

Abstract
Cholesterol (Chol) is vital for cell function as it is essential to a myriad of biochemical and biophysical processes. The atomistic details of Chol’s interactions with phospholipids and proteins is therefore of fundamental interest, and NMR offers unique opportunities to interrogate these properties at high resolution. Towards this end, here we describe approaches for examining the structure and dynamics of Chol in lipid bilayers using high levels of 13C enrichment in combination with magic-angle spinning (MAS) methods. We quantify the incorporation levels and demonstrate high sensitivity and resolution in 2D 13C-13C and 1H-13C spectra, enabling de novo assignments and site-resolved order parameter measurements obtained in a fraction of the time required for experiments with natural abundance sterols. We envision many potential future applications of these methods to study sterol interactions with drugs, lipids and proteins.
Keywords: Solid-State NMR, Cholesterol, 13C-labeling, Biosynthesis, Membrane, Dipolar Couplings
1. Introduction
Sterols are essential in numerous biochemical and biophysical processes inside the cell. Cholesterol (Chol), the major sterol present in mammalian cells, is a key regulator of membrane order, permeability, thickness and lateral organization [1–3] and ultimately membrane protein function [1,4,5]. The regulatory roles of Chol depend directly upon its atomistic interactions with other sterols and phospholipid molecules [6–8]. In fact, Chol has also recently been implicated to have an essential role in HIV-mediated viral fusion [9] and ligand binding leading to apoptosis [10]. The orientation and dynamics of Chol were demonstrated to be essential for its regulatory roles [11,12], although molecular details of the mechanism are still lacking. Additionally, the atomistic specificity of sterol interactions with a variety of proteins is of increased interest, especially because Chol is a major component of mammalian membranes (typically 50 mol%) [13]. For example, Chol interacts with the amyloid precursor protein and may play a key role in amyloidogenesis related to Alzheimer’s disease [14]. These many applications underscore the broad significance of impactful methods to study this ubiquitous molecule in its native bilayer environment.
Much of what is known about the detailed structure and dynamics of Chol in lipid bilayers comes from experimental NMR studies, combined with molecular dynamics (MD). Extensive 2H and 13C studies of Chol have reported order parameters and restraints on the orientation in the bilayer from 2H NMR [15–27] and 13C NMR [28–30]. However, previous NMR dynamics investigations focused on a limited number of labeled sites [15–25] and/or used bulky analogs of Chol [31,32], which are known to behave differently in membranes than the native sterol [2,33]. Additionally, MD simulations of Chol in bilayers have been utilized to ascertain the orientation and fast timescale dynamics (usually <100 ns) at some sites [11,34]. Unfortunately, the rigorous comparison of MD with NMR data has often been limited to the A ring and tail of Chol, due to the relative ease of labeling these portions of the molecule [29]. For example, the most readily available commercial versions of 2H and 13C-Chol are (3,4-13C2), (2,3,4-13C3), (23, 24, 25, 26, 27-13C5), (3-D1), (6-D1), (7-D1), (2,2,3,4,4,6-D6) or (25, 26, 26,26, 27, 27, 27-D7) (from Cambridge Isotope Laboratories, Sigma Aldrich, and Avanti Polar Lipids). Highly enriched Chol would potentially enable more complete global analyses of structure and dynamics, as has been demonstrated to be critical for the high-resolution determination of membrane protein structures.
Here we provide methodological contributions into this active research field including: (1) a cost-effective production of highly 13C-enriched Chol on the >10 mg scale; (2) quantitative analysis of labeling patterns for uniform and 2-13C-acetate forms; (3) collection and assignment of multidimensional scalar and dipolar magic-angle spinning (MAS) solid-state NMR (SSNMR) spectra; and (4) determination of 1H-13C dipolar order parameters.
2. Materials and Methods
2.1. Cholesterol Biosynthesis
2.1.1. Growth
Several YPD plates were prepared using 10 g/L of yeast extract, 20 g/L of peptone, and 20 g/L of agar. Water was then added to this mixture and autoclaved for 30 minutes. The mixture was cooled for about 20 minutes and 50 mL of 40% dextrose solution was added. The plates were then poured and left to cool and solidify. Once the plates solidified, the Rh6829 strain was streaked onto several plates and left to incubate for 24–48 hours until single colonies were visible. Approximately 1 L of media was prepared for the inoculation of the cell colonies. The media consisted of 950 mL milliQ water, 40 mg uracil, 40 mg leucine, 0.90 g 13C-sodium acetate, 7 g yeast nitrogen base with amino acids, and 5 g yeast extract. The mixture was autoclaved for 30 minutes and 50 mL of 40% sterile-filtered dextrose solution was subsequently added to the mixture. 5 colonies of the Rh6829 strain were added to 1 mL of media. 1 mL of the inoculated media was poured back into the 1 L cultures. The 1 L cultures were then incubated at 30 °C for 48–72 hours until the mixture was confluent.
2.1.2. Harvesting
Once confluent, the cells were spun down at 3000g for 30 minutes, 950 mL of the media were poured off and the cells were re-suspended in approximately 50 mL of the remaining media. The re-suspended cell mixtures were transferred to 50 mL conical vials and were then spun down in a centrifuge at 3000g for 5 minutes. The supernatant was discarded and the cells were re-suspended in 50 mL milliQ water (repeated 3 times). After pouring off the water from the final wash, 15 mL of 0.1 M HCl was were added to each vial. The mixtures were vortexed and added to a 1 L round bottom flask. The cells were heated at 90 °C and stirred at 700 rpm for 1 hour. The mixture was then transferred to a 5 L 3-neck round bottom flask. The 1 L round bottom flask was rinsed with 300 mL of 200 proof ethanol and the solution was transferred to the 5 L 3-neck flask. Additionally, the 1 L round bottom flask was rinsed with 1 L of 50% aqueous KOH and transferred to the 5 L flask. The mixture was stirred at 150 rpm and refluxed for 1 hour. After allowing the flask to cool to ambient temperatures, the mixture was transferred to a 4 L separatory funnel. The 3-neck flask was rinsed with 400 mL petroleum ether and poured into the separatory funnel. The organic layer was collected and aqueous layer was rinsed with petroleum ether (4 × 400 mL). The organic layers were combined, dried with sodium sulfate and filtered into a round bottom flask. The solvent was removed under reduced pressure to ca. 5 mL and transferred to a 40 mL IChem vial and the volatiles were removed in vacuo and the sample was dried overnight.
