

Abstract
The discovery of the nitric oxide/cGMP pathway was the basis for our understanding of many normal physiological functions and the pathophysiology of several diseases. Since the discovery and introduction of sildenafil, inhibitors of PDE5 have been the first‐line therapy for erectile dysfunction (ED). The success of sildenafil in the treatment of ED stimulated research in the field of PDE5 inhibition and led to many new applications, such as treatment of lower urinary symptoms, and pulmonary arterial hypertension, which are now approved indications. However, PDE5 inhibitors have also been used in several other disorders not discussed in this review, and the fields of clinical use are increasing. In the present review, the pharmacological basis of the NO/cGMP pathway and the rationale and clinical use of PDE5 inhibitors in different diseases are discussed.
Abbreviations
- ED
erectile dysfunction
- LUTS
lower urinary tract symptoms
- NANC
non‐adrenergic non‐cholinergic
- PAH
pulmonary arterial hypertension
- PKG
cGMP‐dependent protein kinase
Introduction
The discovery in the late 1980s of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509 as the endothelium‐derived relaxing factor was the key to understanding the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=939#1288)‐http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2347 pathway. This discovery led to intensive research on the physiology, pharmacology and pathophysiology of NO (Moncada et al., 1991; Denninger and Marletta, 1999; Murad, 2006), which resulted in Furchgott, Ignarro and Murad receiving the Nobel Prize for Physiology or Medicine in 1998.
NO mediates its biological effects by activating sGC and increasing cGMP synthesis which, in turn, activates certain proteins resulting in different actions. cGMP actions are terminated by http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1304 and PDE5 inhibitors, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2919 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4743, proved vital in the elucidation of the widespread role of cGMP in nitrergic transmission. The synthesis of sildenafil and the (serendipity) discovery of its effects on penile erection provided a major breakthrough in the treatment of erectile dysfunction (ED) and opened new fields of clinical application for this class of drug. Although many PDE5 inhibitors have since been synthesized and developed, most of the information available is for sildenafil, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7299 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7320.
Literature search
A literature search of the electronic sources of different databases such as PubMed Central, Google Scholar and Scopus was used. Keywords used for search included, but were not limited to, PDE5, PDE5 inhibitors, NO, cGMP, ED, male lower urinary tract symptoms (LUTS) and pulmonary arterial hypertension (PAH). In addition, articles from previous reviews on NO, ED and male LUTS by the author were restudied. Sixty articles, considered to cover the field, were selected and referred to in this review.
The NO/soluble GC/cGMP pathway
The NO/sGC/cGMP pathway has been described in detail elsewhere (Moncada et al., 1991; Denninger and Marletta, 1999; Murad, 2006) (Figure 1). NO is synthesized by the oxidation of L‐arginine catalysed by http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=253 that utilizes NADPH and O2 as substrates. Three isoforms of NOS have been identified: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1251 (nNOS or NOS1), http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (iNOS or NOS2) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249 (eNOS or NOS3). NO mediates its biological effects by activating sGC and increasing cGMP synthesis from GTP. The cGMP formed activates http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=287), which, in turn, activates certain proteins resulting in different effects on, for example, growth, viability, smooth muscle relaxation, secretion, ion transport, endothelial permeability and gene transcription. cGMP's effects are terminated by PDE5, which breaks down its phosphodiester bond, an effect prevented by PDE5 inhibitors. It should be noted that the effects of PDE5 inhibitors are crucially dependent on the activity of the NO/sGC/cGMP pathway.
Figure 1.

Cellular signalling with NO and cGMP (modified from Murad, 2006). NO is synthesized by the oxidation of L‐arginine catalysed by NOS. NO mediates its biological effects by activating sGC and increasing cGMP synthesis from GTP. cGC can also be activated by non‐NO‐dependent activators and stimulators (BAY compounds): NO‐independent, haem‐dependent stimulators of sGC (BAY 41–2272, BAY 41–8523, BAY 63–2521 and BAY 60–4552) and NO‐independent, haem‐independent sGC activators (HMR 1766, BAY 58–2667 and BAY 60–2770). The cGMP formed activates cation channels but also cGMP‐dependent protein kinases (cGKs), which, in turn, activate certain proteins (myosin phosphatase, inositol 1,4,5‐trisphosphate receptor I (IP(3)RI)‐associated protein = IRAG, and vasodilator‐stimulated phosphoprotein = VASP), resulting in, for example, smooth muscle relaxation, platelet inhibition and cell growth/differentiation. THe effects of cGMP are terminated by PDE5, which breaks down its phosphodiester bond, an effect prevented by PDE5 inhibitors. It should be emphasized that the effects of PDE5 inhibitors are crucially dependent on the activity of the NO/GC/cGMP pathway.
As indicated in Figure 1, sCG can also be influenced by drugs (‘BAY compounds’) that are able to act directly on the enzyme in an NO‐independent manner (Mónica and Antunes, 2018). Two classes of drugs have been developed to treat cardiovascular diseases: NO‐independent, haem‐dependent stimulators of sGC (BAY 41–2272, BAY 41–8523, BAY 63–2521 and BAY 60–4552) and NO‐independent, haem‐independent sGC activators (HMR 1766, BAY 58–2667 and BAY 60–2770). Some of these drugs, which are in clinical use [e.g. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5257 (BAY 63–2521)], may also be used for urogenital disorders (Mónica and Antunes, 2018).
Historical background of PDE5 inhibitors – discovery of sildenafil
Zaprinast was synthesized in 1974 and later characterized as the first selective PDE5 inhibitor (Gibson, 2001). Even though later studies revealed that zaprinast was not selective for PDE5, it became an important tool for inhibiting PDE5. Thus, zaprinast was shown to enhance NO‐induced relaxation of isolated corpus cavernosum from animals (Ignarro et al., 1990) and humans (Rajfer et al., 1992).
When exploring PDE5 as a target for a range of cardiovascular disorders, particularly hypertension and angina pectoris, Terrett et al. (1996) found that PDE5 was the predominant hydrolysing enzyme in the cytosolic fraction from human corpus cavernosum and suggested that one of their inhibitors synthesized, sildenafil, which was a potent and highly selective PDE5 inhibitor, could be useful as an orally active treatment for male ED. That this was the case was confirmed in a double‐blind, randomized, placebo‐controlled, crossover study on 12 patients (aged 36–63 years) with ED (Boolell et al., 1996), and this initiated rapid progress in the development of selective PDE5 inhibitors for this indication (Andersson, 2001; 2011). Since the introduction of sildenafil, the approved indications for PDE5 inhibitors include not only ED but also male LUTS and PAH. However, PDE5 inhibitors have also been used to treat an increasing number of conditions, but with varying degrees of effectiveness (Gur et al., 2012; Ribaudo et al., 2016).
Distribution of PDE5 enzymes
PDE5 isoenzymes have now been identified in a wide variety of tissues, both in animals and man. They have been demonstrated in, for example, the smooth muscle cells of the corpus cavernosum, vascular and visceral smooth muscle, skeletal muscle, platelets, kidney, lung, spinal cord, cerebellum, pancreas, prostate, urethra and bladder (Lin et al., 2006; Francis et al., 2011; Uckert and Kuczyk, 2011).
