

Abstract
Background and Purpose
In this study, we examined the possibility that 4‐hydroxynonenal (4‐HNE) acting as a ligand for the HCA2 receptor (GPR109A) elicits both anti‐inflammatory and cell death responses.
Experimental Approach
Agonistic activity of 4‐HNE was determined by observing the inhibition of cAMP generation in CHO‐K1‐GPR109A‐Gi cell line, using surface plasmon resonance (SPR) binding and competition binding assays with [3H]‐niacin. 4‐HNE‐mediated signalling pathways and cellular responses were investigated in cells expressing GPR109A and those not expressing these receptors.
Key Results
Agonistic activity of 4‐HNE was stronger than that of niacin or 3‐OHBA at inhibiting forskolin‐induced cAMP production and SPR binding affinity. In ARPE‐19 and CCD‐841 cells, activation of GPR109A by high concentrations of the agonists 4‐HNE (≥10 μM), niacin (≥1000 μM) and 3‐OHBA (≥1000 μM) induced apoptosis accompanied by elevated Ca2+ and superoxide levels. This 4‐HNE‐induced cell death was blocked by knockdown of GPR109A or NOX4 genes, or treatment with chemical inhibitors of Gβγ (gallein), intracellular Ca2+ (BAPTA‐AM), NOX4 (VAS2870) and JNK (SP600125), but not by the cAMP analogue 8‐CPT‐cAMP. By contrast, low concentrations of 4‐HNE, niacin and 3‐OHBA down‐regulated the expression of pro‐inflammatory cytokines IL‐6 and IL‐8. These 4‐HNE‐induced inhibitory effects were blocked by a cAMP analogue but not by inhibitors of Gβγ‐downstream signalling molecules.
Conclusions and Implications
These results revealed that 4‐HNE is a strong agonist for GPR109A that induces Gαi‐dependent anti‐inflammatory and Gβγ‐dependent cell death responses. Moreover, the findings indicate that specific intracellular signalling molecules, but not GPR109A, can serve as therapeutic targets to block 4‐HNE‐induced cell death.
Abbreviations
- 3‐OHBA
β‐D‐hydroxybutyric acid
- 4‐HNE
4‐hydroxynonenal
- 6‐CFDA
6‐carboxyfluroscein diacetate
- 8‐CPT‐cAMP
8‐(4‐chlorophenylthio)adenosine‐cAMP
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐(3‐diphenyl)tetrazolium bromide
- PTx
pertussis toxin
- RPE
retinal pigment epithelium
Introduction
The hydroxycarboxylic acid http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=312 (GPR109A) was first discovered as an orphan GPCR that is primarily expressed in adipocytes and immune cells and was later identified as the niacin (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1588) receptor (Soga et al., 2003; Tunaru et al., 2003; Wise et al., 2003). Previous studies on GPR109A investigated the mechanisms by which high doses of niacin can cause side effects such as flushing. Since then, functional GPR109A have been identified in various types of cells in the spleen, colon and retinal pigment epithelium (RPE) (Wise et al., 2003; Martin et al., 2009; Thangaraju et al., 2009). Individuals who do not take niacin for the treatment of hyperlipidaemia have low levels of circulating niacin, which are not sufficient to activate GPR109A. 3‐Hydroxybutyric acid (3‐OHBA, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1593), the first ketone produced in the fasting state, has been identified as the endogenous ligand for GPR109A and is known to activate the receptor at relatively high concentrations (Taggart et al., 2005).
Niacin‐induced GPR109A activation has been demonstrated to elicit anti‐inflammatory effects by down‐regulating the expression of inflammatory cytokines and chemokines in epithelial cells (Digby et al., 2012; Gambhir et al., 2012). Studies using gene knockdown of GPR109A in the RPE, which is a monolayer of postmitotic cells underlying the photoreceptors in the retina, confirmed the anti‐inflammatory action of GPR109A and suggested that it is a promising target for the treatment of inflammation in diabetic retinopathy (Gambhir et al., 2012; Zimmerman et al., 2012). In contrast to its protective effects, activation of GPR109A has also been observed to induce apoptosis in neutrophils (Kostylina et al., 2008), while patients administered a high dose of niacin developed maculopathy (Gass, 1973; Millay et al., 1988; Domanico et al., 2015). These reports indicate that GPR109A activation by different concentrations of niacin may exert dual effects, namely, induce apoptosis and anti‐inflammatory responses. An anti‐inflammatory role of the GPR109A via Gβγ/PKC‐dependent ERK1/2 activation is shown in macrophages (Shi et al., 2017). However, in RPE, although an anti‐inflammatory role of GPR109A is mediated via suppression of NF‐κB activity, it is not clear such activity is mediated through the Gα or Gβγ pathway (Gambhir et al., 2012). In addition, the molecular mechanisms of how GPR109A exerts dual responses (anti‐inflammatory and cell death) in RPE have not been investigated in detail, or which response is mediated through which component of the receptor, Gαi or Gβγ signalling.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6274) can be produced endogenously via membrane lipid peroxidation (Poli et al., 2008) or exogenously via the peroxidation of plasma low‐density lipoproteins (Stadtmanl, 1994) and dietary polyunsaturated lipids (Pierre et al., 2006). 4‐HNE is produced in the RPE owing to its strong metabolic activity during high oxygen consumption and high levels of polyunsaturated fatty acids during the phagocytosis of photoreceptor outer segments, which is required to facilitate the proper turnover of the photoreceptors (Bok, 1993; Vingerling et al., 1996; Ethen et al., 2007). High 4‐HNE levels lead to the apoptotic degeneration of RPE, which is one of the molecular basis for age‐related macular degeneration, and eventually results in the formation of adducts with photoreceptor outer segment proteins, causing an accumulation of non‐degradable proteins (Krohne et al., 2010; Sparrow and Boulton, 2005). In addition, 4‐HNE is associated with various disease conditions, such as chronic inflammation, cardiac ischaemia, atherogenesis, neurodegenerative diseases and cancer (Dalleau et al., 2013; Csala et al., 2015). Importantly, 4‐HNE produces different cellular responses depending on its concentration. 4‐HNE appears to be beneficial to cells at low concentrations (around 0.1–5.0 μM) by inducing cell proliferation and differentiation, as well as the activation of antioxidant defence mechanisms. By contrast, at high concentrations (around 10–20 μM), 4‐HNE triggers apoptosis via both the intrinsic and extrinsic pathways. Similarly to other inducers of intrinsic apoptosis, 4‐HNE disrupts mitochondrial ATPase activity (Ji et al., 2001; Siddiqui et al., 2012). Extrinsic apoptosis induced by 4‐HNE is reported to be mediated through the induction of http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1875 signalling (Li et al., 2006; Salomoni and Khelifi, 2006; Sharma et al., 2008), possibly due to the formation of 4‐HNE adducts with cell surface Fas receptors, which mimics ligand–cell surface receptor binding (Negre‐Salvayre et al., 2003; Sharma et al., 2008). Considering that 4‐HNE is produced at relatively high levels in the RPE, where a wide range of niacin concentrations can activate GPR109A to trigger opposite responses, the dual action of 4‐HNE suggests the possible involvement of GPR109A ; however, this possibility has not been studied.
In the present study, we demonstrated that GPR109A mediates the two different responses induced by 4‐HNE by comparing the effects of 4‐HNE with those of niacin and 3‐OHBA. In addition, we investigated whether specific signalling pathways and signalling molecules downstream of GPR109A are responsible for eliciting anti‐inflammatory or apoptotic responses depending on the concentration of its agonist.
Methods
Cell culture
The human retinal pigmented epithelial cell line ARPE‐19 and the normal colon epithelial cell line CCD‐841 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The HEK cell line HEK‐293 and CHO cell line CHO‐K1 were obtained from the Korean cell line bank (Seoul, Korea). ARPE‐19 cells were maintained in DMEM/Ham's nutrient mixture F‐12 supplemented with 10% FBS and 1% penicillin/streptomycin (P/S). CCD‐841 and HEK‐293 cells were cultured in minimal essential medium, whereas CHO‐K1 cells were grown in RPMI 1640 medium. All media were supplemented with 10% FBS and 1% P/S. The cAMP Hunter™ CHO‐K1‐GPR109A‐Gi cell line was purchased from DiscoverX (Cat. No. 95‐0122C2; San Diego, CA, USA). Cells were maintained in AssayComplete™ CHO‐K1 medium supplemented with AssayComplete™ serum and antibiotics mix (P/S/glutamine and Geneticin®), which were procured from DiscoverX. Cells were cultured according to the manufacturer's protocol and incubated at 37°C in a 5% CO2 humidified incubator. The culture medium was changed every other day. After they had reached confluence, cells were subcultured after trypsinization with 0.25% trypsin/EDTA solution (ARPE‐19 and CCD‐841 cells) or AssayComplete™ cell detachment reagent (CHO‐K1‐GPR109A‐Gi cells) by splitting at a ratio of 1:3.
Western blotting
For the preparation of whole‐cell lysates, radioimmunoprecipitation assay buffer (150 mM sodium chloride, 1% Triton™ X‐100, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris, adjusted to pH 8.0) was added along with 1× protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA, USA) in cell pellets. Cytoplasmic and nuclear proteins were extracted using an NE‐PER® nuclear and cytoplasmic extraction reagent kit (Thermo Fisher Scientific). After lysis, proteins were quantified using a BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of proteins were loaded and separated on SDS polyacrylamide gels and transferred onto nitrocellulose membranes. Afterwards, the membranes were blocked for 1 h using 5% (w.v‐1) BSA diluted in Tris‐buffered saline containing 0.1% Tween® 20 (TBST). Then, membranes were incubated overnight with the specific primary antibody at 1:1000 dilution. After being washed three times with 1× TBST, membranes were incubated at room temperature for 1 h with a specific secondary antibody conjugated with HRP at 1:5000 dilution. After being washed three times with 1× TBST, membranes were treated with enhanced chemiluminescence reagents (Thermo Fisher Scientific) and subsequently visualized using a luminescence image analyzer (LAS‐4000 Mini; Fujifilm, Tokyo, Japan).
Cell viability assay
Cell viability was measured by the colorimetric 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐(3‐diphenyl)tetrazolium bromide (MTT) staining method. Cells were seeded in 96‐well plates at a density of 2 × 104 cells per well. After 24 h, cells were incubated with the indicated reagent for the indicated duration at the indicated concentrations. Then, 10 μL of MTT (5 g MTT L−1 in H2O) was added to each well, and the cells were incubated for 4 h at 37°C in a 5% CO2 humidified incubator. Finally, 200 μL of DMSO was added to each well and mixed to dissolve the reduced MTT crystals. Relative cell viability was determined by measuring the absorbance using a 540 nm filter (Molecular Devices, Sunnyvale, CA, USA).
Nuclear staining with Hoechst 33258 and 6‐carboxyfluroscein diacetate
Apoptotic cells were stained with DNA‐specific fluorescence probe Hoechst 33258. Cells were seeded at 5 × 104 cells per well in six‐well plates and treated with 4‐HNE for the indicated time periods. Next, cells were stained with Hoechst 33258 and 6‐carboxyfluroscein diacetate (6‐CFDA) for 20 min and subsequently washed with 1× PBS. Stained cells were visualized under a fluorescence microscope (IX73; Olympus, Tokyo, Japan).
