cell Biol , 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges AA, Zhang H, Mehta SB, Occhipinti P, Tani T, Gladfelter AS. (2014). Septin assemblies form by diffusion-driven annealing on membranes. Proc Natl Acad Sci USA , 2146–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YT, Koh JLY, Friesen H, Duffy SK, Cox MJ, Moses A, Moffat J, Boone C, Andrews BJ. (2015). Yeast proteome dynamics from single cell Imaging and automated analysis. Cell , 1413–1424 [DOI] [PubMed] [Google Scholar]
- DeMay BS, Meseroll RA, Occhipinti P, Gladfelter AS. (2009). Regulation of distinct septin rings in a single cell by Elm1p and Gin4p kinases. Mol Biol Cell , 2311–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CM, al-Awar O, Rosenblatt J, Wong ML, Alberts B, Mitchison TJ. (1996). A purified Drosophila septin complex forms filaments and exhibits GTPase activity. J Cell Biol , 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnigan GC, Takagi J, Cho C, Thorner J. (2015). Comprehensive genetic analysis of paralogous terminal septin subunits Shs1 and Cdc11 in Saccharomyces cerevisiae. Genetics , 821–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford SK, Pringle JR. (1991). Cellular morphogenesis in the Saccharomyces cerevisiae cell cycle: localization of the CDC11 gene product and the timing of events at the budding site. Dev Genet , 281–292 [DOI] [PubMed] [Google Scholar]
- Garcia G, III, Bertin A, Li Z, Song Y, McMurray MA, Thorner J, Nogales E. (2011). Subunit-dependent modulation of septin assembly: budding yeast septin Shs1 promotes ring and gauze formation. J Cell Biol , 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittes F, Mickey B, Nettleton J, Howard J. (1993). Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J Cell Biol , 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. (2010). A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res , W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Rodríguez Y, Masuo S, Johnson D, Orlando R, Smith A, Couto-Rodriguez M, Momany M. (2014). Distinct septin heteropolymers co-exist during multicellular development in the filamentous fungus Aspergillus nidulans. PLoS One , e92819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-W, Surka MC, Reynaud D, Pace-Asciak C, Trimble WS. (2006). GTP binding and hydrolysis kinetics of human septin 2. Febs J , 3248–3260 [DOI] [PubMed] [Google Scholar]
- Jain A, Ramani B, Raghunathan K, Jena P, Xiang Y, Ha T. (2009). Single molecule immuno pull down assay (SiMPull) for studying protein-protein interactions. Biophys J , 25a. [Google Scholar]
- Kaplan C, Jing B, Winterflood CM, Bridges AA, Occhipinti P, Schmied J, Grinhagens S, Gronemeyer T, Tinnefeld P, Gladfelter AS, et al. (2015). Absolute arrangement of subunits in cytoskeletal septin filaments in cells measured by fluorescence microscopy. Nano Lett , 3859–3864 [DOI] [PubMed] [Google Scholar]
- Kim MS, Froese CD, Estey MP, Trimble WS. (2011). SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J Cell Biol , 815–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrington R, Small E, Rodger A, Dafforn TR, Addinall SG. (2004). FtsZ fiber bundling Is triggered by a conformational change in bound GTP. J Biol Chem , 48821–48829 [DOI] [PubMed] [Google Scholar]
- Nagaraj S, Rajendran A, Jackson CE, Longtine MS. (2008). Role of nucleotide binding in septin-septin interactions and septin localization in Saccharomyces cerevisiae. Mol Cell Biol , 5120–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby JM, Schaefer AM, Lee PT, Forest MG, Lai SK. (2017). Deep neural networks automate detection for tracking of submicron scale particles in 2D and 3D. arXiv:1704.03009. [DOI] [PMC free article] [PubMed]
- Schindelin Jl, Ignacio A, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Reuden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods , 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Sandblad L, Stenmark S, Gullberg M. (2011). Deciphering the rules governing assembly order of mammalian septin complexes. Mol Biol Cell , 3152–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield PJ, Oliver CJ, Kremer BE, Sheng S, Shao Z, Macara IG. (2003). Borg/Septin interactions and the assembly of mammalian septin heterodimers, trimers, and filaments. J Biol Chem , 3483–3488 [DOI] [PubMed] [Google Scholar]
- Sirajuddin M, Farkasovsky M, Zent E, Wittinghofer A. (2009). GTP-induced conformational changes in septins and implications for function. Proc Natl Acad Sci USA , 16592–16597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang JX, Janmey PA. (1996). The polyelectrolyte nature of F-actin and the mechanism of actin bundle formation. J Biol Chem , 8556–8563 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Higgins DG. (2002). Multiple Sequence Alignment Using ClustalW and ClustalX, Hoboken, NJ: John Wiley & Sons. [DOI] [PubMed] [Google Scholar]
- Versele M, Gullbrand B, Shulewitz MJ, Cid VJ, Bahmanyar S, Chen RE, Barth P, Alber T, Thorner J. (2004). Protein-protein interactions governing septin heteropentamer assembly and septin filament organization in Saccharomyces cerevisiae. Mol Biol Cell , 4568–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrabioiu AM, Gerber SA, Gygi SP, Field CM, Mitchison TJ. (2004). The majority of the Saccharomyces cerevisiae septin complexes do not exchange guanine nucleotides. J Biol Chem , 3111–3118 [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. (2009). Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics , 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems A, McMurray M. (2017). The step-wise pathway of septin hetero-octamer assembly in budding yeast. eLife , 20007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weems AD, Johnson CR, Argueso JL, McMurray MA. (2014). Higher-order septin assembly is driven by GTP-promoted conformational changes: evidence from unbiased mutational analysis in Saccharomyces cerevisiae. Genetics , 711–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L. (2011). ggplot2: elegant graphics for data analysis by WICKHAM. H. Biometrics , 678–679 [Google Scholar]
- Zander S, Baumann S, Weidtkamp-Peters S, Feldbrügge M. (2016). Endosomal assembly and transport of heteromeric septin complexes promote septin cytoskeleton formation. J Cell Sci , 2778–2792 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.