

SUMMARY
Immunoglobulin heavy chain (IgH) genes are assembled by DNA rearrangements that juxtapose a variable (VH), a diversity (DH), and a joining (JH) gene segment. Here we report that in the absence of intergenic control region 1 (IGCR1) the intronic enhancer (Eμ) associates with the next available CTCF binding site located close to VH81X, via putative heterotypic interactions involving YY1 and CTCF. The alternate Eμ/VH81X loop leads to formation of a distorted recombination center, altered DH rearrangements, and disrupts chromosome conformation that favors distal VH recombination. Cumulatively, these features drive highly skewed, Eμ-dependent recombination of VH81X. Sequential deletion of CTCF binding sites on IGCR1-deleted allele suggests that they influence recombination of single proximal VH gene segments. Our observations demonstrate that Eμ interacts differently with IGCR1- or VH- associated CTCF binding sites and, thereby, identify distinct roles for insulator-like elements in directing enhancer activity.
Graphical abstract
INTRODUCTION
Immunoglobulin heavy chain (IgH) genes are assembled by DNA rearrangements that juxtapose a variable (VH), a diversity (DH), and a joining (JH) gene segment into an exon that encodes the antigen-binding domain of antibody heavy chains. The order of rearrangements is fixed. DH to JH rearrangements occur first to create DJH recombined alleles, followed by VH rearrangements to DJH junctions to produce VDJH recombined alleles (Jung et al., 2006; Kumari and Sen, 2015). Approximately 194 VH gene segments, 8–12 DH gene segments, and 4 JH gene segments are distributed over 2.8 Mb of the mouse genome (Figure 1A). These gene segments can be assorted randomly during recombination to generate the large number of different antigen-binding specificities required for optimal immunity. To facilitate generation of diversity, the IgH locus undergoes developmentally-regulated epigenetic changes and alteration of 3D chromatin conformation (Bossen et al., 2012). Based on live cell imaging studies, a current view is that the variable domain consists of several rosettes that form a ‘cloud’ close to the DJH part of the locus to permit comparable utilization of VH gene segments during recombination (Lucas et al., 2014). We previously proposed that the IgH locus conformation is generated by a hierarchy of structural changes that involve different transcription factors (Gerasimova et al., 2015).
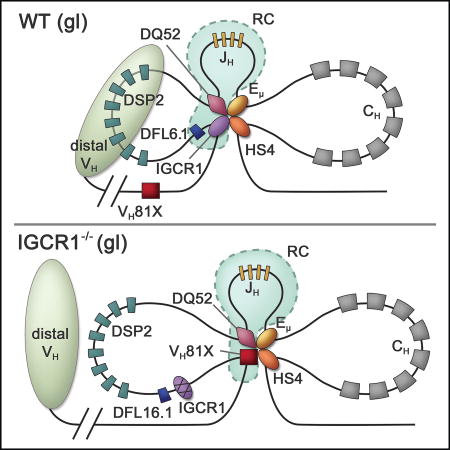
Figure 1. Altered looping of IGCR1-mutated IgH alleles.

A. Schematic representation of the murine germline (unrearranged) IgH locus. Variable gene segments (VH) occupy much of the locus starting at the 5’ end (left). VH gene segments are broadly categorized into three gene clusters: distal (comprising VHJ558 and VH3609 genes), mid (comprising several gene families) and proximal (comprising VH7183 and VHQ52 genes). A gap of 90 kb separates the 3’-most VH gene segments from the 5’-most diversity (DH) gene segment, DFL16.1. A family of closely-related DSP2 gene segments and a unique 3’ DQ52 gene segment spread over 60 kb complete the DH genes. Four joining gene segments (JH) are located within 1 kb 3’ of DQ52, followed by exons that encode the constant parts of antibody heavy chains (labeled CH). IgH gene assembly by recombination puts together one VH, one DH and one JH gene segment (highlighted in red) to encode the variable part of antibody heavy chains. Three key regulatory sequences are indicated as ovals. The IGCR1 is located just 5’ of DFL16.1 (purple; yellow dots represent 2 functional CTCF binding elements (CBEs)), the enhancer Eμ is located in the intron between JH gene segments and Cμ exons (red), and the 3’ regulatory region (HS3a-4) is a cluster of DNase1-hypersensitive sites located beyond the last CH exons (gray).
B. Chromosome conformation capture (3C) analyses of wild-type (WT) or IGCR1-mutated alleles in which both CBEs have been mutated. Pro-B cell lines carrying normal (WT) or IGCR1-mutated IgH alleles (CBE−/−(1) and CBE−/−(2)), and a non-B cell line of endothelial origin (EOMA), were subject to 3C analysis using anchor primers located at Eμ (left) or close to the 3’-most functional VH gene, VH81X (right). All pro-B cells were recombinase (RAG2) deficient to maintain IgH alleles in germline configuration. The region of interest is expanded below a scale view of the IgH locus (in which VH gene families are shown, the part marked D-J contains all DH and JH gene segments, and the part marked C contains all CH exons). Locations of anchor primers (arrows) and sites queried for interactions are indicated. Relative interaction frequency (Y axis) was calculated as described (Guo et al., 2011a) after normalization for ligation efficiency using α-amylase-specific primers. Amplification efficiency for different primer pairs was calculated based on restriction and re-ligation of 3 bacterial artificial chromosomes (BACs) that span the region of interest. Positions and sequences of primers are provided in Table S3. Data are shown as mean ± standard error of measurement (SEM) of two biological replicate 3C assays.
C. Fluorescent in situ hybridization (FISH) analyses of WT and IGCR1-mutated alleles. FISH was carried out as previously described (Guo et al., 2011a) using short probes located as shown in the schematic. Representative nuclei containing WT or IGCR1-mutated (CBE−/−(1)) alleles are shown; probes were color-coded as indicated to the left of the micrograph. BAC RP23-201H14 was used to mark IgH alleles (blue) in selected FISH analyses. It is located 200 kb 3’ of HS4. Spatial distances between probes were measured after deconvolution of images from 100 nuclei. Bar graphs show the percentage of IgH alleles in which the distance between indicated probe pairs fell in the ranges shown in different colors. The cumulative frequency distribution of spatial distances for each color-coded probe combination is shown to the right of the bar graph. D-statistics and P-values for differences between WT and IGCR1-mutated alleles were calculated using a two-sample Kolmogorov-Smirnov test (Table S1). Data from biological replicate experiments are shown in Figure S3B.
See also Figure S1–3 and Table S1, 3.
Three transcription factors have been linked to IgH locus structure. Pax5 and YY1 are required for locus compaction that juxtaposes 5’ and 3’ parts of the locus (Ebert et al., 2011; Fuxa et al., 2004; Liu et al., 2007; Verma-Gaur et al., 2012). The third transcription factor, CTCF, binds to about 80 sites that are mostly in the VH part of the locus (Degner et al., 2011; Lin et al., 2012). Additionally, a cluster of CTCF binding sites mark the 3’ end of the IgH locus (Volpi et al., 2012), and 2 prominent CTCF-binding elements (CBEs) are located within the intergenic control region 1 (IGCR1, Figure 1A). The IGCR1 CBEs have a critical role in regulating VDJH recombination as described below. CTCF has also been shown to regulate V(D)J recombination at other antigen receptor loci. Both TCRβ and TCRδ loci have CTCF binding sites positioned between V and J gene segments, analogous to the location of IGCR1 in the IgH locus (Chen et al., 2015; Majumder et al., 2015; Zhao et al., 2016). Deletion or mutation of these sites changes the frequency of V gene segment utilization at these loci (Chen et al., 2015; Majumder et al., 2015; Zhao et al., 2016). Conversely, introduction of new CTCF binding sites into TCRβ locus reduces rearrangements to varying degrees depending on their location (Chen et al., 2016; Rawat et al., 2017).
The tissue-specific endonucleases RAG1 and RAG2 initiate IgH gene assembly by introducing DNA breaks at recombination signal sequences (RSSs) associated with each rearranging gene segment (Schatz and Ji, 2011). RAG1/2 load onto antigen receptor loci at spatially restricted regions known as recombination centers (RCs) (Ji et al., 2010). The IgH RC forms over a 3.5 kb region between the 3’-most DH gene segment (DQ52) and Eμ, and includes all four JH gene segments. Alt and colleagues have recently proposed that RAG1/2 track from the RC to locate complementary RSSs for synapsis and DNA cleavage (Hu et al., 2015). At the IgH locus, RAG1/2 tracking is limited to a 60 kb chromatin loop between Eμ and IGCR1 (Hu et al., 2015). Spatial proximity of 5’-most (DFL16.1) and 3’-most (DQ52) gene segments to the RC in this configuration may explain, in part, the high frequency of DFL16.1 and DQ52 rearrangements (Choi et al., 2013; Lin et al., 2016). Finally, absence of VH gene segments from this configuration provides a plausible explanation for DH to JH recombination preceding VH recombination.
Eμ and IGCR1 cis-DNA elements (Figure 1A) have been implicated in regulating recombination. Both steps of IgH gene assembly are reduced on Eμ-deleted alleles, with VH rearrangements being affected especially strongly (Afshar et al., 2006; Perlot et al., 2005). This is likely mediated by Eμ’s effects on epigenetic features of unrearranged and DJH rearranged IgH alleles (Chakraborty et al., 2009; Subrahmanyam et al., 2012). Deletion or mutation of CBEs within IGCR1, dramatically dysregulates VH recombination. This is reflected in premature recombination of the 3’-most VH gene segments to unrearranged DQ52 in a breakdown of the normal sequence of IgH rearrangements, sharply reduced use of distal VHJ558 gene segments in VDJH junctions, and suppressed feedback inhibition of proximal VH recombination (Guo et al., 2011b). Mutation of either CBE partially recapitulates recombination phenotypes of doubly-mutated alleles (Lin et al., 2015).