Measurement of apoptosis
Cell apoptosis was detected using a FITC‐annexin V apoptosis detection kit I (BD Biosciences, San Jose, CA, USA) following the manufacturer's protocol. Briefly, ARPE‐19 cells were treated with 4‐HNE for the indicated times and concentrations. Then, the cells were trypsinized, pelleted by centrifugation and washed twice with cold PBS. Cells were suspended in 1× binding buffer at a density of 1 × 106 cells mL−1. The cell suspension (100 μL) was transferred to a culture tube, followed by staining with 5 μL of FITC annexin V and 5 μL of propidium iodide (PI). Cells were further incubated for 15 min at room temperature (25°C) in the dark. Then, 400 μL of 1× binding buffer was further added before the analysis of each sample by flow cytometry on a BD FACSVerse® flow cytometer (BD Biosciences).
Caspase 3/7 activity
A Caspase‐Glo® 3/7 assay kit (Promega, Madison, WI, USA) was used to determine http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619/http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1623 activity according to the manufacturer's instructions. Briefly, ARPE‐19 cells were seeded in a 96‐well plate at a seeding density of 2 × 104 cells per well. After overnight incubation, cells were treated with the indicated concentrations of 4‐HNE for the indicated time periods. Caspase 3/7 reagent was added to each well and incubated for 30 min. Finally, luminescence from the cleaved luminogenic substrate Ac‐DEVD‐pNA was measured on a FLUOstar® Optima microplate reader (BMG Labtech, Offenburg, Germany).
siRNA transfection
ARPE‐19 and CCD‐841 cells were seeded in 60 mm dishes at a density of 3.3 × 104 cells cm−2. After overnight incubation, transfection was performed using DharmaFECT™ 4 transfection reagent with 100 nM of GPR109A, NOX4 or non‐targeting (NT) siRNA (Bioneer, Daejeon, Korea), following the manufacturer's instructions. After transfection, cells were subjected to cell viability assay, cAMP assay, ROS measurement, caspase activity and Western blot analysis. The transfection efficiencies of the respective siRNAs were assessed via Western blotting. Sequences of the siRNA oligos for the target genes were as follows: human GPR109A seq. 1 (GPR109A‐1): sense‐5′‐GGACAACUAUGUGAGGCGU‐3′ and antisense‐5′‐ACGCCUCACAUAGUUGUCC‐3′; GPR109A seq. 2 (GPR109A‐2): sense‐5′‐CAGAUUCAGAGAAUGCGAU‐3′ and antisense‐5′‐AUCGCAUUCUCUGAAUCUG‐3′; human GPR109B (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=313) seq. 1 (GPR109B‐1): sense‐5′‐CUCACAUGCUUUGGUUAGU‐3′ and antisense‐5′‐ACUAACCAAAGCAUGUGAG‐3′; GPR109B seq. 2 (GPR109B‐2): sense‐5′‐CUACUAUGUGCGGCGUUCA‐3′ and antisense‐5′‐UGAACGCCGCACAUAGUAG‐3′; and human NOX4: sense‐5′‐CAGAGUUUACCCAGCACAA‐3′ and antisense‐5′‐UUGUGCUGGGUAAACUCUG‐3.
cAMP measurement
ARPE‐19 cells (1 × 105 cells cm−2 in 24‐well plates) were stimulated with the indicated concentrations of 4‐HNE with or without http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190 for the indicated time periods after transient transfection of the cells with GPR109A or NT siRNA. Dishes were kept on ice and washed with ice‐cold PBS to terminate the reaction. A cAMP kit (R&D Systems, Minneapolis, MN, USA) was used to measure intracellular cAMP concentrations according to the manufacturer's instructions.
Intracellular Ca2+ measurement
Intracellular Ca2+ levels were monitored using a calcium‐sensitive fluorescence indicator, Fura‐2/AM (the membrane‐permeable acetoxymethyl ester of Fura‐2). Cells were seeded on black 96‐well plates at a density of 2 × 104 cells per well. After overnight incubation, cells were washed with warm PBS, and 3 μM Fura‐2/AM was added. Cells were then incubated for 50 min at 37°C. Afterwards, unloaded Fura‐2/AM was removed from cells, followed by washing with Locke's solution. To prevent the leakage of Fura‐2/AM, 250 μM sulfinpyrazone was added to each well and further incubated for 30 min at 37°C. Next, the cells were pretreated with the drugs in serum‐free media with or without 4‐HNE. Fura‐2 fluorescence was measured at the excitation wavelengths of 340 and 380 nm and the emission wavelength of 510 nm using a FLUOstar Optima microplate reader (BMG Labtech). Changes in the 340/380 nm absorbance ratios reflect the changes in Ca2+ ion concentrations.
Lucigenin chemiluminescence assay
A lucigenin chemiluminescence assay was performed following a previously published method (Banskota et al., 2017). Briefly, cells were seeded in white 96‐well plates at a density of 5 × 104 cells per well and treated with 4‐HNE, niacin or 3‐OHBA in assay buffer (pH 7.4) containing 130 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.5 mM CaCl2, 35 mM phosphoric acid and 20 mM HEPES in the absence or presence of SOD for an indicated time period. Chemiluminescence was measured after the addition of 400 μM lucigenin at 37°C using a FLUOstar Optima microplate reader (BMG Labtech). Chemiluminescence produced by lucigenin alone was subtracted from the chemiluminescence values obtained from the treated group.
mRNA copy number determination
Transcript numbers for NOX1, NOX2 and NOX4 were determined based on previously described methods (Banskota et al., 2015). Briefly, human NOX1, NOX2 and NOX4 cDNAs (Invitrogen, Carlsbad, CA, USA) were cloned into the pcDNA5/FRT/TO vector (Invitrogen). The number of transcripts was calculated using the following equation:
Standard curves for NOX1, NOX2 and NOX4 were generated using serially diluted plasmids (copy numbers from 101 to 105). After treating the cells with or without 4‐HNE for 3 h, cDNA was isolated from total RNA using TRIzol reagent (Invitrogen) and subsequently subjected to PCR using a QuantiTect® SYBR® Green PCR kit (Qiagen, Hilden, Germany) with primer sequences specific to NOX1, NOX2 and NOX4. The following primer sequences were used: NOX1 (sense 5′‐GTTTTACCGCTCCCAGCAGA‐3′ and antisense 5′‐GGATGCCATTCCAGGAGAGA‐3′); NOX2 (sense 5′‐CCTAAGATAGCGGTTGATGG‐3′ and antisense 5′‐GACTTGAGAATGGATGCGAA‐3′); and NOX4 (sense 5′‐CAGAAGGTTCCA AGCAGG AG‐3′ and antisense 5′‐GTTGAGGGCATTCACCAG AT‐3′).
Measurement of IL‐6 and IL‐8 levels
ARPE‐19 and CCD‐841 cells were seeded and treated with 4‐HNE at the indicated concentrations for the indicated time periods. Following treatment, cell culture supernatants were collected and centrifuged to remove particulates. Supernatants were analysed for the determination of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998 or IL‐8 ( http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=821 ) levels using specific elisa kits (R&D Systems).
GPR109A receptor functional assay using CHO‐K1‐GPR109A‐Gi cell line
To determine the activities of 4‐HNE, niacin and 3‐OHBA binding towards the GPR109A receptor, we used a HitHunter® cAMP Assay detection kit for small molecules (Cat. No. 90‐0075SM2; DiscoverX). The experiment was performed according to the manufacturer's protocol. Briefly, cAMP Hunter CHO‐K1‐GPR109A‐Gi cells were seeded at a density of 30 000 cells per well in 96‐well plates. After 24 h of seeding, serial dilutions of the agonists (4‐HNE, niacin or 3‐OHBA) were prepared in 1× PBS containing http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5190 (15 μM). Serial dilutions of the agonist were added to the respective wells of the assay plate and incubated for 30 min at 37°C in a 5% CO2 humidified incubator. Following incubation with the agonist, cAMP antibody reagent and cAMP working detection solutions were added and incubated for 1 h at room temperature under dark conditions for the immunocompetition reaction. Then, cAMP solution A was added to the assay plate. After 3 h of incubation at room temperature, luminescence signals were detected on a FLUOstar Optima microplate reader (BMG Labtech).
Surface plasmon resonance binding assay
Surface plasmon resonance (SPR) binding assay was performed using ProteOn XPR36 instrument (Bio‐Rad Laboratory, Hercules, CA, USA) in Gyeonggi Biocenter (Suwon, Korea). The GLH biosensor chip was selected for the experiment. Purified GPR109A (OriGene, Rockville, MD, USA) was directly immobilized onto the biosensor chip surface through amine coupling. After ligand immobilization, solution of analyte (4‐HNE or niacin) at an indicated concentration was injected at a speed of 100 μL·min−1 and allowed to flow over the immobilized GPR109A surface. PBS solution containing 0.05% TBST (pH 7.4) was used as running and analyte‐binding buffer. The binding response of analyte was recorded as response units. Binding kinetics and data analysis were performed using BioRad ProteOn manager software. The K D (equilibrium K D) value was calculated by using the formula:
[3H]‐niacin binding assay
Membranes of HEK‐293T cells stably expressing human GPR109A were used to perform [3H]‐niacin (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1594) equilibrium displacement assay. At first, the membranes were incubated with 15 nM of [3H]‐niacin (specific activity: 50 Ci·mmol−1), (Biotrend, Cologne, Germany) for 1 h at 25°C. Then, niacin or 4‐HNE at different concentrations in assay buffer (50 mM Tris HCL and 1 mM MgCl2, pH 7.4 at 25°C) was added using a HP D300 digital dispenser (Tecan Group Ltd, Männedorf, Switzerland). Non‐specific binding was determined with 10 μM unlabelled niacin. Finally, the reaction was terminated by rapid vacuum filtration, which separates free and bounded radioligand through 96‐well GF/B filter plates using a PerkinElmer Filtermate‐harvester (PerkinElmer, Groningen, The Netherlands). Then the radioactivity retained in the filter was measured by P‐E 1450 Microbeta Wallac Trilux scintillation counter (PerkinElmer).
Mutant GPR109A cloning and mutagenesis
The genes encoding human GNA15 (GenBank accession number AAA35860.1) and human GPR109A (GenBank accession number AAN71621.1) were artificially synthesized (Bionics, Seoul, Korea). Human GNA15 gene digested by HindIII/XhoI restriction enzymes (Thermo Fisher Scientific) was inserted into pcDNA4/TO vector (Invitrogen) for the expression of wild‐type GNA15 protein. GPR109A gene was digested by NotI/XbaI and was then inserted into pcDNA4/TO‐FLAG vector for the expression of GPR109A having an N‐terminal FLAG epitope. Point mutations were introduced into GPR109A by site‐directed mutagenesis method. The pcDNA4/TO‐GPR109A plasmid was amplified using two primers containing a point mutation. After elimination of the template plasmid with DpnI, the PCR product was transformed into Escherichia coli NEB‐turbo competent cell [New England Biolabs (NEB), Ipswich, MA, USA]. Point mutations (R111A, R253A, W256A, F277A and L280A in GPR109A) were confirmed by DNA sequencing. All the transfection‐grade plasmids were prepared using NucleoBond‐Xtra Midi kit (Macherey‐Nagel, Duren, Germany).
Overexpression of GPR109A mutant receptors in CHO‐K1 cells
CHO‐K1 cells seeded in six‐well plates (2 × 104 cells cm−2) were co‐transfected with pcDNA4‐hGNA15 and N‐terminally FLAG‐tagged WT or mutant GPR109A receptors (R111A, R253A, W256A, F277A or L280A) using Lipofectamine 2000 (Invitrogen) as transfection reagent. After 24 h of transfection, CHO‐K1 cells were either subjected to FLAG staining or trypsinized and seeded to perform MTT or HitHunter cAMP assay.