2.1.3. Purification
The crude material after harvesting is light tan (~70% pure). Preparative HPLC yields a >95% pure, colorless solid. The labeled cholesterol was dissolved in 3:1 ethyl acetate:acetone. The sterol was filtered into an HPLC vial. A mixture of isocratic :Ethanol (70:30) was used as the mobile phase to purify the sample using HPLC. The retention time was about 20 minutes. Once the sample has been retained after purification, the solvent was removed under reduced pressure yielding a colorless solid. The sample vial was stored at −20 °C.
2.2. SSNMR Data Collection & Analysis
2.2.1. 13C-13C Constant-time uniform-sign cross-peak (CTUC-COSY) [35] NMR Spectra
750 MHz (1H frequency) spectra were acquired at 20 °C. Spectra were processed using NMRPipe [36] and analyzed using the Sparky program (3.115) for peak-picking [37].
2.2.2. R48318–Symmetry NMR Experiments
600 MHz (1H frequency) 2D 1H-13C dipolar recoupling spectra were acquired at 20 °C, 13.051 kHz, 36 points in the dephasing dimension, for a maximal dephasing period of 1.38 ms. Spectra were processed using NMRPipe [36] and analyzed in the Sparky program for peak picking. NMRPipe was used to extract out the time-domain trajectories.
2.2.3. R48318–Symmetry Data Analysis
The time-domain dipolar coupling dephasing trajectories were analyzed using an in-house model-free fitting program, Iota_MF (written in Python), which Fourier transforms the data to the frequency domain prior to fitting. The Iota_MF program analyzes the data by Average Liouvillian Theory (ALT). The resulting simulated fits yield a scaling factor and angle for each coupling as well as an overall relaxation value. Line shapes were first fitted using only directly attached protons.
2.3. Lipid Bilayer Reconstitution for Solid-State NMR
SSNMR samples were prepared in approximately 20 mg batches of either 10:3 or 40:1 (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC): U-13C Chol. 13C-Chol was added to the lipid (Avanti, 850457C, 10 mg/mL in chloroform), swirled until dissolved, and dried under a flow of nitrogen or argon gas. The lipid-sterol film was then placed under high vacuum overnight. Freshly prepared HEPES buffer of pH 7.0 and water were added to the dry film, which was subsequently vortexed (1 min) and sonicated (3 min). The solution then underwent 5 freeze-thaw steps that included freezing the sample vial in liquid nitrogen and thawing under running water. The solution was frozen a sixth time and immediately placed in the lyophilizer overnight to yield a white powder. After lyophilization, the sample container was flushed with argon before removing the powder and recording its final mass, and the powders were stored under argon. The powders were packed into 3.2 mm standard rotors and hydrated to ~33% by mass with deionized water.
2.4. Determining 13C Labeling Efficiency Using Quantitative Solution NMR
To ascertain the labeling percentage, quantitative proton and carbon spectra were collected. The quantitative 1H NMR spectrum was collected without 13C decoupling, in order to observe the proton multiplicity due to the 13C-1H scalar coupling. The central peak represents the population of protons attached to 12C, while the satellite multiplets depict the population of protons attached to a 13C-labeled atom. The ratio of the satellite peaks over all peaks yields the labeling percentage. Integration bounds for the proton spectra were 25 times the half maximal linewidth. This labeling percentage was related to the quantitative carbon spectrum. Integration bounds for the carbon spectra were 25 times the half maximal linewidth, when possible. For example, the [2-13C-acetate] Chol spectrum did not have any overlapping carbon resonances so it was possible to use 25 times the half maximal linewidth for all peaks. However, for the U-13C-Chol carbon spectrum, due to spectral overlap it is only possible to integrate using that range for 6 out of the 16 resolved resonances.
3. Results and Discussion
3.1. Biosynthesis and Determination of Labeling Efficiency of 13C-Chol
We employed a biosynthetic procedure utilizing a genetically modified S. cerevisiae strain capable of producing isotopically labeled Chol [38,39], instead of its native sterol (ergosterol), from 13C-glucose. Here we utilized 13C-labeled sodium acetate, which enables uniform labeling (with 1,2-13C-acetate) or fractional (“skip” or “checkerboard”) labeling with 1-13C- or 2-13C-acetate, and found the yields to be more cost effective than the fractionally labeled glucose variants. Specifically, the acetate biosynthesis yields approximately 30 mg of Chol from ~0.9 g U-13C, 1-13C or 2-13C-acetate, whereas the reported yield from 1.0 g of U-13C-glucose was ~1 mg in 100 mL of culture medium.
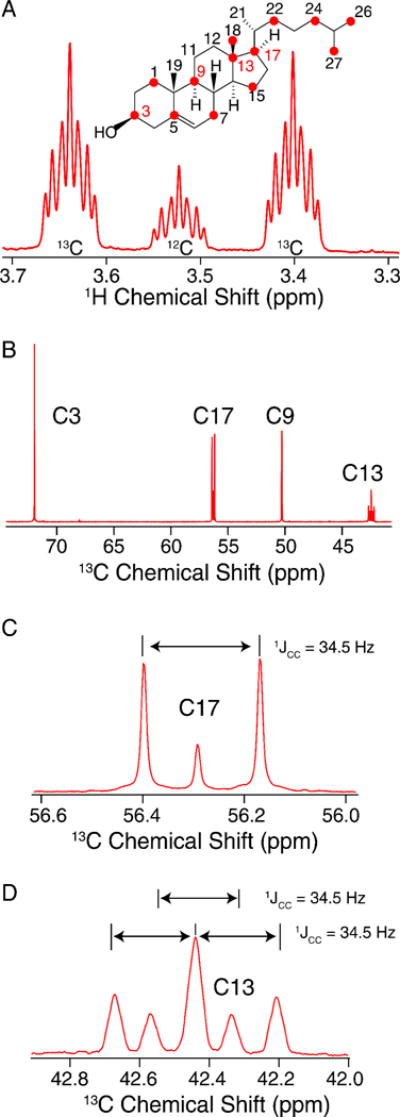
In order to determine the percentage of isotopic enrichment site-specifically, we utilized quantitative solution NMR (qNMR) experiments to determine the labeling efficiency. First, we analyzed the relative intensity of the Chol H3 signal (Figure 1A) 1H{13C} doublet to singlet (without 13C decoupling), to obtain the percent incorporation for that site. (Due to two- and three-bond 1H-13C J-couplings, each 1H{13C} doublet signal has additional, partially resolved fine structure.) In a similar manner, the C6 labeling percentage was determined by analysis of the H6 doublet to singlet ratio. The relative labeling percentages for other sites in the 13C spectrum (Figure 1B) were then determined by comparison of the area of the C3 peak, and validated using the C6 peak. This resulted in an overall labeling of U-13C-Chol to be 72 ± 2%, globally (Table 1). Within the measurement error of ~3%, the site-specific determinations were equivalent throughout the molecule. The maximal labeling percentage is determined by the presence of unlabeled carbon sources, especially dextrose (2 g/L), that are necessary for cell growth.