Our studies identify mechanisms by which CTCF-lacking Eμ and CTCF-binding IGCR1 work together to direct optimal recombination of diverse VH gene segments. Whereas Eμ and IGCR1 are spatially clustered on WT IgH alleles, Eμ loops to an alternate CTCF binding site on IGCR1-mutated alleles. In this configuration VH81X lies close to the RC whereas DFL16.1 moves away from the RC. The nature of the RC itself changes substantially on IGCR1-mutated alleles as evidenced by considerable RAG1/2 recruitment to the vicinity of VH81X. We suggest that a dysregulated RC, incorporation of VH81X into a RAG1/2 tracking domain, and spatial proximity of VH81X to the JH-associated RC combine to induce premature recombination on IGCR1-deleted alleles. Our observations lead to model whereby sequestration of Eμ by IGCR1 interferes with its ability to form highly specific loops with VH region CTCF binding sites, thereby restricting RAG protein to a focused RC and generating a chromatin configuration that attenuates VS recombination while enhancing VH diversity.
RESULTS
Altered looping on IGCR1-mutated IgH alleles
Eμ interacts with IGCR1 on wild-type (WT) IgH alleles (Guo et al., 2011a; Guo et al., 2011b). We hypothesized that Eμ may loop to the next available 5’ CTCF binding site on IGCR1-mutated IgH alleles that lack both CBEs (Figure S1). We tested this hypothesis using chromosome conformation capture (3C) in RAG2-deficient pro-B cells that carry IgH alleles lacking both IGCR1 CBEs (Guo et al., 2011b). RAG2-deficiency was essential to restrict the locus to its unrearranged configuration, thereby permitting analysis of structurally homogeneous alleles. Eμ-IGCR1 interactions were evident on WT IgH alleles but not on IGCR1-mutated IgH alleles (Figure 1B). Instead, Eμ interacted with sequences close to VH81X on IGCR1-mutated alleles (Figure 1B, compare red and blue bars to green bars). Eμ interaction with HS4, a DNase1-hypersensitive site located at the 3’ end of the IgH locus, was not affected by the IGCR1-mutation (Figure 1B, labeled HS4). As a negative control we used EOMA cells of endothelial origin derived from the 129 mouse strain (Figure 1B, yellow bars). Conversely, a 3C anchor near VH81X revealed strong interactions with Eμ as well as HS4 on IGCR1-mutated, but not WT IgH alleles in pro-B cell lines. Lower levels of VH81X to HS4 interactions in non-B cells could reflect cell lineage non-specific chromatin compaction; however, we have not pursued this further in the present study. We conclude that the Eμ/HS4 domain interacts with the next available looping site close to VH81X in the absence of CBEs within IGCR1, resulting in increased VH81X transcription and localized histone modification (Guo et al., 2011b) (Figure S2 and 3A).
To further substantiate these results at a single cell level, we carried out high resolution 3D-fluorescent in situ hybridization (FISH) using short probes to accurately locate specific regions. We found that spatial proximity of IGCR1 to Eμ on WT IgH alleles was disrupted in IGCR1-mutated alleles (Figure 1C, Figure S3B). Conversely, probes close to Eμ and VH81X were spatially distant on WT alleles but closely juxtaposed on IGCR1-mutated alleles. This new Eμ interaction did not extend to the 5’ end of the VH7183 gene family (see below). The combined 3C and FISH studies indicated that Eμ loops to an alternate site close to the 3’-most functional VH gene segment, VH81X, upon disruption of its interaction with IGCR1.
Altered looping requires Eμ, YY1 and CTCF
To test the interpretation that increased VH81X gene transcription on IGCR1-mutated alleles resulted from association with Eμ, we used CRISPR/Cas9 to delete Eμ sequences from a pro-B cell line with IGCR1-mutated IgH alleles. We obtained two independent lines in which Eμ was deleted on both alleles in the context of a mutated IGCR1 (Figure S4A). We found that VH81X transcription induced by IGCR1-mutation was reduced in both Eμ-deleted lines (Figure 2A, left panel). As controls, we assayed transcription from the Eμ-dependent DQ52 promoter. DQ52 transcripts were reduced by Eμ deletion regardless of the status of IGCR1 (Figure 2A, middle panel). Ctcf transcription was comparable in all five cell lines (Figure 2A, right panel). As additional controls, transcription of Yy1 and PAIR elements in the distal VH region were not affected by manipulation of Eμ or IGCR1 (Figure S4A). These observations corroborate the idea that proximity to Eμ directs VH81X transcription and histone modifications in the absence of IGCR1.
Figure 2. Features of the Eμ-VH81X loop on IGCR1-mutated alleles.

A. Eμ-dependent activation of VH81X transcription on IGCR1-mutated alleles. CBE−/−(2)/Eμ−/− (1) and CBE−/−(2)/Eμ−/−(2), are two cell clones derived from the CBE−/−(2) pro-B cell line in which Eμ was deleted by CRISPR/Cas9 technology. Pro-B cells with wild-type and Eμ-deficient IgH alleles are indicated as WT and Eμ−/−, respectively. Total RNA was assayed by quantitative RT-PCR using amplicons indicated on the top line. IgH locus transcript levels were normalized to γ-actin mRNA and are represented relative to levels in CBE−/−(2) cells (Y axis). Ctcf mRNA levels served as an additional out-of-locus control. Data are shown as mean ± SEM of three independent RNA preparations and analyses.
B. CTCF- and YY1-dependence of Eμ-VH81X loop. CTCF and YY1 expression was reduced in CBE−/−(2) cells by lentiviral expression of shRNA directed against the respective mRNAs (Figure S4B). Prevalence of the Eμ-VH81X loop was assessed by 3D-FISH using probes close to these sites. Cells expressing a scrambled shRNA (Scr) served as controls. Representative nuclei from YY1-shRNA, CTCF-shRNA#1 and control (Scr) cells are shown. Spatial distance between probes was measured after deconvolution of images from 100 nuclei. Bar graph shows the percentage of IgH alleles in which the distance between probes fell in the ranges shown in different colors. The cumulative frequency distribution of spatial distances is shown to the right. D-statistics and P-values for differences between KD and control cells were calculated as described in the Figure 1 legend (Table S1). Data from biological replicate experiments are shown in Figure S4C.
C. Transcriptional analyses in YY1- and CTCF-knockdown cells. Total RNA isolated from cells with indicated shRNA expression was assayed by quantitative RT-PCR using amplicons indicated on the top line. IgH locus transcripts (VH81X and DQ52) were normalized to γ-actin mRNA and are represented relative to levels in control cells (Y axis). Additional IgH locus transcripts from Eμ (5’Eμ and 3’Eμ) levels are shown in Figure S4B. Data are shown as mean ± SEM of two independent knock-down experiments followed by RNA analyses.
See also Figure S4 and Table S1, 3.
Eμ-dependent interactions on WT IgH alleles require the transcription factor YY1 (Gerasimova et al., 2015). To determine whether the newly-identified Eμ-VH81X interaction also had the same requirement, we reduced YY1 expression in IGCR1-mutated pro-B cells by introducing a Yy1-specific shRNA, and assayed this interaction by 3D-FISH. We achieved approximately 60% reduction of Yy1 mRNA and protein with the Yy1-specific shRNA compared to a scrambled control (Figure S4B). We found that Eμ-VH81X interactions were reduced in YY1-knockdown (KD) cells (Figure 2B; Figure S4C). Loop disruption was accompanied by reduced transcription of VH81X (Figure 2C). As controls, Eμ-dependent transcripts that initiated at the DQ52 promoter (μ0 transcript) and within Eμ itself (5’Eμ and 3’Eμ) were not affected by YY1 KD (Figure 2C, S4B).
The proposed Eμ interaction site close to VH81X contains a prominent CTCF binding site, but no YY1 binding sites (Figure S1) (Verma-Gaur et al., 2012). We hypothesized that this interaction may arise from heterotypic interactions between YY1 bound at Eμ and CTCF bound at VH81X, such as those have been proposed to occur during X chromosome inactivation (Donohoe et al., 2007). To test this, we knocked-down CTCF expression with three shRNAs (Figure S4B) and assayed the effects on VH81X germline transcription on IGCR1-mutated alleles. We found that the VH81X transcription was reduced by all three CTCF-specific shRNAs but not by a scrambled control (Figure 2C). As seen with YY1-KD, DQ52 and Eμ-associated transcripts were largely unaffected in CTCF-KD cells (Figure 2C, S4B). Additionally, FISH analysis showed that CTCF-KD disrupted the Eμ-VH81X loop (Figure 2B). We conclude that formation of the new Eμ-VH81X loop on IGCR1-mutated alleles requires both CTCF and YY1.
Functional consequences of altered looping on IGCR1-mutated alleles
The high recombination frequency of DFL16.1 and DQ52 on WT alleles has been proposed to be due to their spatial proximity to the RAG1/2-rich RC via Eμ-IGCR1 interaction (Kumari and Sen, 2015). A key prediction of this model is that alterations in the WT loop configuration should change the frequency of DH gene segment usage. In the new configuration on IGCR1-mutated alleles, VH81X replaces DFL16.1 near the RC. Therefore, DFL16.1 recombination frequency should be reduced compared to other DH gene segments. We tested this hypothesis by quantifying DH rearrangements in primary bone marrow pro-B cells from recombinase-sufficient IGCR1-deleted mice.