Determination of mutant receptor expression in CHO‐K1 cells
After 24 h of transfection with the indicated N‐terminally FLAG‐tagged GPR109A mutant receptors, CHO‐K1 cells were washed once with cold PBS and fixed with 4% formaldehyde solution in PBS at room temperature for 20 min. Then the cells were washed again with PBS and blocked for 40 min in 4% BSA in PBS at room temperature followed by the incubation with the FLAG antibody (Sigma‐Aldrich) (at 1:200 dilution in PBS) for 3 h at room temperature. After incubation, cells were washed three times with PBS and were labelled with anti‐mouse secondary antibody (Alexa Fluor® 647) (Abcam) for 1 h. Then, the cells were washed once with PBS, and stained cells were visualized with a fluorescence microscope (IX73; Olympus).
Statistical analysis
Data were obtained from more than five independent experiments and expressed as mean ± SEM. Values were statistically evaluated by Student's t‐test or one‐way ANOVA, followed by the Newman–Keuls comparison method, using Prism 5.0 software (GraphPad, San Diego, CA, USA). P values of less than 0.05 were considered statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
All reagents were procured from Sigma‐Aldrich (St. Louis, MO, USA) unless otherwise specified. Reagents used for the cell culture of all cells used in the experiments were obtained from HyClone Laboratories (Logan, UT, USA). 4‐HNE was purchased from Cayman Chemical Co. (CAS No. 75899‐68‐2; Ann Arbor, MI, USA). FBS, antimycin A, penicillin/streptomycin (P/S) and TRIzol® were purchased from Life Technologies (Carlsbad, CA, USA). DharmaFECT™ 4 was purchased from Dharmacon (Lafayette, CO, USA). Gallein and SR11302 were purchased from Tocris Bioscience (Bristol, UK). c‐Fos antibody was procured from Santa Cruz Biotechnology (Dallas, TX, USA). Monoclonal antibody against human GPR109A was obtained from Novus Biological (Cat. No. NBP2‐12358AF488; Littleton, CO, USA). Anti‐IL‐6 and anti‐IL‐8 antibodies were obtained from Abbiotec (San Diego, CA, USA). Antibodies against Bcl‐2, Bax, phospho‐p38 MAPK, p38 MAPK, phospho‐JNK, JNK, c‐Jun, ERK and NF‐κB were purchased from Cell Signaling Technology, Inc. (Boston, MA, USA). NOX4 (Cat. No. ab109225), lamin B and β‐actin antibodies were purchased from Abcam (Cambridge, UK).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2017), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b, 2017c).
Results
GPR109A activation by 4‐HNE, niacin or 3‐OHBA induces apoptosis in ARPE‐19 and CCD‐841 cells
To investigate whether GPR109A activation induces bipolar responses, we first examined the dose‐dependent cytotoxic responses to GPR109A activation by its known agonists (niacin and 3‐OHBA) in cells that exhibit strong (ARPE‐19), moderate (CCD‐841) and nearly undetectable (HEK‐293) GPR109A expression (Figure 1A). The viability of ARPE‐19 cells exhibited differential dose‐ and time‐dependent responses to niacin (Figure 1B). ARPE‐19 cells did not show changes in viability upon treatment with low niacin concentrations (up to 100 μM) but showed significant decreases in viability at niacin concentrations higher than 300 μM. 3‐OHBA induced stronger decreases in cell viability than niacin (Figure 1B). The inhibitory effects of niacin and 3‐OHBA on cell viability were also observed in CCD‐841 cells (Figure 1C). 4‐HNE induced a stronger inhibitory effect than niacin or 3‐OHBA, and its effective cytotoxic concentration was 100 times lower than that of niacin and 3‐OHBA in both ARPE‐19 (Figure 1D) and CCD‐841 (Figure 1E) cells. In contrast to the cytotoxic effects of 4‐HNE in ARPE‐19 and CCD841 cells, 4‐HNE did not induce any cytotoxic effect in HEK‐293 cells that do not express GPR109A (Figure 1F). In contrast, 4‐HNE was cytotoxic in HEK‐293‐GPR109A cells overexpressing human GPR109A (Figure 1F), indicating the important role of GPR109A in 4‐HNE‐induced cell death. A similar response was observed in CHO‐K1 parental and GPR109A‐overexpressing cells (Figure 1F). The observed decrease in cell viability upon 4‐HNE treatment in ARPE‐19 cells was consistent with the observed apoptosis in the cells, which was determined based on the increased uptake of Hoechst 33258 (a DNA‐binding fluorescent probe) and a reduction in 6‐CFDA accumulation in viable cells (Figure 1G). Cell apoptosis was also determined by an increase in the annexin V/PI double‐positive cell fraction (Figure 1H and Supporting Information Figure S1A, B) and an increase in caspase‐3/7 activity (Figure 1I). We also examined whether the differentiation status of ARPE‐19 cells, such as confluent (differentiated) or subconfluent (less differentiated), influences GPR109A expression and cytotoxic response to 4‐HNE. Cytotoxic response to 4‐HNE (Supporting Information Figure S2A) or 3‐OHBA (Supporting Information Figure S2B) in confluent ARPE‐19 cells was slightly lower than that in subconfluent cells (Figure 1B, D). Similarly, mRNA (Supporting Information Figure S2C) and protein (Supporting Information Figure S2D) expression levels of GPR109A were slightly lower than that in subconfluent ARPE‐19 cells.
Figure 1.
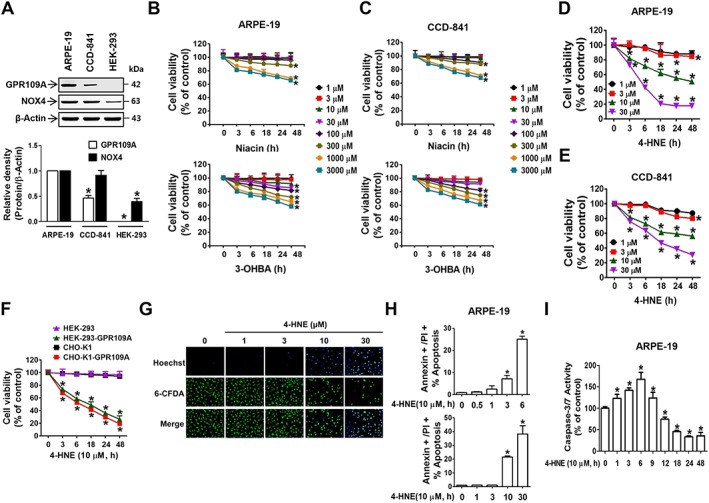
GPR109A activation by 4‐HNE, niacin and 3‐OHBA induces apoptotic death in ARPE‐19 and CCD‐841 cells. (A) GPR109A and NOX4 expression levels were analysed by immunoblotting. Bar graphs represent averaged quantitations of immunoblots from six independent experiments. (B–F) Cell viability was measured via MTT assay. (G) ARPE‐19 cells were stained with Hoechst 33258, a water‐soluble DNA‐binding compound, and 6‐carboxyfluroscein diacetate, a secondary dye that accumulates in viable cells. (H) Annexin V‐positive and PI‐positive ARPE‐19 cell populations were determined via flow cytometry. (I) Caspase‐3/7 activity was measured in 4‐HNE‐treated ARPE‐19 cells using a Caspase‐Glo® 3/7 assay kit. * P < 0.05 versus vehicle‐treated controls.
Next, we examined whether 4‐HNE‐induced cell death was mediated via GPR109A. The 4‐HNE‐induced decrease in cell viability was abolished upon siRNA‐mediated GPR109A knockdown in ARPE‐19 (Figure 2A, B) and CCD‐841 (Figure 2C, D) cells, whereas the 4‐HNE‐induced cytotoxicity was unchanged upon knockdown of GPR109B, a receptor that shares about 90% homology with GPR109A (Figure 2E, F). The specificity of siRNAs for GPR109A and GPR109B was confirmed by detecting mRNA levels of both genes after transfection via qRT‐PCR using specific probe PCR primers (Supporting Information Figure S3A). Two different sequences of siRNAs were used for targeting each receptor, and both sequences exerted the same effects. In all the subsequent experiments, we used the GPR109A‐1 siRNA sequence. Similar to the effects observed in 4‐HNE‐treated cells, GPR109A knockdown suppressed the niacin‐ and 3‐OHBA‐induced decrease in viability in both ARPE‐19 (Supporting Information Figure S3B) and CCD‐841 (Supporting Information Figure S3C) cells.
Figure 2.
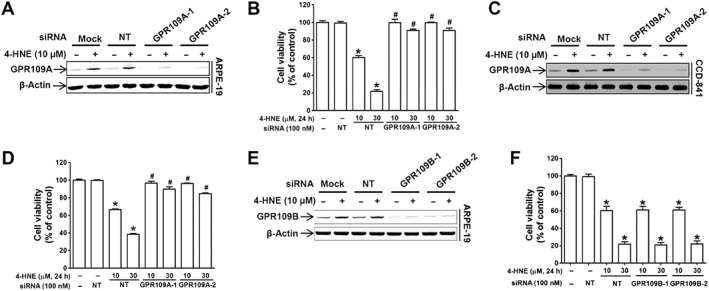
4‐HNE is an agonist for GPR109A. (A, C and E) Transfection efficiency of two different sequences of the indicated siRNAs measured by use of Western blots. (B, D and F) ARPE‐19 cell viability in the absence or presence of 4‐HNE (10 or 30 μM) for 24 h was measured in cells transfected with two different siRNAs targeting GPR109A or GPR109B. * P < 0.05 versus non‐targeting siRNA‐transfected controls. # P < 0.05 versus cells treated with 10 or 30 μM 4‐HNE.
4‐HNE‐induced ARPE‐19 cell death is mediated through Gβγ activation but not through the Gαi signalling pathway
We further examined the role of 4‐HNE in GPR109A activation by investigating the signalling pathways responsible for cell death in ARPE‐19 cells. As shown in Figures 3A and 4, HNE‐induced cell death was blocked by pretreatment with pertussis toxin (PTx), which is an inhibitor of Gi, but not by cholera toxin, which is a Gs activator. In addition, the 4‐HNE‐induced suppression of cAMP production by forskolin was reversed by GPR109A knockdown (Figure 3B). However, treatment with 8‐(4‐chlorophenylthio)adenosine‐cAMP (8‐CPT‐cAMP, a cAMP analogue) did not block 4‐HNE‐induced cell death (Figure 3C), whereas treatment with gallein (Gβγ inhibitor), BAPTA‐AM (intracellular Ca2+ chelator), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5283 (PLC inhibitor) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5193 (PKC inhibitor) blocked 4‐HNE‐induced cell death (Figure 3D). As indicated by the observed inhibitory effect of BAPTA‐AM on 4‐HNE‐induced cell death, 4‐HNE induced a significant increase in intracellular Ca2+ levels at a minimum concentration of 10 μM (Figure 3E). Similarly, at concentrations above 300 μM, niacin and 3‐OHBA both induced dose‐dependent increases in intracellular Ca2+ levels (Supporting Information Figure S4). The observed 4‐HNE‐induced elevation in Ca2+ levels was inhibited by transfection with a siRNA targeting GPR109A (Figure 3F) and pretreatment with gallein (Figure 3G) or U73122 (Figure 3H).
Figure 3.