Figure 1. Quantitative Solution NMR of U-13C Cholesterol.
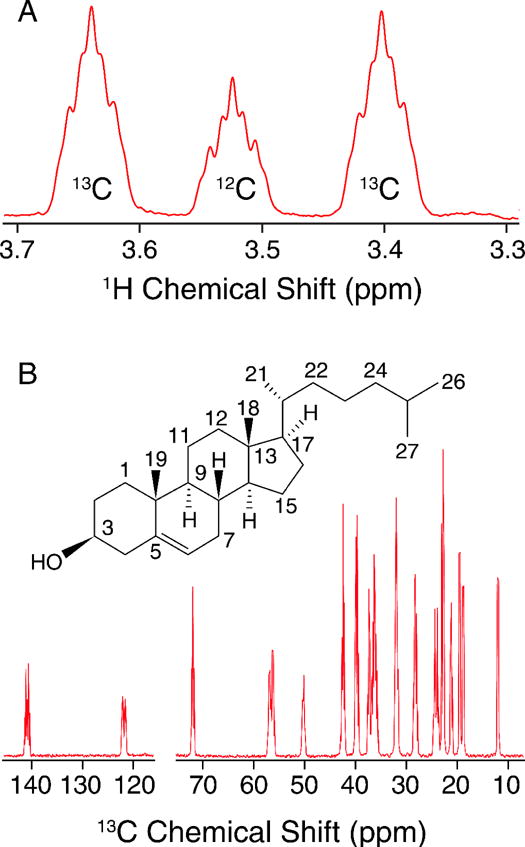
A) 600 MHz Quantitative 1H NMR spectrum showing the C3 region used to interpret labeling incorporation in B) 600 MHz quantitative 13C NMR spectrum.
Table 1. 13C Incorporation of U-13C-labeled Chol.
Asterisks denote an average labeling percentage for overlapped peaks. The error for each measurement is 3%, as determined by integrating a region with no peaks.
| Carbon Number | 13C % Incorporation |
|---|---|
| C1 | 73* |
| C2 | 72* |
| C3 | 73 |
| C4 | 73* |
| C5 | 75 |
| C6 | 76 |
| C7 | 72* |
| C8 | 72* |
| C9 | 69 |
| C10 | 73* |
| C11 | 72 |
| C12 | 74* |
| C13 | 73* |
| C14 | 72* |
| C15 | 71* |
| C16 | 72* |
| C17 | 72* |
| C18 | 66 |
| C19 | 68 |
| C21 | 68 |
| C22 | 73* |
| C23 | 71* |
| C24 | 74* |
| C25 | 72* |
| C26 | 69* |
| C27 | 69* |
| Average | 72 |
| Stdev | 2 |
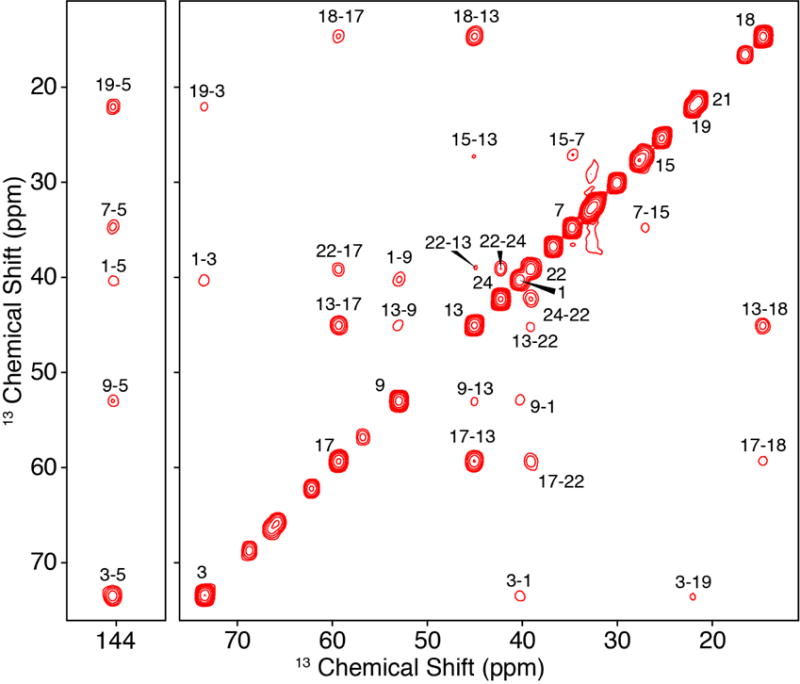
With U-13C Chol on hand, we next proceeded to prepare a sample of Chol in POPC and collect MAS 13C-13C correlation spectra. We utilized the scalar-based CTUC COSY [35] experiment (Figure 2) to unambiguously identify the one-bond correlations throughout the structure and arrive at complete resonance assignments. In particular, the U-13C labeling and correlation spectra enable a de novo assignment that did not rely upon solution NMR chemical shifts and uniquely separated the degenerate signals (C7 v. C8, C10 v. C20 v. C22, C14 v. C17, C15 v. C23).
Figure 2. CTUC COSY of U-13C-Chol in POPC Bilayers.
