

Abstract
The differences among individual eicosanoids in eliciting different physiological and pathological responses are largely unknown because of the lack of valid and simple analytical methods for the quantification of individual eicosanoids and their metabolites in serum, sputum and bronchial alveolar lavage fluid (BALF). Therefore, a simple and sensitive LC–MS/MS method for the simultaneous quantification of 34 eicosanoids in human serum, sputum and BALF was developed and validated. This method is valid and sensitive with a limit of quantification ranging from 0.2 to 3 ng/mL for the various analytes, and has a large dynamic range (500 ng/mL) and a short run time (25 min). The intra- and inter-day accuracy and precision values met the acceptance criteria according to US Food and Drug Administration guidelines. Using this method, detailed eicosanoid profiles were quantified in serum, sputum and BALF from a pilot human study. In summary, a reliable and simple LC–MS/MS method to quantify major eicosanoids and their metabolites was developed and applied to quantify eicosanoids in human various fluids, demonstrating its suitability to assess eicosanoid biomarkers in human clinical trials.
Keywords: biomarker, COPD, eicosanoids, LC-MS/MS
1 | INTRODUCTION
Polyunsaturated fatty acids are precursors of oxylipins, a large family of metabolites involved in various physiological roles such as regulation of cell proliferation, tissue repair, coagulation and immune functions. The eicosanoids are a large subclass of oxylipins, which includes over 100 lipid mediators such as prostaglandins, thromboxanes, leukotrienes (LTs), hydroxyeicosatetraenoic acids (HETEs), dihydroxyeicosatetraenoic acids, hydroxyeicosapentaenoic acids, lipoxins (LXs), reolvins and epoxyeicosatrienoic acid (Shimizu, 2009; Wang & DuBois, 2007). Eicosanoids are synthesized from dihomo γ-linolenic acid, arachidonic acid (AA) and eicosapentaenoic acid via various enzymes such as cyclooxygenase enzymes, lipoxygenase enzymes and cytochrome P450, as well as by nonenzymatic oxidation (Milne, Yin, Hardy, Davies, & Roberts, 2011; Roman, 2002).
Disruption of the homeostasis of eicosanoids is closely related to a range of inflammatory pathological conditions including asthma and chronic obstructive pulmonary disease (COPD), fever, pain, nephritis, cardiovascular diseases, Crohn’s disease and cancer (Dong et al., 2009; Eikelboom et al., 2002; Gainer et al., 2005; Johnson et al., 2006; Ong, Zhang, & Whitworth, 2008). PGE2 regulates tumor angiogenesis in prostate cancer (Jain, Chakraborty, Raja, Kale, & Kundu, 2008), whereas LTs and LXs regulate vasoconstriction and vascular permeability (Stephenson, Lonigro, Hyers, Webster, & Fowler, 1988; Weiss et al., 1983). 20-HETE regulates cerebral microvessel constriction (Miyata & Roman, 2005); conversely, epoxyeicosatrienoic acid metabolites increase cerebral blood flow (Spector, Fang, Snyder, & Weintraub, 2004).
Given the clinical interest in eicosanoids and the complexity of their responses to biological stimuli, it is necessary to systematically monitor the changes in their concentrations in various tissues and biological fluids. This requires sensitive, selective and reproducible methods for their quantification. Quantification of eicosanoids in biological matrices is associated with numerous challenges, including their low concentrations (pM to nM range) in biological fluids. Some eicosanoids are unstable and can also be formed artificially ex vivo after sample collection and during sample preparation. This could be overcome by measuring more stable metabolites as surrogates for their unstable parent compounds. For example, TxB2 and 6-keto-PGF1α are measured as surrogates for TxA2 and PGI2, respectively (Aprikian et al., 2007; Liu et al., 2004; Virtue et al., 2015). Other challenges include the presence of multiple isomeric forms that share the same mass and fragmentation pattern, which makes it difficult to resolve them by mass spectrometry and by chromatography (Tsikas & Zoerner, 2014).
A broad range of techniques have been employed for the separation, detection and quantification of eicosanoids, including HPLC-UV (Terragno & Terragno, 1981; Carrier et al., 1988; Huwyler & Gut, 1990; Lee & DeLuca, 1991; Chavis, Fraissinet, Chanez, Thomas, & Bousquet, 1999), enzyme immunoassays (Gandhi, Budac, Khayrullina, Staal, & Chandrasena, 2017; Shono et al., 1988), LC-fluorescence detection (Aghazadeh-Habashi, Asghar, & Jamali, 2015; Yue et al., 2004), electrophoresis (Herrmann, Steinhilber, & Roth, 1987; VanderNoot & VanRollins, 2002), immunoaffinity chromatography (Tsikas, Suchy, Tödter, Heeren, & Scheja, 2016), gas chromatography–mass spectrometry (GC–MS) (Nithipatikom et al., 2001; Rivera et al., 2004; Tsikas & Zoerner, 2014; Watzer, Reinalter, Seyberth, & Schweer, 2000) and liquid chromatography–mass spectrometry (LC–MS) (Fu et al., 2016; Gachet, Rhyn, Bosch, Quednow, & Gertsch, 2015; Long et al., 2015; Song et al., 2013; Sterz, Scherer, & Ecker, 2012; Strassburg et al., 2012; Wang, Armando, Quehenberger, Yan, & Dennis, 2014). HPLC-UV requires active chromophores to quantify eicosanoids. The main disadvantages of HPLC-UV are the limited sensitivity and specificity of UV detection in complex biological matrices, which typically require long run times (Carrier et al., 1988; Chavis et al., 1999; Huwyler & Gut, 1990; Lee & DeLuca, 1991; Terragno, Rydzik, & Terragno, 1981). Moreover, not all eicosanoids have active chromophores that absorb UV light at appropriate wavelengths (Masoodi & Nicolaou, 2006; Terragno et al., 1981). Disadvantages of UV detection can be overcome by using fluorescence detection. However, eicosanoids do not have an inherent fluorescence signal and require derivatization with fluorescent agents. This process is labor intensive, expensive and time consuming, and produces interfering peaks from side reactions (Aghazadeh-Habashi et al., 2015; Puppolo, Varma, & Jansen, 2014). Immunoassays were also used to quantify eicosanoids, but they are limited to one analyte per assay and they suffer from high cross-reactivity between the numerous eicosanoid isomers (Gandhi et al., 2017; Shono et al., 1988). GC–MS/MS provides high sensitivity and resolution of isomeric eicosanoids but this technique is limited by complex sample preparation and derivatization (Puppolo et al., 2014; Tsikas & Zoerner, 2014; Yang, Chiang, Oh, & Serhan, 2011). The high sensitivity and selectivity of LC–MS/MS can overcome most of the above-mentioned limitations, which makes it the method of choice for the quantification of eicosanoids in biological matrices. Many LC–MS/MS methods, which have been reviewed recently (Kortz, Dorow, & Ceglarek, 2014; Puppolo et al., 2014; Tsikas & Zoerner, 2014; Willenberg, Ostermann, & Schebb, 2015), have been reported for the quantification of a variety of eicosanoids in plasma (Gachet et al., 2015; Strassburg et al., 2012; Wang et al., 2014), serum (Ferreiro-Vera, Mata-Granados, Priego-Capote, Quesada-Gomez, & Luque de Castro, 2011; Long et al., 2015), blood (Song et al., 2013), urine (Fu et al., 2016; Medina et al., 2012; Sterz et al., 2012), tissues (Blewett, Varma, Gilles, Libonati, & Jansen, 2008; Yue et al., 2004, 2007), lung cells (Lee et al., 2016), cell culture media (Furugen, Yamaguchi, & Mano, 2015), sputum (Jian et al., 2013; Yang, Eiserich, Cross, Morrissey, & Hammock, 2012) and bronchial alveolar lavage fluid (BALF) (Yang, Schmelzer, Georgi, & Hammock, 2009). The long-term goal of this project is to support a clinical study that aims to identify eicosanoid-based biomarkers for the prognosis of COPD. Despite, the plethora of available eicosanoid LC–MS methods as cited above, we needed a sensitive method for the simultaneous quantification of specific eicosanoids in several matrices of interest to support our biomarker study. Therefore, we have developed and validated a sensitive and simple LC–MS/MS method for the simultaneous quantification of 34 eicosanoids in human serum, sputum and BALF.
2 | EXPERIMENTAL
2.1 | Chemicals and reagents
Prostaglandin J2 (PGJ2), 20-hydroxy prostaglandin E2 (20-OH-PGE2), prostaglandin B2 (PGB2), prostaglandin D2 (PGD2), prostaglandin E2 (PGE2), AA, 15-hydroxyeicosatetraenoic acid (15-HETE), 12-hydroxyeicosatetraenoic acid (12-HETE), 11-hydroxyeicosatetraenoic acid (11-HETE), 8-hydroxyeicosatetraenoic acid (8-HETE), 5-hydroxyeicosatetraenoic acid (5-HETE), leukotriene E4 (LTE4), leukotriene D4 (LTD4), leukotriene C4 (LTC4), leukotriene B4 (LTB4), 13,14-dihydro-15-keto-prostaglandin E2 (13,14-dihydro-15-keto-PGE2), 11-β prostaglandin F2α (11-β-PGF2α), 8-iso-prostaglandin F2α (8-iso-PGF2α), prostaglandin F2α (PGF2α), 15-keto-prostaglandin E2 (15-keto-PGE2), 6-keto-prostaglandin F1α (6-keto-PGF1α), thromboxane B2 (TXB2), 13,14-dihydro-prostaglandin F2α (13,14-DiOH-PGF2α), prostaglandin F1α (PGF1α), 13,14-dihydro-15-keto-prostaglandin F2α (13,14-DiOH-15-keto-PGF2α), 13,14-dihydro-15-keto-prostaglandin E1 (13,14-DiOH-15-k-PGE1), prostaglandin D1 (PGD1), 13,14-dihydroprostaglandin E1 (13,14-DiOH-PGE1), thromboxane B3 (TXB3), 15-deoxy-delta 12,14 prostaglandin J2 (15-deoxy-delta 12,14 PGJ2), prostaglandin E1 (PGE1), prostaglandin E3 (PGE3), prostaglandin D3 (PGD3), prostaglandin F3α (PGF3α), 13,14-leukotriene C4, tetranor-prostaglandin E metabolite, tetranor-prostaglandin F metabolite, 11-dehydro-thromboxane B3, 2,3-dinor-8-iso prosta-glandin F2α and deuterated compounds (PGE2-d4, TXB2-d4, AA-d8, 15-HETE-d8,and LTB4-d8) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). HPLC-grade methanol (MeOH), acetonitrile (ACN), water, ammonium acetate, aqueous ammonia, formic acid and acetic acid were obtained from Fisher Scientific (Fair Lawn, NJ).