Eleven different http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=260 (PDE 1–11) have been described (Francis et al., 2011; Keravis and Lugnier, 2012). More than 50 isoforms have been characterized, all differing in their primary structures, specificity for cAMP and cGMP, cofactor requirements, kinetic properties, mechanisms of regulation and tissue distributions (Francis et al., 2011). Some PDEs are selective for the hydrolysis of cAMP (PDE 4, 7 and 8) or cGMP (PDE 5, 6 and 9), while others can hydrolyse both cAMP and cGMP (PDE1, 2, 3, 10 and 11). Two PDE5 isoforms, PDE5A1 and PDE5A2, have been demonstrated in humans, but a third isoform, PDE5A3, has also been identified. The variants may allow for differential control of PDE5A gene expression in various cells. The three human PDE5 isoforms differ only in the 5′ end of the mRNA and the corresponding N‐terminal of the protein. PDE5A1, PDE5A2 and PDE5A3 share similar cGMP‐catalytic activities that are differently inhibited by sildenafil or zaprinast, with PDE5A1 being more resistant to such inhibition than PDE5A2 or PDE5A3. PDE5A1 and PDE5A2 are found in nearly all tissues, but PDE5A2 is more widespread, and the distribution of PDE5A3 is limited to smooth muscle (Lin et al., 2006).
General pharmacology of PDE5 inhibitors
As mentioned, inhibition of the PDE5 isoenzymes results in intracellular accumulation of cGMP, which activates cGMP‐dependent protein kinase with subsequent phosphorylation of specific substrate proteins. As a second messenger, cGMP plays a central role in signal transduction and regulates a number of physiological responses, such as smooth muscle relaxation, platelet and cardiac functions through a number of downstream mechanisms (Murad, 2006; Stasch et al., 2011).
Relaxation of smooth muscle
It is well established that NO, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4869 and several other endogenous vasodilators regulate smooth muscle tone through activation of sGC, elevation of cGMP and activation of PKG (cGK), which in turn phosphorylates several proteins. Three different PKGs (PKGIa, PKGIb and PKGII) have been identified in mammals (Figure 1). The NO/cGMP effects on contraction in smooth muscle appear to be mediated specifically by PKG, but not http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=284, because inactivation of PKGI in mice abolished both NO/cGMP‐dependent relaxation of vascular and intestinal smooth muscle and inhibition of platelet aggregation, causing hypertension, intestinal dysmotility and abnormal haemostasis (Pfeifer et al., 1998). There are several specific physiological substrates for PKG in smooth muscle including the regulatory myosin‐binding subunit of myosin phosphatase, calcium‐activated maxi K+ (BKCa; http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=387) channels and IRAG (IP3 receptor‐associated cGMP kinase substrate). Phosphorylation of all of these targets contributes to a reduction in intracellular Ca2+ concentration or reduction in sensitivity to Ca2+ and thereby decreased smooth muscle tone.
Platelet aggregation
The role of the NO/cGMP pathway for platelet function is well established (Mellion et al., 1981; Radomski et al., 1990; Smolenski, 2012). http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1915 and NO, released from the endothelium, are potent inhibitors of platelet function. Their actions are mediated by both PKA and PKG synthesizing cAMP and cGMP respectively. Platelets have been shown to express several PDEs: PDE2A, PDE3A and PDE5A. These three isozymes account for the majority (more than 90%) of platelet PDE activity. PDE2 and PDE3 are able to degrade both cAMP and cGMP; however, they mainly regulate cAMP degradation, whereas PDE5 specifically degrades cGMP. PDE5 is an important regulator of platelet function (Rondina and Weyrich, 2012); knockout of the PKGI gene in mice led to a prothrombotic phenotype. The main isoform expressed in human platelets is PKGIb. In platelets of PKGI knockout mice, the effects of a cGMP analogue on platelet shape change, granule release and aggregation were abolished, whereas the effects of a cAMP analogue were maintained (Pfeifer et al., 1998).
PDE inhibitors in clinical use for stroke prevention (dipyramidole, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7148) mainly act via cAMP, even if they also increase cGMP levels (Rondina and Weyrich, 2012; Yeung and Holinstat, 2012). However, PDE5 inhibitors used to treat ED, including sildenafil, vardenafil and tadalafil, potentiate the effect of nitrovasodilators on platelets. Sildenafil and tadalafil accumulated in platelets with an up to fourfold higher accumulation when platelets were pretreated with an NO donor (Bajraktari et al., 2017). Sildenafil also prolonged bleeding time in healthy volunteers after acute administration (Berkels et al., 2001).
Cardiac function
Both NOS1 and NOS3 are constitutively expressed in the cardiovascular system. NOS3 is mostly found in coronary vascular and endocardial endothelial cells and, to a lesser extent, in cardiac myocytes, whereas NOS1 is predominantly localized to the sarcoplasmic reticulum. In the myocardium, constitutive NO production affects the function and phosphorylation state of several proteins that are involved in excitation‐contraction coupling, for example, the http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=80, troponin I and phospholamban, and inhibits both oxygen consumption and http://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=4 mediated inotropy; abnormal NOS signalling plays a key role in many cardiac disorders (Carnicer et al., 2013; Kim and Kass, 2017). An important hallmark of cardiac failure is abnormal second messenger signalling due to impaired synthesis and catabolism of cAMP and cGMP. Experimental findings using NO donors and NOS inhibitors, or gene deletion, clearly implicate dysfunctional NOS as a critical contributor to many cardiovascular disease states, including diabetic cardiomyopathy, ischaemia/reperfusion injury and atrial fibrillation (Simon et al., 2014). The regulation of cardiac functions by endogenous NO has been suggested to be dependent on the distinct subcellular locations of nNOS and eNOS, through vascular‐dependent and vascular‐independent effects (Massion and Balligand, 2003). The heart expresses seven of the 11 major PDE sub‐types – PDE1, 2, 3, 4, 5, 8 and 9 (Kim and Kass, 2017) – and their different effects on cAMP and cGMP signalling in various cell types, including cardiomyocytes, provides intriguing therapeutic opportunities to counter heart disease. An abundant expression of PDE5 has been demonstrated in isolated canine or murine ventricular cardiomyocytes (Das et al., 2005), and PDE5 is highly expressed in both experimental and human heart disease. There should thus be possibilities to influence cardiac function by PDE5 inhibition, and there is currently a tremendous interest in identifying new clinical uses of PDE5 inhibitors for treatment of a variety of cardiovascular diseases (Kim and Kass, 2017; Sevil Korkmaz‐Icöz et al., 2018).
Approved clinical applications of PDE5 inhibitors
There are currently three approved indications for PDE5 inhibitors: ED, LUTS and PAH.
General rationale for use of PDE5 inhibitors
In the corpora cavernosa, relaxation of the smooth muscle of the sinusoids is induced by NO release from endothelial cells and nitrergic neurons surrounding the arteries and sinusoids (Figure 2). By inhibiting PDE5 hydrolytic activity, PDE5 inhibitors produce a higher rate of accumulation of cGMP in response to the NO, thus enhancing the erectile response.
Figure 2.

Mechanisms of penile erection (from Andersson, 2011). In the penile vessels and the smooth muscle of the corpora cavernosa, the balance between contractile and relaxant factors controls the degree of tone of the penile vasculature and of the smooth muscle. This in turn determines the functional state of the penis: detumescence and flaccidity, tumescence and erection. NO is released from nitrergic nerves, from the endothelium in response to the release of acetylcholine by parasympathetic endothelial nerve endings and by the shear stress produced by increased blood flow in the corporeal sinusoids. This dilates the helicinal arteries, fills the sinusoids, which eventually compresses the circumflex veins against the tunica albuginea, finally resulting in erection.
In the LUT, there is an abundance of PDE5 isoenzymes and their inhibition reduces smooth muscle cell proliferation, relaxes smooth muscle cells, increases tissue oxygenation and modulates afferent nerve activity (Figure 3).
Figure 3.