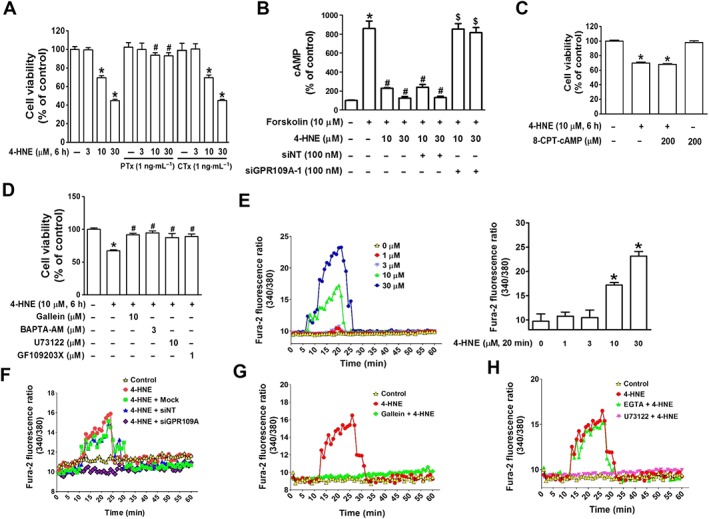
Gβγ‐mediated increase in intracellular Ca2+ levels is associated with 4‐HNE‐induced cell death in ARPE‐19 cells. (A) Cell viability was measured in cells pretreated with PTx or cholera toxin prior to 4‐HNE treatment for 6 h. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus cells treated with 10 or 30 μM 4‐HNE. (B) cAMP was measured using a cAMP detection kit in the presence or absence of non‐targeting or GPR109A siRNA. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus forskolin‐treated cells. $ P < 0.05 versus cells treated with forskolin and 10 or 30 μM 4‐HNE. Cell viability measurement in cells pretreated with either (C) 8‐CPT‐cAMP or (D) inhibitors of Gβγ‐downstream molecules * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus 4‐HNE‐treated cells. (E) The increase in intracellular Ca2+ levels was measured using a Fura‐2 probe. Bars represent means ± SEM from six independent experiments. * P < 0.05 versus vehicle‐treated controls. The 4‐HNE‐induced increase in intracellular Ca2+ levels was blocked by treatment with (F) GPR109A siRNA transfection, (G) gallein or (H) U73122.
Figure 4.
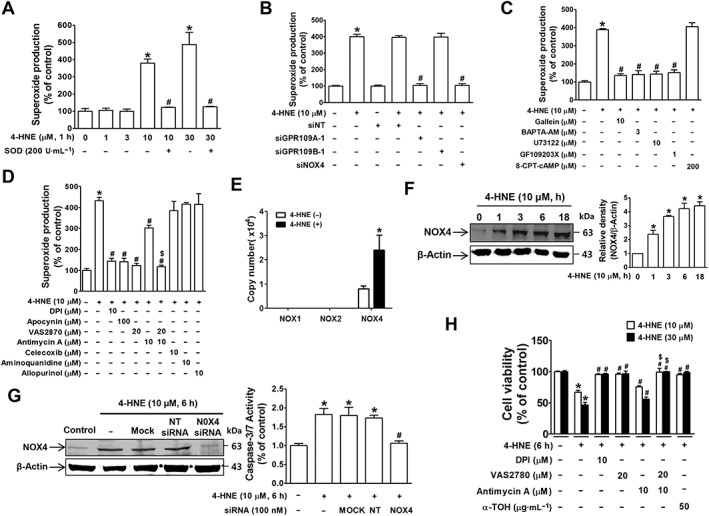
NOX4 mediates 4‐HNE‐induced superoxide production and cell death in ARPE‐19 cells. 4‐HNE induced ROS production, which was quenched by (A) pretreatment with SOD, (B) siRNA knockdown of GPR109A, GPR109B or NOX4, (C) treatment with inhibitors of Gβγ‐downstream molecules and (D) treatment NADPH oxidase inhibitors. * P < 0.05 versus vehicle‐treated or non‐targeting siRNA‐transfected controls, # P < 0.05 versus cells treated with 10 or 30 μM 4‐HNE. (E) Copy numbers of the NADPH oxidase subunits NOX1, NOX2 or NOX4 in ARPE‐19 cells were measured by qRT‐PCR. * P < 0.05 versus vehicle‐treated controls. (F) Western blotting results for NOX4 induction in 4‐HNE‐treated cells. * P < 0.05 versus vehicle‐treated controls. (G) Caspase‐3/7 activity was measured in ARPE‐19 cells pretreated with NOX4 siRNA prior to 4‐HNE treatment. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus non‐targeting siRNA‐transfected cells. (H) Cell viability measurements in cells treated with 4‐HNE in the presence of NOX4 and mitochondrial inhibitors or antioxidants. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus 4‐HNE‐treated cells. $ P < 0.05 versus 4‐HNE plus antimycin A‐treated cells.
Consistent with a previous report, 4‐HNE produced SOD‐quenchable superoxide anion (Dianzani et al., 1996) in a concentration‐dependent manner at a minimum concentration of 10 μM in ARPE‐19 cells (Figure 4A). Similar to differences in cytotoxic effects on confluent and subconfluent ARPE‐19 cells, 4‐HNE‐induced superoxide production in confluent cells (Supporting Information Figure S2E) was increased to threefold, which was smaller than the fourfold increase in subconfluent cells (Figure 4A). 4‐HNE‐induced superoxide anion production was significantly blocked by GPR109A knockdown (Figure 4B) and BAPTA‐AM treatment (Figure 4C) but not by GPR109B knockdown (Figure 4B), indicating that the GPR109A‐Gβγ‐dependent intracellular Ca2+ signalling pathway is linked to 4‐HNE‐induced ROS production. Similarly, gallein, U73122 and GF109203X blocked the 4‐HNE‐induced increase in ROS generation, whereas 8‐CPT‐cAMP had no effect (Figure 4C). Furthermore, we found that 4‐HNE‐induced superoxide anion production was almost completely blocked by treatment with diphenyleneiodonium chloride (inhibitor of flavin‐containing oxidases, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3041 oxidase), apocynin (superoxide scavenging antioxidant), VAS2870 (NOX2/4 inhibitor) or a combined treatment with VAS2870 and antimycin A (mitochondrial electron transport inhibitor), while antimycin A alone partially suppressed the increase in superoxide anion levels (Figure 4D). However, inhibitors of other enzymes, such as COX (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2892), NOS (aminoquinidine) and xanthine oxidase (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6795), had no effect. The expression of NOX4, the NADPH oxidase isoform predominantly expressed in ARPE‐19 cells (Figure 4E), was up‐regulated by 4‐HNE treatment in a time‐dependent manner (Figure 4F). In addition, NOX4 knockdown completely suppressed 4‐HNE‐induced superoxide generation (Figure 4B) and caspase‐3/7 activity (Figure 4G). Similar to the effects of inhibitors on superoxide anion inhibition, 4‐HNE‐induced cell death was more effectively suppressed by a combined treatment with VAS2870 and antimycin A than with either VAS2870 or antimycin A alone (Figure 4H).
JNK‐induced activation of AP‐1, but not NF‐κB, is responsible for 4‐HNE‐induced cell death
In 4‐HNE‐treated ARPE‐19 cells, p38‐MAPK and JNK were activated, while ERK1/2 was not (Figure 5A), and only treatment with a JNK inhibitor (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5273) blocked 4‐HNE‐induced cell death (Figure 5B). JNK activation was blocked by treatment with U73122, BAPTA‐AM, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7802 and apocynin (Figure 5C). While exploring the downstream signalling molecules that are activated following JNK activation, we found that the nuclear levels of c‐Fos and c‐Jun, which comprise AP‐1, were both increased upon treatment with 10 μM 4‐HNE but not with 1 μM 4‐HNE in both ARPE‐19 and CCD‐841 cells (Figure 5D). By contrast, 4‐HNE treatment inhibited NF‐κB nuclear translocation in a concentration‐dependent manner in both cell lines (Figure 5D), and the changes in the nuclear levels of c‐Fos, c‐Jun and NF‐κB were blocked by transfection with a siRNA targeting GPR109A (Figure 5D). In addition, treatment with an AP‐1 inhibitor (SR11302), but not an NF‐κB inhibitor [pyrrolidine dithiocarbamate (PDTC)], blocked 4‐HNE‐induced cell death (Figure 5B, E).
Figure 5.
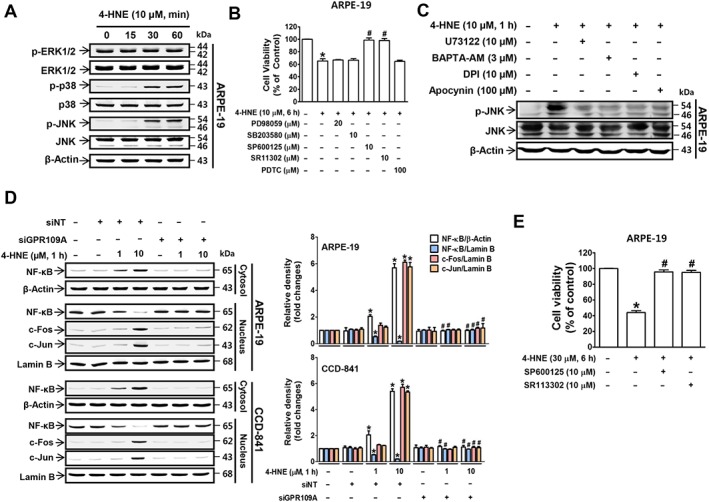
JNK‐activated AP‐1 mediates 4‐HNE‐induced cell death. (A) Immunoblotting results for MAPKs, ERK1/2, p38 and JNK. (B) 4‐HNE‐induced changes in cell viability in cells pretreated with MAPK inhibitors. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus 4‐HNE‐treated cells. (C) Immunoblotting results of JNK phosphorylation. (D) Nuclear translocation of c‐Fos, c‐Jun and NF‐κB was analysed by immunoblotting. * P < 0.05 versus non‐targeting siRNA‐transfected controls. # P < 0.05 versus cells treated with 1 or 10 μM 4‐HNE. (E) 4‐HNE‐induced changes in cell viability in cells pretreated with an AP‐1 inhibitor. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus 4‐HNE (10 or 30 μM)‐treated cells.
4‐HNE and other GPR109A agonists suppress the expression of inflammatory cytokines through Gαi‐dependent NF‐κB inhibition
Previous studies have shown that GPR109A elicits an anti‐inflammatory response by down‐regulating the expression of inflammatory cytokines. Thus, we also examined the effects of 4‐HNE on the expression levels of the inflammatory cytokines IL‐6 and IL‐8. 4‐HNE suppressed IL‐6 and IL‐8 expression in a concentration‐ and time‐dependent manner in both ARPE‐19 (Figure 6A) and CCD‐841 (Figure 6B) cells similarly to niacin (Supporting Information Figure S5A) and 3‐OHBA (Supporting Information Figure S5B). The 4‐HNE‐induced reduction in IL‐6 and IL‐8 levels was blocked by the siRNA knockdown of GPR109A, which was contrasted by increased expression of Bax and GPR109A by 4‐HNE (Figure 6C). Unlike the observed involvement of AP‐1 in 4‐HNE‐induced cell death, treatment with the AP‐1 inhibitor SR11302 did not affect the 4‐HNE‐induced changes in the expression levels of inflammatory cytokines, whereas treatment with PDTC (NF‐κB inhibitor) potentiated the 4‐HNE‐induced down‐regulation of IL‐6 and IL‐8 levels (Figure 6D). Treatment of the cells with the cAMP analogue 8‐CPT‐cAMP significantly blocked the 4‐HNE‐induced inhibition of NF‐κB translocation to the nucleus (Figure 6E) and the down‐regulation of IL‐6 and IL‐8 expression (Figure 6F). However, treatment with inhibitors of Gβγ signalling molecules (BAPTA‐AM, VAS2870, SP600125, PD98059 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5269) did not have those effects (Figure 6E, F). In addition, the 4‐HNE‐induced up‐regulation of GPR109A and NOX4 protein expression was significantly inhibited by treatment with SR11302 (Figure 6D), BAPTA‐AM, VAS2870 and SP600125 (Figure 6F). These inhibitory effects of 4‐HNE on IL‐6 and IL‐8 expressions were not altered by GPR109B knockdown (Supporting Information Figure S5C), and were reversed by PTx treatment (Supporting Information Figure S5E). Compared with subconfluent cells (Figure 6C, D), effects of 4‐HNE on IL‐6 and IL‐8 expression in confluent ARPE‐19 cells (Supporting Information Figure S2F) were slightly reduced similar to its cytotoxic effects (Supporting Information Figure S2A), and the expression of receptors and NOX4 (Supporting Information Figure S2C, D).