To amplify DFL16.1, DSP2 and DQ52 rearrangements from pro-B cell genomic DNA, we used 5’ primers that recognize either DFL16.1, or the 8 DSP2 gene segments (in 129 mouse strain), or DQ52 and a 3’ primer close to JH4 (Figure 3A). We found that the proportion of DFL16.1 compared to DSP2 and DQ52 rearrangements was reduced on IGCR1-deleted alleles (Figure 3A, quantitated in Figure S5A). We also quantitated DH to JH rearrangements in cell lines carrying WT and IGCR1-mutated IgH alleles. Because these lines were deficient in RAG2, we induced rearrangements by lentiviral transduction of Rag2. Though rearrangement levels were variable between lines, the observed trends were similar to those in primary pro-B cells, with the proportion of DFL16.1 to DSP2 and DQ52 recombination being reduced in both IGCR1-mutated lines (Figure S5B).
Figure 3. DH gene segment utilization on IGCR1-deleted IgH alleles in bone marrow pro-B cells.

A. Analysis of DJH junctions. Genomic DNA purified from pro-B cells (B220+IgM−CD43+) from WT and IGCR1-deleted IgH alleles(Guo et al., 2011b) was used in amplification reactions with 5’ primers specific for either DFL16.1 (blue arrow), 8 DSP2 (green arrow) or DQ52 gene segments (orange arrow) together with a 3’ primer located beyond JH4 (red arrow). Amplification products were analyzed by agarose gel electrophoresis. 5-fold increasing amounts of genomic DNA starting at 4ng (lanes 3, 6) are shown for each genotype. The non-rearranging ROSA26 locus served as a loading control. Rearrangement of each DH gene segment to the four JH gene segments is indicated to the left of the gel. Lane 7 is an amplification reaction with no genomic DNA. Two biological replicate purifications of pro-B cells were performed. Data shown is a representative rearrangement analysis from one experiment. Averaged data from both experiments is provided in Figure S5A.
B. DH utilization in VDJH junctions in pro-B cells from mice carrying WT and IGCR1-deleted IgH alleles. Pro-B cell genomic DNA was amplified using a 5’ primer specific for 19 out of 49 rearrangeable VH7183 gene segments (blue arrows and detailed in Figure S5C), including the 3’-most VH7183 gene segment indicated in the schematic, and a 3’ primer located just after JH1 (red arrow). Amplification products were sub-cloned into pGEM®-T, and 55 clones from each of two pro-B cell preparations from WT and IGCR1-deleted mice were sequenced to estimate VH and DH usage (Table S2). DFL16.1 and DSP2 utilization in each genotype ranged between 39 and 50 subclones per experiment; DH gene segment identity could not be established in the remaining clones due to end processing during VDJH recombination. Numbers of clones with DFL16.1 and DSP2 rearrangements from each genotype are shown in the graph. Data are shown as mean ± SEM of two experiments carried out with independently sorted pro-B cell preparations.
See also Figure S5 and Table S2, 3.
We previously proposed that DH recombination brings the associated 5’-recombination signal sequence (RSS) into the RC for VH to DJH recombination (Subrahmanyam et al., 2012). Thus, DH recombination frequency should also be reflected in DH utilization in VDJH junctions. To test this we amplified VH7183-DJH1 junctions from bone marrow pro-B cells containing WT and IGCR1-deleted IgH alleles followed by cloning and sequencing (Figure 3B, top line; Figure S5C). From two samples each of WT and IGCR1-deleted primary pro-B cells, we obtained sequence information from approximately 90 clones from each genotype (Figure 3B; Table S2). Cumulatively, we found that DFL16.1 was utilized in 28 clones and DSP2 in 61 clones from pro-B cells carrying WT IgH alleles. (The small size of DQ52 precluded identification of this gene segment in the VDJH clones analyzed). We estimated that any single DSP2 gene segment of 8 was used 7–8 times on average amongst our clones. This distribution reflects the prevailing idea that DFL16.1 rearranges more frequently than DSP2 gene segments on WT alleles (Choi et al., 2013; Lin et al., 2016). In contrast, we obtained only 6 DFL16.1-containing VDJH junctions and 85 DSP2-containing VDJH junctions from IGCR1-mutated pro-B cells, resulting in 5-fold reduced DFL16.1 utilization in the absence of IGCR1 (Figure 3B). As expected (Guo et al., 2011b), several VH7183 segments recombined on WT alleles, whereas VH81X was used most frequently on IGCR1-deleted alleles (Table S2). Our interpretation is that movement of DFL16.1 away from the recombination center leads to its reduced utilization in DH recombination and, thereby, in reduced availability of DFL16.1 JH1 junctions as substrates for VH recombination.
VH81X recombination in the absence of IGCR1
Proximity of VH81X close to the RC on IGCR1-mutated alleles also predicts that it should be utilized more efficiently in VDJH recombination. To test this, we compared kinetics of VH recombination on WT and IGCR1-mutated IgH alleles 4, 5, or 14 days after Rag2 transduction into IGCR1-mutated cell lines. In two different cell lines we observed high levels of VDJH and VDQ52 rearrangements at d4/5 or d14 after transduction (Figure 4A, B; Figure S6A). Amongst VH7183 and VHQ52 genes, VH81X and VHQ52.2.4 were used almost exclusively on IGCR1-mutated alleles, with no detectable rearrangements of the distal VHJ558 gene segments (Figure S6B). Both forms of VH recombination on IGCR1-mutated alleles were Eμ-dependent (Figure 4C). In contrast, WT IgH alleles underwent only DH-JH recombination in the same time frame. As noted previously Eμ deletion reduced but did not eliminate DH recombination (Figures S5B, S6A and C). Expression of the introduced Rag2 gene did not differ more than 2-fold amongst the three cell lines (Figure S6D). We conclude that VH recombination of germline IgH alleles is greatly increased when IGCR1 is inactive, likely due to placement of VH81X close to the RC. This could also explain the greater efficiency of VH rearrangements to a pre-existing DJH junction on IGCR1-mutated IgH alleles (Hu et al., 2015).
Figure 4. Eμ-dependent recombination efficiency of IGCR1-mutated IgH alleles.

Top line shows a schematic of the IgH locus. 5’ primers that score for VHJ558 (blue), VH7183 (orange), VHQ52 (green) or specifically VH81X (light blue) gene segments were used with a 3’ primer located just after DQ52 (black) or JH4 (red) to quantitate VDH or VDJH recombination, respectively, on WT and IGCR1-mutated IgH alleles. Pro-B cell lines with wild-type (WT), IGCR1-mutated alleles (CBE−/−(1) and (2)), Eμ and IGCR1 double-mutated alleles (CBE−/−(2)/Eμ−/−(1) and CBE−/−(2)/Eμ−/−(2)) and were infected with Rag2-expressing lentivirus. After 4–5 days (A and B) or 14 days (C) selection with puromycin, genomic DNA was prepared to assay rearrangements.
A, B. Representative rearrangement profiles for each IGCR1-mutated line assayed using primers indicated to the left of the gel. For the top 4 panels, the 3’ primer was located beyond JH4, thereby scoring for VDJH rearrangements to each JH gene segment. Lanes 7 and 15 are positive controls using total bone marrow genomic DNA from WT mice; lanes 8 and 16 contain no genomic DNA. The next 3 panels use VH primers as indicated, with a 3’ primer located just after DQ52; these products score for VH to DQ52 rearrangements. An amplicon from the ROSA26 locus was used as a DNA loading control. The three lanes associated with each cell type use 5-fold increasing genomic DNA concentration starting at 8ng (lanes 3, 6, 11, 14).
C. Representative rearrangement profiles of IGCR1- plus Eμ- deficient IgH alleles. Data shown is representative of 2 independent Rag2 transduction experiments into each cell line. Analyses of DH recombination after 4–5 days selection and analyses of VH and DH recombination after 14 days selection are shown in Figures S5B, S6A and S6C, respectively.
See also Figure S6 and Table S3.
Altered RC on IGCR1-mutated alleles
To directly investigate the RC of IGCR1-deleted alleles, we carried out chromatin immunoprecipitation (ChIP) with antibodies directed against RAG1 and RAG2. Earlier studies show that the IgH locus is distinct from other antigen receptor loci in recruiting RAG1 only when cells co-express RAG2 (Ji et al., 2010). Because the cells we worked with lacked RAG2, we expressed this protein by lentiviral transduction prior to ChIP. Consistent with earlier observations, RAG1 did not bind to IgH locus in RAG2-deficient control cells with WT IgH alleles (Figure 5B, yellow bars). In RAG2-expressing cells we found highest levels of RAG1 at DQ52 and JH gene segments corresponding to the RC on both WT (Figure 5B, green bars) and IGCR1-mutated alleles (Figure 5B, red and blue bars). Amplicons located near Cγ3 and at the γ-actin promoter served as negative controls. Lower levels of RAG1 were detected at DFL16.1, and hardly any was present at the DSP2 gene segments in both genotypes. We did not detect RAG1 binding to remaining IgH locus amplicons on WT alleles (Figure 5B, green bars; precise locations of VH amplicons are shown in Figure S7A). In contrast, IGCR1-mutated cell lines had relatively high levels of RAG1 over a 25 kb region that encompassed 5 proximal VH gene segments extending from VH7183.4.6 to VH7183.1.1 (Figure S7A).