2.2 | Instrumentation
A Waters Acquity ultra performance liquid chromatography (UPLC) system (Waters, Milford, MA, USA) coupled to an Applied Biosystem 6500 Q TRAP® quadrupole linear ion trap hybrid mass spectrometer with an electrospray ionization (ESI) source (Applied Biosystems, MDS Sciex, Foster City, CA, USA) was used throughout. The UPLC and MS systems were controlled by Empower 3.0 and Analyst 1.6.2 software, respectively. All chromatographic separations were performed with an Acquity UPLC® BEH shield RP18 column (1.7 μm, 150 × 2.1 mm) equipped with an Acquity UPLC C18 guard column (Waters, Milford, MA, USA).
2.3 | Liquid chromatographic and mass spectrometric conditions
The mobile phase consisted of 0.1% acetic acid in water (mobile phase A) and 0.1% acetic acid in ACN–MeOH (90:10; mobile phase B), at total flow rate of 0.3 mL/min. The chromatographic separation was achieved using 25 min gradient elution. The initial mobile phase composition was 20% B for the first 3.0 min, gradually increasing to 65% B in 13 min, gradually increasing to 95% B in 3.0 min, then held constant at 95% B for 4.0 min, and finally brought back to the initial condition of 20% B in 0.20min followed by 2 min re-equilibration. The injection volume of all samples was 10 μL.
The mass spectrometer parameters, such as temperature, voltage and gas pressure, were optimized by infusing each analyte and the internal standard (IS) using a 5 μg/mL solution in 50% MeOH via a Harvard ‘22’ standard infusion syringe pump (Harvard Apparatus, South Natick, MA, USA) at 10 μL/min. All eicosanoids were detected in the negative ionization mode and deprotonated molecules were used as the precursors for selected reaction monitoring (SRM) with the following mass spectrometer source settings: ion spray voltage, −4000 V; source temperature, 500°C, curtain gas, 15 AU; gas 1, 40 AU, gas 2, 40 AU, collision gas pressure, high; Q1/Q3 resolution, high; and interface heater, on. SRM transitions for each analyte and IS, as well as their respective optimum MS parameters, such as declustering potential and collision energy, are shown in Table 1.
TABLE 1.
Summary of selected reaction monitoring (SRM), precursor and product ions (Q1, Q3), internal standard (IS), declustering potential, collision energy and retention time used for eicosanoids in negative ESI mode
| Analytes ID | Analytes | IS used | Selected reaction monitoring (Q1/Q3) | Declustering potential | Collision energy | Retention time |
|---|---|---|---|---|---|---|
| 1 | PGJ2 | PGE2-d4 | 333.1/315.2 | −60 | −12 | 13.60 |
| 3 | 20-OH-PGE2 | PGE2-d4 | 367.1/349.3 | −58 | −16 | 6.68 |
| 4 | PGB2 | PGE2-d4 | 333.0/235.0 | −90 | −28 | 13.85 |
| 6 | PGD2 | PGE2-d4 | 351.1/271.0 | −60 | −20 | 11.77 |
| 7 | PGE2 | PGE2-d4 | 351.1/271.0 | −60 | −20 | 11.39 |
| 8 | AA | AA-d8 | 303.0/258.9 | −75 | −18 | 20.30 |
| 9 | 15-HETE | 15-HETE-d8 | 319.0/175.1 | −80 | −20 | 18.10 |
| 10 | 12-HETE | 15-HETE-d8 | 319.0/179.0 | −80 | −20 | 18.40 |
| 11 | 11-HETE | 15-HETE-d8 | 319.0/167.2 | −95 | −22 | 18.30 |
| 12 | 8-HETE | 15-HETE-d8 | 319.0/154.8 | −80 | −22 | 18.30 |
| 13 | 5-HETE | 15-HETE-d8 | 319.0/114.7 | −65 | −20 | 18.50 |
| 14 | LTE4 | LTB4-d4 | 438.1/333.0 | −70 | −26 | 13.20 |
| 15 | LTD4 | LTB4-d4 | 495.1/176.8 | −80 | −28 | 11.50 |
| 16 | LTC4 | LTB4-d4 | 624.1/271.8 | −110 | −32 | 15.10 |
| 17 | LTB4 | LTB4-d4 | 335.0/194.8 | −75 | −22 | 14.90 |
| 18 | 13,14-DiOH-15-Keto-PGE2 | PGE2-d4 | 351.0/333.0 | −75 | −20 | 12.50 |
| 19 | 11-β-PGF2α | PGE2-d4 | 353.2/309.1 | −95 | −28 | 10.50 |
| 20 | 8-iso-PGF2α | PGE2-d4 | 353.2/309.1 | −95 | −28 | 10.31 |
| 21 | PGF2α | PGE2-d4 | 353.2/309.1 | −95 | −28 | 10.97 |
| 22 | 15-Keto-PGE2 | PGE2-d4 | 349.0/331.1 | −45 | −14 | 12.00 |
| 23 | 6-Keto-PGF1α | PGE2-d4 | 369.0/163.0 | −95 | −38 | 8.89 |
| 24 | TXB2 | TXB2-d4 | 369.0/168.8 | −75 | −26 | 10.50 |
| 28 | Ibuprofen | PGE2-d4 | 205.0/161.0 | −30 | −10 | 15.50 |
| 29 | 13,14-DiOH-PGF2α | PGE2-d4 | 355.0/311.3 | −94 | −34 | 11.80 |
| 30 | PGF1α | PGE2-d4 | 355.0/311.3 | −94 | −34 | 11.23 |
| 31 | 13,14-DiOH-15-Keto-PGF2α | PGE2-d4 | 353.2/112.8 | −102 | −38 | 12.30 |
| 32 | 13,14-DiOH-15-Keto-PGE1 | PGE2-d4 | 353.2/335.1 | −47 | −17 | 13.00 |
| 33 | PGD1 | PGE2-d4 | 353.2/317.1 | −55 | −18 | 12.00 |
| 34 | 13,14-DiOH-PGE1 | PGE2-d4 | 355.2/337.1 | −30 | −20 | 12.3 |
| 35 | TXB3 | TXB2-d4 | 367.1/168.6 | −90 | −24 | 9.48 |
| 36 | 15-deoxy-delta 12,14 PGJ2 | PGE2-d4 | 315.1/271.2 | −40 | −18 | 17.20 |
| 37 | PGE1 | PGE2-d4 | 353.2/317.1 | −55 | −18 | 11.77 |
| 38 | PGE3 | PGE2-d4 | 349.1/331.2 | −30 | −13 | 10.47 |
| 39 | PGD3 | PGE2-d4 | 349.1/331.2 | −30 | −13 | 10.77 |
| 40 | PGF3α | PGE2-d4 | 351.2/307.0 | −120 | −26 | 10.07 |
| 41 | 14,15-LTC4 | LTB4-d4 | 624.1/272.1 | −50 | −30 | 13.58 |
| 42 | Tetranor-PGEM | PGE2-d4 | 327.1/309.1 | −50 | −15 | 2.49 |
| 43 | Tetranor-PGFM | PGE2-d4 | 329.0/311.1 | −50 | −18 | 2.25 |
| 44 | 11-De TXB3 | TXB2-d4 | 365.1/303.2 | −100 | −22 | 10.60 |
| 45 | 2,3 Dinor 8-iso PGF2 | PGE2-d4 | 325.1/237.2 | −50 | −15 | 8.51 |
| 25 | TXB2-d4 | NA | 373.1/172.8 | −27 | −22 | 10.45 |
| 26 | PGE2-d4 | NA | 355.1/192.9 | −46 | −27 | 11.40 |
| 27 | AA-d8 | NA | 311.0/267.1 | −75 | −20 | 20.20 |
| 46 | 15 -HETE-d8 | NA | 327.2/226.2 | −60 | −18 | 18.05 |
| 47 | LTB4-d4 | NA | 339.1/197.1 | −85 | −10 | 14.80 |
NA, Not applicable.
2.4 | Preparation of charcoal-stripped serum for calibration curves
Serum was stripped with activated charcoal to remove endogenous eicosanoids. Twelve milliliters of charcoal suspension (0.66 g of dextran-coated charcoal in 100 mL of Dulbecco’s phosphate-buffered saline) was transferred into a glass tube, centrifuged at 4000 g for 15 min at 4°C, and the supernatant Dulbecco’s phosphate-buffered saline was discarded. Serum (6.0 mL) was then added on to the charcoal pellet under continuous stirring at 37 ± 1°C for 2 h, centrifuged at 13,000 g for 15 min, and the supernatant was collected. The process was repeated a second time for maximal removal of endogenous eicosanoids. This stripped serum was used to construct serum calibration curves.
