

Here, Newman et al. provide insight into the role of mRNA splicing in C. elegans RNAi and show that viable null mutations in U1 and U2 snRNP-specific splicing factor genes cause defects in RNAi. They also demonstrate that RNAi targeted transcripts are tightly bound to spliceosomes, which suggests multiple layers of regulation by the spliceosome at early steps of small RNA-mediated gene silencing.
Keywords: RNAi, mRNA splicing, small RNA biogenesis, spliceosome
Abstract
RNAi pathways detect and silence foreign nucleic acids such as viruses as well as endogenous genes in many species. The phylogenetic profile across eukaryotes of proteins that mediate key steps in RNAi is correlated with the profiles of multiple mRNA splicing proteins and with intron number, suggesting that RNAi may surveil mRNA splicing to detect the divergent or absent introns of viruses. Here we examine the role of mRNA splicing in Caenorhabditis elegans RNAi. We found that viable null mutations in U1 and U2 small nuclear ribonucleic protein (snRNP)-specific splicing factor genes cause defects in RNAi. The U1A ortholog rnp-2 is required for normal ERGO-1 Argonaute class 26G siRNA biogenesis, trans-splicing of the eri-6/7 transcript, and targeting of poorly conserved gene transcripts by WAGO Argonaute class 22G siRNAs. We found that gene transcripts engaged by the siRNA-generating machinery are poorly conserved, possess few introns, and often have introns that are divergent from introns with strong consensus splicing sites found in highly conserved genes. We present biochemical evidence that RNAi targeted transcripts are tightly bound to spliceosomes. These findings suggest multiple layers of regulation by the spliceosome at early steps of small RNA-mediated gene silencing.
RNAi was initially discovered via the surprising potency of introduced dsRNA to silence homologous genes, particularly in nematodes, plants, and fungi. The potent response to introduced dsRNA is a dividend from a rich world of endogenous siRNAs (endo-siRNAs) generated from genomic transcripts by a complex small RNA-presenting protein machinery. These endo-siRNAs mediate a wide range of gene regulatory processes, including the surveillance and silencing of foreign nucleic acids such as viruses.
Genetic screens for defects in RNAi revealed that particular suites of eukaryotic proteins, especially the Argonaute class of RNA-binding proteins, present 21- to 26-nucleotide (nt) small RNAs processed from dsRNA either introduced via viral or experimental interventions or from endogenous genomic locations to then cleave a complementary mRNA or initiate more complex transcriptional silencing events at the homologous genetic locus. Argonaute proteins are guided to particular target mRNAs by the antisense complementarity of particular siRNAs or microRNAs (miRNAs), which weave into their PIWI and PAZ domains (Song et al. 2004). Animal and plant genomes encode multiple Argonaute proteins that mediate specialized small RNA functions (for example, miRNA vs. siRNA), whereas many fungal species encode only one Argonaute protein. Strikingly, a number of fungal and protist species—notably, Saccharomyces cerevisiae—have lost their Argonaute orthologs in evolution (Drinnenberg et al. 2011; Tabach et al. 2013). Correlating with the loss of Argonaute homologs in those fungi and protists is a massive diminution in intron number (from tens of thousands of introns in many fungal species that encode an Argonaute to, for example, ∼100 remaining introns, mostly in ribosomal genes, in S. cerevisiae) (Tabach et al. 2013).