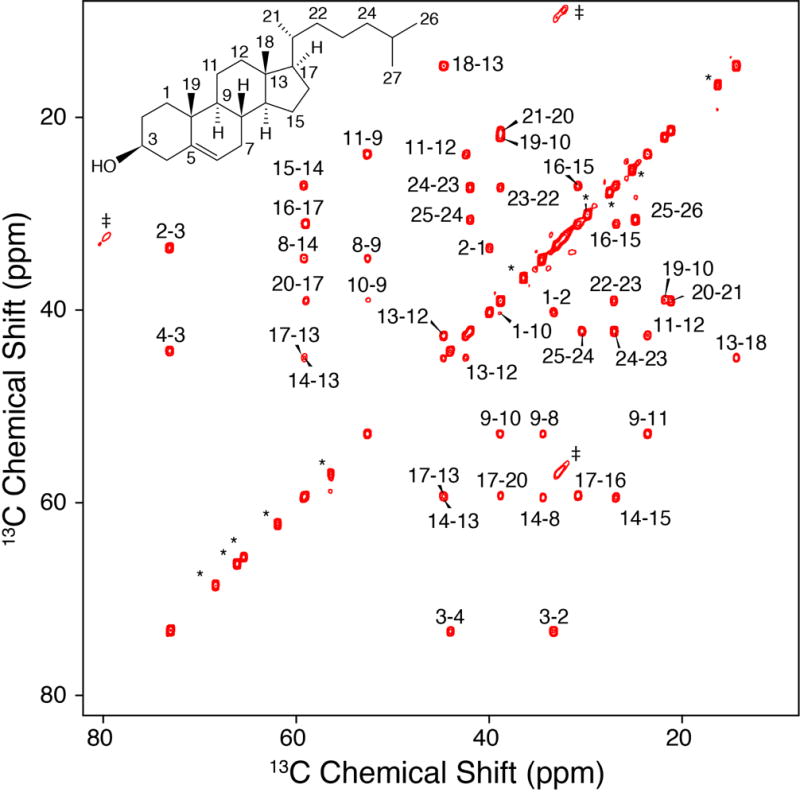
750 MHz, 8.928 kHz MAS SSNMR 13C-13C 2D CTUC COSY spectrum of 40:1 POPC: U-13C. The asterisks in the figure represent POPC peaks, the daggers represent artifacts arising from the subharmonics of the spinning [35].
3.2. Recoupling Experiments to Determine Averaged 1H-13C Dipolar Couplings
Next we proceeded to collect 2D separated local field spectra with the R48318-symmetry based pulse sequence [40,41]. With the high levels of 13C enrichment, sensitivity for these experiments was enhanced more than an order of magnitude, relative to samples with natural 13C abundance. Experiments were carried out with 3.2 mm rotors and an active volume of ~18 uL, accommodating a sample of 11.6 mg 10:3 POPC:U-13C Chol, hydrated with 5.7 mg H2O. Of this 17.3 mg sample, the signal from the ~2.8 mg of U-13C Chol yielded a high-resolution 1D spectrum (Figure 3) with resolution sufficient to observe multiplicity due to J-couplings in only 64 scans (Figure 3).
Figure 3. High Resolution 1D 13C Spectrum.
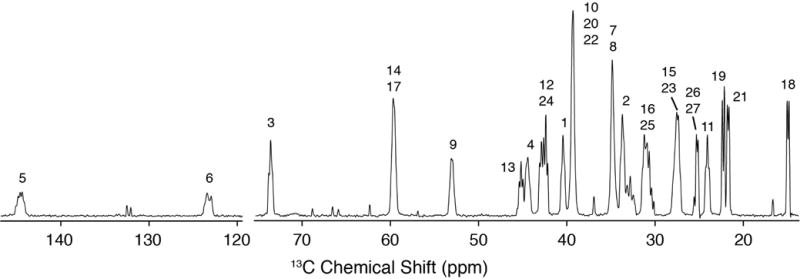
600 MHz MAS SSNMR 13C cross polarization spectrum (46 ms, 64 scans) of 10:3 POPC: U-13C-Chol with multiplicity due to J-couplings. The additional peaks not labeled are POPC peaks.
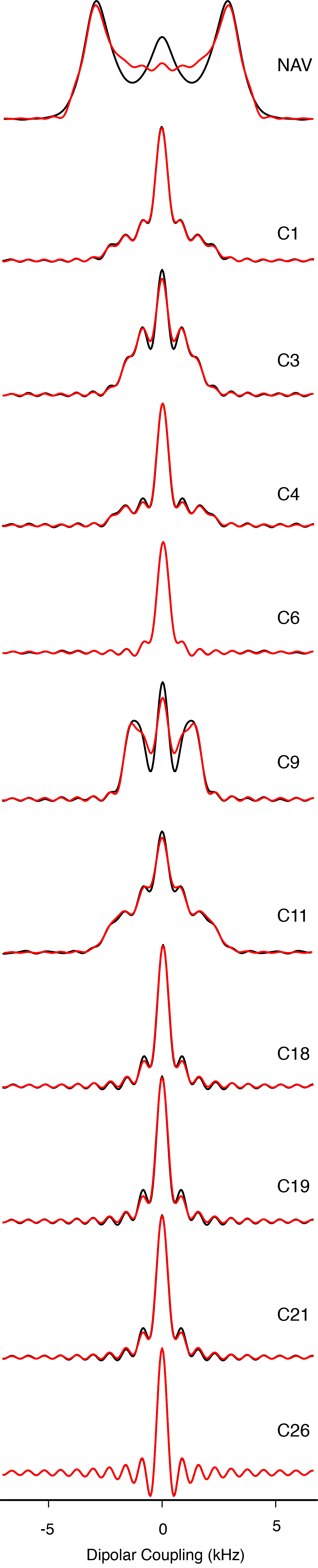
The high sensitivity also enabled 2D 1H-13C dipolar recoupling data sets to be collected in 20 minutes or less. The separated local field line shapes resulting from the 1H-13C dipolar couplings (Figure 4) were fit according to the average Liouvillian theory (ALT) approximations described in Hohwy et al. [42] and Rienstra et al. [43]. Data sets were normalized against the 1Hα-13Cα scaled dipolar coupling of N-acetyl valine (NAV), which is representative of a one-bond rigid lattice coupling (Figure 1S).
Figure 4. Dipolar Spectra.
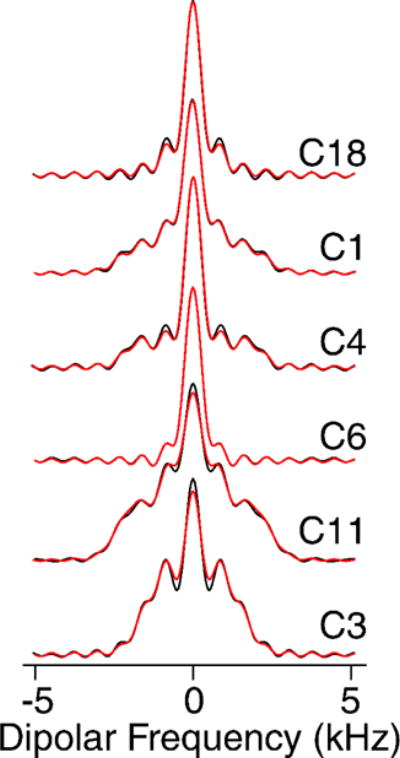
600 MHz MAS SSNMR R48318 dipolar spectra. Black lines are experimental data and red lines represent the fit.
Remarkably, the Chol data fit with higher quality than small molecule model compounds, as a consequence of the better convergence of the ALT model for fast limit motionally-averaged dipolar couplings that are small compared to the MAS rate and multiple pulse cycle time [43]. A representative set of dipolar coupling line shapes arising from CH, CH2 and CH3 groups are depicted in Figure 4.