2.5 | Preparation of standard solutions and calibration curves
Aliquots from original stock solutions of every analyte were mixed to prepare spiking solution mixtures, which were stored at −80°C. Blank serum (pooled, n = 10) was purchased from Equitech Enterprises Inc. (Kerriville, TX, USA) and stripped from endogenous eicosanoids as described above. Blank sputum and BALF were collected from healthy control subjects. Stripped serum was used to construct serum calibration curves, whereas sputum and BALF calibration curves were prepared in untreated matrices.
The calibration ranges of the various eicosanoids were divided into three categories: 0.2–500, 1–500 and 3–500 ng/mL. Five hundred microliters of stripped blank serum, 10 × −diluted stripped blank serum, untreated blank sputum and BALF were spiked with spiking analyte (10×) and IS (10×) solutions, 10 μL each, and vortexed for 30 s. Samples were then extracted as described below and reconstituted in 100 μL of 50% ACN in deionized water. Five stable-labeled eicosanoids were used as internal standards (IS) for the different analytes as described in Table 1. The final concentration of all five ISs was 100 ng/mL and the final concentrations of analytes in standards and QC samples are listed in Table 2.
TABLE 2.
Dynamic range, linearity, accuracy, and precision of eicosanoids in human serum
| Analytes ID | Dynamic range (ng/mL) | LLOQ (0.2 ng/mL) | LQC (1 ng/mL) | MQC (30 ng/mL) | HQC (100 ng/mL) | ULOQ (500 ng/mL) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy | RSD (%) | Accuracy | RSD (%) | Accuracy | RSD (%) | Accuracy | RSD (%) | Accuracy | RDS. (%) | ||
| 1 | 0.2–500 | 111 | 9 | 95 | 15 | 90 | 1 | 98 | 4 | 112 | 4 |
| 4 | 0.2–500 | 101 | 10 | 98 | 4 | 91 | 3 | 92 | 4 | 90 | 3 |
| 6 | 0.2–500 | 99 | 14 | 101 | 10 | 94 | 3 | 94 | 4 | 94 | 4 |
| 7 | 0.2–500 | 109 | 12 | 94 | 6 | 101 | 2 | 104 | 3 | 113 | 2 |
| 14 | 0.2–500 | 104 | 10 | 88 | 9 | 92 | 6 | 94 | 5 | 114 | 3 |
| 17 | 0.2–500 | 110 | 8 | 105 | 11 | 97 | 5 | 99 | 6 | 115 | 5 |
| 18 | 0.2–500 | 110 | 8 | 87 | 3 | 85 | 1 | 87 | 9 | 100 | 2 |
| 20 | 0.2–500 | 99 | 6 | 103 | 9 | 92 | 3 | 97 | 4 | 97 | 1 |
| 21 | 0.2–500 | 105 | 9 | 101 | 13 | 99 | 4 | 109 | 4 | 115 | 1 |
| 22 | 0.2–500 | 103 | 13 | 88 | 7 | 91 | 6 | 103 | 5 | 116 | 1 |
| 24 | 0.2–500 | 94 | 11 | 109 | 3 | 100 | 1 | 99 | 1 | 106 | 1 |
| 28 | 0.2–500 | 106 | 12 | 92 | 13 | 91 | 4 | 95 | 2 | 103 | 4 |
| 29 | 0.2–500 | 104 | 12 | 100 | 10 | 103 | 5 | 108 | 3 | 108 | 3 |
| 30 | 0.2–500 | 105 | 8 | 101 | 11 | 96 | 2 | 101 | 2 | 108 | 2 |
| 31 | 0.2–500 | 90 | 11 | 90 | 10 | 93 | 5 | 89 | 6 | 85 | 1 |
| 32 | 0.2–500 | 103 | 7 | 86 | 5 | 86 | 3 | 86 | 5 | 115 | 3 |
| 33 | 0.2–500 | 103 | 11 | 97 | 11 | 96 | 3 | 97 | 8 | 93 | 3 |
| 34 | 0.2–500 | 107 | 14 | 79 | 6 | 88 | 3 | 85 | 5 | 90 | 3 |
| 35 | 0.2–500 | 93 | 12 | 92 | 9 | 100 | 3 | 103 | 11 | 96 | 10 |
| 36 | 0.2–500 | 110 | 9 | 96 | 14 | 88 | 3 | 90 | 2 | 114 | 3 |
| 37 | 0.2–500 | 105 | 15 | 90 | 5 | 94 | 4 | 99 | 3 | 98 | 1 |
| 38 | 0.2–500 | 105 | 13 | 94 | 8 | 90 | 3 | 97 | 1 | 104 | 3 |
| 39 | 0.2–500 | 96 | 14 | 103 | 5 | 95 | 4 | 95 | 6 | 107 | 2 |
| 44 | 0.2–500 | 103 | 5 | 102 | 7 | 102 | 4 | 100 | 2 | 112 | 2 |
| 45 | 0.2–500 | 96 | 13 | 105 | 10 | 90 | 1 | 94 | 3 | 113 | 2 |
| LLOQ (1 ng/mL) | LQC (3 ng/mL) | MQC (30 ng/mL) | HQC (100 ng/mL) | LOW (500 ng/mL) | |||||||
| 19 | 1–500 | 104 | 13 | 10.2 | 10 | 90 | 2 | 93 | 4 | 85 | 1 |
| 23 | 1–500 | 105 | 11 | 6.0 | 6 | 97 | 8 | 96 | 8 | 107 | 3 |
| 40 | 1–500 | 102 | 7 | 2.4 | 2 | 89 | 4 | 100 | 4 | 89 | 3 |
| LLOQ (3 ng/mL) | LQC (10 ng/mL) | MQC (100 ng/mL) | HQC (350 ng/mL) | ULOQ (500 ng/mL) | |||||||
| 9 | 3–500 | 115 | 4 | 4.7 | 5 | 115 | 4 | 103 | 3 | 100 | 5 |
| 10 | 3–500 | 110 | 10 | 6.5 | 6 | 115 | 3 | 105 | 7 | 99 | 6 |
| 11 | 3–500 | 112 | 6 | 5.4 | 5 | 107 | 3 | 103 | 7 | 102 | 5 |
| 12 | 3–500 | 97 | 15 | 6.2 | 6 | 114 | 7 | 106 | 9 | 99 | 5 |
| 13 | 3–500 | 110 | 12 | 7.4 | 7 | 105 | 5 | 98 | 5 | 94 | 4 |
| 43 | 3–500 | 95 | 12 | 11.6 | 12 | 105 | 9 | 110 | 11 | 106 | 12 |
2.6 | Sample preparation
For serum, sputum and BALF samples, Oasis® HLB 3 cm3 (60 mg) SPE cartridges (Waters, Milford, MA, USA) were used for sample extraction. A 500 μL sample was spiked with 10 μL IS and diluted with 1500 μL 5% acetic acid in water, vortexed and loaded onto SPE cartridges pre-conditioned with 2 mL MeOH, followed by 2 mL 0.1% acetic acid in H2O. Loaded cartridges were washed with 2 mL 0.1% acetic acid in H2O and eluted with 2 mL MeOH. Eluates were evaporated under vacuum at room temperature and reconstituted in 100 μL of 50% ACN in water, i.e. samples were concentrated 5-fold after evaporation and reconstitution.
2.7 | Extraction recovery
Recoveries of analytes and labeled ISs from charcoal-stripped serum, 10× diluted charcoal-stripped serum, original serum, 10× diluted serum, sputum and BALF were determined by dividing the peak area ratio of analyte to IS (after subtracting any endogenous background) from blank samples spiked before extraction by those from neat unextracted standards for both the low and high QCs (n = 5).
2.8 | Method validation
The ratios of analyte to IS and the 1/x2 weighting scheme were used in all calibration curves. The method was validated using five QC points for each calibration curve and the concentrations of the QC points are shown in Table 2. Five replicates of each QC point were analyzed each day to determine the intra- and inter-day accuracy and precision. This process was repeated three times over 3 days in order to determine the inter-day accuracy and precision using freshly prepared calibration curves. Intra-day accuracy and precision were calculated from the bias (%) [% (measured – theoretical)/measured concentrations] and relative standard deviation [RSD (%) = % standard deviation/mean], respectively, for the five replicates of each QC point. Inter-day accuracy and precision were calculated similarly using the 15 replicates of each QC point from the three validation runs.
2.9 | Stability studies
Stability experiments were carried out to examine the analyte stability in stock solutions, original matrices (samples spiked, stored at different conditions, then extracted before analysis) and extracted matrices (samples spiked, extracted, then stored at different conditions after extraction) under different conditions. Stability studies included autosampler stability (at 4°C for 48 h), bench-top stability (at room temperature for 8 h), freeze–thaw stability (three freeze–thaw cycles) and long-term stability (at −20°C and at −80°C for 6 months), for both the low and high QCs (n = 3).
2.10 | Human subjects
This work was performed as part of a clinical trial that aims to determine the role of the inhibition of PGE production in restoring lung repair processes and thus improving outcomes of COPD. This study was approved by the institutional review board at the clinical sites where the samples were collected and written informed consent was obtained from all individuals. In the healthy control arm of this study, healthy subjects, age > 45, with no medical conditions that would place them at untoward risk for bronchoscopy and broncho alveolar lavage were recruited after obtaining written consents. Healthy nonsmoking controls were recruited locally either from prior study participants or de novo and, other than smoking history, they met the same criteria as smoking controls and had no emphysema, defined as <3% of lung voxels with density < −950 Hounsfield units on quantitative CT scan. In addition, control subjects had post bronchodilator forced expiratory volume in one second (FEV1) ≥80% predicted and an FEV1/forced vital capacity ratio of at least 0.7. Serum, BALF and sputum samples were collected and stored at −80°C until the time of LC–MS/MS analysis.