Figure 5. RAG1 and RAG2 recruitment to WT and IGCR1-mutated alleles.

A. Scale schematic of the IgH locus indicating VH gene families (J558/3609, mid, and 7183/Q52), and the 3’ part of the locus containing DH and JH gene segments (D–J), and CH exons (CH). An expanded view of the portion of the locus assayed for RAG1/2 binding by ChIP. ChIP amplicon VH81X (2) scores for unrearranged VH81X only; intergenic amplicons IG1 and IG2 are located approximately 30 kb and 60 kb from VH81X in the 90 kb region between DFL6.1 and VH81X. DSP2 ChIP primers identify 8 DSP2 gene fragments in the unrearranged configuration.
B. RAG1 ChIP. Anti-RAG1 antibody was used to co-precipitate RAG1-associated genomic DNA as described by (Ji et al., 2010) from RAG2-deficient pro-B cell lines that contain IGCR1-mutated (CBE−/−(1) and CBE−/−(2)) or WT IgH alleles (red, purple and green bars, respectively). RAG2 was expressed in these lines by lentiviral transduction. The pro-B cell line with WT IgH alleles was transduced with a puromycin-only lentivirus serving as a control (yellow bars). Co-precipitated genomic DNA and input DNA were assayed by quantitative (real time) PCR and RAG1 enrichment calculated as described by Ji et al. (Y axis). Cγ3 and γ-actin amplicons served as negative controls for RAG1 binding. Two independent RAG2 transductions were carried out, data are shown as mean ± SEM of three independent ChIP experiments analyzed in duplicate for each amplicon.
C. RAG2 ChIP. Anti-RAG2 antibody was used to co-precipitate associated genomic DNA. Cell lines and amplicons are as described in part B. In this case only Cγ3 amplicon served as a negative control, since RAG2 is known to be recruited to the γ-actin promoter in the absence of RAG1. Two independent RAG2 transductions were carried out, data are shown as mean ± SEM of two independent ChIP experiments.
D. RAG1 ChIP in IGCR1-deleted D345 cells. CRISPR/Cas9 was used to delete IGCR1 in D345 pro-B cell line that expresses a catalytically inactive RAG1 and endogenous RAG2. Two cell clones, D345/IGCR1−/−(1) and D345/IGCR1−/−(2), were used for ChIP with anti-RAG1 antibodies. Co-precipitated DNA was analyzed as described in parts B/C with indicated primers. Cγ3 and γ-actin amplicons served as negative controls for RAG1 binding. Data are shown as mean ± SEM of two independent ChIP experiments from each cell line.
See also Figure S7 and Table S3.
RAG2 binding to WT and IGCR1-mutated alleles showed a similar pattern (Figure 5C), with levels at the proximal VH genes reaching 25–35% of those seen at the RC (represented by DQ52 and JH2 amplicons). As expected from earlier studies, the γ-actin promoter scored positive in RAG2 ChIP, presumably because of H3K4me3 binding by the RAG2 plant homeodomain. We also noted somewhat higher levels of RAG2 binding at unrearranged DFL16.1 and DSP2 regions than previously detected (Ji et al., 2010). At present, we cannot rule out that this is due to some spreading of RAG2 from the RC due to ectopic expression of this protein. However, close coincidence of our observations with all other aspects of published studies of RAG recruitment to WT IgH alleles (Ji et al., 2010) strongly supports the idea that substantial levels of RAG1/2 accumulate over proximal VH gene segments on IGCR1-mutated alleles.
A caveat to this experiment was that expression of RAG2 that was necessary to evaluate the RC also led to ongoing V(D)J recombination. That is, ChIP signals could have arisen from rearranged VH7183 and VHQ52 gene segments present in the cell population. To unequivocally rule out involvement of rearranged IgH alleles to RAG1/2 ChIP signals, we deleted the IGCR1 in D345 pro-B cells that express a catalytically inactive RAG1 together with endogenous RAG2 (Ji et al., 2010). In these cells IgH alleles are uniformly in unrearranged configuration. To determine the distribution of RAG proteins, we carried out ChIP with anti-RAG1 antibodies in two independently derived D345 clones that lack IGCR1 (Figure S7B). We found increased association of RAG1 over approximately 15 kb encompassing the 3’-most VH7183 and VHQ52 gene segments in both lines that lacked IGCR1, but not in parental D345 cells (Figure 5D). The levels of RAG1 in the VH region in the D345 derivatives were approximately 20% of those seen at the JH-associated RC. We conclude that an altered RC is generated over proximal VH gene segments prior to initiation of VDJH recombination on IgH alleles that lack IGCR1.
Reduced distal VH looping in the absence of IGCR1
Identification of the Eμ-VH81X loop on IGCR1-mutated IgH alleles provides plausible mechanisms for the hyper-recombinogenic state of VH81X and VHQ52.2.4. Generation of this loop per se does not readily explain the vastly reduced utilization of distal VHJ558 gene segments as well as gene segments located in the 5’ part of the VH7183 region on IGCR1-mutated alleles (Guo et al., 2011b) (Figure 6A). A possible explanation proposed for skewed VH gene segment usage was that premature VH81X rearrangements used up substrates to which distal VH gene segments could otherwise recombine (Guo et al., 2011b).
Figure 6. Re-directing VDJH recombination on IGCR1-mutated IgH alleles.

A. Scale schematic of the IgH locus indicating locations of FISH probes used to assay conformation of IGCR1-mutated alleles. Probes labeled 3’J558, 5’7183, IGCR1 and Eμ range in length from 4–10 kb, and were generated by amplification of corresponding regions (Table S3) from BACs or genomic DNA. Distances between selected probes are indicated based on mm10.
B. Analysis of VH-DJH interactions by FISH. Probes located in the VH part of the locus (5’7183, 3’J558) and in the DH-JH part of the locus (Eμ and IGCR1) were hybridized to pro-B cell lines containing WT or IGCR1-mutated (CBE−/−(1)) IgH alleles. BAC RP23-201H14 (blue) was used to mark IgH alleles. Representative nuclei are shown, with probe combinations indicated on the left. Spatial distances between probes were measured after image deconvolution from 100 nuclei. Bar graphs show the percentage of IgH alleles in which inter-probe distance fell in the ranges shown by different colors. Cumulative frequency distribution of spatial distances for each color-coded probe combination is shown to the right of the bar graph. D-statistics and P-values for differences between WT and IGCR1-mutated alleles were calculated using the two-sample Kolmogorov-Smirnov test (Table S1). At least 2 independent FISH experiments were carried out with each probe combination (Figure S8B).
C. Sequential activation of CTCF-proximal VH gene segments on IGCR1-mutated alleles. The 3’ part of the VH region is expanded below the schematic to show the location of several proximal VH gene segments and CTCF binding sites. Sites C1–C5 represent the first to fifth CTCF binding sites beyond IGCR1 (labeled CBE1 and CBE2). Sequences indicated by blue bars around C1 and C2 were deleted using CRISPR/Cas9 in the context of the IGCR1 mutated cell line CBE−/−(1) to generate cell lines CBE−/−(1)/C1−/−, CBE−/−(1)/C2−/−, and CBE−/−(1)/C1−/−C2−/−. All cell lines were infected with RAG2-expressing lentivirus to assay recombination. After 10 days selection with puromycin, genomic DNA was prepared to analyze VH utilization by deep sequencing (Hu et al., 2016). Total reads in each cell line were aligned to 128 VH genes in 129 strain mice (Retter et al., 2007). Sequences of the first 10 proximal VH accounted for 99% of reads. Proportion of VH utilization (Y axis) was calculated as a fraction of reads of a specific gene fragment for each cell line. Genomic DNA from 2 independent RAG2 infection experiments were combined for sequencing. Further details are provided in Table S4 and S5.
See also Figure S8–10 and Tables S1, 3–5.
We have previously proposed that Eμ- and YY1-dependent interactions juxtapose distal VH gene segments to the 3’ IgH domain (Gerasimova et al., 2015; Guo et al., 2011a). It was possible, therefore, that altered 3D configuration of IGCR1-mutated IgH alleles precluded this association. To test this hypothesis we carried out 3D-FISH with probes located close to Eμ, at regions approximately 400 kb away near the 5’ end of the VH7183 region (5’7183), and approximately 1 Mb away at the 3’ end of the VHJ558 region (3’J558) (Figure 6A). We found that Eμ-5’7183 as well as Eμ-3’J558 interactions were reduced on IGCR1-deleted alleles compared to WT IgH alleles in RAG2-deficient pro-B cell lines (Figure 6B; Figure S8A, B). We also used a FISH probe located at IGCR1 together with the 3’J558 probe as another measure of the spatial proximity of distal VH gene segments to the 3’ IgH domain. We found that these probes were closely associated on WT IgH alleles but not on IGCR1-mutated alleles (Figure 6B; Figure S8A, B). As a control we assayed a CTCF-dependent, but Eμ-independent, interaction that compacts the VH region (Gerasimova et al., 2015). This 300 kb interaction was not affected by the IGCR1 mutation (Figure S8C), consistent with earlier indications that IGCR1does not regulate VH structure (Medvedovic et al., 2013). We conclude that the pre-folded distal VH region does not gain spatial proximity to the DH part of the locus on IGCR1-deleted alleles. This configuration is very similar to that of Eμ-deficient IgH alleles, suggesting that both these regulatory sequences work together to direct distal VH recombination. Similarity between Eμ- and IGCR1-deficient alleles was also evident in shared transcriptional responses in the distal VH region in these genotypes (Figure S9).