The order parameters we measured (Table 1S) are consistent with values reported by Ferreira et al. [28] for 34% cholesterol in POPC above the liquid crystal phase transition: for C1 0.39±0.07 (versus ~0.375 for Ferreira), C3 0.38±0.03 (0.4), for C4 0.42±0.01 (0.35), C9 0.47±0.01 (0.45), C11 0.43±0.07 (0.45), and C18 0.21±0.01 (0.15). Additionally, we were able to measure the C6 (0.12±0.01) and C26/C27 (0.03±0.01) order parameters, which had too low a signal-to-noise ratio at natural abundance to be measured accurately.
3.3. Skip-Labeled 13C-Chol
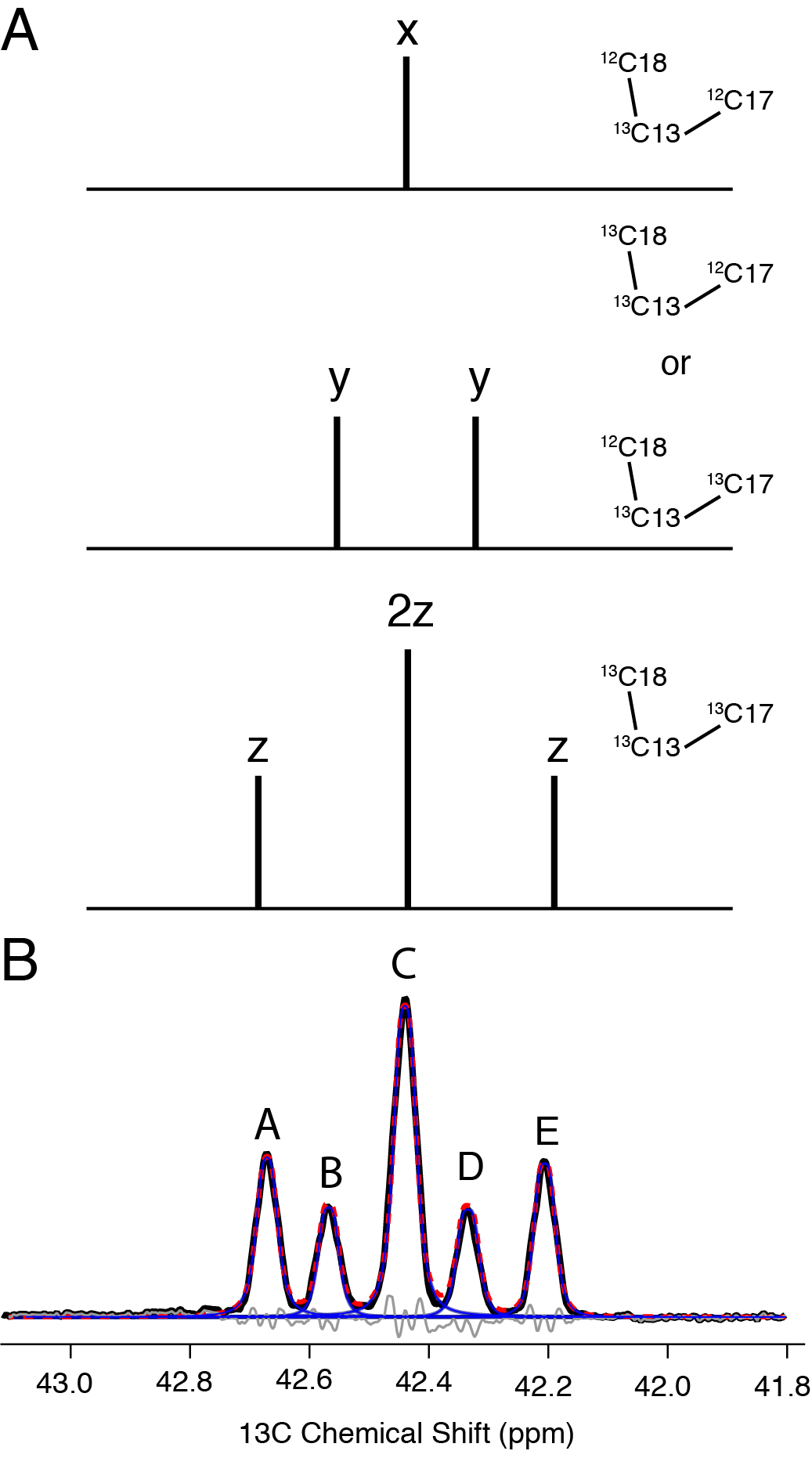
Fractional 13C-labeling has been demonstrated to be indispensable for structural studies of proteins [44–48], particularly to improve the resolution and sensitivity of 2D 13C-13C spectra. Thus, we were intrigued by the potential for skip-labeling to yield similar benefits for studies of Chol, to enable future applications where spectral overlap may be problematic for U-13C-labeled samples. The biosynthetic approach presented here is compatible with either 1-13C or 2-13C-acetate, allowing for the production of complementary skip-13C-Chol patterns. Here we have evaluated the incorporation percentage for [2-13C-acetate]-Chol as described in Methods 2.4 and results summarized in Table 2. As a result of using skip-13C-Chol, the multiplet structure in the 13C 1D spectrum is considerably sharper due to the absence of most two-bond 1H-13C scalar couplings (Figure 5A). The reduction in linewidth is also observed in the quantitative 13C spectrum due to the elimination of most one-bond 13C-13C scalar couplings (Figure 5B). Additionally, in cases where 13C-13C couplings are observed due to neighboring 13C atoms (Figures 5C and 5D), we were able to leverage this information to analyze the multiplet structures and further validate the percentage of 13C incorporation. We also utilized the 13C 1D spectrum (Figure 5D) to quantify the ratios of isotopomers. For example, the labeling of C13, C17 and C18 is positively correlated, with all three sites 13C-labeled at ~73% (whereas only 60% would be expected as the product of the individual labeling percentages). In addition, for another 24% population, both C13 and either C17 or C18 are labeled. These results are consistent the reported Chol biosynthetic pathway [39], and validated with a dipolar correlation (DARR) 13C-13C 2D spectrum of [2-13C-acetate]-Chol in POPC bilayers (Figure 6) which shows strong cross peaks among the C13, C17 and C18 sites. The C5 olefin peak appears at ~144 ppm and shows strong correlations to C3, C19, C7 and C9 sites. In addition, two-bond correlations are observed for the mostly odd-numbered carbons that are labeled in the sterol rings. Linewidths are approximately 60 Hz for the DARR 13C-13C 2D spectra. These resolution and sensitivity of the 40:1 POPC: [2-13C-acetate]-Chol spectrum demonstrate their potential benefits in future applications where spectral overlap is problematic.