3 | RESULTS
3.1 | LC–MS/MS method development
In this study, an LC–MS/MS method for the quantification of eicosanoids from different classes including PGs, TBXs, HETE, AA and LTs in human serum, sputum and BALF was developed and validated. All eicosanoids have a free carboxylic acid functional group, which ionized efficiently in the negative ionization mode. Table 1 summarizes the MS/MS conditions used to quantify all 34 eicosanoids. Figure 1 shows a representative LC–MS/MS chromatogram of all eicosanoid standards. Mass spectrometer parameters were optimized during method development to maximize not only sensitivity but also selectivity. For example, several HETEs shared the same precursor as well as fragment masses; therefore, the most selective rather than the most sensitive SRM transitions were used for the quantification of these analytes (Figure 2).
FIGURE 1.

Representative chromatograms of all eicosanoid standards at 10 ng/mL under final chromatography and detection conditions. Peaks are labeled with analytes IDs and retention times as given in Table 1
FIGURE 2.
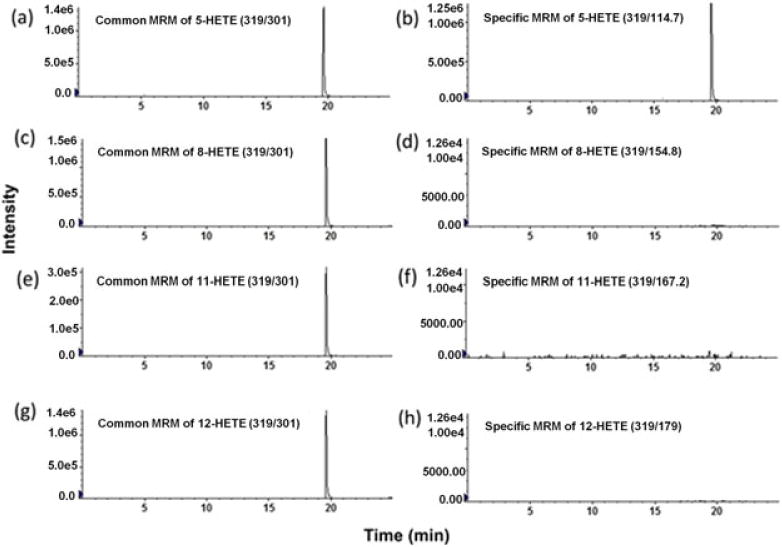
Isobaric compounds that also share fragmentation patterns as well as retention times were distinguished via specific selected reaction monitoring (SRMs). LC–MS/MS chromatograms of various hydroxyeicosatetraenoic acids (HETEs) after the injection of a 5-HETE standard at 500 ng/mL. Isobaric HETEs including 5-HETE (13), 8-HETE (12), 11-HETE (11) and 12-HETE (10) were not resolved chromatographically, and they produced both common and selective fragments in MS/MS. The 319/301 transition was the most sensitive but was shared by all HETEs (a, c, e, g). In contrast, the less sensitive but more selective transitions of HETEs were used, including 319/114.7 for 5-HETE (b), 319/154.8 for 8-HETE (d), 319/167.2 for 11-HETE (f) and 319/179 for 12-HETE (h)
LC conditions were optimized to separate all eicosanoids of interest with a desirable peak shape and signal intensity using an Acquity UPLC®BEH shield RP18 column (1.7 μm, 150 × 2.1 mm). Various mobile phases with a pH range of 3–9 were screened to optimize LC conditions. The less hydrophobic eicosanoids including PGs, TXs and LTs eluted earlier and largely independent of the mobile phase pH. In contrast, acidic pH mobile phases resulted in better peak shape and longer retention of the more hydrophobic eicosanoids including HETEs and AA. Therefore, acetic acid was used as an aqueous and organic mobile phase modifier.
Many eicosanoids are isobaric compounds that share the same parent mass and also the same fragmentation pattern, such as PGE2, PGD2 and 13,14-dihydro-15-k-PGE2. Therefore, these compounds have to be chromatographically resolved (Figure 3). Moreover, eicosanoids can undergo in-source fragmentation into other eicosanoids; therefore, even some analytes with different masses have to be resolved chromatographically to distinguish in-source fragments from other analytes (Figure 3). Therefore, both chromatographic separation and MS/MS specificity were required to quantify all eicosanoids of interest. Under final chromatography conditions, >34 eicosanoids in human serum, sputum and BALF were separated in 25 min.
FIGURE 3.
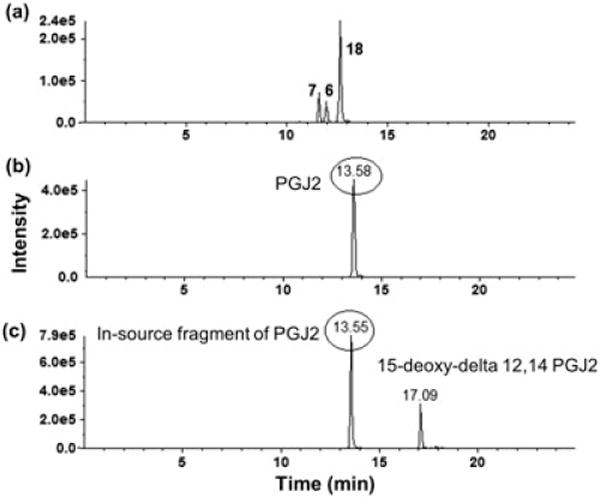
Isobaric eicosanoids that undergo in-source fragmentation were separated chromatographically. (a) The isobaric compounds PGE2 (7), PGD2 (6) and 13,14-dihydro-15-k-PGE2 (18) share the same SRM transition of 351 → 333 and had to be separated chromatographically [retention time (RT) = 11.58, 11.77, and, 12.5 min, respectively]. (b) PGJ2 (333.1 → 315.2) and (c) 15-dexoxy-delta 12,14- PGJ2 (315.2 → 271.2) have different precursors as well as fragment masses. However, PGJ2 produces an in-source fragment (315.2) with the same mass as the parent 15-dexoxy-delta 12,14-PGJ2. Therefore, the two compounds had to be separated chromatographically
Some eicosanoids (HETEs) had residual peak areas in serum after charcoal stripping. Therefore, calibration curves for these eicosanoids were constructed using 10× diluted charcoal stripped serum to decrease the residual peak areas after matrix stripping. Extraction recoveries were similar (90–115%) for these eicosanoids in diluted and undiluted serum and subsequently the method was validated with two sets of calibration curves, with and without 10× diluted stripped serum.
3.2 | Method validation
The method was validated for each analyte using three calibration curves prepared on 3 days. Table 2 shows the validation results in human serum including dynamic ranges and inter-day accuracy and precision values. Three dynamic ranges were used to cover all eicosanoids at relevant physiological concentrations in the various matrices, namely 0.2–500, 1–500 and 3–500 ng/mL.
The method of background subtraction was used to account for the background/endogenous concentrations in blank matrices before spiking with analyte standards. Therefore, the differences in the lower limit of quantification of the various eicosanoids are not necessarily due to differences in the sensitivity of the analytes, but rather to the differences in the endogenous background levels in the blanks used for building the calibration curves. R2 was >0.998 for all eicosanoids in all matrices, confirming the linearity of the assay in the selected calibration ranges.
Intra- and inter-day accuracy and precision were determined to evaluate the reliability and reproducibility of this method. Table 2 shows the inter-day accuracy and precision of standards prepared in human serum. Validation data for all other matrices are shown in Tables S1–3 in the Supporting Information. Accuracy and precision were ≤20% at LLOQ and ≤15% at the other four QC concentrations for all eicosanoids in serum, sputum and BALF.
3.3 | Recovery
Several protein precipitation and SPE methods were investigated to increase extraction recovery and decrease matrix effect. The large variation in the physicochemical properties between different classes of eicosanoids resulted in different extraction recoveries of these compounds. The average extraction recoveries of all analytes were 57–115% in serum, 69–115% in 10× dilute serum, 41–115% in BALF and 34–115% in sputum (data not shown). Our result confirms that charcoal stripped serum to mimic human serum with similar recovery rates using analyte/IS peak area ratios.
3.4 | Stability studies
Stability of eicosanoids in stocks and biological matrices was studied under various conditions as outlined in Section 2.10. Table 3 lists unstable analytes with >20% loss in peak area under the different storage conditions. Eicosanoids not listed in Table 3 were stable under all storage conditions. All eicosanoids were stable in stock solutions and extracted serum in the autosampler at 4°C for up to 24 h except compounds 8 and 27. However, by 48 h the peak areas of some eicosanoids decreased markedly (52–97%). Eicosanoids were also stable in stock solution, original matrix, and extracted matrix samples at room temperature on the bench up to 8 h except for compounds 3, 18, 22, 27, 33, 39 and 41–44 in serum, which were stable only for 2 h on the bench. In addition, compounds 15, 16 and 41 were stable in serum only for few minutes after spiking.
TABLE 3.
