

Abstract
Anaphylaxis is a life‐threatening type I allergic reaction. Antigen–antibody complexes induce mast cells, basophils and neutrophils to release large amounts of histamine and/or PAF. These mediators induce hypotension and vascular hyper‐permeability and subsequent anaphylaxis dependent on the endothelial production of NO. Here, we have summarized previous studies reporting the mechanisms underlying the functional changes within the vasculature, specifically focusing on vascular permeability triggered by histamine or PAF. In addition to these pro‐inflammatory factors, PGD2 is abundantly released in anaphylaxis, mainly from mast cells. We recently demonstrated that mast cell‐derived PGD2 attenuates anaphylactic responses by inhibiting vascular hyper‐permeability in mouse models. Our findings suggest that pro‐ and anti‐inflammatory factors concurrently regulate vascular permeability in anaphylaxis. In this mini‐review, we discuss the multifactorial mechanisms underlying vascular hyper‐permeability in anaphylaxis.
Abbreviations
- NO
nitric oxide
- PGD2
prostaglandin
Introduction
Anaphylaxis is an acute, severe and potentially fatal systemic allergic reaction with hypotension and vascular hyper‐permeability as underlying symptoms. Approximately 1 in 300 Europeans have experienced anaphylaxis at some point in their lives (Panesar et al., 2013). Antigens, mostly contained in foods, drugs or insect venom, and their specific antibodies trigger anaphylaxis. Experimental studies show that antigen–antibody complexes stimulate mast cells as well as basophils and neutrophils to release http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1204 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1831 within 10 min (Lowell, 2011). Upon activation, mast cells initially release a large amount of histamine (about 1.5 μg·mL−1 in mouse plasma), and basophils and neutrophils subsequently release about 10 ng·mL−1 of PAF (Figure 1, Choi et al., 1998). These mediators are potent inducers of vascular hyper‐permeability in both mice and humans (Michel et al., 1987; Korhonen et al., 2009). There is accumulating evidence demonstrating that pharmacological inhibition or deficiency of endothelial NOS (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1249) abolishes vascular hypotension/hyper‐permeability in experimental models of anaphylaxis (Cauwels et al., 2006; Ashina et al., 2015). Thus, histamine or PAF‐induced production of NO by endothelium is assumed to be a critical factor in development of anaphylaxis.
Figure 1.
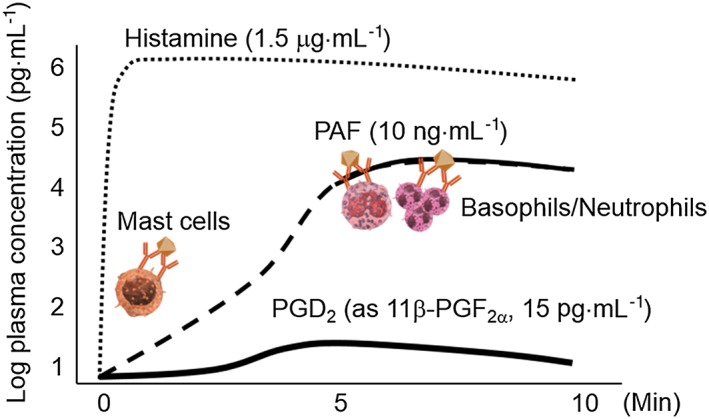
Estimated plasma concentrations of histamine, PAF and PGD2 in anaphylaxis. The stimulation of antigen and its specific IgE initially activates mast cells and releases a large amount of histamine. A few minutes later, activated mast cells de novo synthesize and secrete PAF and PGD2. Following mast cell activation, the antigen and its specific IgG stimulate basophils and/or neutrophils to synthesize PAF de novo and release it in greater concentrations than that of mast cells.
A few minutes after histamine release, mast cells de novo synthesize and release PGD2, achieving an estimated circulating concentration of 15 pg·mL−1 in human plasma (Figure 1, Bochenek et al., 2004). Although the role of mast cell‐derived PGD2 in anaphylaxis has not been determined, we recently demonstrated that PGD2 attenuates anaphylactic responses by inhibiting vascular hyper‐permeability (Nakamura et al., 2017). Our findings suggest the presence of multifactorial mechanisms underlying the modulation of anaphylactic vascular function.
Here, we summarize and discuss the mechanisms underlying anaphylaxis focusing on the modulation of vascular permeability by histamine, PAF and PGD2.