Table 2. 13C Incorporation of [2-13C-acetate]-Chol.
Error of these measurements is 3% as determined by integrating a region of the spectrum with no peaks.
| Carbon Number | 13C % incorporation |
|---|---|
| C1 | 85 |
| C3 | 83 |
| C5 | 80 |
| C7 | 86 |
| C9 | 86 |
| C13 | 90 |
| C15 | 92 |
| C17 | 80 |
| C18 | 82 |
| C19 | 83 |
| C21 | 86 |
| C22 | 89 |
| C24 | 88 |
| C26 | 87 |
| C27 | 87 |
| Average | 86 |
| Stdev | 4 |
Figure 5. 600 MHz Solution spectra of Skip 13C-Chol.
A) Carbon 3 region of 1H quantitative NMR spectrum used to obtain a 13C labeling ratio applied to B) the quantitative 13C Carbon NMR. C) and D) represent labeling percentages of C17 split by is 13C labeled neighbor C18 and C13 is split by C17 and C18 neighbors, respectively.
Figure 6. 600 MHz MAS SSNMR of [2-13C-acetate]-Chol in POPC.
DARR 13C-13C 2D (250 ms mixing time) of 40:1 POPC: [2-13C-acetate]-Chol, where unlabeled peaks on or near the diagonal are from the lipid, POPC.
4. Conclusions and Outlook
We have obtained a highly incorporated 13C-labeled Chol by feeding a genetically modified S. Cerevisiae strain [39] 13C-sodium acetate. Using qNMR, we determined the 13C-enrichment for U-13C Chol and 2-13C-acetate Chol to be 72 and 86%, respectively. Using labeled acetate instead of glucose produced a higher degree of incorporation of skip-labeled 13C-Chol in comparison to previous studies [39]. We anticipate that the feeding and growth schedule as well 13C-acetate to dextrose ratio could be further optimized to yield even higher rates of 13C incorporation. Nevertheless, the current incorporation levels already enable highly sensitive measurements. For example, we prepared a sample of U-13C-Chol in POPC, collected several MAS 13C-13C correlation spectra and completed resonance assignments. The high level of enrichment allowed us to measure order parameters in a 20-minute experiment using less than 3 mg of labeled material, whereas prior determinations of equivalent order parameters with natural abundance cholesterol required ~4.8 hrs for a 34 mol% Chol/POPC sample in a 4 mm HR-MAS rotor [28].
We envision that 13C-enriched Chol will be useful for a variety of studies by SSNMR in order to examine structure, dynamics and interactions with membrane proteins and drugs. For example, we have previously demonstrated binding of the antifungal drug amphotericin B (AmB) to labeled ergosterol [49], resulting in changes in the chemical shifts, order parameters, paramagnetic relaxation effects and lipid/water correlations. In similar manner, we anticipate that studies with 13C-Chol will yield insight into the structural basis of the sterol-specificity of AmB analogs with reduced toxicity [50–52]. A related approach has been demonstrated recently by Gronenborn [53] and colleagues, who used a 13C labeled carbohydrate probe with a 13C labeled protein to investigate intermolecular distances in a sugar-protein complex in order to elucidate not only the binding site of the sugar to the protein, but also the conformation of the bound sugar. These studies underscore the potential of using 13C-labeled small molecules to elucidate their key role in biology.
Supplementary Material
{kind=link}
{kind=link}
Highlights.
Production of cholesterol with three 13C labeling patterns
Reconstitution of 13C-cholesterol into lipid bilayer samples
Magic-angle spinning to yield high resolution spectra
Resonance assignments of membranous cholesterol
Rapid order parameter measurements
Acknowledgments
We gratefully acknowledge S. Phinney for help during the biosynthesis, and J. Heredia for preliminary analysis of the quantitative solution NMR data. This work was supported by the NIH (R01GM112845, R01GM123455 and GM118185). L.D.R. is supported by the NIH-supported Chemical Biology Interface Training Program (F30DK081272).
Abbreviations
- Chol
cholesterol
- MAS
magic angle spinning
- MD
molecular dynamics
- SSNMR
solid-state nuclear magnetic resonance
- CTUC COSY
constant-time uniform-sign cross-peak correlation spectroscopy
- ALT
average Liouvillian theory
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- NAV
N-acetyl valine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kulig W, Olżyńska A, Jurkiewicz P, Kantola AM, Komulainen S, Manna M, Pourmousa M, Vazdar M, Cwiklik L, Rog T, Khelashvili G, Harries D, Telkki V-V, Hof M, Vattulainen I, Jungwirth P. Cholesterol under oxidative stress—How lipid membranes sense oxidation as cholesterol is being replaced by oxysterols. Free Radic Biol Med. 2015;84:30–41. doi: 10.1016/j.freeradbiomed.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Róg T, Vattulainen I. Cholesterol, sphingolipids, and glycolipids: What do we know about their role in raft-like membranes? Chem Phys Lipids. 2014;184:82–104. doi: 10.1016/j.chemphyslip.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Róg T, Pasenkiewicz-Gierula M, Vattulainen I, Karttunen M. Ordering effects of cholesterol and its analogues. Biochim Biophys Acta - Biomembr. 2009;1788:97–121. doi: 10.1016/j.bbamem.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Ohvo-Rekila H. Cholesterol interactions with phospholipids in membranes. Prog Lipid Res. 2002;41:66–97. doi: 10.1016/S0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- 5.Yao H, Lee MW, Waring AJ, Wong GCL, Hong M. Viral fusion protein transmembrane domain adopts β-strand structure to facilitate membrane topological changes for virus–cell fusion. Proc Natl Acad Sci. 2015;112:10926–10931. doi: 10.1073/pnas.1501430112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai J, Alwarawrah M, Huang J. Instability of Cholesterol Clusters in Lipid Bilayers and The Cholesterol’s Umbrella Effect. J Phys Chem B. 2010;114:840–848. doi: 10.1021/jp909061h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simons K, Vaz WLC. Model Systems, Lipid Rafts, and Cell Membranes. Annu Rev Biophys Biomol Struct. 2004;33:269–295. doi: 10.1146/annurev.biophys.32.110601.141803. [DOI] [PubMed] [Google Scholar]