Unstable eicosanoids in different matrices under various storage conditionsa
| Storage condition | Autosampler at 4 °C | Freeze-thaw three cycles at −20°C | Bench-top at room temperature | Long-term stability at −20°C | Long-term stability at −80°C | ||||
|---|---|---|---|---|---|---|---|---|---|
| Matrix | Extracted matrix | Original matrix | Original matrix | Extracted matrix | Original matrix | Extracted matrix | Original matrix | ||
| Time | Up to 24 h | Up to 3 cycles | Up to 2 h | Up to 8 h | Up to 4h | Up to 180 days | Up to 3 days | Up to 7 days | Up to 180 days |
| Stock solution | 8 and 27 | 17 and 42 | All stable | 16 and 41 | NA | 16 and 41 | NA | NA | 16 and 41 |
| Serum | 8 and 27 | 3, 15, 16, 18, 33, 36, 41, 42, 43 and 44 | 3, 15, 16, 41 and 42 | 3,15, 16, 18, 22, 33, 39, 41, 42, 43, 27 and 44 | All stable | 15,16, 18, 22, 33, 36, 38, 41, 42, 43 and 44 | 15, 16 | 1, 14, 15, 16, 33, 34 and 41 | NA |
Analytes with >20% loss by the reported time intervals under different storage conditions are presented.
Under long-term storage conditions, eicosanoids were stable in stock solution and original matrices at −20°C for up to 6 months except for compounds 18, 22, 33, 38, 43 and 44 in serum. In addition, compounds 15, 16, 41 and 42 in serum were stable for only 7 days after spiking. In contrast, in extracted matrices, all eicosanoids except compounds 15 and 16 were stable for only 3 days at −20°C and many started degrading after 7 days. At −80°C, all eicosanoid stocks were stable for up to 6 months except for compounds 16 and 41, which were stable until 2 months.
3.5 | Human eicosanoids profiles
Eicosanoid profiles in serum, sputum and BALF of healthy human subjects were characterized using this LC–MS/MS method (Table 4). In accordance with previous reports, 12-HETE (22 ng/mL) and 8-HETE (0.6 ng/mL) were the HETEs with highest and lowest concentrations in serum, respectively (Hennessy et al., 2017; Schuchardt et al., 2013). Among serum prostaglandins, PGD2 and PGE2 had the highest concentrations (0.4 ng/mL), whereas PGJ2 (0.06 ng/mL) showed the lowest concentration. Among the three thromboxanes of interest, only TXB2 (6.4 ng/mL) was detected in serum. Eicosanoids concentrations in sputum and BALF were on average more than 10× lower than serum. In sputum and BALF, highest concentrations were observed for HETEs (0.5–2 ng/mL). The concentrations reported in this manuscript were comparable with recent reports from healthy human subjects using LC–MS/MS analyses in serum (Hennessy et al., 2017; Schuchardt et al., 2013; Song et al., 2013) and sputum (Jian et al., 2013).
TABLE 4.
Concentrations (ng/mL) of eicosanoids in serum (n = 5), sputum (n = 2), bronchial and alveolar fluids (n = 5) of healthy human subjects
| Matrix | PGJ2 | PGB2 | PGD2 | PGE2 | 15-HETE | 12-HETE | 11-HETE | 8-HETE | 5-HETE | LTE4 | LTB4 | PGF2α | 13,14-DiOH-PGF2α | TXB2 | TXB3 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Serum | Mean | 0.06 | 0.1 | 0.39 | 0.43 | 1.35 | 22 | 1.3 | 0.55 | 9.4 | 0.3 | 0.44 | — | 0.10 | 6.4 | 0.05 |
| SD | 0.03 | 0.03 | 0.19 | 0.32 | 0.63 | 14 | 0.6 | 0.28 | 5.5 | 0.16 | 0.10 | — | 0.02 | 5.2 | 0.08 | |
|
| ||||||||||||||||
| Sputum | Mean | — | — | — | 0.09 | 1.05 | 2.13 | 0.29 | 0.27 | 2.71 | — | 0.72 | 0.04 | — | 0.23 | — |
| SD | — | — | — | 0.01 | 0.09 | 0.3 | 0.03 | 0.01 | 0.27 | — | 0.65 | 0.0 | — | 0.01 | — | |
|
| ||||||||||||||||
| Bronchial fluid | Mean | — | — | — | — | 0.43 | 0.32 | 0.24 | 0.18 | 0.48 | — | 0.09 | 0.04 | — | — | — |
| SD | — | — | — | — | 0.25 | 0.18 | 0.17 | 0.19 | 0.24 | — | 0.13 | 0.06 | — | — | — | |
|
| ||||||||||||||||
| Alveolar fluid | Mean | — | — | — | — | — | — | — | — | 0.18 | — | 0.1 | 0.08 | — | — | — |
| SD | — | — | — | — | — | — | — | — | 0.09 | — | 0.09 | 0.03 | — | — | — | |
—, Not detected.
4 | DISCUSSION
Owing to matrix effects on the ionization of analytes in the ESI MS source, it is critical to prepare calibration curves in the same or equivalent matrices as the study samples. This becomes a problem for endogenous analytes including eicosanoids, where analyte-free blank matrices are not available to spike with analyte standards of known concentrations for the construction of calibration curves. Various approaches are followed to solve the problem of endogenous background in blank matrices for the construction of calibration curves, which were reviewed recently (Thakare, Chhonker, Gautam, Alamoudi, & Alnouti, 2016). These approaches including background subtraction (Gachet et al., 2015), standard addition (Prasain et al., 2013; Strassburg et al., 2012; Yang et al., 2009), surrogate analytes (Deems, Buczynski, Bowers-Gentry, Harkewicz, & Dennis, 2007; Gouveia-Figueira & Nording, 2015; Levison et al., 2013) and surrogate matrix (Idborg et al., 2014; Jian et al., 2013; Kortz, Dorow, Becker, Thiery, & Ceglarek, 2013; Massey & Nicolaou, 2013; Montuschi, Martello, Felli, Mondino, & Chiarotti, 2004; Ogawa, Tomaru, Matsumoto, Watanabe, & Higashi, 2016; Squellerio et al., 2014; Yoshida, Kodai, Takemura, Minamiyama, & Niki, 2008; Zhang et al., 2011), which were used for the quantification of eicosanoids in various biological matrices.
As we discussed previously (Thakare et al., 2016), every one of these approaches has advantages and disadvantages. Therefore, we applied and compared the various approaches for the quantification of eicosanoids in serum and found that activated charcoal was the most accurate and convenient method for this application. Activated charcoal is an efficient adsorbent; consequently, blank serum free of eicosanoids was prepared by stripping serum from endogenous eicosanoids using activated charcoal. This eicosanoid-free serum was used to construct the calibration curves for the analyses of serum samples. The charcoal-stripping conditions were optimized to maximize eicosanoids depletion from serum. Most eicosanoids were completely depleted, but some eicosanoids (HETEs and LTB4) had trace residual peaks in serum after stripping with charcoal. For these eicosanoids, the background peak area of the remaining trace levels was subtracted from the peak area of the calibration curve standards, which allowed the construction of calibration curves with high accuracy and precision. Using analyte/IS peak area ratios, the recoveries of eicosanoids in the charcoal-stripped serum were similar to those in unstripped serum (data not shown), which indicates that matrix effect was the same for the study samples (unstripped serum) and calibration curve (stripped serum).
Three dynamic ranges were used to cover all analytes in serum, BALF, and sputum at relevant physiological concentrations, namely 0.2–500, 1–500 and 3–500 ng/mL. The different dynamic ranges were used because the various eicosanoids had different sensitivities, endogenous concentrations and/or signal linearity. For example, the LLOQ of 5-HETE, 8-HETE, 11-HETE, 13-HETE and 15-HETE was 3 ng/mL in serum, not owing to limitations in detection sensitivity (limit of detection 0.1 ng/mL), but rather because of the relatively high residual background of these eicosanoids in the blank matrix used to construct the calibration curve after matrix stripping, which did not allow consistent subtraction from the peak areas of spiked standards <3 ng/mL. To quantify levels <3 ng/mL, calibration curves were constructed using 10-fold diluted charcoal-stripped serum to decrease the residual peak areas after matrix stripping. Consequently, the method was validated with two sets of calibration curves, one set in 10× diluted stripped plasma and another in stripped undiluted plasma. Recoveries of these eicosanoids in undiluted and 10× diluted serum were similar.
On the other hand, no matrix stripping was applied for BALF and sputum because we were able to obtain batches of blank matrices ranging from undetectable to trace levels for most eicosanoids of interest, and this background was subtracted from the peak areas of calibration standards.
Eicosanoids comprise a large family of endogenous compounds, and many members of this family are isobaric with very similar physio-chemical properties including isomers and stereoisomers. Many eicosanoids not only share the same mass, but also have the same fragmentation pattern, resulting in the same SRM transitions. Moreover, many eicosanoids undergo in-source fragmentation, which results in fragments with similar masses to other eicosanoids. In addition, interfering peaks could arise from other unknown endogenous components of the matrix. Therefore, MS/MS specificity by itself is not always adequate to separate all eicosanoids, and chromatographic resolution is required for their separation in time. For example, the isobaric compounds PGE2, PGD2 and 13,14-dihydro-15-k-PGE2 are identified through the same 351 → 333 SRM transition, but were separated chromatographically [retention time (RT) = 11.39, 11.77 and 12.5 min, respectively; Figure 3]. Similarly, the isobaric compounds PGF2α, 11-β PGF2α and 8-iso-PGF2α are identified through the same 353.2 → 309.1 SRM transition but were separated chromatographically (RT = 11.7, 12.0 and 13.0 min, respectively; Figure 1). Variation in RT over the period of 12 months of utilization of this method for all analytes was <10%.
Under our final LC–MS/MS conditions, all eicosanoids of interest were resolved from each other in <25 min and all standards produced single peaks. One exception was TXB2 and TXB3 and their d4-labeled IS (TXB2-d4), each of which produced two peaks (completely chromatographically resolved) that belonged to their anomers. Both anomers for both compounds were detected in standards as well as biological samples. The peak areas for both anomers were summed.