Cell‐types and mediators contributing to anaphylaxis
There are two main types of murine models for anaphylaxis frequently used in experiments; passive systemic anaphylaxis (PSA) and active systemic anaphylaxis (ASA). The PSA model is generated in naïve mice by the adoptive transfer of antigen specific IgE followed by the injection of antigen. Genetically modified mast cell‐deficient or IgE receptor (FcεRI)‐deficient mice totally lack anaphylactic hypotension and vascular hyper‐permeability in PSA (Dombrowicz et al., 1993; Miyajima et al., 1997). Also, the genetic deletion of histamine synthase, histidine decarboxylase, suppressed the PSA response of mice (Makabe‐Kobayashi et al., 2002). Thus, antigen/IgE‐induced PSA depends mainly on the FcεRI/mast cells/histamine axis which is referred to as the Classical pathway.
On the other hand, the ASA model is generated by immunizing with, and subsequently rechallenging with, the same antigen. Antigen immunization elicits abundant production of IgG rather than IgE. Murine ASA produces stronger anaphylactic reactions than PSA and are fatal in many cases, which is a model closer to to human anaphylaxis. In ASA, the deficiency of mast cells or IgE receptor (FcεRI) decreased anaphylactic reactions to some extent, but these deficiencies could not prevent the fatal consequences. However, the depletion of basophils or neutrophils, gene deficiency of IgG receptor (FcγR) was sufficient to block the fatal consequences of ASA (Miyajima et al., 1997; Tsujimura et al., 2008; Jönsson et al., 2011). Pharmacological inhibition of the PAF receptor exhibits similar protective effects (Arias et al., 2009). Collectively, the IgG/FcγR/basophil, neutrophil/PAF axis, referred to as the Alternative pathway, is involved in lethal ASA, while the IgE‐dependent Classical pathway also partly contributes to non‐lethal ASA.
Mechanisms underlying histamine‐ and PAF‐induced vascular hyper‐permeability in anaphylaxis
Rapid and marked vascular leaks in peripheral tissues results in tissue oedema, a hallmark of anaphylaxis (Simons and Sheikh, 2013). At the same time, rapid and large increases in blood flow to peripheral tissue lead to a decrease in the volume of circulating blood and subsequent hypotension and shock. Thus, investigators have recognized vascular hyper‐permeability as a major causative factor of anaphylaxis.
Vascular leaking occurs mainly in post‐capillary venules where the tightness of adherence junctions (AJs) between vascular endothelial cells (ECs) determines the extent of vascular leakiness. VE‐cadherin forms a complex with p120‐catenin, β‐catenin and plakoglobin to stabilize AJs. Since VEGF was identified as a potent vascular permeability factor by Senger in 1983, there have been many studies focusing on the molecular mechanisms of AJ disruption by VEGF (Senger et al., 1983). Yang et al. originally showed that VEGF disrupts the endothelial barrier via NO production by using a pharmacological eNOS inhibitor in vivo (Yang et al., 1996). Gene deficiency of eNOS also abolished VEGF‐induced vascular hyper‐permeability. In detail, Lorenzo and Lin et al. showed that VEGF‐induced NO disrupts endothelial adherence junctions by the enhanced activation of a Rac GTPase and the stabilization of cortical actin (Di Lorenzo et al., 2014). Other groups have highlighted the importance of tyrosine phosphorylation of VE‐cadherin in the disruption of the endothelial barrier in vitro (Esser et al., 1998).
Histamine and PAF also stimulate endothelial NO production with an increase in intracellular Ca2+ concentration (Lantoine et al., 1998; Zhu and He, 2005). Histamine is derived from the decarboxylation of the amino acid histidine, a reaction that is catalysed by the enzyme L‐histidine decarboxylase. It induces physiological action by binding to G protein‐coupled histamine receptors, designated H1, H2, H3 and H4. Mayhan showed that pharmacological inhibition of NOS or NO‐sensitive GC decreased histamine‐induced vascular leakage in hamster cheek pouches (Mayhan, 1994). Mikelis et al. showed that PSA or histamine‐induced vascular leakage through the H1 receptor and H1 receptor activation leads to endothelial barrier disruption through a RhoA and ROCK‐mediated VE‐cadherin rearrangement (Mikelis et al., 2015). Korhonen et al. demonstrated that a Gq/11 deficiency specific to endothelial cells did not elicit histamine or PAF‐induced local vascular hyper‐permeability, hypothermia and fatal shock (Korhonen et al., 2009).