RNAi mechanisms silence viral infections in many species; for example, the PIWI-interacting RNAs (piRNAs) of Drosophila silence retrotransposons that are the remnants of past viral infections (Malone and Hannon 2009), and RNAi pathways of Caenorhabditis elegans mediate resistance to viruses (Lu et al. 2005; Felix et al. 2011). C. elegans has additional specialized Argonaute proteins that specifically detect recently acquired and duplicated genes that are likely to be the result of horizontal gene transfer from viral infection (Fischer et al. 2011). Consistent with the correlation between loss of RNAi pathways and loss of introns in some fungi and protists, recently acquired foreign genes in many organisms, including C. elegans, that arrive most likely by viral infection have fewer introns (Fischer et al. 2011), and those introns are less likely to be optimized for a particular species, in contrast to introns in germline-transmitted genes inherited and optimized over millions to billions of years. Also consistent with the model that RNA splicing is surveilled by RNAi pathways, the phylogenetic pattern of conservation of a number of protein-coding genes that mediate RNA splicing mirrors the phylogenetic profile of Argonaute proteins, and some of these splicing genes have been shown to function in RNAi (Kim et al. 2005; Tabach et al. 2013). The coincident pattern of loss of splicing genes and RNA silencing genes also suggests that they act in a common pathway (Kim et al. 2005; Tabach et al. 2013). In addition, C. elegans piRNAs (21U RNAs) mediate the sorting of poorly conserved gene transcripts to endogenous RNAi (endo-RNAi) pathways (de Albuquerque et al. 2015; Phillips et al. 2015). However, because the piRNA-specific Argonaute (PRG-1) resides in perinuclear P granules (Billi et al. 2014), it is possible that regulatory processes between the points of transcription and splicing and nuclear export play roles in the sorting of foreign-looking RNAs into RNA silencing pathways. Indeed, single-stranded piRNA transcripts are distinguished from normal essential transcripts in the germline of Drosophila melanogaster by active repression of transcript splicing (Zhang et al. 2012, 2014). In support of the model that RNAi pathways surveil poor introns for foreign signatures, an abundant class of endo-siRNAs is generated from poorly spliced transposons in the Argonaute-bearing fungus Cryptococcus neoformans (Dumesic et al. 2013). Transposons spread between genetically isolated species, and many of them have viral features (Kidwell and Lisch 1997).
Here, we assessed the roles of pre-mRNA splicing in RNAi-mediated surveillance. We found that the disruption of particular splicing factors causes a marked depletion of siRNAs that target poorly conserved genes harboring few introns (i.e., ERGO-1-specific 26G siRNAs) (Fischer et al. 2011). These poorly conserved genes are likely to have entered C. elegans via viral infection of ancestral nematode genomes. siRNAs from these foreign genes are generated from both spliced and unspliced transcripts, indicating that the siRNA-generating machinery of C. elegans encounters unspliced transcripts. We present biochemical evidence that siRNA targeted foreign gene transcripts are selectively stalled on spliceosomes.
Results
Viable mutations in the core splicing gene rnp-2 disrupts endo-RNAi pathways
Inactivation of C. elegans mRNA-splicing genes causes defects in RNAi, but the molecular mechanism of this coupling has not been established (Kim et al. 2005). Null mutations in many of the genes that mediate steps in mRNA splicing are lethal, but there are several viable mRNA-splicing mutants that we could test in phenotypic assays for RNAi-defective phenotypes (Fig. 1A). Three such viable splicing gene-null mutants are rnp-2/U1A, rnp-3/U2B′′, and mog-2/U2A′. rnp-2(mg582) is a hypomorphic mutant generated by CRISPR to create an in-frame deletion of two amino acid residues that are critical for binding to the U1 snRNA (Supplemental Fig. S1; Law et al. 2005). rnp-3(ok1424), mog-2(ok1221), and a second rnp-2 mutant used in this study (tm4058) contain large deletions within the coding regions of each respective gene and are presumed to be null mutations. rnp-2 and rnp-3, two genes encoding RNA recognition motif (RRM) domain proteins, were shown previously to act redundantly despite normally residing within the U1 and U2 small nuclear ribonucleic protein (snRNP) complexes, respectively (Saldi et al. 2007). RNP-2 and RNP-3 are closely paralogous (49% identity over ∼200 amino acids) but are assembled by unknown mechanisms into distinct U1 and U2 snRNPs. To gain insight into rnp-2- and rnp-3-specific functions (and query whether these genes genetically interact with other splicing genes implicated previously in RNAi pathways), we screened a gene inactivation sublibrary encoding spliceosome components (representing 124 genes) for synthetic lethality and/or sterility. Many splicing component gene inactivations are lethal in wild type; therefore, their synthetic lethality with the viable spliceosome mutants could not be tested. However, 24 gene inactivations showed weak or no phenotypes on wild type but strong synthetic-lethal phenotypes in either the rnp-2(mg582) or rnp-3(ok1424) mutant backgrounds; for example, rnp-2(mg582) was synthetic-lethal with inactivation of tcer-1/TCERG1 transcriptional elongation as well as splicing factor and rnp-5/RNPS1 exon-junction complex protein (Supplemental Table S1), two splicing factors shown previously to enhance transgene silencing, a hallmark of the enhanced RNAi (“eri”) phenotype (Fischer et al. 2013). Some of the genetic loci that can mutate to an Eri phenotype, including the ergo-1 Argonaute and the eri-6/7 helicase, disable the endo-RNAi-based silencing of genes that are present in multiple duplicated copies in C. elegans but are poorly conserved even with closely related nematodes; the genes targeted by these eri pathways are recently acquired, probably viral, genes (Fischer et al. 2011). eri mutant animals that no longer silence these recently acquired genes are now enabled to silence transgenes or, with dsRNA, triggers of RNAi more robustly than wild type, suggesting that much of the bandwidth of C. elegans RNAi is saturated by the silencing of these recently acquired genes. A major mystery is how these foreign genetic elements are detected to be targeted by RNAi for silencing.