- 8.Fantini J, Barrantes FJ. How cholesterol interacts with membrane proteins: an exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol. 2013;4:31. doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang ST, Kiessling V, Simmons JA, White JM, Tamm LK. HIV gp41–mediated membrane fusion occurs at edges of cholesterol-rich lipid domains. Nat Chem Biol. 2015;11:424–431. doi: 10.1038/nchembio.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis AK, Valley CC, Peery SL, Brummel B, Braun AR, Karim CB, Sachs JN. Death Receptor 5 Networks Require Membrane Cholesterol for Proper Structure and Function. J Mol Biol. 2016;428:4843–4855. doi: 10.1016/j.jmb.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khelashvili G, Pabst G, Harries D. Cholesterol orientation and tilt modulus in DMPC bilayers. J Phys Chem B. 2010;114:7524–7534. doi: 10.1021/jp101889k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aittoniemi J, Róg T, Niemelä P, Pasenkiewicz-Gierula M, Karttunen M, Vattulainen I. Tilt: Major Factor in Sterols’ Ordering Capability in Membranes. J Phys Chem B. 2006;110:25562–25564. doi: 10.1021/jp064931u. [DOI] [PubMed] [Google Scholar]
- 13.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science (80-) 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaikh SR, Cherezov V, Caffrey M, Soni SP, LoCascio D, Stillwell W, Wassall SR. Molecular Organization of Cholesterol in Unsaturated Phosphatidylethanolamines: X-ray Diffraction and Solid State 2 H NMR Reveal Differences with Phosphatidylcholines. J Am Chem Soc. 2006;128:5375–5383. doi: 10.1021/ja057949b. [DOI] [PubMed] [Google Scholar]
- 16.Brzustowicz MR, Stillwell W, Wassall SR. Molecular organization of cholesterol in polyunsaturated phospholipid membranes: a solid state 2 H NMR investigation. FEBS Lett. 1999;451:197–202. doi: 10.1016/S0014-5793(99)00567-0. [DOI] [PubMed] [Google Scholar]
- 17.Vermeer LS, de Groot BL, Réat V, Milon A, Czaplicki J. Acyl chain order parameter profiles in phospholipid bilayers: computation from molecular dynamics simulations and comparison with 2H NMR experiments. Eur Biophys J. 2007;36:919–931. doi: 10.1007/s00249-007-0192-9. [DOI] [PubMed] [Google Scholar]
- 18.Matsumori N, Kasai Y, Oishi T, Murata M, Nomura K. Orientation of Fluorinated Cholesterol in Lipid Bilayers Analyzed by 19 F Tensor Calculation and Solid-State NMR. J Am Chem Soc. 2008;130:4757–4766. doi: 10.1021/ja077580l. [DOI] [PubMed] [Google Scholar]
- 19.Murari R, Murari MP, Baumann WJ. Sterol orientations in phosphatidylcholine liposomes as determined by deuterium NMR. Biochemistry. 1986;25:1062–7. doi: 10.1021/bi00353a017. http://www.ncbi.nlm.nih.gov/pubmed/3754460. [DOI] [PubMed] [Google Scholar]
- 20.Marsan MP, Muller I, Ramos C, Rodriguez F, Dufourc EJ, Czaplicki J, Milon A. Cholesterol Orientation and Dynamics in Dimyristoylphosphatidylcholine Bilayers: A Solid State Deuterium NMR Analysis. Biophys J. 1999;76:351–359. doi: 10.1016/S0006-3495(99)77202-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dufourc EJ, Smith ICP. A detailed analysis of the motions of cholesterol in biological membranes by 2H-NMR relaxation. Chem Phys Lipids. 1986;41:123–135. doi: 10.1016/0009-3084(86)90004-6. [DOI] [PubMed] [Google Scholar]
- 22.Dufourc EJ, Parish EJ, Chitrakorn S, Smith ICP. Structural and dynamical details of cholesterol-lipid interaction as revealed by deuterium NMR. Biochemistry. 1984;23:6062–6071. doi: 10.1021/bi00320a025. [DOI] [Google Scholar]
- 23.Vogel A, Scheidt HA, Baek DJ, Bittman R, Huster D. Structure and dynamics of the aliphatic cholesterol side chain in membranes as studied by 2 H NMR spectroscopy and molecular dynamics simulation. Phys Chem Chem Phys. 2016;18:3730–3738. doi: 10.1039/C5CP05084G. [DOI] [PubMed] [Google Scholar]
- 24.Taylor MG, Akiyama T, Smith ICP. The molecular dynamics of cholesterol in bilayer membranes: A deuterium NMR study. Chem Phys Lipids. 1981;29:327–339. doi: 10.1016/0009-3084(81)90066-9. [DOI] [Google Scholar]
- 25.Oldfield E, Meadows M, Rice D, Jacobs R. Spectroscopic studies of specifically deuterium labeled membrane systems. Nuclear magnetic resonance investigation of the effects of cholesterol in model systems. Biochemistry. 1978;17:2727–2740. doi: 10.1021/bi00607a006. [DOI] [PubMed] [Google Scholar]
- 26.Siminovitch DJ, Ruocco MJ, Olejniczak ET, Das Gupta SK, Griffin RG. Anisotropic 2H-nuclear magnetic resonance spin-lattice relaxation in cerebroside- and phospholipid-cholesterol bilayer membranes. Biophys J. 1988;54:373–381. doi: 10.1016/S0006-3495(88)82970-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonmatin JM, Smith ICP, Jarrell HC, Siminovitch DJ. Use of a comprehensive approach to molecular dynamics in ordered lipid systems: cholesterol reorientation in oriented lipid bilayers. A deuterium NMR relaxation case study. J Am Chem Soc. 1990;112:1697–1704. doi: 10.1021/ja00161a007. [DOI] [Google Scholar]
- 28.Ferreira TM, Coreta-Gomes F, Ollila OHS, Moreno MJ, Vaz WLC, Topgaard D. Cholesterol and POPC segmental order parameters in lipid membranes: solid state 1 H–13 C NMR and MD simulation studies. Phys Chem Chem Phys. 2013;15:1976–89. doi: 10.1039/c2cp42738a. [DOI] [PubMed] [Google Scholar]
- 29.Higinbotham J, Beswick PH, Malcolmson RJ, Reed D, Parkinson JA, Sadler IH. 13C-NMR determination of the molecular dynamics of cholesterol in dimyristoylphosphatidylcholine vesicles. Chem Phys Lipids. 1993;66:1–11. doi: 10.1016/0009-3084(93)90025-X. [DOI] [PubMed] [Google Scholar]