While the above studies were focused on EC barrier modulation, we recently demonstrated, using intravital microscopy, that histamine dilated vasculature and increased blood flow, which induced hyper‐permeability in post‐capillary venulae. Vasoconstriction by phenylephrine or NOS inhibition strongly attenuated the histamine‐induced blood flow increase and the subsequent vascular hyper‐permeability without changing the localization of VE‐cadherin. NO‐dependent vascular dilation is assumed to be at least one major factor in histamine‐induced vascular leakage (Ashina et al., 2015).
PAF is a circulating lipid mediator released by activated neutrophils and monocytes under inflammatory conditions. PAF not only mediates platelet aggregation but also exerts a wide range of biological functions in innate and adaptive immunity via a single receptor (Hanahan, 1986). Cauwels et al. have demonstrated that a deficiency of eNOS, but not iNOS, diminishes PAF‐ or ASA‐induced hyper‐permeability, hypotension and fatal responses in mice. They also found that PI3K/Akt signalling mediates the PAF‐induced eNOS activation in anaphylactic shock (Cauwels et al., 2006). Several in vitro studies have also demonstrated that PAF promotes eNOS translocation from the plasma membrane to the cytosol, which preferentially induces hyper‐permeability (Sánchez et al., 2008, 2009, 2006). Thus, PAF‐induced fatal anaphylaxis relies upon NO‐dependent vascular hyper‐permeability. However, the details of its downstream mechanism remain unclear. NO‐mediated signalling activated by VEGF‐ or histamine stimulation may also be involved in PAF‐induced vascular hyper‐permeability.
Vascular mural and endothelial cells work together to control a range of vascular functions. The structure and cellular components of the vasculature vary by tissue type and site. Activated mast cells produce VEGF and TNF‐α in addition to histamine and PAF. Inflamed tissue secretes various types of bioactive substances including prostanoids. We cannot exclude the possibility that these mediators are included in the disruption of the vascular barrier during anaphylaxis. Comprehensive in vivo studies focusing on both functions are required to fully elucidate the pathophysiological implications of vascular permeability in anaphylaxis.
Role of PGD2 in anaphylaxis
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1881 was identified by Hamberg in 1973 (Hamberg and Fredholm, 1976). A haematopoietic PGD synthase (H‐PGDS) metabolizes COX‐derived PGH2 to PGD2. H‐PGDS is expressed in haematopoietic lineage cells, such as mast cells and dendritic cells, and is involved in the regulation of peripheral inflammation. PGD2 exerts its biological functions through two types of receptors, the Gs‐coupled http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=338 and the Gi‐coupled DP2 receptor (also known as CRTH2, chemoattractant receptor‐homologous molecule expressed on Th2 cells).
PGD2 is the major lipid mediator that is released from mast cells. We recently demonstrated that mast cell‐derived PGD2 inhibits murine anaphylaxis (Nakamura et al., 2017). Systemic H‐PGDS deficiency strongly enhanced PSA and mast cell activator‐induced vascular hyper‐permeability, hypotension and hypothermia. This hyperreactivity was also seen in mast cell specific H‐PGDS‐deficient mice. We also found that deficiency of the DP1 receptor enhanced anaphylactic responses while activation of these receptors was inhibitory. Pretreatment with the NOS inhibitor L‐NAME suppressed the excessive reactions seen in H‐pgds −/− mice suggesting that PGD2 inhibits NO production. Activation of DP1 receptors decreases intracellular Ca2+ and NO production in ECs. The decrease in NO production may be partially responsible for the DP1 receptor‐mediated anti‐anaphylactic reaction. In addition, we have shown that deletion of the DP1 receptor enhanced croton oil‐induced vascular hyper‐permeability in vivo (Kobayashi et al., 2013) and that stimulation of DP1 receptors rearranged the endothelial cytoskeleton during PKA/Rac1 activation in vitro. This signalling is also involved in the endothelial barrier enhancement mediated by DP1 receptors in anaphylaxis.
Concluding remarks
As summarized and discussed above, the sequential release of histamine and PAF and PGD2 concurrently govern vascular permeability via several signalling cascades, centred on the NO axis (Figure 2A, B). However, their overlapping roles and pathophysiological significance remain unknown. Further studies are required for a more detailed and comprehensive understanding of anaphylaxis.
Figure 2.