Sequential utilization of CTCF binding sites
Our observations are consistent with the hypothesis that Eμ interacts with the next available CTCF binding site in the locus upon loss of CTCF binding to IGCR1. To directly test the sequential nature of Eμ interaction with CTCF sites, we used CRISPR/Cas9 to delete one or more CTCF sites in the proximal VH genes. The first 5 CTCF sites in this region are shown in Figure 6C (C1–C5); as previously noted, each of these lies close to a functional VH gene segment (Lin et al., 2012). We deleted a 2 kb region containing C1 (C1−/−), or a 1 kb region containing C2 (C2−/−), or a 12 kb region containing both C1 and C2 (C1−/−C2−/−) (Figure S10A), and assessed the consequences on VDJ in the context of WT or IGCR1-mutated IgH alleles.
Analysis of VH gene repertoire by deep sequencing (Hu et al., 2016) revealed substantial differences. As previously noted, IGCR1-mutated alleles recombined VH81X almost exclusively (Figure 6C, labeled CBE−/−(1) and CBE−/−(2)). However, loss of C1 (together with a part of VH81X) resulted in predominant use of VHQ52.2.4 along with much lower levels of VH7183.1.1 (a pseudogene with functional RSS) and VH7183.4.6. That is the gene segment closest to the next available CTCF site (C2) dominated the repertoire. Loss of only C2 by itself recapitulated the phenotype of IGCR1-mutated alleles. We infer that absence of C2 is ‘invisible’ when C1 is intact. However, when both C1 and C2 were lost, albeit in the context of a larger deletion, we observed dominant use of VH7183.4.6 which is located close to C3. As noted with C1 deletion, the adjacent VHQ52.3.8 gene located approximately 12 kb further 5’ also recombined at low levels (Figure 6C). Overall recombination efficiency was comparable among all cell lines (Figure S10B).
We note the following caveats in interpreting these observations. First, because we deleted 1–2 kb of genomic DNA in generating C1 and C2 deficiencies, the observed recombination effects could have arisen by changes in gene spacing rather than loss of specific CTCF binding sites. We consider this is unlikely because individual C1 and C2 mutations removed similar amounts of genomic DNA but resulted in very different recombination phenotypes. Second, because each of the C1 and C2 mutations also removed a part of a nearby VH gene segment, the observed repertoire shift could be because of the loss of the gene segment rather than CTCF binding. However, loss of C1 greatly accentuated only VHQ52.2.4 recombination but not VH7183.1.1 recombination, though both gene segments lie comparable distances from C1. The major difference between them is presence of a CTCF peak near VHQ52.2.4 but not VH7183.1.1. Our interpretation is that Eμ sequentially and selectively activates the VH gene segment closest to the first CTCF binding site on IGCR1-mutated alleles.
DISCUSSION
Consequences of altered Eμ looping
Our observations highlight dynamic interactions of Eμ, an enhancer that does not bind CTCF, with CTCF binding elements in the IgH locus to regulate VDJ recombination. Before rearrangements start Eμ interacts with CTCF-binding IGCR1 and with regulatory regions at the 3’ end of the locus on WT IgH alleles (Figure 7). The Eμ/IGCR1 loop constrains RAG1/2 tracking from the JH-associated RC to a domain that contains DH and JH gene segments but no VH gene segments. Accordingly, DH gene segments rearrange first. Loss of Eμ-IGCR1 interactions on IGCR1-mutated IgH alleles results in Eμ interacting with the next available CTCF site located approximately 90 kb further 5’ near VH81X (Figures 1, 2 and 7 lower line). In this configuration VH81X displaces DFL16.1 from its RC-proximal position. We propose that reduced frequency of DFL16.1 rearrangements that we observed on IGCR1-mutated alleles (Figure 3) reflects this spatial displacement, even though the relative disposition of DFL16.1 and DSP2 gene segments on linear DNA remains unchanged. To the best of our knowledge this is the first analysis of recombination efficiency in the context of different chromatin loops.
Figure 7. Model for Eμ- and IGCR1-dependent configuration of the 3’ IgH domain.

Top and bottom panels show proposed structures of WT and IGCR1-deficient IgH alleles, respectively, in unrearranged (left) and DJH recombined (right) configurations.
Left: Altered structure and recombination center formation on IGCR1-mutated IgH alleles prior to initiation of recombination (gl = germline alleles). Interactions between IGCR1 (blue oval), the DQ52 promoter (purple diamond), Eμ (yellow oval) and HS4 of the 3’ regulatory region (orange oval) nucleate a 3-loop configuration (top). RAG1/2-rich recombination center (RC) is shown within dotted lines (light blue). The intermediate-sized loop contains all the DH gene segments. The nearest functional VH gene segment, VH81X, is located 90 kb from IGCR1. The compacted domain of distal VH gene segments is shown as a light green oval. Mutation of IGCR1 (bottom) leads to looping of Eμ to the next available CTCF binding site close to VH81X. DFL16.1 lies near the middle of the new 160 kb loop, VH81X is located spatially close to DQ52 and the RC, and the distal VH domain moves away. High levels of RAG1/2 at VH81X reflect an altered RC on IGCR1-mutated alleles.
Right: Proposed structure of WT (top) and IGCR1-deficient (bottom) DJH recombined IgH alleles as discussed in the text.
The altered topological domain of IGCR1-mutated alleles permits RAG1/2 to track to proximal VH gene segments, enabling greatly increased recombination efficiency and mis-timed VH recombination. Eμ is essential for these aberrant rearrangements, most of which involve VH81X. The exquisite specificity of RAG activity on IGCR1-mutated alleles is evident from the much lower levels of recombination of the adjacent functional gene segment, VHQ52.2.4, that is located only 10 kb 5’of VH81X ((Guo et al., 2011b), Figure 6). However, deleting the VH81X-associated CTCF site, C1, redirects recombination to VHQ52.2.4 (Figure 6). While recognizing the caveats associated with deleting rather than mutating CTCF sites as discussed in the results section, our interpretation is that Eμ loops specifically to the first available CTCF binding site in the absence of IGCR1 and directs recombination almost exclusively to the associated VH gene segment. This interpretation is further corroborated by dominant use of VH7183.4.6, that is located closest to the third CTCF binding site (C3), when the first two sites C1 and C2 are deleted within context of IGCR1-deficiency (Figure 6). Conversely, the recombination phenotype of C2-deleted alleles is indistinguishable from a locus with only the IGCR1 mutation since the first available Eμ looping site C1 remains intact. The specificity of Eμ interactions with CTCF sites in the VH region has interesting general implications as discussed below.
The nature of Eμ/CTCF interactions
Eμ interaction with IGCR1-associated CBEs or VH region-associated CBEs has substantially different functional consequences. On WT alleles both Eμ and IGCR1 are necessary to bring a pre-folded VH region into proximity of the 3’ IgH domain that contains DH and JH gene segments. Mutation of either of these regulatory sequences disrupts distal VH to D/JH association as well as increases distal VH sterile transcription (Figures 6B, S9, (Gerasimova et al., 2015; Verma-Gaur et al., 2012)). By contrast, Eμ interaction with proximal VH region CBEs (on IGCR1-mutated alleles) leads to highly specific activation of the nearest VH gene and loss of distal VH proximity to the D/JH domain (Figures 4 and 6). We infer that interaction of Eμ with IGCR1 permits flexibility that is essential to generate a diverse repertoire, whereas Eμ interaction with proximal VH CBEs is functionally precise.
Our observations are consistent with the idea that Eμ and IGCR1 together present a composite interface that serves as a bridge, or linker, to distal VH genes, whereas proximal VH CBEs serve only as classical boundaries to limit Eμ activity. One possibility for the different outcomes of Eμ interaction may be the organization of CBEs, with IGCR1 containing two oppositely-orientated CBEs compared to single CBEs associated with each proximal VH gene segment. The highly specific functional interaction of Eμ with the closest CTCF site on IGCR1-deficient alleles evokes the recently proposed loop-extrusion model for choice of CTCF interacting sites (Goloborodko et al., 2016; Rao et al., 2017). It will be interesting to ascertain whether Eμ orientation plays a role in the site selection process as has been proposed for CTCF sites. The functional complexity of CTCF associations with enhancers revealed here may apply more broadly in specifying tissue-specific gene expression.
Further implications for Eμ-dependent V(D)J recombination
Eμ/IGCR1 interactions affect VH recombination by at least two mechanisms. First, altered Eμ-dependent looping in the absence of IGCR1 brings specific VH sequences, and exclude others, from spatial proximity to the JH-associated RC. Second, distribution of RAG1/2 proteins change markedly on IGCR1-mutated alleles compared to the well-defined localized RC on WT alleles (Figure 5). It is possible that VH-associated RAG1/2 can initiate recombination on IGCR1-mutated alleles in addition to the normal JH-associated RAG1/2. For example, RAG1/2 bound near the VH81X RSS on IGCR1-mutated alleles could readily track to the 5’ DQ52 RSS to promote VH81X to germline DQ52 rearrangements. Conversely, RAG1/2 bound to the 5’ DQ52 RSS could track to a spatially proximal VH81X RSS on IGCR1-mutated alleles, but not on WT alleles where its movements would be constrained by Eμ-IGCR1 association. Our working hypothesis is that availability of a few VH gene segments within the RAG1/2 tracking domain, generation of an ectopic RC, and spatial proximity of VH gene segments to the JH-associated RC jointly increase recombination efficiency on IGCR1-deficient alleles resulting in premature recombination.