Figure 6.
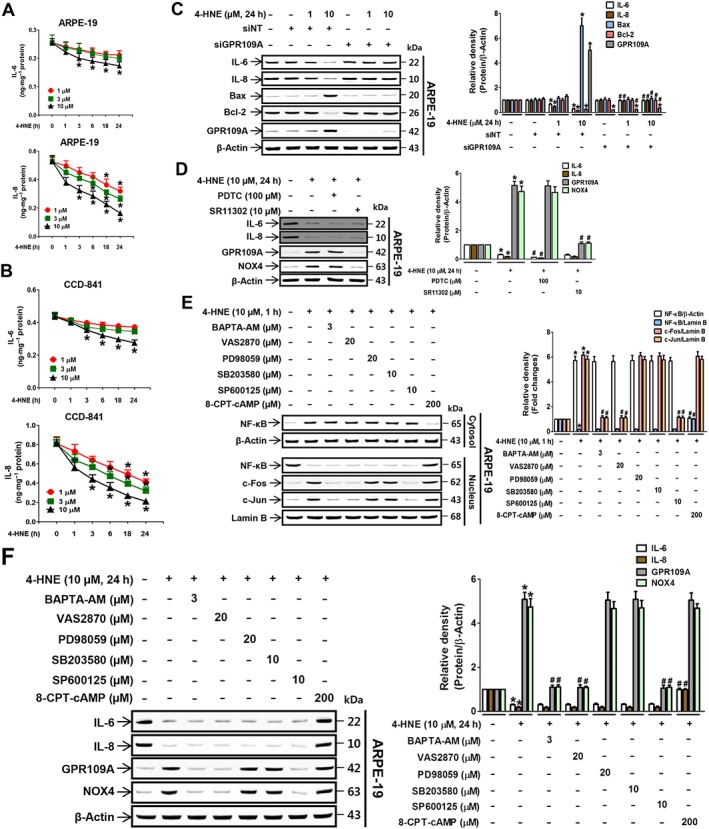
4‐HNE inhibits IL‐6 and IL‐8 expression through the Gαi‐dependent NF‐κB pathway. Secretion of the pro‐inflammatory cytokines IL‐6 and IL‐8 in (A) ARPE‐19 and (B) CCD‐841 cells was measured by elisa. * P < 0.05 versus vehicle‐treated controls. 4‐HNE‐induced expression of IL‐6, IL‐8, Bax, Bcl‐2 and GPR109A was measured in ARPE‐19 cells in the presence of (C) non‐targeting or GPR109A siRNA or (D) inhibitors of NF‐κB and AP‐1. * P < 0.05 versus vehicle‐targeting or non‐targeting siRNA‐transfected controls. # P < 0.05 versus cells treated with 1 or 10 μM 4‐HNE. (E) Nuclear translocation of the transcription factors NF‐κB, c‐Fos and c‐Jun, and (F) expression levels of IL‐6, IL‐8, GPR109A and NOX4 were measured in ARPE‐19 cells pretreated with 8‐CPT‐cAMP or inhibitors of intracellular Ca2+ (BAPTA‐AM), NOX4 (VAS2870), ERK1/2 (PD98059), p38 (SB203580) and JNK (SP600125) prior to 4‐HNE treatment. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus 4‐HNE‐treated cells.
4‐HNE is not competing with niacin for binding sites in GPR109A
Considering that the cytotoxic and anti‐inflammatory effects of 4‐HNE were better than niacin or 3‐OHBA, and these effects were blocked by GPR109A knockdown, we examined the possibility that 4‐HNE is a strong agonist for GPR109A by comparing its effects with those of niacin and 3‐OHBA in a receptor function study and receptor‐binding assay. The inhibition of forskolin‐induced cAMP production by niacin, 3‐OHBA and 4‐HNE were assayed using th eCHO‐K1‐GPR109A‐Gi cell line (Figure 7A) that was specifically engineered to detect the change in cAMP generation upon the binding of a small molecule to GPR109A. The EC50 values of niacin, 3‐OHBA and 4‐HNE were 105.9 (±1.9), 86.7 (±3.4) and 61.4 (±1.8) nM, respectively, indicating that 4‐HNE may be the most potent ligand for GPR109A. To exclude the possibility that the inhibitory effect of 4‐HNE on cAMP might be due to decrease in cell viability, we measured the cytotoxicity of 4HNE in CHO‐K1‐GPR109A‐Gi cells. Treatment of CHO‐K1‐GPR109A‐Gi cells with the drugs for the same period of time as in cAMP measurement did not induce cytotoxicity (Figure 7B). To confirm direct binding of 4‐HNE to GPR109A, SPR binding assay was performed. The SPR binding affinity of 4‐HNE (0.84 ± 0.1 mM) to purified GPR109A immobilized onto the biosensor chip surface was about sixfold stronger than that of niacin (5.28 ± 0.83 mM) (Figure 7C). To examine whether 4‐HNE competes with niacin for a binding site in GPR109A, we performed a competitive receptor binding assay of 4‐HNE with [3H]‐niacin. 4‐HNE did not inhibit or replace the specific binding of [3H]‐niacin to GPR109A (Figure 7D), indicating that 4‐HNE did not compete with niacin for a binding site in GPR109A. To confirm this binding site difference, we prepared plasmids of five different GPR109A mutant substituting tentative interacting amino acid residues with alanine (R111A, R253A, W256A, F277A and L280A) based on a previous report (Tunaru et al., 2005), and the mutant genes were introduced into CHO‐K1 cells. The transfection efficiency of the FLAG‐tagged plasmids was confirmed via FLAG staining in CHO‐K1 cells (Figure 7E). For functional analysis on the mutants (Figure 7F), the changes in forskolin‐induced cAMP level upon stimulation with 4‐HNE or niacin were measured. R253A and W256A mutation did not alter the receptor response to either niacin or 4‐HNE, while the F277A mutation suppressed the receptor response, by half, to both the niacin and 4‐HNE treatment. The most significant change in receptor response to 4‐HNE was observed in the L280A mutation, which was followed by the R111A mutation. However, in the case of the receptor response to niacin, the R111A mutation totally blocked the response, while it was minimally altered in the L280A mutant. Because the competitive receptor binding assay with [3H]‐niacin and functional study on GPR109A and its mutants with 4‐HNE and niacin showed that the binding site of 4‐HNE and niacin is not the same, we further confirmed that these chemicals have different binding sites by examining the additive activity of the combination treatment with 4‐HNE in the presence of niacin or 3‐OHBA. Using CHO‐K1‐GPR109A‐Gi cells, it was found that co‐treatment of the cells with combination of either 4‐HNE plus niacin or 4‐HNE plus 3‐OHBA induced significant enhancement of cAMP activity compared with each chemical treatment given alone, while co‐treatment with niacin plus 3‐OHBA produced the same activity as that of 3‐OHBA alone (Figure 7G). From intracellular Ca2+ (Figure 7H) and cell viability (Figure 7I) measurements using ARPE‐19 cells, 4‐HNE in combination with niacin or 3‐OHBA tended to enhance its activity, but no enhanced activity was observed for the combination of niacin plus 3‐OHBA.
Figure 7.
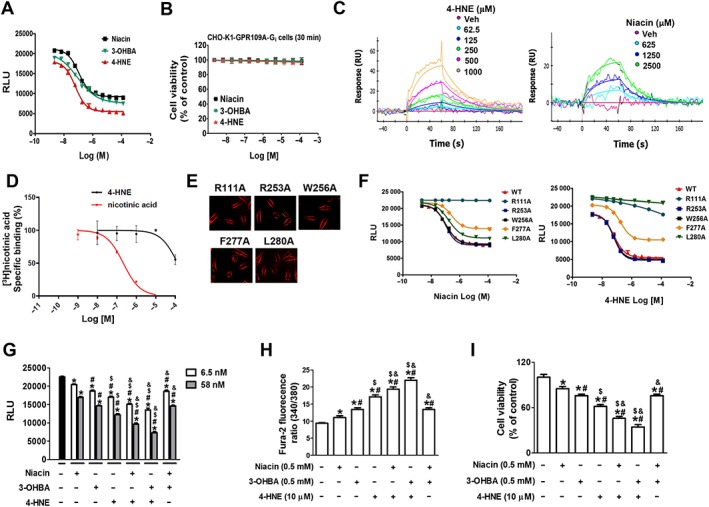
4‐HNE is a stronger agonist than niacin, not competing with niacin for binding sites in GPR109A. (A) A functional activity assay using the CHO‐K1‐GPR109A‐Gi cell line, which was designed to detect the inhibition of cAMP generation, was performed in the cells pre‐stimulated with forskolin prior to treatment with the other agonists. (B) 4‐HNE, niacin, 3‐OHBA did not induce cytotoxicity in CHO‐K1‐GPR109A‐Gi cells. (C) SPR binding assay showing 4‐HNE and niacin bind to GPR109A in a concentration‐dependent manner. (D) [3H]‐niacin binding competition assay with 4‐HNE and niacin showing their effects are concentration‐dependent in HEK‐293T cells stably expressing human GPR109A. (E and F) CHO‐K1 cells were transfected with five different FLAG‐tagged plasmids of GPR109A mutants (R111A, R253A, W256A, F277A and L280A). (E) Indicates their transfection efficiency as demonstrated by FLAG staining and (F) shows the results of the functional activity assay indicating that 4‐HNE and niacin decreased the forskolin‐induced production of cAMP in a concentration‐dependent manner. (G–I) Effects of combination treatments with the GPR109A ligands (niacin, 3‐OHBA or 4‐HNE). (G) CHO‐K1‐GPR109A cells were treated with a combination of two from three ligands, and forskolin‐induced cAMP levels were measured. In ARPE‐19 cells, effects of combination treatments on changes in (H) intracellular Ca2+ levels and (I) cell viability were determined. * P < 0.05 versus vehicle‐treated controls. # P < 0.05 versus niacin‐treated cells. $ P < 0.05 versus 3‐OHBA‐treated cells. & P < 0.05 versus 4‐HNE‐treated cells. RU, response units.
Discussion
4‐HNE elicited stronger cellular responses than niacin or 3‐OHBA, which are known exogenous and endogenous GPR109A agonists respectively. More importantly, GPR109A activation was shown to elicit dual responses in the same cells, namely, anti‐inflammatory responses and cell apoptosis. These results provide evidence that GPR109A may not be a suitable therapeutic target for the management of retinal inflammation (Gambhir et al., 2012). Rather, our present study identified the signalling molecules that mediate the 4‐HNE‐induced anti‐inflammatory response without causing cell death. The results further suggest that not GPR109A, but intracellular signalling molecules can serve as therapeutic targets to block retinal inflammation or to inhibit the development of 4‐HNE‐mediated cell death.