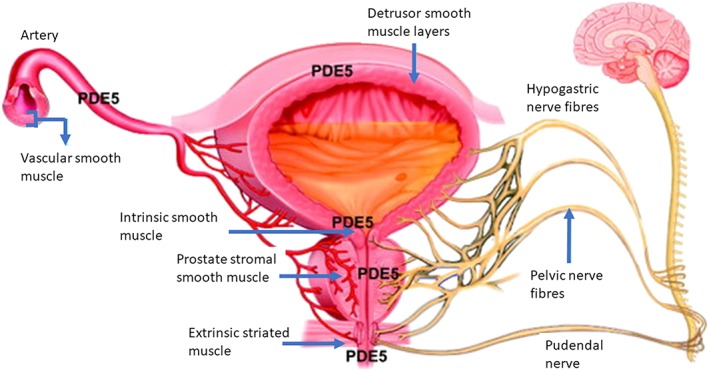
Distribution of PDE5 enzymes in the lower urinary tract. PDE5 isoenzymes are widely distributed in the LUT: the smooth muscle of the detrusor and the internal urethral sphincter, prostatic stroma, external urethral sphincter and the vasculature. Each of these locations can be involved in the pathophysiology of LUTS, resulting in afferent and efferent signalling in the pelvic, hypogastric and pudendal nerves. PDE5 inhibitors may improve LUTS by improving LUT oxygenation, relaxation of smooth muscle, negative regulation of proliferation and trans‐differentiation of prostate stroma, down‐regulation of prostate inflammation and reduction of bladder afferent nerve activity.
In the pulmonary vasculature, NO plays an important role as a vasorelaxant, and a high level of PDE5 has been demonstrated (Figure 4). This provides a strong molecular basis for using a PDE5 inhibitor to treat PAH.
Figure 4.
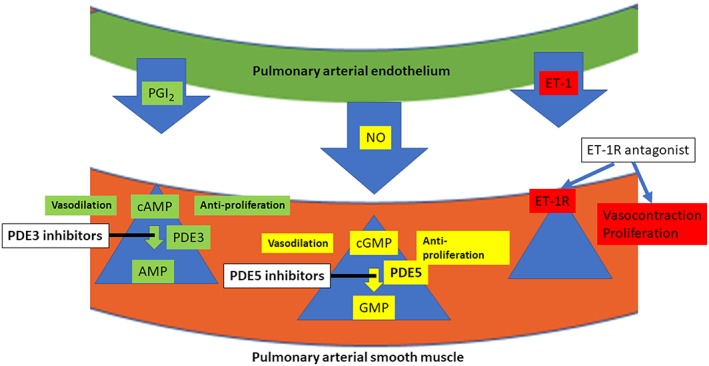
The principal targets for current therapies for PAH PGI2 (prostacyclin), relaxes smooth muscle cells via increases in intracellular cAMP levels. Inhibitors of PDE3, such as milrinone, stabilize the cAMP concentrations. NO dilates smooth muscle cells by increasing intracellular cGMP levels, and these levels are maintained by inhibition of PDE5 by, for example, sildenafil. For PDE5 inhibitors to be effective, there must be activity in the NO/sGC/cGMP pathway. ET‐1 is a vasoconstrictor, and blockers of both http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=219 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=220 receptor subtypes, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3494, decrease smooth muscle tone.
Erectile function and dysfunction
Historical background
To maintain the penis in the flaccid state, the smooth muscles of penile arteries and trabeculae are kept contracted, probably by the release of noradrenaline (NA) acting on postjunctional http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=4. Dilatation of penile arteries and sinusoids with the resultant increase in penile blood flow is considered a primary haemodynamic event in erection (Figure 2). Stimulation of the parasympathetic sacral nerves innervating the penis produces erection, and several findings have suggested that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=294 participates in the induction of erection (Andersson and Wagner, 1995; Andersson, 2001). However, experiments on isolated erectile tissues showed that muscarinic receptor stimulation had no or only minor inhibitory effects on relaxations induced by electrical field stimulation, and the fact that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=320 was without effect on erections provoked by visual stimulation or by local vibration (Wagner, 1981 1981) favoured the occurrence of an non‐adrenergic non‐cholinergic (NANC) dilator substance. The finding of Furchgott and Zawadzki (1980) that acetylcholine‐induced dilatation of rabbit aorta was mediated by the release of a relaxant agent from the endothelial cells, which they later termed the endothelium‐derived relaxing factor, focused interest on NANC mechanisms in penile erectile tissue. The reaction of penile erectile tissue to different autonomic drugs and electrical field stimulation was a starting point for the development of drugs that could influence penile erection. Hedlund and Andersson (1985) found that in NA‐contracted human corpus cavernosum preparations acetylcholine, but particularly the muscarinic receptor agonist, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=298, had a potent relaxant action, and Saenz de Tejada et al. (1988) showed that the relaxing effect of exogenous acetylcholine is dependent on an intact endothelium. Subsequent studies have indicated that other agents beside acetylcholine also cause vasodilatation by an endothelium‐dependent mechanism, but that the most important relaxing factor is NO. This was shown in both rabbit (Ignarro et al., 1990) and human (Holmquist et al., 1991, 1992; Rajfer et al., 1992) corpus cavernosum. These studies and the observation that the PDE5 inhibitor, sildenafil, caused penile erection were the basis for the development and use of number of selective PDE5 inhibitors for treatment of ED.
Physiology of penile erection
Sexual arousal through visual, tactile, olfactory or imaginative stimuli stimulates neural pathways in the brain that result in the release of NO from nerves and endothelial cells directly into the penis (Andersson and Wagner, 1995; Andersson, 2001, 2011). Erectogenic stimuli may also originate in the sacral spinal erection centre in response to direct tactile stimulation of the penis, and efferent neurons from this centre reach the pelvic plexus, from which postganglionic nitrergic fibres pass in the cavernous nerves to the corpora cavernosa. NO is released from nitrergic nerve terminals in the corpus cavernosum and also from the endothelium in response to the release of acetylcholine by parasympathetic endothelial nerve endings and by the shear stress produced by increased blood flow in the corporeal sinusoids. NO penetrates into the cytoplasm of smooth muscle cells and binds to the haem moiety of sGC. The interaction of NO with sGC causes a conformational change in this enzyme, activating it, and this will catalyse the breakdown of GTP into cGMP which is the intracellular trigger for penile erection. cGMP activates PKG leading to a cascade of events. Even if other pathways with cAMP as a second messenger (activating PKA) are involved, the cGMP and PKG pathway is the most important. As mentioned above, PKGI‐deficient mice showed deficient NO/cGMP‐dependent relaxation of vascular smooth muscle. These mice have a very low ability to reproduce and relaxant responses to neuronally or endothelially released or exogenously administered NO in corpus cavernosum tissue were markedly reduced (Hedlund et al., 2000). The activation of PKG and PKA initiates a series of cellular events via phosphorylation of various targets in the smooth muscle cell: inhibition of the noradrenergic pathway, opening of BKCa channels and activation of the Ca2+ ATPase transporter in the membrane of the sarcoplasmic reticulum. Eventually, these processes lead to a reduction in intracellular calcium levels and a consequent relaxation of arterial and trabecular smooth muscle. This results in vasodilatation and enhancement of blood flow into the cavernosal sinusoids and eventually penile erection. Blood becomes trapped in the corporal bodies by compression of subtunical venules against the tunica albuginea (full erection phase; Figure 2) and contraction of the voluntary ischiocavernosus muscle (rigid erection phase).