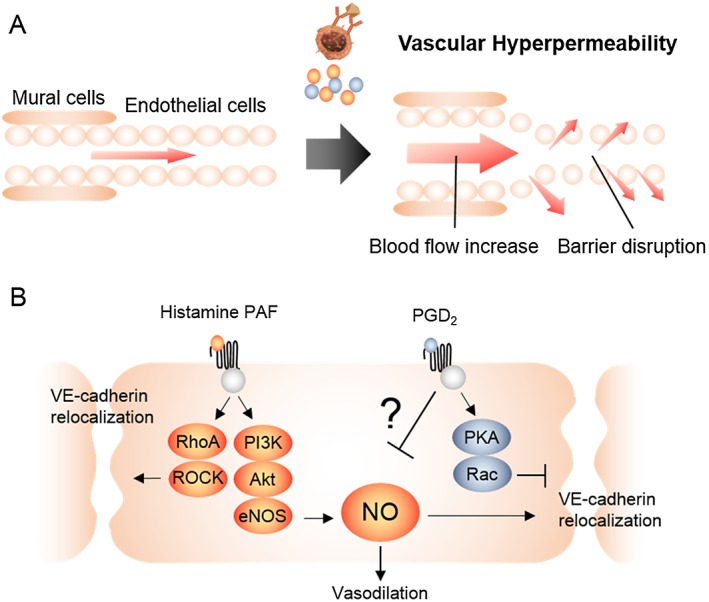
The mechanism of anaphylactic vascular hyper‐permeability. (A) Anaphylactic mediators induce vascular hyper‐permeability through the increase of blood flow and/or endothelial barrier disruption. (B) Histamine and PAF activate Gq‐coupled receptor and lead to eNOS activation through PI3K/Akt signalling followed by NO release. Released NO causes vasodilation and/or VE‐cadherin re‐localization. Histamine and PAF receptors activate RhoA/ROCK and mediates the disruption of adherence junctions. In contrast, the Gs‐coupled PGD2 receptor DP1, enhances the formation of endothelial barriers via PKA/Rac1 activation.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study is supported by the Japan Society for the Promotion of Science, Futaba Electronics Memorial Foundation, Hoyu Science Foundation, the Naito Foundation, the Nipponham Foundation and the Skylark Food Science Institute.
Nakamura, T. , and Murata, T. (2018) Regulation of vascular permeability in anaphylaxis. British Journal of Pharmacology, 175: 2538–2542. doi: 10.1111/bph.14332.
References
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Other proteins. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias K, Baig M, Colangelo M, Chu D, Walker T, Goncharova S et al (2009). Concurrent blockade of platelet‐activating factor and histamine prevents life‐threatening peanut‐induced anaphylactic reactions. J Allergy Clin Immunol 124: 307–314.e2. [DOI] [PubMed] [Google Scholar]
- Ashina K, Tsubosaka Y, Nakamura T, Omori K, Kobayashi K, Hori M et al (2015). Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS One 10: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochenek G, Nizankowska E, Gielicz A, Swierczynska M, Szczeklik A (2004). Plasma 9alpha,11beta‐PGF2, a PGD2 metabolite, as a sensitive marker of mast cell activation by allergen in bronchial asthma. Thorax 59: 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauwels A, Janssen B, Buys E, Sips P, Brouckaert P (2006). Anaphylactic shock depends on PI3K and eNOS‐derived NO. J Clin Invest 116: 2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi IH, Shin YM, Park JS, Lee MS, Han EH, Chai OH et al (1998). Immunoglobulin E‐dependent active fatal anaphylaxis in mast cell‐deficient mice. J Exp Med 188: 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A, Lin MI, Murata T, Landskroner‐Eiger S, Schleicher M, Kothiya M et al (2014). eNOS‐derived nitric oxide regulates endothelial barrier function through VE‐cadherin and Rho GTPases. J Cell Sci 127: 2120–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowicz D, Flamand V, Brigman KK, Koller BH, Kinet JP (1993). Abolition of anaphylaxis by targeted disruption of the high affinity immunoglobulin E receptor alpha chain gene. Cell 75: 969–976. [DOI] [PubMed] [Google Scholar]
- Esser S, Lampugnani MG, Corada M, Dejana E, Risau W (1998). Vascular endothelial growth factor induces VE‐cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci 111 (Pt 1): 1853–1865. [DOI] [PubMed] [Google Scholar]
- Hamberg M, Fredholm BB (1976). Isomerization of prostaglandin H2 into prostaglandin D2 in the presence of serum albumin. Biochim Biophys Acta 431: 183–189. [DOI] [PubMed] [Google Scholar]
- Hanahan DJ (1986). Platelet Activating Factor : A BIOLOGICALLY ACTIVE PHOSPHOGLYCERIDE. Annu Rev Biochem 55: 483–509. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson F, Mancardi DA, Kita Y, Karasuyama H, Iannascoli B, Van Rooijen N et al (2011). Mouse and human neutrophils induce anaphylaxis. J Clin Invest 121: 1484–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Tsubosaka Y, Hori M, Narumiya S, Ozaki H, Murata T (2013). Prostaglandin D2‐DP signaling promotes endothelial barrier function via the cAMP/PKA/Tiam1/Rac1 pathway. Arterioscler Thromb Vasc Biol 33: 565–571. [DOI] [PubMed] [Google Scholar]