Moreover, eicosanoids can undergo in-source fragmentation into other eicosanoids, which means that sometimes even analytes with different masses have to be resolved chromatographically to distinguish in-source fragments from other analytes. For example, PGJ2 (333.1 → 315.2) and 15-dexoxy-delta 12,14-PGJ2 (315.2 → 271.2) have different parent as well as fragment masses, yet PGJ2 produces a shadow peak with the same SRM as 15-dexoxy-delta 12,14-PGJ2, i.e. 315.2 → 271.2. Therefore, PGJ2 and 15-dexoxy-delta 12,14-PGJ2 had to be chromatographically resolved (Figure 3).
In addition to chromatographic resolution, isobaric compounds with similar SRMs can be distinguished if they produce specific SRMs, which may not be the most sensitive ones. For example, isobaric HETEs such as 8-HETE, 11-HETE, 5-HETE and 12-HETE were not resolved chromatographically, but every isomer produced unique fragments that were not produced by the other isomers, namely 319 → 154.8, 319 → 167.2, 319 → 114.7 and 319 → 179, respectively, which was also shown previously (Gomolka et al., 2011; Kempen, Yang, Felix, Madden, & Newman, 2001; Strassburg et al., 2012; Willenberg et al., 2015). However, these specific SRMs were less sensitive than common SRMs such as 319 → 301.1 (Figure 2).
Although the detection and quantification of eicosanoids concentrations have become a routine analysis in many biomedical laboratories, only a few reports have addressed eicosanoid stability under different storage and analysis conditions (Gouveia-Figueira & Nording, 2014; Maddipati & Zhou, 2011; Squellerio et al., 2014; Sterz et al., 2012; Wang et al., 2014; Zhang, Yang, Ai, & Zhu, 2015). Most of these studies have reported issues related to eicosanoid instability in original matrices or stock solutions for some eicosanoids. In this report, stability studies were carried out in stock solution, original matrices (serum) and extracted matrices. In the autosampler, by 48 h the peak area of some eicosanoids had decreased markedly (52–97%). This could be a result of degradation and/or precipitation owing to evaporation of organic solvent over time. Therefore, samples were not stored in the autosampler longer than 24 h. In contrast, in extracted matrices, all eicosanoids except compounds 15 and 16 were stable for only 3 days at −20°C and many started degrading after 7 days. Therefore, extracted samples should be run or re-run within 3 days from the time of sample preparation. Sample preparation, analyses and storage conditions were adjusted in this method to ensure eicosanoid stability under these conditions.
Accordingly, we excluded some eicosanoids from this method because they were not stable on the bench or in long-term storage (3, 15, 16, 41 and 42), in the autosampler (8, 27) or after storage for longer than 2 h.
For valid quantitative analysis, analytical standards of high purity are always required. Some commercially available standards of eicosanoids contained impurities that were detected by LC–MS/MS at the time of purchase. For example, PGJ2 (1) contained 15-Keto-PGE2 (22) (0.28%), 15-deoxy-delta 12,14 PGJ2 (36) (3.87%), PGE3 (38) (0.60%) and an unknown component (SRM = 222/123) (0.11%). Similarly, PGE3 (38) contained unknown component 1 (0.60%), component 2 (0.65%) and PGF3 (40) (5.67%). Another example is LTC4 (16), which contained an unknown component at SRM similar to 15-HETE (9) (0.11%) but with a different retention time, and LTD4 (15) (0.15%). These components could be impurities formed during the synthesis process, or degradants that formed after synthesis, during shipping or during the 3 days’ during which stocks were stored at −80°C, from the time standards arrived to the time they were analyzed. Carryover and/or LC–MS system contamination was excluded by the lack of any of these impurities in injected blanks. The analytes were still included in the method because none of the validation criteria were compromised.
5 | CONCLUSION
In summary, an LC–MS/MS method was developed for the simultaneous quantification of eicosanoids in human serum, BALF and sputum. The method was sensitive, selective, accurate and precise with a wide dynamic range. This method was successfully applied to the study of eicosanoid in healthy human subjects. The characterization of the detailed eicosanoids profile in healthy and COPD subjects will facilitate a better understanding of the pathological and physiological role of eicosanoids in humans.
Supplementary Material
Abbreviations
- 11-β-PGF2α
11-b prostaglandin F2α
- 13
14-dihydro-15-keto-PGE2, 13,14-dihydro-15-keto-prostaglandin E2
- 13
14-DiOH-15-keto-PGF2α, 13,14-dihydro-15-keto-prostaglandin F2α
- 13
14-DiOH-15-k-PGE1, 13,14-dihydro-15-keto-prostaglandin E1
- 13
14-DiOH-PGE1, 13,14-dihydro-prostaglandin E1
- 13
14-DiOH-PGF2α, 13,14-dihydro-prostaglandin F2α
- 15-deoxy-delta 12
14 PGJ2, 15-deoxy-delta 12,14 Prostaglandin J2
- 15-keto-PGE2
15-keto-prostaglandin E2
- 20-OH-PGE2
20-hydroxy prostaglandin E2
- 6-keto-PGF1α
6-keto-prostaglandin F1α
- 8-iso-PGF2α
8-iso-prostaglandin F2α
- AA
arachidonic acid
- ACN
acetonitrile
- BALF
bronchial alveolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- ESI
electrospray ionization
- FEV1
forced expiratory volume in one second
- FVE
forced vital capacity
- HETE
hydroxyeicosatetraenoic acid
- LT
leukotriene
- LTB4
leukotriene B4
- LTC4
leukotriene C4
- LTD4
leukotriene D4
- LTE4
leukotriene E4
- LX
lipoxin
- MeOH
methanol
- PGB2
prostaglandin B2
- PGD1
prostaglandin D1
- PGD2
prostaglandin D2
- PGD3
prostaglandin D3
- PGE
prostaglandin E
- PGE1
prostaglandin E1
- PGE2
prostaglandin E2
- PGE3
prostaglandin E3
- PGF1α
prostaglandin F1α
- PGF2α
prostaglandin F2α
- PGF3α
prostaglandin F3α
- PGJ2
prostaglandin J2
- SRM
selected reaction monitoring
- TXB2
thromboxane B2
- TXB3
thromboxane B3
Footnotes
ORCID
Rhishikesh Thakare http://orcid.org/0000-0002-8219-6830
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- Aghazadeh-Habashi A, Asghar W, Jamali F. Simultaneous determination of selected eicosanoids by reversed-phase HPLC method using fluorescence detection and application to rat and human plasma, and rat heart and kidney samples. Journal of Pharmaceutical and Biomedical Analysis. 2015;110:12–19. doi: 10.1016/j.jpba.2015.02.041. [DOI] [PubMed] [Google Scholar]
- Aprikian O, Reynaud D, Pace-Asciak C, Leone P, Blancher F, Monnard I, Mace K. Neonatal dietary supplementation of arachidonic acid increases prostaglandin levels in adipose tissue but does not promote fat mass development in guinea pigs. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2007;293:R2006–R2012. doi: 10.1152/ajpregu.00382.2007. [DOI] [PubMed] [Google Scholar]
- Blewett AJ, Varma D, Gilles T, Libonati JR, Jansen SA. Development and validation of a high-performance liquid chromatography–electrospray mass spectrometry method for the simultaneous determination of 23 eicosanoids. Journal of Pharmaceutical and Biomedical Analysis. 2008;46:653–662. doi: 10.1016/j.jpba.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Carrier DJ, Bogri T, Cosentino GP, Guse I, Rakhit S, Singh K. HPLC studies on leukotriene A4 obtained from the hydrolysis of its methyl ester. Prostaglandins Leukotrienes and Essential Fatty Acids. 1988;34:27–30. doi: 10.1016/0952-3278(88)90021-x. [DOI] [PubMed] [Google Scholar]
- Chavis C, Fraissinet L, Chanez P, Thomas E, Bousquet J. A method for the measurement of plasma hydroxyeicosatetraenoic acid levels. Analytical Biochemistry. 1999;271:105–108. doi: 10.1006/abio.1999.4113. [DOI] [PubMed] [Google Scholar]
- Deems R, Buczynski MW, Bowers-Gentry R, Harkewicz R, Dennis EA. Detection and quantitation of eicosanoids via high performance liquid chromatography–electrospray ionization–mass spectrometry. In: Brown HA, editor. Methods in Enzymology. New York: Academic Press; 2007. pp. 59–82. [DOI] [PubMed] [Google Scholar]
- Dong LM, Shu XO, Gao YT, Milne G, Ji BT, Yang G, Abnet CC. Urinary prostaglandin E2 metabolite and gastric cancer risk in the shanghai women’s health study. Cancer Epidemiology Biomarkers and Prevention. 2009;18:3075–3078. doi: 10.1158/1055-9965.EPI-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelboom JW, Hirsh J, Weitz JI, Johnston M, Yi Q, Yusuf S. Aspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular events. Circulation. 2002;105:1650–1655. [Google Scholar]
- Ferreiro-Vera C, Mata-Granados JM, Priego-Capote F, Quesada- Gomez JM, Luque de Castro MD. Automated targeting analysis of eicosanoid inflammation biomarkers in human serum and in the exometabolome of stem cells by SPE-LC-MS/MS. Analytical and Bioanalytical Chemistry. 2011;399:1093–1103. doi: 10.1007/s00216-010-4400-6. [DOI] [PubMed] [Google Scholar]
- Fu J, Schoeman JC, Harms AC, van Wietmarschen HA, Vreeken RJ, Berger R, Hankemeier T. Metabolomics profiling of the free and total oxidised lipids in urine by LC-MS/MS: Application in patients with rheumatoid arthritis. Analytical and Bioanalytical Chemistry. 2016;408:6307–6319. doi: 10.1007/s00216-016-9742-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furugen A, Yamaguchi H, Mano N. Simultaneous quantification of leukotrienes and hydroxyeicosatetraenoic acids in cell culture medium using liquid chromatography/tandem mass spectrometry. Biomedical Chromatography. 2015;29:1084–1093. doi: 10.1002/bmc.3395. [DOI] [PubMed] [Google Scholar]