The RPE consumes large amounts of oxygen and generates endogenous 4‐HNE during the physiological degradation of photoreceptor outer segments, whereas the intestinal epithelium is a target organ of the paracrine action of 4‐HNE, which is generated during food digestion or during phagocytosis by macrophages (Goicoechea et al., 2011). Our present study revealed that low levels of 4‐HNE did not exert cytotoxic effects on ARPE‐19 (a representative RPE cell line) and CCD‐841 (a representative colon epithelial cell line), whereas high levels of 4‐HNE induced cell death. Consistent with our present results using ARPE and CCD‐841 cells, human neuronal and fibroblast cells were demonstrated to be sensitive to 4‐HNE at concentrations greater than 5 μM (Kutuk et al., 2004; Abarikwu et al., 2012). In these cells, 4‐HNE‐induced apoptosis has been linked to an enhanced mRNA and protein expression of Bax and caspase‐3 due to increased mitochondrial oxidative stress (Abarikwu et al., 2012; Raza and John, 2006). In addition, our results showed that cell death and the increased Bax/Bcl‐2 ratio and caspase‐3 activity induced by 4‐HNE were blocked by GPR109A knockdown, suggesting that 4‐HNE activates the extrinsic apoptosis pathway via GPR109A. Moreover, our results showing that 4‐HNE did not induce cytotoxicity in HEK‐293 cells, which do not express GPR109A, further confirm 4‐HNE‐induced cell death is mediated through GPR109A. Notably, treatment with inhibitors of Gβγ downstream molecules (gallein, BAPTA‐AM, U73122 and GF109203X) blocked 4‐HNE‐induced cell death, while treatment with a cAMP analogue that inhibits the Gαi signalling pathway did not have that effect, indicating that Gβγ signalling mediates 4‐HNE‐induced cell death.
Although 4‐HNE is generated under conditions of oxidative stress, our present findings showed that 4‐HNE‐activated GPR109A induced superoxide production via NOX4, consistent with previous reports showing that NADPH oxidase present in ARPE cells plays a role in age‐related macular degeneration (Li et al., 2008). Previous studies have shown that an increase in the cytosolic Ca2+ level stimulates ROS production (Xu et al., 2011; Crnkovic et al., 2012). Concordantly, our results showing that cell death and superoxide production were completely blocked by an intracellular Ca2+ chelator (BAPTA‐AM) and by the knockdown of GPR109A or NOX4 suggest that 4‐HNE regulates NOX4 activation via the Gβγ‐cytosolic Ca2+ signalling pathway downstream of GPR109A. In turn, Gβγ‐cytosolic Ca2+‐NOX4 signalling induces mitochondrial superoxide production. Consistent with previous observations, although p38 and JNK activation are both dependent on superoxide production, only JNK was found to be involved in 4‐HNE‐induced cell death (Jang et al., 2009; Kutuk and Basaga, 2007; Song et al., 2001). JNK activates the transcription factor AP‐1, which mediates 4‐HNE‐induced GPR109A expression and cell death (Kutuk et al., 2006). A previous report also suggested that JNK is involved in 4‐HNE‐induced Fas induction and cell death in human lens epithelial cells, although the study did not elucidate the mechanisms through which 4‐HNE‐induced signalling leads to JNK activation (Li et al., 2006). Our present results showed the detailed mechanism by which 4‐HNE‐activated GPR109A induced JNK activation, that is, via a pathway consisting of Gβγ, cytosolic Ca2+, NOX4, superoxide and JNK. Furthermore, because the enhancer and promoter regions of Fas contain AP‐1 binding sites (Lasham et al., 2000; Rudert et al., 1998), the present results could help answer the unsolved question of the mechanisms by which 4‐HNE increases the transcriptional induction of Fas.
In accordance with previous studies using endothelial cells (Herbst et al., 1999), Kupffer cells (Luckey et al., 2002) and monocytes (Digby et al., 2012), the results of our present study indicated that low levels of 4‐HNE did not exert cytotoxic effects on ARPE‐19 and CCD‐841 cells but instead inhibited the IL‐6 and IL‐8 production in a concentration‐dependent manner. Given that cells undergoing aerobic metabolism generate a basal level of ROS, which in turn, generate a basal level of 4‐HNE (Parola et al., 1999), the physiological function of GPR109A under low‐dose 4‐HNE stimulation is to induce protective effects by eliciting the anti‐inflammatory response. Although 4‐HNE is known to alter protein expression via post‐translational modification, our results showed that the decrease in IL‐6 and IL‐8 levels induced by low doses of 4‐HNE was blocked by GPR109A knockdown or treatment with a cAMP analogue, indicating that this activity of 4‐HNE was mediated through receptor‐dependent modifications during gene expression. Consistent with cAMP being proposed as a selective modulator of the pro‐inflammatory transcription factor NF‐κB (Gerlo et al., 2011), the 4‐HNE‐induced inhibition of NF‐κB was blocked by treatment with a cAMP analogue. In addition, the expression of pro‐inflammatory cytokines was further suppressed by treatment with an NF‐κB inhibitor but was not affected by treatment with an AP‐1 inhibitor, thereby indicating that cAMP‐dependent NF‐κB is the primary regulator mediating 4‐HNE‐induced changes in IL‐6 and IL‐8 expression. Similarly to niacin and 3‐OHBA, the action of 4‐HNE was mediated by GPR109A and more specifically by Gαi signalling, which is linked to the suppression of the nuclear translocation of NF‐κB. Such protective in vitro and in vivo effects of GPR109A have also been demonstrated with niacin treatment, which resulted in the suppression of colitis (Singh et al., 2014) and the induction of anti‐inflammatory/anti‐atherosclerotic effects (Lukasova et al., 2011). Similarly, the anti‐inflammatory effects of 3‐OHBA, an endogenous agonist generated under ketoacidosis conditions, are also mediated by GPR109A in the RPE (Gambhir et al., 2012) and colon (Hamer et al., 2008). Previously, the EC50 of 3‐OHBA in GPR109A‐mediated effects was reported to be approximately 700 μM (Taggart et al., 2005), and the required concentration of niacin or butyrate for exerting anti‐inflammatory effects was demonstrated to be between 0.5 and 25 mM in cells and mice (Singh et al., 2014). By contrast, the results of our present study showed that the same degree of anti‐inflammatory activity can be achieved by 4‐HNE at 100‐fold lower concentrations than those of niacin and 3‐OHBA. These findings, in conjunction with the results of the receptor‐binding assay study, confirm that 4‐HNE may be strong endogenous ligand for GPR109A at concentrations of less than 10 μM (Esterbauer et al., 1991; Parola et al., 1999). Furthermore, our results suggest that strong GPR109A agonists, if developed, may have a narrow therapeutic dose range for anti‐inflammatory activity without causing cell death. Therefore, we suggest that the signalling pathway of GPR109A activation, particularly the cAMP‐dependent NF‐κB pathway, is a suitable target for the management of retinal inflammation.
The results of the functional study and receptor‐binding studies using SPR and competition binding assay with [3H]‐niacin demonstrated that 4‐HNE is a potent endogenous ligand of GPR109A, but scarcely competes with niacin or 3‐OHBA. The results generated questions: which region will be the binding site for 4‐HNE, and 4‐HNE shares the site with niacin or 3‐OHBA. To answer the questions, we used the modelled structure of GPR109A from GPCR‐I‐TASSER server (https://zhanglab.ccmb.med.umich.edu/GPCR-HGmod/models/Q8TDS4) and FTMap to predict the potential binding site of 4‐HNE. GPCR‐I‐TASSER proved its performance in the blind tests as well as the tests with the known experimental structures. FTMap searches the ligand binding sites by letting small organic molecules bind to all the pockets of a target protein and suggests the potential binding sites by clustering the docked molecules based on the interaction energies. The results of FTMap indicated two ligand binding sites in GPR109A that consist of several small clusters. Of the two, one includes the site around Arg111 that the previous mutation studies (Tunaru et al., 2005) have identified as the orthogonal site. The other is located at the top of the outer membrane region. However, this site is unlikely to be the region for interacting with 4‐HNE if the mutation data were considered. We then investigated whether the orthogonal site is large enough to include both 4‐HNE, and niacin or 3‐OHBA, simultaneously, using docking simulation with Glide‐XP. The docked models suggest that 4‐HNE can bind to the site without causing steric hindrance with niacin and 3‐OHBA (Supporting Information Figure S6). Interestingly, niacin and 3‐OHBA were in contact with Arg111, and 4‐HNE with Leu280 in the model, which is qualitatively consistent with the mutation data. The results would suggest a useful clue to the understanding of the results with 4‐HNE despite the imperfectness of model structure and docking method.
So far, only http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=485 (TRPA1) has been identified as a direct target of 4‐HNE (Trevisani et al., 2007). However, activation of this channel is unable to explain the various and potent roles of 4‐HNE. Compared with the known GPR109A agonists, the chemical feature of 4‐HNE is distinct. Most strong agonists contain carboxylic acid or tetrazole moieties, where tetrazole is the bioisostere of carboxylate. Pentyl fumarate shows the closest similarity to 4‐HNE with a K i of 700 nM (Peters et al., 2010), but the quantified chemical similarity between the two molecules is 0.26 in terms of Tanimoto coefficient using ECFP4 fingerprint, indicating that the similarity is little shared (Supporting Information Figure S7).
Conclusions
In conclusion, our results demonstrate that 4‐HNE is a strong agonist for GPR109A and that GPR109A activation can elicit two responses depending on the concentration of the agonist. In particular, 4‐HNE can elicit either an anti‐inflammatory response, which is mediated through the Gαi/cAMP/NF‐κB pathway, or cell death, which is mediated through the Gβγ/Ca2+/NOX4/JNK/AP‐1 pathway (Figure 8). By demonstrating the opposing and pathway‐specific effects elicited by GPR109A through either Gαi or Gβγ signalling, our results suggest a strategy to develop therapeutics for the treatment of retinal inflammation and 4‐HNE‐associated chronic diseases.
Figure 8.
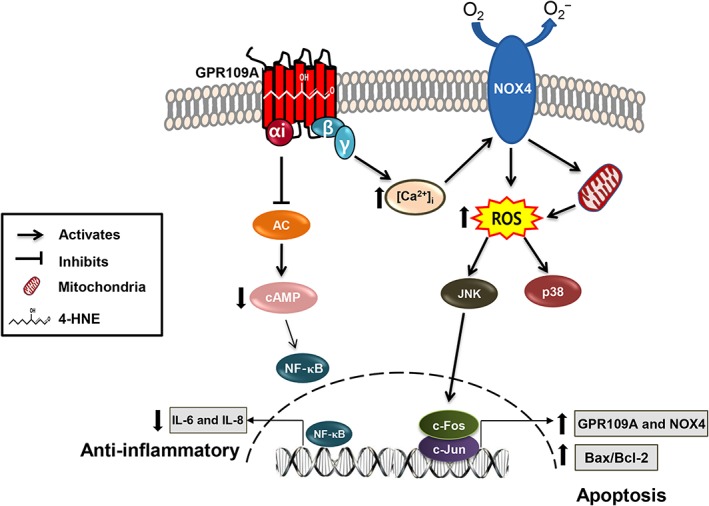
Proposed mechanism by which 4‐HNE activates GPR109A and elicits bipolar responses, Gαi‐mediated anti‐inflammatory and Gβγ‐mediated cell death responses.
Author contributions
J.‐A.K. contributed in the study conception and design; J.G., S.B., S.S., J.‐G.J., E.K. and Y.W. in the acquisition of data; J.G., S.B., J.‐G.J., D.Y.K. and J.‐A.K. in the analysis and interpretation of data; J.G. in the drafting of the manuscript; and J.‐G.J., H.W.C. and J.‐A.K. in the critical revision of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 4‐HNE induces ARPE‐19 cell apoptosis in a concentration‐ and time‐dependent manner. Annexin V‐positive and PI‐positive ARPE‐19 cell populations were determined after the treatment with 4‐HNE 10 μM at an indicated time points (A) and at an indicated concentrations of 4‐HNE at 6 h (B) by using a flow cytometry.