Since cGMP plays a key role in these processes, potential interventions for inadequate smooth muscle relaxation include increasing the level of intracellular cGMP. This can be achieved by PDE5 inhibition, but in the absence of stimulation of the NO pathway, PDE5 inhibition is ineffective. In human cavernosal tissue, at least 13 isoenzymes have been identified, including PDE3 (cGMP‐inhibited cAMP PDE), PDE4 (cAMP‐specific PDE) and PDE5 (cGMP‐specific PDE). Functionally, PDE 3A and 5A seem to be the most important. Three of the PDE families (PDEs 5, 6 and 9) have a 100‐fold substrate preference for cGMP over cAMP as substrate and are, therefore, considered to be cGMP‐specific PDEs. PDE5 and PDE9 are the only cGMP‐specific PDEs that are expressed in peripheral tissues; PDE6 is expressed in the retina (Lin, 2006; Francis et al., 2011).
Clinical efficacy
Sildenafil citrate was the first effective oral treatment for ED, and its introduction marked a milestone in the ED history (Goldstein et al., 1998; Osterloh, 2004). Current guidelines recommend PDE5 inhibitors as first‐line therapy for most men with ED who do not have a specific contraindication to their use; they state that PDE5 inhibitors are effective, safe and well‐tolerated therapies, and that there are no significant differences in efficacy, safety and tolerability between the approved drugs (Hatzimouratidis et al., 2016). These recommendations are based on numerous trials, which have established both the efficacy and the safety profile. Sildenafil was the first (in 1998) compound introduced clinically followed by vardenafil and tadalafil (in 2003) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7448 (in 2013). These drugs are available in most countries of the world. Other PDE5 inhibitors such as udenafil, mirodenafil and lodenafil are also available in some countries (Hatzimouratidis et al., 2016).
The structural differences between these PDE5 inhibitors are small, but they have different pharmacokinetic (Table 1) and dynamic properties (Table 2). In clinical practice, this is reflected in their duration of action. The extended plasma half‐time of tadalafil provides a longer therapeutic effect (up to 36 h), and this also might be the case for udenafil (up to 12 h). The duration of action for avanafil is longer than 6 h, that for sildenafil is at least 12 h and that for vardenafil is at least 10 h. The limited published data have shown that the onset and duration of action for mirodenafil and lodenafil are similar to those for sildenafil.
Table 1.
Pharmacokinetics of PDE5 inhibitors (from Hatzimouratidis et al., 2016)
| PDE5 inhibitor | Tmax (h) | t 1/2 (h) | Cmax (ng·mL−1) | AUC (ng × h·mL−1) |
|---|---|---|---|---|
| Avanafil 200 mg | 0.75 | 5.1 | 2920 | 8490 |
| Lodenafil 160 mg | 1.2 | 2.4 | 157 | 530 |
| Mirodenafil 100 mg | 1.4 | 2.5 | 2989 | 7907 |
| Sildenafil 100 mg | 0.95 | 3.98 | 514 | 1670 |
| Tadalafil 20 mg | 2 | 17.5 | 378 | 8066 |
| Vardenafil FCT 20 mg | 0.66 | 3.9 | 20.9 | 74.5 |
| Vardenafil ODT 10 mg | 1.5 | 4.23 | 7.34 | 30.39 |
| Udenafil 200 mg | 0.76 | 9.88 | 1137 | 7898 |
AUC, area under the curve; Cmax, maximum plasma concentration; FCT, film‐coated tablet;
ODT, oro‐dispersible tablet; PDE5, phosphodiesterase type 5; t 1/2, time required for elimination of one half of the inhibitor from plasma; Tmax, time required for attaining maximum plasma concentration.
PK data were obtained after single‐dose oral administration of the different PDE5 inhibitors.
Table 2.
Pharmacodynamics of PDE5 inhibitors (modified from Hatzimouratidis et al., 2016)
| PDE5 inhibitor | MW g·mol−1 | MF | IC50 for PDE5 (nmol·L−1) | PDE selectivity |
|---|---|---|---|---|
| Avanafil | 483.957 | C23H26CIN7O3 | 5.2 | Highly selective for PDE5 |
| Lodenafil | 1035.206 | C47H62N12O11S2 | 0.015 | Low activity against PDE1 and PDE6 |
| Mirodenafil | 531.672 | C26H37N5O5S | 0.33 | Comparable to sildenafil for PDE5 |
| Sildenafil | 474.580 | C22H30N6O4S | 3.7 | Low activity against PDE6 Very low activity against PDE1 |
| Tadalafil | 389.411 | C22H19N3O4 | 1.8 | Low activity against PDE11 Very low activity against PDE6 |
| Vardenafil | 408.607 | C23H32N6O4S | 0.091 | Low activity against PDE6 Very low activity against PDE1 |
| Udenafil | 516.661 | C25H36N6O4S | 8.25 | Comparable to sildenafil for PDE5 |
MF, molecular formula; IC50, concentration that inhibits the effect by 50%.
The following sources/species were used for the determination of IC50 values: avanafil: canine lung (Kotera et al., 2012); lodenafil: human platelets (Toque et al., 2008); mirodenafil: source of PDE5 not indicated (Shin et al., 2006); sildenafil, tadalafil and vardenafil: bovine PDE (Blount et al., 2004); udenafil: human platelets (Doh et al., 2002).
Overall, efficacy for the different PDE5 inhibitors rates is 60–70% with on‐demand treatment regimens (Albersen et al., 2010). Of the patients that initially do not respond to PDE5 inhibitors, between 30 and 50% may be converted to responders by counselling the patient and his partner. Some patients who fail to achieve an erection when taking PDE5 inhibitors on‐demand benefit from a daily dosing regimen. Furthermore, in the male suffering from ED in the context of late onset hypogonadism, addition of testosterone supplementation might enhance PDE5 inhibitor therapy.
As the efficacy of PDE5 inhibitors depends on the integrity of the NO pathway in producing cGMP, it is evident that patients in whom this pathway is disturbed or defective will benefit far less than the general population from PDE5 inhibitors. Disease states that diminish NO availability include denervation of the erectile tissue following radical prostatectomy; severe diabetes with neuropathy and endothelial dysfunction; metabolic syndrome; and down‐regulation of NOS expression as may be seen in atherosclerosis, ageing and hypogonadism.
Adverse effects
The safety profile of the currently available PDE5 inhibitors is excellent, based on post‐marketing data and further demonstrated by the recent FDA approvals for daily use of PDE5 inhibitors. These drugs are contraindicated in patients with unstable angina pectoris, recent myocardial infarction, certain arrhythmias and poorly controlled hypertension. Furthermore, patients who are treated with nitrates or nitrate‐donors should not take PDE5 inhibitors, and use of PDE5 inhibitors with α1‐adrenoceptor antagonists may result in postural hypotension. The most common adverse events from PDE5 inhibitors are attributable to specific inhibition of PDE5 and vasodilatation in tissues other than the penis and include headache, facial and ocular hyperaemia, nasal congestion, myalgia and back pain. Adverse events account for about 25% of cases in which PDE5 inhibitors are discontinued, but the most common reason for discontinuation of PDE5 inhibitors being lack of efficacy. There have been rare reports of serious adverse events such as seizures, non‐arteritic ischaemic optic neuritis and acute hearing loss. Some adverse events can be attributed to cross‐reactivity with other PDE‐isoforms (Table 3). Vision disturbances, which are believed to result from cross‐reactivity of PDE5 inhibitors with PDE6 (an isoform of PDE that is abundantly present in the cones of the retina), have been reported with PDE5 inhibitor use. Tadalafil has been shown to cross‐react with PDE11 to some extent, although no consequences of this cross‐reactivity are currently known. None of the PDE5 inhibitors available has shown clinically significant cross‐reactivity with PDE isoforms other than PDE6.
Table 3.