Mechanistic features of Eμ-IGCR1 identified in this study also apply to DJH recombined alleles (Figure 7, right). After DH recombination of WT alleles Eμ would still be sequestered by IGCR1, the RC would be restricted to the DJH region, and distal VH genes would be spatially proximal to the DJH part of the locus. This configuration would permit diverse VH genes to recombine, but with low efficiency. By contrast, in the absence of IGCR1 Eμ would loop to VH81X, the RC would spread to the proximal VH gene segments, and distal VH gene segments would move away from the 3’ IgH domain. This configuration would yield highly efficient VH81X recombination to the DJH junction, but relatively poor use of distal VH gene segments. We conjecture that in order to gain diversity the immune system evolved away from efficiency by undercutting the ability of Eμ to loop to very specific sites.
How does IGCR1 enforce VH diversity?
There is ample evidence that proximal and distal VH recombination is regulated differently. Most prominently, loss of either transcription factor Pax5 or YY1 significantly reduces distal VH recombination without affecting proximal VH recombination (Fuxa et al., 2004; Hesslein et al., 2003; Liu et al., 2007). It is likely, therefore, that mechanisms by which IGCR1 enforces diverse VH gene segment choice are different for proximal versus distal gene segments. As discussed above, distal VH gene may achieve regulated access to DJH junctions via a composite Eμ/IGCR1 interface. This does not explain how IGCR1 over-rules the tendency of Eμ to loop to the nearest CTCF binding site. Our working model draws upon our earlier observation that sequences 5’ of DFL16.1 (that are very likely to be IGCR1) interact in a CTCF-dependent manner with proximal VH region (Gerasimova et al., 2015; Guo et al., 2011a) and the proposal that CBE1 of IGCR1 interacts with proximal VH-associated CTCF binding sites (Lin et al., 2015). We propose that IGCR1 contacts one, or a subset, of CTCF sites in individual cells. Eμ is brought to each of these different CTCF sites via Eμ/IGCR1 interaction, thereby activating different VH gene segments in individual cells. The key distinction between WT and IGCR1-deficient alleles is how Eμ gets close to VH gene segments. Our hypothesis is that this happens via CTCF/CTCF interactions between IGCR1 and proximal VH CTCF sites on WT alleles, whereas it happens by loop extrusion from Eμ to the nearest CTCF site on IGCR1-deficient alleles. By sequestering Eμ’s tendency to form highly specific loops with proximal CTCF binding sites, IGCR1 ensures generation of a diverse IgH repertoire.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Ranjan Sen (Senranja@grc.nia.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
WT 129 strain mice were purchased from Charles River Laboratories International. IGCR1-deleted mice were as described previously (Guo et al., 2011b). All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of Boston Children’s Hospital.
Cell lines
Abelson virus transformed pro-B cell lines CBE−/−(1) and CBE−/−(2) are deficient for RAG2 and homozygous for IgH alleles in which both CBEs in IGCR1 are mutated (Guo et al., 2011b). RAG2-deficient pro-B cells contain WT IgH alleles, and Eμ−/− pro-B cells have a 220bp deletion of the intronic enhancer on both IgH alleles. Both lines lack RAG2 (Chakraborty et al., 2009). EOMA, an endothelial cell line from 129 strain of mice, was purchased from ATCC. CBE−/− (2)/Eμ−/−(1), CBE−/−(2)/Eμ−/−(2), D345/IGCR1−/−(1), D345/IGCR1−/−(2), CBE−/−(1)/C1−/−, WT/C1−/−, CBE−/−(1)/C2−/−, WT/C2−/−, CBE−/−(1)/C1−/−C2−/− and WT/C1−/−C2−/− were generated with CRISPR/Cas9 system, described below. Cells were cultured in RPMI medium with 10% fetal bovine serum (FBS) and 56μM 2-mercaptoethanol at 37°C in a 5% CO2 humidified atmosphere.
METHOD DETAILS
Isolation of pro-B cells from bone marrow
Bone marrow derived pro-B cells, marked as B220+IgM−CD43+, were purified from WT 129 or IGCR1−/− mice.
Chromosome conformation capture (3C)
Chromosome conformation capture (3C) assays were performed as described (Wuerffel et al., 2007) using HindIII to digest crosslinked chromatin. Briefly, cells were crosslinked with 1% (v/v) formaldehyde solution in HBSS buffer. After nuclear extraction, cross-linked DNA was digested overnight with HindIII overnight. Reaction was terminated with SDS, followed by neutralization with Triton™ X-100 solution. Digested DNA was treated with T4 DNA ligase overnight. RNA and protein were digested by RNAse A and proteinase K. 2-Propanolprecipitated DNA was further purified by extraction with phenol and chloroform, followed by 0.3M sodium acetate (ph 5.2) and 65% ethanol. Quant-iT™ PicoGreen® dsDNA Assay Kit was used to quantify DNA. 3C ligation products were measured by Taqman quantitative PCR technology (Hagege et al., 2007). 3C results between experiments were first normalized using the α-amylase gene. Calculation was Xspecific primer = 2Ct (α-amylase - specific primer). To determine primer efficiency, equal moles of bacterial artificial chromosomes covering the genomic region under study, RP23-351J19 (231.6 kb, 5.55 µg), RP23-269D13 (202.8 kb, 4.86 µg), and RP23-363G23 (152.2 kb, 3.65µg) were mixed, digested with HindIII, re-ligated, and measured by Taqman quantitative PCR technology. Calculation of primer efficiency relative to the Eμ-VH81X was Especific primer = 2Ct (Eμ-VH81X - specific primer). 3C results between experiments were additionally normalized with respect to primer efficiency. Calculation was Relative Interaction= Xspecific primer / Especific primer.
Fluorescent in situ hybridization (FISH)
FISH was performed as described (Guo et al., 2011a). Briefly, cells were fixed on poly-L-lysine coated slides at a concentration of 5×106 cells/ml using 200,000 cells per slide. Slides were kept for 15 minutes at 37°C, washed in PBS, followed by fixation with 4% paraformaldehyde in PBS. Cells were further washed with 0.1 M Tris-HCl (pH 7.4), followed by PBS. Fixed cells were treated with 100 µg/ml RNase A in PBS, permeabilized using 0.5% saponin/0.5 % Triton-X 100/PBS for 30 min at room temperature, and rinsed in PBS. DNA probes were denatured at 75°C for 5 min and applied to cells that were denatured in formamide at 73°C followed by incubation in a dark humid chamber.
Position-specific 3-10kb probes were generated by PCR using BAC templates or genomic DNA with the primers listed in Table S3. Images were acquired using a Nikon T200 microscope equipped with a 100× lens and motorized 100μm Piezo Z-axis stage (Applied Scientific Instrumentation). Depending on the size of the nucleus, 30–40 serial optical sections spaced by 0.2μm were acquired. The data sets were deconvolved using NIS-Elements software (Nikon). Statistical analyses of spatial distance measurements were carried out using a two-sample Kolmogorov-Smirnov test in R as previously described (Gerasimova et al., 2015). 100 or more distance measures were analyzed by Kolmogorov-Smirnov statistical analyses (https://www.wessa.net/rwasp_Reddy-Moores%20K-S%20Test.wasp).
YY1 and CTCF knockdown
YY1 and CTCF shRNA lentiviral plasmids were purchased from Sigma-Aldrich. The empty vector pLKO.1 was used as the control. Virus preparation and infection were carried out according to Addgene protocol (https://www.addgene.org/tools/protocols/plko/). Briefly, shRNA and packaging plasmid (psPAX and pMD2.G) were added into 293T with Bio-T. Lentivirus were harvested by high speed centrifugation (25000rpm, 2 hour) after three days of culture. After infection, cells were cultured in puromycin (2µg/ml) for 5 days, and then harvested for RNA, protein and FISH analyses.
Rag2 expression
Lentiviral particles expressing Rag2 were generated as described (Kutner et al., 2009) by transiently transfecting 293T cells with lentiviral plasmid containing Rag2 and puromycin resistance DNA fragment (pHIV-RAG2-IRES-puro) along with helper plasmids pMD2.G and psPAX2 using Bio-T reagent. pHIV-IRES-puro was used as control. The supernatant containing the virus was collected at 72h after transfection and concentrated by ultracentrifugation for 2h at 25,000 rpm and 20°C over a 20% sucrose cushion. Supernatant was removed after ultracentrifugation, and 200µl PBS were added to the tube. Fresh virus was prepared to transduction. All procedures involving lentiviruses were performed under BSL2 conditions.
DJH/VDJH recombination assays
Genomic DNA was purified from sorted bone marrow pro-B (IgM−B220+CD43+) pro-B cells from WT or IGCR1−/− mice. Fivefold serial dilutions of genomic DNA (200ng, 40ng, 8ng) were used to perform PCR to analyze DJH rearrangements. Primers used in this assay are listed in Table S3. Primers flanking the ROSA26 gene were used as a loading control under the same conditions. GeneRuler 100bp Plus, 100 bp DNA Ladder, or 1 Kb Plus DNA Ladder were used to confirm PCR product size. VH7183-DJH1 rearrangements in pro-B cells were amplified with LongAmp® Taq DNA Polymerase, cloned into pGEM®-T vector, transformed into MAX Efficiency® DH5α competent cells and sequenced. The DH family was identified according to NCBI Genbank (http://www.ncbi.nlm.nih.gov/igblast/showGermline.cgi?organism=mouse&chainType=DH&seqType=nucleotide&functionClass=1). For quantitation, ethidium bromide-stained agarose gels were scanned and analyzed for gray density with GeneTools software from Syngene.