Figure S2 Reduced cytotoxicity and anti‐inflammatory effect of 4‐HNE in confluent ARPE‐19 cells correlates with lower level expression of receptors and NOX4. After 24 hours of cell seeding at a density of 1*105 cells/cm2, cells were sub‐confluent, whereas after 48 hours of cell seeding with same seeding density, cells were confluent. (A, E and F) Confluent cells were treated with or without 4‐HNE at an indicated concentration for an indicated period of time and examined cytotoxicity (A), superoxide production (E) and Il‐6 and IL‐8 protein expressions (F). *P < 0.05 vs. vehicle‐treated controls. (B) Confluent ARPE‐19 cells were treated with or without 3‐OHBA in a concentration and time‐dependent manner and examined cytotoxicity. *P < 0.05 vs. vehicle‐treated controls. (C, D) Comparison of basal mRNA (C) and protein (D) expressions in sub‐confluent and confluent ARPE‐19 cells. *P < 0.05 vs. sub‐confluent ARPE‐19 cells.
Figure S3 Niacin and 3‐OHBA induces cell death in GPR109A–dependent manner. (A) Transfection efficiency of siRNA targeted against GPR109A or GPR109B was measured by qRT‐PCR. After RNA extraction from the cells, cDNA was synthesized using the Goscript reverse transcription system (Promega Corporation, WI, USA). qRTPCR was performed using Quantitect Probe PCR kit (Qiagen, CA, USA) following the manufacturers protocol using probe PCR primers specific for GPR109A (Hs02341584_s1) or GPR109B (Hs02341102_s1) obtained from Applied Biosystems (Thermo Fisher Scientific corporation, CA, USA). *P < 0.05 vs. vehicle‐treated controls. # P < 0.05 vs. 4‐HNE‐treated groups. (B and C) Cell viability was measured after the treatment with niacin or 3‐OHBA in GPR109A siRNA‐transfected ARPE‐19 (B) and CCD‐841 (C) cells. *P < 0.05 vs. vehicle‐treated controls. # P < 0.05 vs. niacin or 3‐OHBA‐treated groups.
Figure S4 Increase in intracellular Ca2+ levels after treatment of ARPE‐19 cells with indicated concentrations of niacin or 3‐OHBA. Bars represent means ± SEM from six independent experiments. *P < 0.05 vs. vehicle‐treated controls.
Figure S5. Niacin, 3‐OHBA and 4‐HNE inhibits the pro‐inflammatory cytokines IL‐6 and IL‐8 expression. (A‐B) Secretion of the IL‐6 and IL‐8 in ARPE‐19 (A) and CCD‐841 (B) cells was measured via ELISA. *P < 0.05 vs. vehicle‐treated controls. (C and D) 4‐HNE‐induced expression of IL‐6 and IL‐8 was measured in GPR109B siRNA‐transfected ARPE‐19 cells (C) and GPR109A siRNA‐transfected CCD‐841 cells (D). *P < 0.05 vs. vehicle‐treated controls # P < 0.05 vs. 4‐HNE‐treated groups. (E) 4‐HNE induced IL‐6 and IL‐8 protein expression was recovered by PTx treatment. *P < 0.05 vs. vehicle‐treated controls.
Figure S6 Model structure of GPR109A in complex with niacin, 3‐OHBA, and 4‐HNE. The structure was extracted from GPCR‐I‐TASSER server (https://zhanglab.ccmb.med.umich.edu/GPCR-HGmod/models/Q8TDS4) (Zhang et al., 2015). The predicted two ligand binding sites by FTMap (Kozakov et al., 2015) are circled in blue and orange colours. Arg‐111 and Leu‐280 that were identified as the critical residues by mutational studies are represented with spheres. The niacin, 3‐OHBA, and 4‐HNE were docked into the structure using Glide‐XP (Friesner et al., 2004). The figure was prepared by Pymol (Schrodinger, 2010).
Figure S7 The closest chemicals to 4‐HNE of the known GPR109A agonists. ZINC IDs and parentheses enclosed corresponding Tanimoto coefficients are written.
Acknowledgements
We greatly appreciate Dr Rongfang Liu and Dr Adriaan IJzerman in Leiden Academic Centre for Drug Research (Leiden, The Netherlands) for performing and providing the competitive receptor binding assay of 4‐HNE with [3H]‐niacin and Ms Dong Hwa Choi in Gyeonggi Biocenter (Suwon, Korea) for helping in the SPR binding assay. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Ministry of Science, ICT, and Future Planning (NRF‐2014R1A4A1071040).
Gautam, J. , Banskota, S. , Shah, S. , Jee, J.‐G. , Kwon, E. , Wang, Y. , Kim, D. Y. , Chang, H. W. , and Kim, J.‐A. (2018) 4‐Hydroxynonenal‐induced GPR109A (HCA2 receptor) activation elicits bipolar responses, Gαi‐mediated anti‐inflammatory effects and Gβγ‐mediated cell death. British Journal of Pharmacology, 175: 2581–2598. doi: 10.1111/bph.14174.
References
- Abarikwu SO, Pant AB, Farombi EO (2012). 4‐Hydroxynonenal induces mitochondrial‐mediated apoptosis and oxidative stress in SH‐SY5Y human neuronal cells. Basic Clin Pharmacol Toxicol 110: 441–448. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banskota S, Regmi SC, Gautam J, Gurung P, Lee Y‐J, Ku SK et al (2017). Serotonin disturbs colon epithelial tolerance of commensal E. coli by increasing NOX2‐derived superoxide. Free Radic Biol Med 106: 196–207. [DOI] [PubMed] [Google Scholar]
- Banskota S, Regmi SC, Kim J‐A (2015). NOX1 to NOX2 switch deactivates AMPK and induces invasive phenotype in colon cancer cells through overexpression of MMP‐7. Mol Cancer 14: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bok D (1993). The retinal pigment epithelium: a versatile partner in vision. J Cell Sci 1993: 189–195. [DOI] [PubMed] [Google Scholar]
- Crnkovic S, Riederer M, Lechleitner M, Hallstrom S, Malli R, Graier WF et al (2012). Docosahexaenoic acid‐induced unfolded protein response, cell cycle arrest, and apoptosis in vascular smooth muscle cells are triggered by Ca2+‐dependent induction of oxidative stress. Free Radic Biol Med 52: 1786–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csala M, Kardon T, Legeza B, Lizak B, Mandl J, Margittai E et al (2015). On the role of 4‐hydroxynonenal in health and disease. BBA Mol Basis Dis 1852: 826–838. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalleau S, Baradat M, Guéraud F, Huc L (2013). Cell death and diseases related to oxidative stress: 4‐hydroxynonenal (HNE) in the balance. Cell Death Differ 20: 1615–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianzani C, Parrini M, Ferrara C, Fantozzi R (1996). Effect of 4‐hydroxynonenal on superoxide anion production from primed human neutrophils. Cell Biochem Funct 14: 193–200. [DOI] [PubMed] [Google Scholar]
- Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M et al (2012). Anti‐inflammatory effects of niacin in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler Thromb Vasc Biol 32: 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domanico D, Verboschi F, Altimari S, Zompatori L, Vingolo EM (2015). Ocular effects of niacin: a review of the literature. Med Hypothesis Discov Innov Ophthalmol 4: 64. [PMC free article] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H (1991). Chemistry and biochemistry of 4‐hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81–128. [DOI] [PubMed] [Google Scholar]
- Ethen CM, Reilly C, Feng X, Olsen TW, Ferrington DA (2007). Age‐related macular degeneration and retinal protein modification by 4‐hydroxy‐2‐nonenal. Invest Ophthalmol Vis Sci 48: 3469–3479. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT et al (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47: 1739–1749. [DOI] [PubMed] [Google Scholar]
- Gambhir D, Ananth S, Veeranan‐Karmegam R, Elangovan S, Hester S, Jennings E et al (2012). GPR109A as an anti‐inflammatory receptor in retinal pigment epithelial cells and its relevance to diabetic retinopathy. Invest Ophthalmol Vis Sci 53: 2208–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass JDM (1973). Drusen and disciform macular detachment and degeneration. Arch Ophthalmol 90: 206–217. [DOI] [PubMed] [Google Scholar]
- Gerlo S, Kooijman R, Beck IM, Kolmus K, Spooren A, Haegeman G (2011). Cyclic AMP: a selective modulator of NF‐κB action. Cell Mol Life Sci 68: 3823–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goicoechea E, Brandon EF, Blokland MH, Guillen MD (2011). Fate in digestion in vitro of several food components, including some toxic compounds coming from omega‐3 and omega‐6 lipids. Food Chem Toxicol 49: 115–124. [DOI] [PubMed] [Google Scholar]
- Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost F, Brummer RJ (2008). Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 27: 104–119. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2017). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46 (D1): D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst U, Toborek M, Kaiser S, Mattson MP, Hennig B (1999). 4‐Hydroxynonenal induces dysfunction and apoptosis of cultured endothelial cells. J Cell Physiol 181: 295–303. [DOI] [PubMed] [Google Scholar]
- Jang YJ, Kang NJ, Lee KW, Lee HJ (2009). Protective effects of red wine flavonols on 4‐hydroxynonenal‐induced apoptosis in PC12 cells. Ann N Y Acad Sci 1171: 170–175. [DOI] [PubMed] [Google Scholar]
- Ji C, Amarnath V, Pietenpol JA, Marnett LJ (2001). 4‐Hydroxynonenal induces apoptosis via caspase‐3 activation and cytochrome c release. Chem Res Toxicol 14: 1090–1096. [DOI] [PubMed] [Google Scholar]
- Kostylina G, Simon D, Fey M, Yousefi S, Simon H (2008). Neutrophil apoptosis mediated by niacin receptors (GPR109A). Cell Death Differ 15: 134–142. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Grove LE, Hall DR, Bohnuud T, Mottarella SE, Luo L et al (2015). The FTMap family of web servers for determining and characterizing ligand‐binding hot spots of proteins. Nat Protoc 10: 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krohne TU, Holz FG, Kopitz J (2010). Apical‐to‐basolateral transcytosis of photoreceptor outer segments induced by lipid peroxidation products in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 51: 553–560. [DOI] [PubMed] [Google Scholar]
- Kutuk O, Adli M, Poli G, Basaga H (2004). Resveratrol protects against 4‐HNE induced oxidative stress and apoptosis in Swiss 3T3 fibroblasts. Biofactors 20: 1–10. [DOI] [PubMed] [Google Scholar]
- Kutuk O, Basaga H (2007). Apoptosis signalling by 4‐hydroxynonenal: a role for JNK‐c‐Jun/AP‐1 pathway. Redox Rep 12: 30–34. [DOI] [PubMed] [Google Scholar]
- Kutuk O, Poli G, Basaga H (2006). Resveratrol protects against 4‐hydroxynonenal‐induced apoptosis by blocking JNK and c‐JUN/AP‐1 signaling. Toxicol Sci 90: 120–132. [DOI] [PubMed] [Google Scholar]
- Lasham A, Lindridge E, Rudert F, Onrust R, Watson J (2000). Regulation of the human fas promoter by YB‐1, Purα and AP‐1 transcription factors. Gene 252: 1–13. [DOI] [PubMed] [Google Scholar]
- Li J, Sharma R, Patrick B, Sharma A, Jeyabal P, Reddy P et al (2006). Regulation of CD95 (Fas) expression and Fas‐mediated apoptotic signaling in HLE B‐3 cells by 4‐hydroxynonenal. Biochemistry 45: 12253–12264. [DOI] [PubMed] [Google Scholar]
- Li Q, Dinculescu A, Shan Z, Miller R, Pang J, Lewin AS et al (2008). Downregulation of p22phox in retinal pigment epithelial cells inhibits choroidal neovascularization in mice. Mol Ther 16: 1688–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckey SW, Taylor M, Sampey BP, Scheinman RI, Petersen DR (2002). 4‐Hydroxynonenal decreases interleukin‐6 expression and protein production in primary rat Kupffer cells by inhibiting nuclear factor‐κB activation. J Pharmacol Exp Ther 302: 296–303. [DOI] [PubMed] [Google Scholar]
- Lukasova M, Malaval C, Gille A, Kero J, Offermanns S (2011). Niacin inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest 121: 1163–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PM, Ananth S, Cresci G, Roon P, Smith S, Ganapathy V (2009). Expression and localization of GPR109A (PUMA‐G/HM74A) mRNA and protein in mammalian retinal pigment epithelium. Mol Vis 15: 362. [PMC free article] [PubMed] [Google Scholar]
- Millay RH, Klein ML, Illingworth DR (1988). Niacin maculopathy. Ophthalmology 95: 930–936. [DOI] [PubMed] [Google Scholar]
- Negre‐Salvayre A, Vieira O, Escargueil‐Blanc I, Salvayre R (2003). Oxidized LDL and 4‐hydroxynonenal modulate tyrosine kinase receptor activity. Mol Aspects Med 24: 251–261. [DOI] [PubMed] [Google Scholar]
- Parola M, Bellomo G, Robino G, Barrera G, Dianzani MU (1999). 4‐Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1: 255–284. [DOI] [PubMed] [Google Scholar]
- Peters JU, Kuhne H, Dehmlow H, Grether U, Conte A, Hainzl D et al (2010). Pyrido pyrimidinones as selective agonists of the high affinity niacin receptor GPR109A: optimization of in vitro activity. Bioorg Med Chem Lett 20: 5426–5430. [DOI] [PubMed] [Google Scholar]
- Pierre F, Peiro G, Taché S, Cross AJ, Bingham SA, Gasc N et al (2006). New marker of colon cancer risk associated with heme intake: 1,4‐dihydroxynonane mercapturic acid. Cancer Epidemiol Biomarkers Prev 15: 2274–2279. [DOI] [PubMed] [Google Scholar]
- Poli G, Biasi F, Leonarduzzi G (2008). 4‐Hydroxynonenal–protein adducts: a reliable biomarker of lipid oxidation in liver diseases. Mol Aspects Med 29: 67–71. [DOI] [PubMed] [Google Scholar]
- Raza H, John A (2006). 4‐Hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S‐transferase A4‐4 and cytochrome P450 2E1 in PC12 cells. Toxicol Appl Pharmacol 216: 309–318. [DOI] [PubMed] [Google Scholar]
- Rudert F, Lindridge E, Lasham A, Wang Y, Grandison P, Watson J (1998). Silencer and enhancer regions in the human CD95 (Fas/APO‐1) gene with sequence similarity to the granulocyte‐macrophage colony‐stimulating factor promoter: binding of single strand‐specific silencer factors and AP‐1 and NF‐AT‐like enhancer factors. DNA Cell Biol 17: 991–1002. [DOI] [PubMed] [Google Scholar]
- Salomoni P, Khelifi AF (2006). Daxx: death or survival protein? Trends Cell Biol 16: 97–104. [DOI] [PubMed] [Google Scholar]
- Schrodinger, LLC (2010). The PyMOL Molecular Graphics System, Version 1.3r1.