- Gachet MS, Rhyn P, Bosch OG, Quednow BB, Gertsch J. A quantitiative LC-MS/MS method for the measurement of arachidonic acid, prostanoids, endocannabinoids, N-acylethanolamines and steroids in human plasma. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2015;976–977:6–18. doi: 10.1016/j.jchromb.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Gainer JV, Bellamine A, Dawson EP, Womble KE, Grant SW, Wang Y, Capdevila JH. Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation. 2005;111:63–69. doi: 10.1161/01.CIR.0000151309.82473.59. [DOI] [PubMed] [Google Scholar]
- Gandhi AS, Budac D, Khayrullina T, Staal R, Chandrasena G. Quantitative analysis of lipids: A higher-throughput LC-MS/MS-based method and its comparison to ELISA. Future Science OA. 2017;3:FSO157. doi: 10.4155/fsoa-2016-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomolka B, Siegert E, Blossey K, Schunck WH, Rothe M, Weylandt KH. Analysis of omega-3 and omega-6 fatty acid- derived lipid metabolite formation in human and mouse blood samples. Prostaglandins and Other Lipid Mediators. 2011;94:81–87. doi: 10.1016/j.prostaglandins.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Gouveia-Figueira S, Nording ML. Development and validation of a sensitive UPLC-ESI-MS/MS method for the simultaneous quantification of 15 endocannabinoids and related compounds in milk and other biofluids. Analytical Chemistry. 2014;86:1186–1195. doi: 10.1021/ac403352e. [DOI] [PubMed] [Google Scholar]
- Gouveia-Figueira S, Nording ML. Validation of a tandem mass spectrometry method using combined extraction of 37 oxylipins and 14 endocannabinoid-related compounds including prostamides from biological matrices. Prostaglandins and Other Lipid Mediators. 2015;121(Pt A):110–121. doi: 10.1016/j.prostaglandins.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Hennessy E, Rakovac Tisdall A, Murphy N, Carroll A, O’Gorman D, Breen L, Sreenan S. Elevated 12-hydroxyeicosatetraenoic acid (12-HETE) levels in serum of individuals with newly diagnosed type 1 diabetes. Diabetes Medicine. 2017;34:292–294. doi: 10.1111/dme.13177. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Steinhilber D, Roth HJ. Determination of leukotriene B4 by high-performance liquid chromatography with electrochemical detection. Journal of Chromatography B: Biomedical Sciences and Applications. 1987;416:170–175. doi: 10.1016/0378-4347(87)80500-5. [DOI] [PubMed] [Google Scholar]
- Huwyler J, Gut J. Single-step organic extraction of leukotrienes and related compounds and their simultaneous analysis by high- performance liquid chromatography. Analytical Biochemistry. 1990;188:374–382. doi: 10.1016/0003-2697(90)90623-h. [DOI] [PubMed] [Google Scholar]
- Idborg H, Pawelzik SC, Perez-Manso M, Bjork L, Hamrin J, Herlenius E, Jakobsson PJ. Evaluation of urinary prostaglandin E2 metabolite as a biomarker in infants with fever due to viral infection. Prostaglandins, Leukottrienes and Essential Fatty Acids. 2014;91:269–275. doi: 10.1016/j.plefa.2014.09.006. [DOI] [PubMed] [Google Scholar]
- Jain S, Chakraborty G, Raja R, Kale S, Kundu GC. Prostaglandin E2 regulates tumor angiogenesis in prostate cancer. Cancer Research. 2008;68:7750–7759. doi: 10.1158/0008-5472.CAN-07-6689. [DOI] [PubMed] [Google Scholar]
- Jian W, Edom RW, Xue X, Huang MQ, Fourie A, Weng N. Quantitation of leukotriene B4 in human sputum as a biomarker using UPLC-MS/MS. Journal of Chromatography B: Analytical Technologies in the Biomedical and Life Sciences. 2013;932:59–65. doi: 10.1016/j.jchromb.2013.06.010. [DOI] [PubMed] [Google Scholar]
- Johnson JC, Schmidt CR, Shrubsole MJ, Billheimer DD, Joshi PR, Merchant NB. Urine PGE-M: A metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clinical Gastroenterology and Hepatology. 2006;4:1358–1365. doi: 10.1016/j.cgh.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Kempen EC, Yang P, Felix E, Madden T, Newman RA. Simultaneous quantification of arachidonic acid metabolites in cultured tumor cells using high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Analytical Biochemistry. 2001;297:183–190. doi: 10.1006/abio.2001.5325. [DOI] [PubMed] [Google Scholar]
- Kortz L, Dorow J, Becker S, Thiery J, Ceglarek U. Fast liquid chromatography–quadrupole linear ion trap–mass spectrometry analysis of polyunsaturated fatty acids and eicosanoids in human plasma. Journal of Chromatography B, Analytical Technologies in Biomedicine and Life Sciences. 2013;927:209–213. doi: 10.1016/j.jchromb.2013.03.012. [DOI] [PubMed] [Google Scholar]
- Kortz L, Dorow J, Ceglarek U. Liquid chromatography–tandem mass spectrometry for the analysis of eicosanoids and related lipids in human biological matrices: A review. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2014;964:1–11. doi: 10.1016/j.jchromb.2014.01.046. [DOI] [PubMed] [Google Scholar]
- Lee KC, DeLuca PP. Simultaneous determination of prostaglandins E1, A1 and B1 by reversed-phase high-performance liquid chromatography for the kinetic studies of prostaglandin E1 in solution. Journal of Chromatography. 1991;555:73–80. doi: 10.1016/s0021-9673(01)87168-5. [DOI] [PubMed] [Google Scholar]
- Lee JW, Mok HJ, Lee DY, Park SC, Ban MS, Choi J, Kim HD. UPLC-MS/MS-based profiling of eicosanoids in RAW264.7 cells treated with lipopolysaccharide. International Journal of Molecular Science. 2016;17:508. doi: 10.3390/ijms17040508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison BS, Zhang R, Wang Z, Fu X, Didonato JA, Hazen SL. Quantification of fatty acid oxidation products using online high-performance liquid chromatography tandem mass spectrometry. Free Radical Biology and Medicine. 2013;59:2–13. doi: 10.1016/j.freeradbiomed.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pestina TI, Berndt MC, Steward SA, Jackson CW, Gartner TK. The roles of ADP and TXA in botrocetin/VWF-induced aggregation of washed platelets. Journal of Thrombosis and Haemostasis. 2004;2:2213–2222. doi: 10.1111/j.1538-7836.2004.01023.x. [DOI] [PubMed] [Google Scholar]
- Long A, Zhong G, Li Q, Lin N, Zhan X, Lu S, Tan L. Detection of 19 types of para-arachidonic acids in five types of plasma/serum by ultra performance liquid chromatography–tandem mass spectrometry. International Journal of Clinical and Experimental Medicine. 2015;8:9248–9256. [PMC free article] [PubMed] [Google Scholar]
- Maddipati KR, Zhou SL. Stability and analysis of eicosanoids and docosanoids in tissue culture media. Prostaglandins and Other Lipid Mediators. 2011;94:59–72. doi: 10.1016/j.prostaglandins.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Masoodi M, Nicolaou A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Communications in Mass Spectrometry. 2006;20:3023–3029. doi: 10.1002/rcm.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey KA, Nicolaou A. Lipidomics of oxidized polyunsaturated fatty acids. Free Radical Biology and Medicine. 2013;59:45–55. doi: 10.1016/j.freeradbiomed.2012.08.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina S, Dominguez-Perles R, Gil JI, Ferreres F, Garcia-Viguera C, Martinez-Sanz JM, Gil-Izquierdo A. A ultra-pressure liquid chromatography/triple quadrupole tandem mass spectrometry method for the analysis of 13 eicosanoids in human urine and quantitative 24 hour values in healthy volunteers in a controlled constant diet. Rapid Communications in Mass Spectrometry. 2012;26:1249–1257. doi: 10.1002/rcm.6224. [DOI] [PubMed] [Google Scholar]
- Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ. Isoprostane generation and function. Chemical Reviews. 2011;111:5973–5996. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. Journal of Smooth Muscle Research. 2005;41:175–193. doi: 10.1540/jsmr.41.175. [DOI] [PubMed] [Google Scholar]
- Montuschi P, Martello S, Felli M, Mondino C, Chiarotti M. Ion trap liquid chromatography/tandem mass spectrometry analysis of leukotriene B4 in exhaled breath condensate. Rapid Communications in Mass Spectrometry. 2004;18:2723–2729. doi: 10.1002/rcm.1682. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, DiCamelli RF, Kohler S, Gumina RJ, Falck JR, Campbell WB, Gross GJ. Determination of cytochrome P450 metabolites of arachidonic acid in coronary venous plasma during ischemia and reperfusion in dogs. Analytical Biochemistry. 2001;292:115–124. doi: 10.1006/abio.2001.5044. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Tomaru K, Matsumoto N, Watanabe S, Higashi T. LC/ESI-MS/MS method for determination of salivary eicosapentaenoic acid concentration to arachidonic acid concentration ratio. Biomedical Chromatography. 2016;30:29–34. doi: 10.1002/bmc.3421. [DOI] [PubMed] [Google Scholar]
- Ong SLH, Zhang Y, Whitworth JA. Reactive oxygen species and glucocorticoid-induced hypertension. Clinical and Experimental Pharmacology and Physiology. 2008;35:477–482. doi: 10.1111/j.1440-1681.2008.04900.x. [DOI] [PubMed] [Google Scholar]