RAG2-deficient pro-B cell lines carrying IGCR1-mutated alleles (CBE−/−(1) and (2)) or WT IgH alleles (WT) were infected with RAG2-expressing lentivirus, and were cultured in complete medium with puromycin (2µg/ml). After 4 days (CBE−/−(2)), 5 days (CBE−/−(1)) or 14 days (CBE−/−(1), CBE−/−(2), CBE−/−(2)/Eμ−/−(1), CBE−/−(2)/Eμ−/−(2), Eμ−/−) selection with puromycin, cells were harvested. Genomic DNA was collected with DNeasy Blood & Tissue Kit, and the DNA was used to analyze DJH/VDJH rearrangements with HotStarTaq DNA Polymerase as described as above.
VDJH deep sequencing assays
VDJH deep sequencing assays were performed as previously described (Hu et al., 2016). Briefly, 40ng genomic DNA from each sample was sonicated to an average size of 750bp. Sonicated DNA was hybridized with Bio-JH1 primer, purified with Dynabeads C1 streptavidin beads and used for library generation as described (Lin et al., 2016). Paired-end reads were generated by Illumina HiSeq 2500 sequencer. Samples were separated using barcodes present in Read-1 (Table S4). Adapters, if present were removed by cutadapt and bad quality bases (< Q33) were trimmed from Read-2 keeping a minimum length of 50 bases. The reads (Read-2 only) were aligned to 600 bases of different V-regions using bowtie2. Reads which aligned to different V-regions with an alignment quality > 20 were counted. Genome build utilized mm9 IgH V-regions.
Chromatin Immunoprecipitation (ChIP)
ChIP analysis of modified histones and RAG1 and RAG2 was done as described (Chakraborty et al., 2007; Ji et al., 2010). 20×106 cells were resuspended in 9ml RPMI containing 2% serum in a 15ml conical flask, followed by 1ml of 10% HCHO and rotated at room temperature for 15 min. Glycine was added to terminate the crosslinking to a final concentration of 0.125M and rocked for 5 min at room temperature. Cells were washed twice with 10 ml cold PBS (2K, 4 min, 4°C). After washes, cell pellet was resuspended in 300 µl RIPA buffer (10 mM Tris pH 7.4, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 0.8M NaCl), and incubated on ice for 10 min. Chromatin was sonicated using a water bath sonicator (Diagenode) (5 × 5 min, high power) resulting in DNA of 300 to 700 bp. Samples were centrifuged (14K, 10 min, 4°C) and the supernatant transferred to a new 1.5ml tube. 40 µg Dynabeads Protein G (Thermofisher cat# 1003D) was washed with 1ml RIPA buffer for preclearing the chromatin. The chromatin was then added to the protein G beads and rotated for 1 hr at 4°C. Beads were removed using magnet and the supernatant was transferred to a new tube containing second batch of protein G beads and rotated for 1 hr at 4°C. Supernatant was transferred to a new tube. 10% chromatin was kept to be used as input. Precleared chromatin was divided equally into two tubes. To each tube heparin was added to final concentration of 500 ng/ml and gelatin from cold water to a final concentration 2%. One tube was incubated with 5µg anti-RAG1/RAG2 and another tube with 5 µg normal rabbit IgG as a control and rocked at 4C overnight. Dynabeads protein G (preblocked with 2% BSA and heparin (500 ng/ml) for 1 hr at RT) were added to the tube and rotated for 3 hr at 4°C, centrifuged (7K, 2 min, 4°C), and the beads were washed sequentially (2×10 min, 4°C) with 1 ml RIPA (0.3 M NaCl), 1 ml RIPA (0.8 M NaCl), LiCl buffer (0.25M LiCl, 0.5% NP-40, 0.5% NaDOC), followed by one wash with 1ml of TE + 0.2 % Triton X-100 and finally once with 1 ml TE. Beads were resuspended in 100 µl TE containing 50 µg Proteinase K and 0.25% SDS and incubated at 65°C for 5h. The “input” chromatin was treated identically. After phenol/chloroform extraction, the aqueous phase was adjusted to 0.2M NaCl and DNA precipitated with 20 µg glycogen and 1 ml ethanol. The DNA pellet was washed with 70% ethanol and resuspended in water. The primers which are used to detect the IgH locus and gamma actin promoter are listed in Table S3. qPCR was performed in duplicates using SYBr green in applied biosystem. A standard curve generated for each primer set was used to quantitate DNA recovered in pull down sample and the input chromatin. IP/Inputcorr was calculated as ((IPsp – IPrIg)/Input) × 1000, where IPsp and IPrig are the amount of DNA recovered in IPs with the specific antibody and rabbit IgG as described (Ji et al., 2010).
Histone ChIP (anti-H3K4me3, anti-H3K9ac, anti-H3K9me2), were carried out as described above. After DNA precipitation, real time PCR was carried out with 200pg of immunoprecipitated or input DNA. The abundance of specific sequences in the immunoprecipitate relative to that in input DNA was calculated as previously described (Chakraborty et al., 2007). Calculation is Relative Abundance (specific amplicon) = 2Ct (input-IP). Oligonucleotides used for real-time PCR analysis are provided in Table S3.
CTCF ChIP-Seq
CTCF ChIP-Seq was extracted from (Lin et al., 2012) (GEO: GSM987805). Direction of CTCF was analyzed with software designed by Yan Cui at University of Tennessee Health Science Center (http://insulatordb.uthsc.edu/), and determined by using higher score and better match as criteria.
RNA isolation, RT–PCR and RNA-Seq
Total RNA was isolated using RNeasy Plus Mini Kit. 1µg RNA was used to generate cDNA with Superscript III with random hexamers according to manufacturer’s protocols. Approximately 1/40 of the reverse-transcription-generated cDNA was analyzed with iTaq™ Universal SYBR. Primers that were used for PCR are provided in Table S3. Briefly, the RNA-Seq library was made with the Illumina TruSeq Stranded mRNA sample preparation kit following the manufacturer’s recommended procedure, and the resulting libraries were QCed using Fragment Analyzer and pooled together. The pooled library concentration was further quantified using KAPA library quantification qPCR kit, and sequenced on Illumina NextSeq 500 using a High Output 75 cycle sequencing kit targeting 20–30 million single reads of 75bp. For each RNA-seq sample, the sequencing reads were mapped to mouse genome mm9 using TopHat with default settings. Signal tracks were generated using bedtools and UCSC Genome Browser software. Signals in each sample were normalized to total number of reads and then scaled by multiplying a constant N (N=100,000,000).
CRISPR/Cas9 System
pHIV-U6-gRNA-Cas9-Blast plasmid was generated by insertion of U6 promoter and gRNA DNA fragment into lentiCas9-Blast. Two different gRNA sequences, targeting one DNA locus, were separately cloned into plasmid lentiCRISPR v2 and pHIV-U6-gRNA-Cas9-Blast as previously described (Sanjana et al., 2014) with appropriate primer (Table S3), followed by transformation into One Shot® Stbl3™ competent cells. Lentiviral particles were generated and harvested according to protocol (Sanjana et al., 2014). CBE−/−(1), CBE−/−(2) or D345 cell lines were infected with freshly prepared gRNA/Cas9 expressing lentivirus. Cells were cultured with the presence of both puromycin (2µg/ml) and blasticidin (15µg/ml) for 4 days, followed by single cell cloning. Single cell clones were collected, and genomic DNA was purified with Quick-DNA™ Universal 96 Kit. PCR was employed for detection of deletion of Eμ (CBE−/−(2)) or IGCR1 (D345) with appropriate primer (Table S3). CBE−/−(2)/Eμ−/−(1), CBE−/−(2)/Eμ−/−(2), D345/IGCR1−/−(1) and D345/IGCR1−/−(2), were harvested and used for analysis of RNA and or RAG1 ChIP. CBE−/−(1)/C1−/−, CBE−/−(1)/C2−/− and CBE−/−(1)/C1−/−C2−/− were used for VDJH deep sequencing assays.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis, including D-statistics and P-values, were carried out for FISH data (Figure 1C, 2B, 6B). Detailed statistical analysis result for FISH is listed on Table S1.
DATA AND SOFTWARE AVAILABILITY
Raw data of images are deposited in Mendeley (http://dx.doi.org/10.17632/5f23hhtpnj.1). Raw data for RNA-Seq and VDJ-Seq are deposited at NCBI (GSE109631 and GSE110090). We also provide five supplemental data files (Tables S1–5). Software for Kolmogorov-Smirnov test can be used online (https://www.wessa.net/rwasp_Reddy-Moores%20K-S%20Test.wasp). RNA-Seq analyses were carried out using TopHat (https://ccb.jhu.edu/software/tophat/index.shtml), Bedtools (http://bedtools.readthedocs.io/en/latest/) and UCSC Genome Browser software (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/). VDJ-Seq analyses were carried out using Cutadapt (http://cutadapt.readthedocs.io/en/stable/guide.html) and Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml).