Selectivity of the four worldwide available PDE5 inhibitors (Hatzimouratidis et al., 2016)
| Selectivity versus PDE5 (fold difference) | ||||
|---|---|---|---|---|
| PDE5 isoenzyme | Avanafil | Sildenafil | Vardenafil | Tadalafil |
| PDE1 | >10 192 | 375 | 1012 | 10 500 |
| PDE2 | 9808 | 39 375 | 273 810 | >25 000 |
| PDE3 | >19 231 | 16 250 | 26 190 | >25 000 |
| PDE4 | 1096 | 3125 | 14 286 | 14 750 |
| PDE5 | 1 | 1 | 1 | 1 |
| PDE6 | 121 | 16 | 21 | 550 |
| PDE7 | 5192 | 13 750 | 17 857 | >25 000 |
| PDE8 | 2308 | >62 500 | 1 000 000 | >25 000 |
| PDE9 | >19 231 | 2250 | 16 667 | >25 000 |
| PDE10 | 1192 | 3375 | 17 857 | 8750 |
| PDE11 | >19 231 | 4875 | 5952 | 25 |
Symptoms associated with benign prostatic obstruction
Many epidemiological studies in different geographical areas have provided strong evidence that the association between LUTS and ED is not dependent on age. Several biological mechanisms have been implicated in this association, for example, the NO/cGMP pathway, http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=289‐mediated activation, autonomic hyperactivity, pelvic ischaemia/microvascular dysfunction, inflammatory pathways, sex hormones and psychological factors, but further research is required to better understand the molecular pathways involved. Evaluation of the possible impact of metabolic syndrome seems relevant (Cellek et al., 2014; De Nunzio et al., 2017).
PDE5 inhibitors cause relaxation of the bladder detrusor smooth muscle, and this effect is partly independent of NO (Oger et al., 2010). Fusco et al. (2012) found that http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9532 (H2S), which is predominantly formed from L‐cysteine by the enzymes http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=279#1443 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=279#1443, may be involved in the relaxant effect of sildenafil in the human bladder. CBS and CSE were found to be present in the bladder dome and efficiently convert L‐cysteine into H2S, and both NaHS and L‐cysteine relaxed human bladder strips. Sildenafil also caused relaxation of bladder strips and a concentration‐dependent increase in H2S production, and the authors suggested that this effect may account in part for the efficacy of PDE5 inhibitors in LUTS. Whether this opens up new therapeutic approaches remains to be established.
Rationale for use of PDE5 inhibitors
It is well documented that PDE5 inhibitors improve male LUTS, regardless of whether these symptoms are associated with ED (Uckert and Stief, 2011). The effects forming the rationale for use of PDE5 inhibitors in male LUTS may include improvement of LUT oxygenation, relaxation of smooth muscle, negative regulation of proliferation and trans‐differentiation of prostate stroma, reduction of bladder afferent nerve activity and down‐regulation of prostate inflammation (Andersson et al., 2011; Giuliano et al., 2013).
As mentioned above, PDE5 is expressed and has biological activity in all parts of the genitourinary tract (Figure 3) including the vasculature and urethral striated muscle (Lin et al., 2006); however, with regard to its role in LUTS pathophysiology, focus has been on smooth muscle in the prostate, bladder and urethra (Fibbi et al., 2010; Andersson et al., 2011; Gacci et al., 2016). Pathophysiological changes underlying the development of BPH‐LUTS are multifaceted, and structural and functional changes in the bladder, prostate, supporting vasculature and innervation can affect bladder function (Andersson et al., 2011; Gacci et al., 2016).
Nomiya et al. (2013) found, in a rat model of chronic pelvic ischaemia, that treatment with tadalafil reduced bladder overactivity, decreased indicators of bladder ischaemia, normalized changes in NOS activity and prevented the accumulation of collagen. Thus, there might be several mechanisms generating afferent activity in the LUT, and changes in afferent signalling from the smooth muscle in the prostate, bladder and urethra seem to be the common mechanism in the onset of LUTS. This afferent activity is inhibited by PDE5 inhibitors. Interestingly, the mucosal effects of PDE5 inhibitors do not seem to have been specifically investigated.
Clinical efficacy
To date, only tadalafil has been approved for the treatment of LUTS secondary to BPH. The first clinical trial was conducted by Sairam et al. (2002) using sildenafil for treatment of LUTS/BPH/ED patients. After 3 months of treatment, there was a significant inverse relationship between international prostate symptom score (IPSS) and international index of erectile function score suggesting that sildenafil improved both LUTS and ED. Since then, the effects of PDE5 inhibitors, especially tadalafil, on LUTS/BPH have been extensively investigated. Many randomized controlled clinical trials (RCTs) have shown significant improvement in urinary symptoms and that the drug is well tolerated (Stief et al., 2008; Roehrborn et al., 2010; Martínez‐Salamanca et al., 2011; Gacci et al., 2016). Several other PDE5 inhibitors may improve male LUTS; however, only tadalafil (5 mg once daily) has been licensed for the treatment of LUTS with or without ED.
The majority of the studies demonstrated that PDE5 inhibitors alone were efficacious in decreasing IPSS total score, storage subscore and voiding subscore, with the exception of the maximum urinary flow rate (Qmax). Oelke et al. (2014) showed that LUTS/BPH patients treated daily with tadalafil (5 mg) had greater treatment satisfaction compared with daily http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=488 (0.4 mg) or placebo. Gacci et al. (2012) conducted an extensive pair‐wise meta‐analysis on the use of PDE5 inhibitors alone or in combination with α1‐adrenoceptor antagonists for the treatment of LUTS/BPH. They found that the combination of PDE5 inhibitors with α‐adrenoceptor antagonists can significantly improve both LUTS and erectile function in men with BPH. This was confirmed by Wang et al. (2014) who found in a systematic review and network meta‐analysis including 64 RCTs with 28 196 participants that combination of α1‐adrenoceptor antagonists together with PDE5 inhibitors was the most effective therapy.
Adverse effects
Liu et al. (2011) found that the relative risk of adverse events from tadalafil, vardenafil and sildenafil was 2.27, 1.86 and 1.22 respectively: the overall incidence of adverse events was 37.31% for PDE5 inhibitors compared with 24.03% for placebo, while serious adverse events were reported in 1.10% of men treated with tadalafil, 1.85% of those treated with vardenafil and 1.05% of those treated with sildenafil. The first meta‐analysis of adverse events due to PDE5 inhibitors reported that flushing, gastro‐oesophageal reflux, headache and dyspepsia were the most common side effects (odds ratio for occurrence: 4.88; 2.21; 1.88; 1.85; respectively). Moreover, regarding the overall tolerability of the association between PDE5 inhibitors and α1‐adrenoceptor antagonists, Gacci et al. (2012) described seven out of 103 adverse events (6.8%) with combined therapy and five of 99 (5.1%) in men treated with α‐adrenoceptor antagonists alone. Similarly, flushing (4.37%), headache (4.23%), dyspepsia (3.69%), nasopharyngitis (2.27%) and dizziness (1.69%) were the most common treatment‐related adverse events in a comparison network meta‐analytic study on PDE5 inhibitors (Wang et al., 2014).
Pulmonary arterial hypertension
PAH is defined as a group of diseases of the small pulmonary arteries, characterized by vascular narrowing leading to a progressive increase in pulmonary vascular resistance and increased right ventricle afterload. The consequence of this is failure of the afterload‐intolerant right ventricle, eventually leading to premature death (Hampl and Herget, 2000; Humbert et al., 2004). PAH has a multifactorial pathobiology and pathophysiological mechanisms of the disease include pulmonary endothelial dysfunction, which leads to impaired production of vasodilators, such as NO and prostacyclin, and overexpression of vasoconstrictors, such as endothelin‐1 (Figure 4). However, vascular proliferation and remodelling of the bladder wall is the hallmark of the pathogenesis. The multifactorial pathophysiology implies that treatment needs to be individualized and that one therapy may not be optimal for all patients. This is reflected in the number of drugs used for treatment. At present, there are five approved classes of drugs: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=21 antagonists, PDE5 inhibitors, prostacyclin (PGI2) analogues, calcium channel blockers and sGC stimulators (Desai and Desouza, 2017) (Figure 4). Combination therapy may be useful, but therapy should be individualized.