- Sharma R, Sharma A, Dwivedi S, Zimniak P, Awasthi S, Awasthi YC (2008). 4‐Hydroxynonenal self‐limits fas‐mediated DISC‐independent apoptosis by promoting export of Daxx from the nucleus to the cytosol and its binding to Fas. Biochem 47: 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lai X, Ye L, Chen K, Cao Z, Gong W et al (2017). Activated niacin receptor HCA2 inhibits chemoattractant‐mediated macrophage migration via Gβγ/PKC/ERK1/2 pathway and heterologous receptor desensitization. Sci Rep 7: 42279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui M, Kumar V, Kashyap M, Agarwal M, Singh A, Jahan S et al (2012). Short‐term exposure of 4‐hydroxynonenal induces mitochondria‐mediated apoptosis in PC12 cells. Hum Exp Toxicol 31: 336–345. [DOI] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H et al (2014). Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40: 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga T, Kamohara M, Takasaki J, Matsumoto S‐i, Saito T, Ohishi T et al (2003). Molecular identification of niacin receptor. Biochem Biophys Res Commun 303: 364–369. [DOI] [PubMed] [Google Scholar]
- Song BJ, Soh Y, Bae M‐A, Pie J‐E, Wan J, Jeong K‐S (2001). Apoptosis of PC12 cells by 4‐hydroxy‐2‐nonenal is mediated through selective activation of the c‐Jun N‐terminal protein kinase pathway. Chem Biol Interact 130: 943–954. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Boulton M (2005). RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res 80: 595–606. [DOI] [PubMed] [Google Scholar]
- Stadtmanl ER (1994). Michael addition‐type 4‐hydroxy‐2‐nonenal adducts in modified low‐density lipoproteins: markers for atherosclerosis. Biochem 33: 12487–12494. [DOI] [PubMed] [Google Scholar]
- Taggart AK, Kero J, Gan X, Cai T‐Q, Cheng K, Ippolito M et al (2005). (d)‐β‐hydroxybutyrate inhibits adipocyte lipolysis via the niacin receptor PUMA‐G. J Biol Chem 280: 26649–26652. [DOI] [PubMed] [Google Scholar]
- Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD et al (2009). GPR109A is a G‐protein‐coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res 69: 2826–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisani M, Siemens J, Materazzi S, Bautista DM, Nassini R, Campi B et al (2007). 4‐Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci U S A 104: 13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K et al (2003). PUMA‐G and HM74 are receptors for niacin and mediate its anti‐lipolytic effect. Nat Med 9: 352–355. [DOI] [PubMed] [Google Scholar]
- Tunaru S, Lattig J, Kero J, Krause G, Offermanns S (2005). Characterization of determinants of ligand binding to the niacin receptor GPR109A (HM74A/PUMA‐G). Mol Pharmacol 68: 1271–1280. [DOI] [PubMed] [Google Scholar]
- Vingerling JR, Hofman A, Grobbee DE, De Jong PT (1996). Age‐related macular degeneration and smoking: the Rotterdam Study. Arch Ophthalmol 114: 1193–1196. [DOI] [PubMed] [Google Scholar]
- Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M et al (2003). Molecular identification of high and low affinity receptors for niacin. J Biol Chem 278: 9869–9874. [DOI] [PubMed] [Google Scholar]
- Xu B, Chen S, Luo Y, Chen Z, Liu L, Zhou H et al (2011). Calcium signaling is involved in cadmium‐induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PloS One 6: e19052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J, Jang R, Zhang Y (2015). GPCR‐I‐TASSER: a hybrid approach to G protein‐coupled receptor structure modeling and the application to the human genome. Structure 23: 1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman MA, Singh N, Martin PM, Thangaraju M, Ganapathy V, Waller JL et al (2012). Butyrate suppresses colonic inflammation through HDAC1‐dependent Fas upregulation and Fas‐mediated apoptosis of T cells. Am J Physiol Gastrointest Liver Physiol 302: G1405–G1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 4‐HNE induces ARPE‐19 cell apoptosis in a concentration‐ and time‐dependent manner. Annexin V‐positive and PI‐positive ARPE‐19 cell populations were determined after the treatment with 4‐HNE 10 μM at an indicated time points (A) and at an indicated concentrations of 4‐HNE at 6 h (B) by using a flow cytometry.
Figure S2 Reduced cytotoxicity and anti‐inflammatory effect of 4‐HNE in confluent ARPE‐19 cells correlates with lower level expression of receptors and NOX4. After 24 hours of cell seeding at a density of 1*105 cells/cm2, cells were sub‐confluent, whereas after 48 hours of cell seeding with same seeding density, cells were confluent. (A, E and F) Confluent cells were treated with or without 4‐HNE at an indicated concentration for an indicated period of time and examined cytotoxicity (A), superoxide production (E) and Il‐6 and IL‐8 protein expressions (F). *P < 0.05 vs. vehicle‐treated controls. (B) Confluent ARPE‐19 cells were treated with or without 3‐OHBA in a concentration and time‐dependent manner and examined cytotoxicity. *P < 0.05 vs. vehicle‐treated controls. (C, D) Comparison of basal mRNA (C) and protein (D) expressions in sub‐confluent and confluent ARPE‐19 cells. *P < 0.05 vs. sub‐confluent ARPE‐19 cells.
Figure S3 Niacin and 3‐OHBA induces cell death in GPR109A–dependent manner. (A) Transfection efficiency of siRNA targeted against GPR109A or GPR109B was measured by qRT‐PCR. After RNA extraction from the cells, cDNA was synthesized using the Goscript reverse transcription system (Promega Corporation, WI, USA). qRTPCR was performed using Quantitect Probe PCR kit (Qiagen, CA, USA) following the manufacturers protocol using probe PCR primers specific for GPR109A (Hs02341584_s1) or GPR109B (Hs02341102_s1) obtained from Applied Biosystems (Thermo Fisher Scientific corporation, CA, USA). *P < 0.05 vs. vehicle‐treated controls. # P < 0.05 vs. 4‐HNE‐treated groups. (B and C) Cell viability was measured after the treatment with niacin or 3‐OHBA in GPR109A siRNA‐transfected ARPE‐19 (B) and CCD‐841 (C) cells. *P < 0.05 vs. vehicle‐treated controls. # P < 0.05 vs. niacin or 3‐OHBA‐treated groups.
Figure S4 Increase in intracellular Ca2+ levels after treatment of ARPE‐19 cells with indicated concentrations of niacin or 3‐OHBA. Bars represent means ± SEM from six independent experiments. *P < 0.05 vs. vehicle‐treated controls.
Figure S5. Niacin, 3‐OHBA and 4‐HNE inhibits the pro‐inflammatory cytokines IL‐6 and IL‐8 expression. (A‐B) Secretion of the IL‐6 and IL‐8 in ARPE‐19 (A) and CCD‐841 (B) cells was measured via ELISA. *P < 0.05 vs. vehicle‐treated controls. (C and D) 4‐HNE‐induced expression of IL‐6 and IL‐8 was measured in GPR109B siRNA‐transfected ARPE‐19 cells (C) and GPR109A siRNA‐transfected CCD‐841 cells (D). *P < 0.05 vs. vehicle‐treated controls # P < 0.05 vs. 4‐HNE‐treated groups. (E) 4‐HNE induced IL‐6 and IL‐8 protein expression was recovered by PTx treatment. *P < 0.05 vs. vehicle‐treated controls.
Figure S6 Model structure of GPR109A in complex with niacin, 3‐OHBA, and 4‐HNE. The structure was extracted from GPCR‐I‐TASSER server (https://zhanglab.ccmb.med.umich.edu/GPCR-HGmod/models/Q8TDS4) (Zhang et al., 2015). The predicted two ligand binding sites by FTMap (Kozakov et al., 2015) are circled in blue and orange colours. Arg‐111 and Leu‐280 that were identified as the critical residues by mutational studies are represented with spheres. The niacin, 3‐OHBA, and 4‐HNE were docked into the structure using Glide‐XP (Friesner et al., 2004). The figure was prepared by Pymol (Schrodinger, 2010).
Figure S7 The closest chemicals to 4‐HNE of the known GPR109A agonists. ZINC IDs and parentheses enclosed corresponding Tanimoto coefficients are written.