- Prasain JK, Arabshahi A, Taub PR, Sweeney S, Moore R, Sharer JD, Barnes S. Simultaneous quantification of F2-isoprostanes and prostaglandins in human urine by liquid chromatography tandem–mass spectrometry. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2013;913–914:161–168. doi: 10.1016/j.jchromb.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppolo M, Varma D, Jansen SA. A review of analytical methods for eicosanoids in brain tissue. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2014;964:50–64. doi: 10.1016/j.jchromb.2014.03.007. [DOI] [PubMed] [Google Scholar]
- Rivera J, Ward N, Hodgson J, Puddey IB, Falck JR, Croft KD. Measurement of 20-hydroxyeicosatetraenoic acid in human urine by gas chromatography–mass spectrometry. Clinical Chemistry. 2004;50:224–226. doi: 10.1373/clinchem.2003.025775. [DOI] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiological Reviews. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Schuchardt JP, Schmidt S, Kressel G, Dong H, Willenberg I, Hammock BD, Schebb NH. Comparison of free serum oxylipin concentrations in hyper- vs. normolipidemic men. Prostaglandins, Leukottrienes and Essential Fatty Acids. 2013;89:19–29. doi: 10.1016/j.plefa.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T. Lipid mediators in health and disease: Enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annual Review of Pharmacology and Toxicology. 2009;49:123–150. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- Shono F, Yokota K, Horie K, Yamamoto S, Yamashita K, Watanabe K, Miyazaki H. A heterologous enzyme immunoassay of prostaglandin E2 using a stable enzyme-labeled hapten mimic. Analytical Biochemistry. 1988;168:284–291. doi: 10.1016/0003-2697(88)90320-x. [DOI] [PubMed] [Google Scholar]
- Song J, Liu X, Wu J, Meehan MJ, Blevitt JM, Dorrestein PC, Milla ME. A highly efficient, high-throughput lipidomics platform for the quantitative detection of eicosanoids in human whole blood. Analytical Biochemistry. 2013;433:181–188. doi: 10.1016/j.ab.2012.10.022. [DOI] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): Metabolism and biochemical function. Progress in Lipid Research. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Squellerio I, Porro B, Songia P, Veglia F, Caruso D, Tremoli E, Cavalca V. Liquid chromatography–tandem mass spectrometry for simultaneous measurement of thromboxane B2 and 12(S)-hydroxyeicosatetraenoic acid in serum. Journal of Pharmaceutical and Biomedical Analysis. 2014;96:256–262. doi: 10.1016/j.jpba.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Stephenson AH, Lonigro AJ, Hyers TM, Webster RO, Fowler AA. Increased concentrations of leukotrienes in bronchoalveolar lavage fluid of patients with ARDS or at risk for ARDS. American Review of Respiratory Disease. 1988;138:714–719. doi: 10.1164/ajrccm/138.3.714. [DOI] [PubMed] [Google Scholar]
- Sterz K, Scherer G, Ecker J. A simple and robust UPLC-SRM/MS method to quantify urinary eicosanoids. Journal of Lipid Research. 2012;53:1026–1036. doi: 10.1194/jlr.D023739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassburg K, Huijbrechts AM, Kortekaas KA, Lindeman JH, Pedersen TL, Dane A, Vreeken RJ. Quantitative profiling of oxylipins through comprehensive LC-MS/MS analysis: Application in cardiac surgery. Analytical and Bioanalytical Chemistry. 2012;404:1413–1426. doi: 10.1007/s00216-012-6226-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terragno A, Rydzik R, Terragno NA. High performance liquid chromatography and UV detection for the separation and quantitation of prostaglandins. Prostaglandins. 1981;21:101–112. doi: 10.1016/0090-6980(81)90200-8. [DOI] [PubMed] [Google Scholar]
- Thakare R, Chhonker YS, Gautam N, Alamoudi JA, Alnouti Y. Quantitative analysis of endogenous compounds. Journal of Pharmaceutical and Biomedical Analysis. 2016;128:426–437. doi: 10.1016/j.jpba.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Tsikas D, Suchy MT, Tödter K, Heeren J, Scheja L. Utilizing immunoaffinity chromatography (IAC) cross-reactivity in GC–MS/MS exemplified at the measurement of prostaglandin E1 in human plasma using prostaglandin E2-specific IAC columns. Journal of Chromatography B. 2016;1021:101–107. doi: 10.1016/j.jchromb.2015.04.026. [DOI] [PubMed] [Google Scholar]
- Tsikas D, Zoerner AA. Analysis of eicosanoids by LC-MS/MS and GC-MS/MS: A historical retrospect and a discussion. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2014;964:79–88. doi: 10.1016/j.jchromb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- VanderNoot VA, VanRollins M. Capillary electrophoresis of cytochrome P-450 epoxygenase metabolites of arachidonic acid. 1. Resolution of regioisomers. Analytical Chemistry. 2002;74:5859–5865. doi: 10.1021/ac025909+. [DOI] [PubMed] [Google Scholar]
- Virtue S, Masoodi M, de Weijer BA, van Eijk M, Mok CY, Eiden M, Vidal-Puig A. Prostaglandin profiling reveals a role for haematopoietic prostaglandin D synthase in adipose tissue macrophage polarisation in mice and humans. International Journal of Obesity (London) 2015;39:1151–1160. doi: 10.1038/ijo.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Armando AM, Quehenberger O, Yan C, Dennis EA. Comprehensive ultra-performance liquid chromatographic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. Journal of Chromatography, A. 2014;1359:60–69. doi: 10.1016/j.chroma.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, DuBois RN. Methods in Enzymology. Vol. 433. Amsterdam, Netherlands: Elsevier; 2007. Measurement of eicosanoids in cancer tissues; pp. 27–50. [DOI] [PubMed] [Google Scholar]
- Watzer B, Reinalter S, Seyberth HW, Schweer H. Determination of free and glucuronide conjugated 20-hydroxyarachidonic acid. (20-HETE) in urine by gas chromatography/negative ion chemical ionization mass spectrometry. Prostaglandins, Leukottrienes and Essential Fatty Acids. 2000;62:175–181. doi: 10.1054/plef.2000.0138. [DOI] [PubMed] [Google Scholar]
- Weiss JW, Drazen JM, McFadden ER, Jr, Weller P, Corey EJ, Lewis RA, Austen KF. Airway constriction in normal humans produced by inhalation of leukotriene D. Potency, time course and effect of aspirin therapy. Journal of the American Medical Association. 1983;249:2814–2817. [PubMed] [Google Scholar]
- Willenberg I, Ostermann AI, Schebb NH. Targeted metabolomics of the arachidonic acid cascade: Current state and challenges of LC-MS analysis of oxylipins. Analytical and Bioanalytical Chemistry. 2015;407:2675–2683. doi: 10.1007/s00216-014-8369-4. [DOI] [PubMed] [Google Scholar]
- Yang R, Chiang N, Oh SF, Serhan CN. Current Protocols in Immunology. Hoboken, NJ: John Wiley & Sons; 2011. Metabolomics–lipidomics of eicosanoids and docosanoids generated by phagocytes. (Chapter 14, Unit 14 26) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Eiserich JP, Cross CE, Morrissey BM, Hammock BD. Metabolomic profiling of regulatory lipid mediators in sputum from adult cystic fibrosis patients. Free Radical Biology and Medicine. 2012;53:160–171. doi: 10.1016/j.freeradbiomed.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Analytical Chemistry. 2009;81:8085–8093. doi: 10.1021/ac901282n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Kodai S, Takemura S, Minamiyama Y, Niki E. Simultaneous measurement of F2-isoprostane, hydroxyoctadecadienoic acid, hydroxyeicosatetraenoic acid, and hydroxycholesterols from physiological samples. Analytical Biochemistry. 2008;379:105–115. doi: 10.1016/j.ab.2008.04.028. [DOI] [PubMed] [Google Scholar]
- Yue H, Jansen SA, Strauss KI, Borenstein MR, Barbe MF, Rossi LJ, Murphy E. A liquid chromatography/mass spectrometric method for simultaneous analysis of arachidonic acid and its endogenous eicosanoid metabolites prostaglandins, dihydroxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and epoxyeicosatrienoic acids in rat brain tissue. Journal of Pharmaceutical and Biomedical Analysis. 2007;43:1122–1134. doi: 10.1016/j.jpba.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Strauss KI, Borenstein MR, Barbe MF, Rossi LJ, Jansen SA. Determination of bioactive eicosanoids in brain tissue by a sensitive reversed-phase liquid chromatographic method with fluorescence detection. Journal of Chromatography B: Analytical Technology in Biomedicine and Life Sciences. 2004;803:267–277. doi: 10.1016/j.jchromb.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yang N, Ai D, Zhu Y. Systematic metabolomic analysis of eicosanoids after omega-3 polyunsaturated fatty acid supplementation by a highly specific liquid chromatography–tandem mass spectrometry-based method. Journal of Proteome Research. 2015;14:1843–1853. doi: 10.1021/pr501200u. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang G, Clarke PA, Huang JTJ, Takahashi E, Muirhead D, Lin Z. Simultaneous and high-throughput quantitation of urinary tetranor PGDM and tetranor PGEM by online SPE-LC-MS/MS as inflammatory biomarkers. Journal of Mass Spectrometry. 2011;46:705–711. doi: 10.1002/jms.1941. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.