Rationale for use of PDE5 inhibitors
Corbin et al. (2005) demonstrated a high level of PDE5 in lung, approximately as high as that in penile corpus cavernosum, and suggested that the abundance of PDE5 in lung vascular smooth muscle may provide a strong molecular basis for PDE5 inhibitor treatment of PAH. It has been shown that PDE5 is up‐regulated in conditions associated with PAH, and by selectively inhibiting PDE5 with, for example, sildenafil citrate, NO‐mediated vasodilatation in the lung can be obtained; PDE5 inhibition may also have antiproliferative effects on pulmonary vascular smooth‐muscle cells. However, the efficacy of PDE5 inhibitors is dependent on the presence of sufficient amounts of endogenous NO to activate sGC and generate cGMP. Since NO dilates smooth muscle cells by increasing intracellular cGMP levels, low endogenous NO/cGMP production significantly limits or impairs the effects of PDE5 inhibitors. Inhaled NO is the first‐line vasodilator therapy in persistent pulmonary hypertension of the newborn and is commonly used in the intensive care unit (Kim et al., 2016). In term and near‐term infants with hypoxia, oxygenation was improved in approximately 50% of infants receiving inhaled NO (Barrington et al., 2017). PDE5 inhibitor therapy is the most commonly recommended oral treatment option in children with PAH (Kim et al., 2016). For PDE5 inhibitors to be effective, there must be sufficient activity of the NO/ sGC/cGMP pathway. If there is insufficient NO production to stimulate sGC, an alternative way is the use of NO‐independent sCG stimulators (e.g. riociguat: BAY 63–2521). PGI2 (prostacyclin) induces relaxation of vascular smooth muscle by stimulating the production of cAMP and inhibits the growth of smooth‐muscle cells. In addition, it is a powerful inhibitor of platelet aggregation. Inhibitors of PDE3, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5225, stabilize the cAMP concentrations, but milrinone is not commonly used in the treatment of PAH.
Clinical efficacy
Galiè et al. (2005) performed a double‐blind, placebo‐controlled study of the effects of placebo or sildenafil (20, 40 or 80 mg, p.o.) three times a day for 12 weeks in 278 randomly assigned patients with symptomatic PAH. The primary measure of efficacy was the change in exercise capacity, as measured by the total distance walked in 6 min, from baseline to week 12. They found that sildenafil improved exercise capacity, WHO functional class and haemodynamics. Most adverse events were mild to moderate in intensity for all treatment groups. Unegbu et al. (2017) performed a systematic review of the comparative effectiveness and safety of PDE5 inhibitors in the management of paediatric patients with PAH. They found strong evidence that PDE5 inhibitor use improves echocardiography measurements, cardiac catheterization parameters and oxygenation compared with baseline or placebo. There is also evidence that low‐ and moderate‐doses of sildenafil are safe regimens for children. In a meta‐analysis assessing the effects of PDE5 inhibitors in patients suffering from PAH due to left chronic heart failure, De Vecchis et al. (2017) found that among patients (>18 years old) with reduced left ventricular ejection fraction, a significant benefit was conferred by PDE5 inhibitors against the risk of the composite endpoint of death and hospitalization. In contrast, patients with preserved left ventricular ejection fraction had no benefit from PDE5 inhibitor treatment.
Adverse effects
The safety profile has generally been benign with headaches, flushing, dyspepsia, diarrhoea, nasal congestion and tinnitus among the most commonly reported side effects (Galiè et al., 2005; Unegbu et al., 2017).
Conclusions
The NO/cGMP pathway is involved in many normal physiological functions and in the pathophysiology of a wide range of diseases. PDE5 inhibitors have not only been invaluable tools for the study of the functions and dysfunctions of the NO/cGMP pathway but have also been used as therapeutic agents as a result of their effects on, for example, smooth muscle, cardiovascular tissues and platelets. Since the discovery and introduction of sildenafil, inhibitors of PDE5 have been the first‐line therapy for ED, and the success of sildenafil has stimulated research in the field and led to a number of new applications, such as treatment of LUTS and PAH, now approved indications. PDE5 isoenzymes have been identified in a wide variety of tissues, which should imply that PDE5 inhibitors will have an effect on many organs and functions, and they have also been used for many not yet approved indications, such as diabetes and cancer, and the fields of clinical use are increasing (Gur et al., 2012; Ribaudo et al., 2016). Even though PDE5 inhibitors are useful treatments of many diseases, they are by no means a panacea, and new clinical applications should always be guided by the results of randomized clinical trials.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Conflict of interest
The author declares no conflicts of interest.
Andersson, K.‐E. (2018) PDE5 inhibitors – pharmacology and clinical applications 20 years after sildenafil discovery. British Journal of Pharmacology, 175: 2554–2565. doi: 10.1111/bph.14205.
References
- Albersen M, Shindel AW, Mwamukonda KB, Lue TF (2010). The future is today: emerging drugs for the treatment of erectile dysfunction. Expert Opin Emerg Drugs 15: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE, Wagner G (1995). Physiology of penile erection. Physiol Rev 75: 191–236. [DOI] [PubMed] [Google Scholar]
- Andersson KE (2001). Pharmacology of penile erection. Pharmacol Rev 53: 417–450. [PubMed] [Google Scholar]
- Andersson KE (2011). Mechanisms of penile erection and basis for pharmacological treatment of erectile dysfunction. Pharmacol Rev 63: 811–859. [DOI] [PubMed] [Google Scholar]
- Andersson KE, de Groat WC, McVary KT, Lue TF, Maggi M, Roehrborn CG et al (2011). Tadalafil for the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia: pathophysiology and mechanism(s) of action. NeurourolUrodyn 30: 292–301. [DOI] [PubMed] [Google Scholar]
- Bajraktari G, Burhenne J, Bugert P, Haefeli WE, Weiss J (2017). Cyclic guanosine monophosphate modulates accumulation of phosphodiesterase 5 inhibitors in human platelets. Biochem Pharmacol 145: 54–63. [DOI] [PubMed] [Google Scholar]
- Barrington KJ, Finer N, Pennaforte T, Altit G (2017. Jan 5). Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev (1): CD000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkels R, Klotz T, Sticht G, Englemann U, Klaus W (2001). Modulation of human platelet aggregation by the phosphodiesterase type 5 inhibitor sildenafil. J Cardiovasc Pharmacol 37: 413–421. [DOI] [PubMed] [Google Scholar]
- Blount MA, Beasley A, Zoraghi R, Sekhar KR, Bessay EP, Francis SH et al (2004). Binding of tritiated sildenafil, tadalafil, or vardenafil to the phosphodiesterase‐5 catalytic site displays potency, specificity, heterogeneity, and cGMP stimulation. Mol Pharmacol 66: 144–152. [DOI] [PubMed] [Google Scholar]