- Korhonen H, Fisslthaler B, Moers A, Wirth A, Habermehl D, Wieland T et al (2009). Anaphylactic shock depends on endothelial Gq/G11. J Exp Med 206: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantoine F, Iouzalen L, Devynck MA, Millanvoye‐Van Brussel E, David‐Dufilho M (1998). Nitric oxide production in human endothelial cells stimulated by histamine requires Ca2+ influx. Biochem J 330: 695–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell CA (2011). Neutrophils give us a shock. J Clin Invest 121: 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makabe‐Kobayashi Y, Hori Y, Adachi T, Ishigaki‐Suzuki S, Kikuchi Y, Kagaya Y et al (2002). The control effect of histamine on body temperature and respiratory function in IgE‐dependent systemic anaphylaxis. J Allergy Clin Immunol 110: 298–303. [DOI] [PubMed] [Google Scholar]
- Mayhan WG (1994). Nitric oxide accounts for histamine‐induced increases in macromolecular extravasation. Am J Physiol Hear Circ Physiol 266: H2369–H2373. [DOI] [PubMed] [Google Scholar]
- Michel L, Mencia‐Huerta JM, Benveniste J, Dubertret L (1987). Biologic properties of LTB4 and paf‐acether in vivo in human skin. J Invest Dermatol 88: 675–681. [DOI] [PubMed] [Google Scholar]
- Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P et al (2015). RhoA and ROCK mediate histamine‐induced vascular leakage and anaphylactic shock. Nat Commun 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet JP, Galli SJ (1997). Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and FcyRIII. J Clin Invest 99: 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Fujiwara Y, Yamada R, Fujii W, Hamabata T, Lee MY et al (2017). Mast cell–derived prostaglandin D 2 attenuates anaphylactic reactions in mice. J Allergy Clin Immunol 140: 630–632.e9. [DOI] [PubMed] [Google Scholar]
- Panesar SS, Javad S, de Silva D, Nwaru BI, Hickstein L, Muraro A et al (2013). The epidemiology of anaphylaxis in Europe: a systematic review. Allergy Eur J Allergy Clin Immunol 68: 1353–1361. [DOI] [PubMed] [Google Scholar]
- Sánchez FA, Kim DD, Durán RG, Meininger CJ, Durán WN (2008). Internalization of eNOS via caveolae regulates PAF‐induced inflammatory hyperpermeability to macromolecules. Am J Physiol Hear Circ Physiol Oct 295: 1642–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK et al (2009). Internalization of eNOS and NO delivery to subcellular targets determine agonist‐induced hyperpermeability. Proc Natl Acad Sci U S A 106: 6849–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez FA, Savalia NB, Durán RG, Lal BK, Boric MP, Durán WN (2006). Functional significance of differential eNOS translocation. Am J Physiol Hear Circ Physiol 291: 1058–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger D, Galli S, Dvorak A, Perruzzi C, Harvey V, Dvorak H (1983). Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science(80‐. ) 219: 983–985. [DOI] [PubMed] [Google Scholar]
- Simons FER, Sheikh A (2013). Anaphylaxis: the acute episode and beyond. BMJ 346: 1–10. [DOI] [PubMed] [Google Scholar]
- Tsujimura Y, Obata K, Mukai K, Shindou H, Yoshida M, Nishikado H et al (2008). Basophils play a pivotal role in immunoglobulin‐G‐mediated but not immunoglobulin‐E‐mediated systemic anaphylaxis. Immunity 28: 581–589. [DOI] [PubMed] [Google Scholar]
- Yang RH, Thomas GR, Bunting S, Ko A, Ferrara N, Keyt B et al (1996). Effects of vascular endothelial growth factor on hemodynamics and cardiac performance. J Cardiovasc Pharmacol 27: 838–844. [DOI] [PubMed] [Google Scholar]
- Zhu L, He P (2005). Platelet‐activating factor increases endothelial [Ca2+]i and NO production in individually perfused intact microvessels. Am J Physiol Heart Circ Physiol 288: H2869–H2877. [DOI] [PubMed] [Google Scholar]