- 30.Yeagle PL, Albert AD, Boesze-Battaglia K, Young J, Frye J. Cholesterol dynamics in membranes. Biophys J. 1990;57:413–424. doi: 10.1016/S0006-3495(90)82558-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrescu AD, Gallegos AM, Okamura Y, Strauss JF, Schroeder F. Steroidogenic Acute Regulatory Protein Binds Cholesterol and Modulates Mitochondrial Membrane Sterol Domain Dynamics. J Biol Chem. 2001;276:36970–36982. doi: 10.1074/jbc.M101939200. [DOI] [PubMed] [Google Scholar]
- 32.Pucadyil TJ, Mukherjee S, Chattopadhyay A. Organization and Dynamics of NBD-Labeled Lipids in Membranes Analyzed by Fluorescence Recovery after Photobleaching. J Phys Chem B. 2007;111:1975–1983. doi: 10.1021/jp066092h. [DOI] [PubMed] [Google Scholar]
- 33.Robalo JR, Ramalho JPP, Loura LMS. NBD-Labeled Cholesterol Analogues in Phospholipid Bilayers: Insights from Molecular Dynamics. J Phys Chem B. 2013;117:13731–13742. doi: 10.1021/jp406135a. [DOI] [PubMed] [Google Scholar]
- 34.Khelashvili G, Rappolt M, Chiu SW, Pabst G, Harries D. Impact of sterol tilt on membrane bending rigidity in cholesterol and 7DHC-containing DMPC membranes. Soft Matter. 2011;7:10299. doi: 10.1039/c1sm05937h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Olsen RA, Elliott DW, Boettcher JM, Zhou DH, Rienstra CM, Mueller LJ. Constant-Time Through-Bond 13 C Correlation Spectroscopy for Assigning Protein Resonances with Solid-State NMR Spectroscopy. J Am Chem Soc. 2006;128:9992–9993. doi: 10.1021/ja062347t. [DOI] [PubMed] [Google Scholar]
- 36.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 37.Goddard TD, Kneller DG. SPARKY 3. Univ. California; San Fr.: (n.d.) [Google Scholar]
- 38.Souza CM, Schwabe TME, Pichler H, Ploier B, Leitner E, Guan XL, Wenk MR, Riezman I, Riezman H. A stable yeast strain efficiently producing cholesterol instead of ergosterol is functional for tryptophan uptake, but not weak organic acid resistance. Metab Eng. 2011;13:555–569. doi: 10.1016/j.ymben.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Shivapurkar R, Souza CM, Jeannerat D, Riezman H. An efficient method for the production of isotopically enriched cholesterol for NMR. J Lipid Res. 2011;52:1062–1065. doi: 10.1194/jlr.D014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou G, Byeon IJL, Ahn J, Gronenborn AM, Polenova T. 1 H– 13 C/1 H– 15 N Heteronuclear Dipolar Recoupling by R-Symmetry Sequences Under Fast Magic Angle Spinning for Dynamics Analysis of Biological and Organic Solids. J Am Chem Soc. 2011;133:18646–18655. doi: 10.1021/ja203771a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao X, Eden M, Levitt MH, Edén M, Levitt MH. Recoupling of heteronuclear dipolar interactions in solid-state NMR using symmetry-based pulse sequences. Chem Phys Lett. 2001;342:353–361. doi: 10.1016/S0009-2614(01)00593-0. [DOI] [Google Scholar]
- 42.Hohwy M, Rienstra CM, Jaroniec CP, Griffin RG. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. J Chem Phys. 1999;110:7983. doi: 10.1063/1.478702. [DOI] [Google Scholar]
- 43.Rienstra CM, Hohwy M, Mueller LJ, Jaroniec CP, Reif B, Griffin RG. Determination of Multiple Torsion-Angle Constraints in U− 13 C, 15 N-Labeled Peptides: 3D 1 H− 15 N− 13 C− 1 H Dipolar Chemical Shift NMR Spectroscopy in Rotating Solids. J Am Chem Soc. 2002;124:11908–11922. doi: 10.1021/ja020802p. [DOI] [PubMed] [Google Scholar]
- 44.LeMaster DM, Kushlan DM. Dynamical Mapping of E. coli Thioredoxin via 13 C NMR Relaxation Analysis. J Am Chem Soc. 1996;118:9255–9264. doi: 10.1021/ja960877r. [DOI] [Google Scholar]
- 45.Hong M, Jakes K. Selective and extensive 13C labeling of a membrane protein for solid-state NMR investigations. J Biomol NMR. 1999;14:71–4. doi: 10.1023/A:1008334930603. [DOI] [PubMed] [Google Scholar]
- 46.Hong M. Determination of Multiple φ-Torsion Angles in Proteins by Selective and Extensive 13C Labeling and Two-Dimensional Solid-State NMR. J Magn Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 47.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 48.Franks WT, Wylie BJ, Schmidt HLF, Nieuwkoop AJ, Mayrhofer RM, Shah GJ, Graesser DT, Rienstra CM. Dipole tensor-based atomic-resolution structure determination of a nanocrystalline protein by solid-state NMR. Proc Natl Acad Sci U S A. 2008;105:4621–4626. doi: 10.1073/pnas.0712393105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson TM, Clay MC, Cioffi AG, Diaz Ka, Hisao GS, Tuttle MD, Nieuwkoop AJ, Comellas G, Maryum N, Wang S, Uno BE, Wildeman EL, Gonen T, Rienstra CM, Burke MD. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol. 2014;10:400–6. doi: 10.1038/nchembio.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilcock BC, Endo MM, Uno BE, Burke MD. C2′-OH of amphotericin B plays an important role in binding the primary sterol of human cells but not yeast cells. J Am Chem Soc. 2013;135:8488–8491. doi: 10.1021/ja403255s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Endo MM, Cioffi AG, Burke MD. Our Path to Less Toxic Amphotericins. Synlett. 2016;27:337–354. doi: 10.1055/s-0035-1560800. [DOI] [Google Scholar]
- 52.Davis SA, Vincent BM, Endo MM, Whitesell L, Marchillo K, Andes DR, Lindquist S, Burke MD. Nontoxic antimicrobials that evade drug resistance. Nat Chem Biol. 2015;11:481–487. doi: 10.1038/nchembio.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nestor G, Anderson T, Oscarson S, Gronenborn AM. Exploiting Uniformly 13 C-Labeled Carbohydrates for Probing Carbohydrate–Protein Interactions by NMR Spectroscopy. J Am Chem Soc. 2017;139:6210–6216. doi: 10.1021/jacs.7b01929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.