- Boolell M, Allen MJ, Ballard SA, Gepi‐Attee S, Muirhead GJ, Naylor AM et al (1996). Sildenafil: an orally active type 5 cyclic GMP‐specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res 8: 47–52. [PubMed] [Google Scholar]
- Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, Kass DA (2013). Nitric oxide synthases in heart failure. Antioxid Redox Signal 18: 1078–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellek S, Cameron NE, Cotter MA, Fry CH, Ilo D (2014). Microvascular dysfunction and efficacy of PDE5 inhibitors in BPH‐LUTS. Nat Rev Urol 11: 231–241. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Beasley A, Blount MA, Francis SH (2005). High lung PDE5: a strong basis for treating pulmonary hypertension with PDE5 inhibitors. Biochem Biophys Res Commun 334: 930–938. [DOI] [PubMed] [Google Scholar]
- Das A, Xi L, Kukreja RC (2005). Phosphodiesterase‐5 inhibitor sildenafil preconditions adult cardiac myocytes against necrosis and apoptosis. Essential role of nitric oxide signaling. J Biol Chem 280: 12944–12955. [DOI] [PubMed] [Google Scholar]
- Denninger JW, Marletta MA (1999). Guanylate cyclase and the NO/cGMP signaling pathway. Biochim Biophys Acta 1411: 334–350. [DOI] [PubMed] [Google Scholar]
- De Nunzio C, Roehrborn CG, Andersson K‐E, McVary KT (2017). Erectile dysfunction and Lower urinary tract symptoms. Eur Urol Focus 3: 352–363. [DOI] [PubMed] [Google Scholar]
- Desai A, Desouza SA (2017). Treatment of pulmonary hypertension with left heart disease: a concise review. Vasc Health Risk Manag 13: 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vecchis R, Cesaro A, Ariano C, Giasi A, Cioppa C (2017). Phosphodiesterase‐5 inhibitors improve clinical outcomes, exercise capacity and pulmonary hemodynamics in patients with heart failure with reduced left ventricular ejection fraction: a meta‐analysis. J Clin Med Res 9: 488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh H, Shin CY, Son M, Ko JI, Yoo M, Kim SH et al (2002). Mechanism of erectogenic effect of the selective phosphodiesterase type 5 inhibitor, DA‐8159. Arch Pharm Res 25: 873–878. [DOI] [PubMed] [Google Scholar]
- Francis SH, Blount MA, Corbin JD (2011). Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 91: 651–690. [DOI] [PubMed] [Google Scholar]
- Fibbi B, Morelli A, Vignozzi L, Filippi S, Chavalmane A, De Vita G et al (2010). Characterization of phosphodiesterase type 5 expression and functional activity in the human male lower urinary tract. J Sex Med 7: 59–69. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376. [DOI] [PubMed] [Google Scholar]
- Fusco F, di Villa Bianca, Mitidieri E, Cirino G, Sorrentino R, Mirone V (2012). Sildenafil effect on the human bladder involves the L‐cysteine/hydrogen sulfide pathway: a novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur Urol 62: 1174–1180. [DOI] [PubMed] [Google Scholar]
- Gacci M, Corona G, Salvi M, Vignozzi L, McVary KT, Kaplan SA et al (2012). A systematic review and meta‐analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with alpha‐blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol 61: 994–1003. [DOI] [PubMed] [Google Scholar]
- Gacci M, Andersson KE, Chapple C, Maggi M, Mirone V, Oelke M et al (2016). Latest evidence on the use of phosphodiesterase type 5 inhibitors for the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur Urol 70: 124–133. [DOI] [PubMed] [Google Scholar]
- Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D et al (2005). Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 353: 2148–2157. [DOI] [PubMed] [Google Scholar]
- Gibson A (2001). Phosphodiesterase 5 inhibitors and nitrergic transmission‐from zaprinast to sildenafil. Eur J Pharmacol 411: 1–10. [DOI] [PubMed] [Google Scholar]
- Giuliano F, Oelke M, Jungwirth A, Hatzimouratidis K, Watts S, Cox D et al (2013). Tadalafil once daily improves ejaculatory function, erectile function, and sexual satisfaction in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia and erectile dysfunction: results from a randomized, placebo‐ and tamsulosin‐controlled, 12‐week double‐blind study. J Sex Med 10: 857–865. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Lue TF, Padma‐Nathan H, Rosen RC, Steers WD, Wicker PA. (1998). Oral sildenafil in the treatment of erectile dysfunction. Sildenafil Study Group. N Engl J Med 338:1397–404. [DOI] [PubMed] [Google Scholar]
- Gur S, Kadowitz PJ, Serefoglu EC, Hellstrom WJ (2012). PDE5 inhibitor treatment options for urologic and non‐urologic indications: 2012 update. Curr Pharm Des 18: 5590–5606. [DOI] [PubMed] [Google Scholar]
- Hampl V, Herget J (2000). Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev 80: 1337–1372. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzimouratidis K, Salonia A, Adaikan G, Buvat J, Carrier S, El‐Meliegy A et al (2016). Pharmacotherapy for erectile dysfunction: recommendations from the fourth International Consultation for Sexual Medicine (ICSM 2015). J Sex Med 13: 465–488. [DOI] [PubMed] [Google Scholar]
- Hedlund H, Andersson KE (1985). Comparison of the responses to drugs acting on adrenoreceptors and muscarinic receptors in human isolated corpus cavernosum and cavernous artery. J Auton Pharmacol 5: 81–88. [DOI] [PubMed] [Google Scholar]
- Hedlund P, Aszodi A, Pfeifer A, Alm P, Hofmann F, Ahmad M et al (2000). Erectile dysfunction in cyclic GMP‐dependent kinase I‐deficient mice. Proc Natl Acad Sci U S A 97: 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmquist F, Hedlund H, Andersson KE (1991). L‐NG‐nitro arginine inhibits non‐adrenergic, non‐cholinergic relaxation of human isolated corpus cavernosum. Acta Physiol Scand 141: 441–442. [DOI] [PubMed] [Google Scholar]
- Holmquist F, Hedlund H, Andersson KE (1992). Characterization of inhibitory neurotransmission in the isolated corpus cavernosum from rabbit and man. J Physiol 449: 295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G (2004). Treatment of pulmonary arterial hypertension. N Engl J Med 351: 1425–1436. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Bush PA, Buga GM, Wood KS, Fukuto JM, Rajfer J (1990). Nitric oxide and cyclic GMP formation upon electrical field stimulation cause relaxation of corpus cavernosum smooth muscle. Biochem Biophys Res Commun 170: 843–850. [DOI] [PubMed] [Google Scholar]
- Keravis T, Lugnier C (2012). Cyclic nucleotide phosphodiesterase (PDE) isozymes as targets of the intracellular signalling network: Benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol 165: 1288–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GE, Kass DA (2017). Cardiac phosphodiesterases and their modulation for treating heart disease. Handb Exp Pharmacol 243: 249–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, McSweeney J, Lee J, Ivy D (2016). Pediatric cardiac intensive care society 2014 consensus statement: pharmacotherapies in cardiac critical care pulmonary hypertension. Pediatr Crit Care Med 17 (3 Suppl 1): S89–S100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz‐Icöz S, Radovits T, Szabó G (2018). Targeting phosphodiesterase 5 as a therapeutic option against myocardial ischaemia/reperfusion injury and for treating heart failure. Br J Pharmacol 175: 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotera J, Mochida H, Inoue H, Noto T, Fujishige K, Sasaki T et al (2012). Avanafil, a potent and highly selective phosphodiesterase‐5 inhibitor for erectile dysfunction. J Urol 188: 668–674. [DOI] [PubMed] [Google Scholar]