Supplementary Material
Acknowledgments
We thank Frederick W. Alt, Sherry Lin and Suvi Jain (Harvard Medical School) for generously providing IGCR1-mutated cell lines and sorted bone marrow pro-B cells from IGCR1-deleted mice, and for critiquing the manuscript. Antibodies against RAG1 and RAG2 were generously provided by Elizabeth Corbett and David G. Schatz (Yale University), and we thank Amy L. Kenter (University of Illinois College of Medicine) for comments on the manuscript. Marc Raley (NIDA) contributed to preparation of the figures. This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS X.Q. and R.S. conceived and designed the research. X.Q., G.K., T.G., H.D., L.L. and A.S. performed experiments. X.Q., S.D., W.H.W., K.G.B., W.Z. and H.J analyzed the data. X.Q. and R.S. interpreted the data and results, and wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Afshar R, Pierce S, Bolland DJ, Corcoran A, Oltz EM. Regulation of IgH gene assembly: role of the intronic enhancer and 5'DQ52 region in targeting DHJH recombination. J Immunol. 2006;176:2439–2447. doi: 10.4049/jimmunol.176.4.2439. [DOI] [PubMed] [Google Scholar]
- Bossen C, Mansson R, Murre C. Chromatin topology and the regulation of antigen receptor assembly. Annu Rev Immunol. 2012;30:337–356. doi: 10.1146/annurev-immunol-020711-075003. [DOI] [PubMed] [Google Scholar]
- Chakraborty T, Chowdhury D, Keyes A, Jani A, Subrahmanyam R, Ivanova I, Sen R. Repeat organization and epigenetic regulation of the DH-Cmu domain of the immunoglobulin heavy-chain gene locus. Mol Cell. 2007;27:842–850. doi: 10.1016/j.molcel.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Chakraborty T, Perlot T, Subrahmanyam R, Jani A, Goff PH, Zhang Y, Ivanova I, Alt FW, Sen R. A 220-nucleotide deletion of the intronic enhancer reveals an epigenetic hierarchy in immunoglobulin heavy chain locus activation. J Exp Med. 2009;206:1019–1027. doi: 10.1084/jem.20081621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Carico Z, Shih HY, Krangel MS. A discrete chromatin loop in the mouse Tcra-Tcrd locus shapes the TCRdelta and TCRalpha repertoires. Nat Immunol. 2015;16:1085–1093. doi: 10.1038/ni.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhao L, Alt FW, Krangel MS. An Ectopic CTCF Binding Element Inhibits Tcrd Rearrangement by Limiting Contact between Vdelta and Ddelta Gene Segments. J Immunol. 2016;197:3188–3197. doi: 10.4049/jimmunol.1601124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi NM, Loguercio S, Verma-Gaur J, Degner SC, Torkamani A, Su AI, Oltz EM, Artyomov M, Feeney AJ. Deep sequencing of the murine IgH repertoire reveals complex regulation of nonrandom V gene rearrangement frequencies. J Immunol. 2013;191:2393–2402. doi: 10.4049/jimmunol.1301279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci U S A. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell. 2007;25:43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Ebert A, McManus S, Tagoh H, Medvedovic J, Salvagiotto G, Novatchkova M, Tamir I, Sommer A, Jaritz M, Busslinger M. The distal V(H) gene cluster of the Igh locus contains distinct regulatory elements with Pax5 transcription factor-dependent activity in pro-B cells. Immunity. 2011;34:175–187. doi: 10.1016/j.immuni.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Fuxa M, Skok J, Souabni A, Salvagiotto G, Roldan E, Busslinger M. Pax5 induces V-to-DJ rearrangements and locus contraction of the immunoglobulin heavy-chain gene. Genes Dev. 2004;18:411–422. doi: 10.1101/gad.291504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova T, Guo C, Ghosh A, Qiu X, Montefiori L, Verma-Gaur J, Choi NM, Feeney AJ, Sen R. A structural hierarchy mediated by multiple nuclear factors establishes IgH locus conformation. Genes Dev. 2015;29:1683–1695. doi: 10.1101/gad.263871.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloborodko A, Imakaev MV, Marko JF, Mirny L. Compaction and segregation of sister chromatids via active loop extrusion. Elife. 2016;5 doi: 10.7554/eLife.14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Gerasimova T, Hao H, Ivanova I, Chakraborty T, Selimyan R, Oltz EM, Sen R. Two forms of loops generate the chromatin conformation of the immunoglobulin heavy-chain gene locus. Cell. 2011a;147:332–343. doi: 10.1016/j.cell.2011.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, Hansen E, Despo O, Bossen C, Vettermann C, et al. CTCF-binding elements mediate control of V(D)J recombination. Nature. 2011b;477:424–430. doi: 10.1038/nature10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G, de Laat W, Forne T. Quantitative analysis of chromosome conformation capture assays (3C-qPCR) Nat Protoc. 2007;2:1722–1733. doi: 10.1038/nprot.2007.243. [DOI] [PubMed] [Google Scholar]
- Hesslein DG, Pflugh DL, Chowdhury D, Bothwell AL, Sen R, Schatz DG. Pax5 is required for recombination of transcribed, acetylated, 5' IgH V gene segments. Genes Dev. 2003;17:37–42. doi: 10.1101/gad.1031403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Meyers RM, Dong J, Panchakshari RA, Alt FW, Frock RL. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat Protoc. 2016;11:853–871. doi: 10.1038/nprot.2016.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Zhang Y, Zhao L, Frock RL, Du Z, Meyers RM, Meng FL, Schatz DG, Alt FW. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell. 2015;163:947–959. doi: 10.1016/j.cell.2015.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Resch W, Corbett E, Yamane A, Casellas R, Schatz DG. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell. 2010;141:419–431. doi: 10.1016/j.cell.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- Kumari G, Sen R. Chromatin Interactions in the Control of Immunoglobulin Heavy Chain Gene Assembly. Adv Immunol. 2015;128:41–92. doi: 10.1016/bs.ai.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc. 2009;4:495–505. doi: 10.1038/nprot.2009.22. [DOI] [PubMed] [Google Scholar]
- Lin SG, Ba Z, Du Z, Zhang Y, Hu J, Alt FW. Highly sensitive and unbiased approach for elucidating antibody repertoires. Proc Natl Acad Sci U S A. 2016;113:7846–7851. doi: 10.1073/pnas.1608649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SG, Guo C, Su A, Zhang Y, Alt FW. CTCF-binding elements 1 and 2 in the Igh intergenic control region cooperatively regulate V(D)J recombination. Proc Natl Acad Sci U S A. 2015;112:1815–1820. doi: 10.1073/pnas.1424936112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, Murre C. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat Immunol. 2012;13:1196–1204. doi: 10.1038/ni.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Schmidt-Supprian M, Shi Y, Hobeika E, Barteneva N, Jumaa H, Pelanda R, Reth M, Skok J, Rajewsky K, et al. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007;21:1179–1189. doi: 10.1101/gad.1529307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JS, Zhang Y, Dudko OK, Murre C. 3D trajectories adopted by coding and regulatory DNA elements: first-passage times for genomic interactions. Cell. 2014;158:339–352. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder K, Koues OI, Chan EA, Kyle KE, Horowitz JE, Yang-Iott K, Bassing CH, Taniuchi I, Krangel MS, Oltz EM. Lineage-specific compaction of Tcrb requires a chromatin barrier to protect the function of a long-range tethering element. J Exp Med. 2015;212:107–120. doi: 10.1084/jem.20141479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Tamir IM, Schwickert TA, Novatchkova M, Sun Q, Huis In 't Veld PJ, Guo C, Yoon HS, et al. Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity. 2013;39:229–244. doi: 10.1016/j.immuni.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlot T, Alt FW, Bassing CH, Suh H, Pinaud E. Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc Natl Acad Sci U S A. 2005;102:14362–14367. doi: 10.1073/pnas.0507090102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. Cohesin Loss Eliminates All Loop Domains. Cell. 2017;171:305–320 e324. doi: 10.1016/j.cell.2017.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawat P, Jalan M, Sadhu A, Kanaujia A, Srivastava M. Chromatin Domain Organization of the TCRb Locus and Its Perturbation by Ectopic CTCF Binding. Mol Cell Biol. 2017;37 doi: 10.1128/MCB.00557-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retter I, Chevillard C, Scharfe M, Conrad A, Hafner M, Im TH, Ludewig M, Nordsiek G, Severitt S, Thies S, et al. Sequence and characterization of the Ig heavy chain constant and partial variable region of the mouse strain 129S1. J Immunol. 2007;179:2419–2427. doi: 10.4049/jimmunol.179.4.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- Subrahmanyam R, Du H, Ivanova I, Chakraborty T, Ji Y, Zhang Y, Alt FW, Schatz DG, Sen R. Localized epigenetic changes induced by DH recombination restricts recombinase to DJH junctions. Nat Immunol. 2012;13:1205–1212. doi: 10.1038/ni.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma-Gaur J, Torkamani A, Schaffer L, Head SR, Schork NJ, Feeney AJ. Noncoding transcription within the Igh distal V(H) region at PAIR elements affects the 3D structure of the Igh locus in pro-B cells. Proc Natl Acad Sci U S A. 2012;109:17004–17009. doi: 10.1073/pnas.1208398109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpi SA, Verma-Gaur J, Hassan R, Ju Z, Roa S, Chatterjee S, Werling U, Hou H, Jr, Will B, Steidl U, et al. Germline deletion of Igh 3' regulatory region elements hs 5, 6, 7 (hs5-7) affects B cell-specific regulation, rearrangement, and insulation of the Igh locus. J Immunol. 2012;188:2556–2566. doi: 10.4049/jimmunol.1102763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Frock RL, Du Z, Hu J, Chen L, Krangel MS, Alt FW. Orientation-specific RAG activity in chromosomal loop domains contributes to Tcrd V(D)J recombination during T cell development. J Exp Med. 2016;213:1921–1936. doi: 10.1084/jem.20160670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.