- Lin CS, Lin G, Xin ZC, Lue TF (2006). Expression, distribution and regulation of phosphodiesterase 5. Curr Pharm Des 12: 3439–3457. [DOI] [PubMed] [Google Scholar]
- Liu L, Zheng S, Han P, Wei Q (2011). Phosphodiesterase‐5 inhibitors for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a systematic review and meta‐analysis. Urology 77: 123–129. [DOI] [PubMed] [Google Scholar]
- Martínez‐Salamanca JI, Carballido J, Eardley I, Giuliano F, Gratzke C, Rosen R et al (2011). Phosphodiesterase type 5 inhibitors in the management of non‐neurogenic male lower urinary tract symptoms: critical analysis of current evidence. Eur Urol 60: 527–535. [DOI] [PubMed] [Google Scholar]
- Massion PB, Balligand JL (2003). Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol 546 (Pt 1): 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellion BT, Ignarro LJ, Ohlstein EH, Pontecorvo EG, Hyman AL, Kadowitz PJ (1981). Evidence for the inhibitory role of guanosine 3′, 5′‐monophosphate in ADP‐induced human platelet aggregation in the presence of nitric oxide and related vasodilators. Blood 57: 946–955. [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 43: 109–142. [PubMed] [Google Scholar]
- Mónica FZ, Antunes E (2018). Stimulators and activators of soluble guanylate cyclase for urogenital disorders. Nat Rev Urol 15: 42–54. [DOI] [PubMed] [Google Scholar]
- Murad F (2006). Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med 355: 2003–2011. [DOI] [PubMed] [Google Scholar]
- Nomiya M, Burmeister DM, Sawada N, Campeau L, Zarifpour M, Keys T et al (2013). Prophylactic effect of tadalafil on bladder function in a rat model of chronic bladder ischemia. J Urol 189: 754–761. [DOI] [PubMed] [Google Scholar]
- Oelke M, Giuliano F, Baygani SK, Melby T, Sontag A (2014). Treatment satisfaction with tadalafil or tamsulosin vs placebo in men with lower urinary tract symptoms (LUTS) suggestive of benign prostatic hyperplasia (BPH): results from a randomised, placebo‐controlled study. BJU Int 114: 568–575. [DOI] [PubMed] [Google Scholar]
- Oger S, Behr‐Roussel D, Gorny D, Lebret T, Validire P, Cathelineau X et al (2010). Signalling pathways involved in sildenafil‐induced relaxation of human bladder dome smooth muscle. Br J Pharmacol 160: 1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh IH (2004). The discovery and development of Viagra ® (sildenafil citrate) In: Dunzendorter U. (ed). Sildenafil. © 2004 Birkhauser Verlag: Switzerland, pp. 1–13. [Google Scholar]
- Pfeifer A, Klatt P, Massberg S, Ny L, Sausbier M, Hirneiss C et al (1998). Defective smooth muscle regulation in cGMP kinase I‐deficient mice. EMBO J 17: 3045–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski MW, Palmer RM, Moncada S (1990). An L‐arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc Natl Acad Sci U S A 87: 5193–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehrborn CG, Kaminetsky JC, Auerbach SM, Montelongo RM, Elion‐Mboussa A, Viktrup L (2010). Changes in peak urinary flow and voiding efficiency in men with signs and symptoms of benign prostatic hyperplasia during once daily tadalafil treatment. BJU Int 105: 502–507. [DOI] [PubMed] [Google Scholar]
- Rajfer J, Aronson WJ, Bush PA, Dorey FJ, Ignarro LJ (1992). Nitric oxide as a mediator of relaxation of the corpus cavernosum in response to nonadrenergic, noncholinergic neurotransmission. N Engl J Med 326: 90–94. [DOI] [PubMed] [Google Scholar]
- Ribaudo G, Pagano MA, Bova S, Zagotto G (2016). New therapeutic applications of phosphodiesterase 5 inhibitors (PDE5‐Is). Curr Med Chem 23: 1239–1249. [DOI] [PubMed] [Google Scholar]
- Rondina MT, Weyrich AS (2012). Targeting phosphodiesterases in anti‐platelet therapy. Handb Exp Pharmacol 210: 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz de Tejada I, Blanco R, Goldstein I, Azadzoi K, de las Morenas A, Krane RJ et al (1988). Cholinergic neurotransmission in human corpus cavernosum. I. Responses of isolated tissue. Am J Physiol 254 (3 Pt 2): H459–H467. [DOI] [PubMed] [Google Scholar]
- Sairam K, Kulinskaya E, McNicholas TA, Boustead GB, Hanbury DC (2002). Sildenafil influences lower urinary tract symptoms. BJU Int 90: 836–839. [DOI] [PubMed] [Google Scholar]
- Shin H‐I, Lee L, Kim D‐K (2006). Synthesis of 5‐ethyl‐2‐{5‐[4‐(2‐hydroxyethyl)piperazin‐1‐ylsulfonyl]‐2‐n‐propoxyphenyl}‐7‐n‐propyl‐3,5‐dihydro‐4H‐pyrrolo[3,2‐d]‐[2‐14C]pyrimidin‐4‐one·2HCl (14C‐SK3530·2 HCl). J Label Comp Radiopharm 49: 1141–1149. [Google Scholar]
- Simon JN, Duglan D, Casadei B, Carnicer R (2014). Nitric oxide synthase regulation of cardiac excitation‐contraction coupling in health and disease. J Mol Cell Cardiol 73: 80–91. [DOI] [PubMed] [Google Scholar]
- Smolenski A (2012). Novel roles of cAMP/cGMP‐dependent signaling in platelets. J Thromb Haemost 10: 167–176. [DOI] [PubMed] [Google Scholar]
- Stasch JP, Pacher P, Evgenov OV (2011). Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 123: 2263–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stief CG, Porst H, Neuser D, Beneke M, Ulbrich E (2008). A randomised, placebo‐controlled study to assess the efficacy of twice‐daily vardenafil in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. Eur Urol 53: 1236–1244. [DOI] [PubMed] [Google Scholar]
- Terrett NK, Bell AS, Brown D, Ellis P (1996). Sildenafil (Viagra) a potent and selective inhibitor of type 5 cGMP phosphodiesterase with the utility for treatment of male erectile dysfunction. Bioorg Medicinal Chem Lett 6: 1819–1824. [Google Scholar]
- Toque HA, Teixeira CE, Lorenzetti R, Okuyama CE, Antunes E, De Nucci G (2008). Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum. Eur J Pharmacol 591: 189–195. [DOI] [PubMed] [Google Scholar]
- Uckert S, Kuczyk MA (2011). Cyclic nucleotide metabolism including nitric oxide and phosphodiesterase‐related targets in the lower urinary tract. Handb Exp Pharmacol 202: 527–542. [DOI] [PubMed] [Google Scholar]
- Uckert S, Stief CG (2011). Treatment of erectile dysfunction and lower urinary tract symptoms by phosphodiesterase inhibitors. Handb Exp Pharmacol 204: 307–322. [DOI] [PubMed] [Google Scholar]
- Unegbu C, Noje C, Coulson JD, Segal JB, Romer L (2017). Pulmonary hypertension therapy and a systematic review of efficacy and safety of PDE5 inhibitors. Pediatrics 139 pii: e20161450. [DOI] [PubMed] [Google Scholar]
- Wagner G (1981, 1981). Erection: physiology and endocrinology In: Wagner G, Green R. (eds). Impotence: physiological, psychological, surgical diagnosis and treatment. Plenum Press: New York, pp. 25–36. [Google Scholar]
- Wang X, Wang X, Li S, Meng Z, Liu T, Zhang X (2014). Comparative effectiveness of oral drug therapies for lower urinary tract symptoms due to benign prostatic hyperplasia: a systematic review and network meta‐analysis. PLoS One 9: e107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung J, Holinstat M (2012). Newer agents in antiplatelet therapy: a review. J Blood Med 3: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]