Figure 1.

rnp-2 mutants have defects in ERGO-1-dependent 26G siRNA production. (A, top) Schematic of the splicing factor genes examined in this study and their roles in the U1 and U2 snRNPs. (Bottom) Domains of RNP-2, RNP-3, and MOG-2. (B) Desilencing of the siR-1 mCherry reporter transgene (Montgomery et al. 2012) in splicing mutant strains; eri-6(mg379) is presented as a positive control. (C) Small RNA profiles of embryos from viable splicing mutants. Pie charts represent the distribution of genome features to which small RNAs align. The apparent increase in the miRNA fraction in rnp-2(mg582) is due to the decrease in coding genes, pseudogenes, transposons, and intergenic endo-siRNAs. (D) Bar plots representing length and first nucleotide distributions. (Bottom panel) Small (26-nt) RNA populations are plotted alone for better visualization. (E) Genome browser image of E01G4.5 antisense siRNAs, a gene strongly depleted for 26G siRNAs in rnp-2(mg582).
To investigate whether any of the viable splicing mutations affect endo-RNAi pathways, we monitored the response of a ubl-1::mCherry-siR-1-sensor transgene (corresponding to the abundant endogenous 22G siRNA siR-1) (Montgomery et al. 2012) to these mutants. siR-1 is derived from a cluster of 22G siRNAs on the X chromosome that are dependent on ERGO-1 Argonaute class 26G siRNA pathway components for their formation (Montgomery et al. 2012). The ubl-1::GFP-siR-1-sensor contains a single target site for siR-1 embedded in the 3′ untranslated region (UTR) of ubiquitin-like1 (ubl-1) and expressed under the control of the ubiquitous ubl-1 promoter and is single-copy Mos1 inserted into the C. elegans genome so that repetitive transgene silencing does not complicate the analysis (Montgomery et al. 2012). The silencing of this siRNA sensor requires the activity of the ERGO-1 Argonaute, the ERI-6/7 helicase, and other factors essential for ERGO-1 class 26G as well as factors required for 22G siRNA formation and activity, such as mut-16 (Montgomery et al. 2012). Expression of mCherry is therefore observed when the production of siR-1 or the response to that siRNA is abrogated by mutations in the Eri pathway or the Mutator pathway or particular pre-mRNA processing factors that affect siR-1 production or response. A mutation in rnp-2, but not mog-2 or rnp-3, desilenced the siR-1 reporter transgene (Fig. 1B). Thus, rnp-2 gene activity is necessary for the production of or response to the siR-1 siRNA. mog-2 and rnp-3 (and the U2 snRNP in general) do not act in this siR-1 pathway but may function in other RNAi pathways.
endo-siRNA defects in viable splicing mutants revealed by high-throughput sequencing
To observe endo-siRNA phenotypes more generally, we profiled small RNAs using high-throughput sequencing of rnp-2, rnp-3, and mog-2 mutant embryos. We targeted embryos for two reasons: First, 26G siRNAs that trigger many secondary 22G siRNAs are generated early in development as the developing embryo is surveilled for viruses (Fischer et al. 2011; Billi et al. 2014). Second, embryo preparations are easier to compare between strains, since there are no growth rate synchronization issues that confound larval or adult stage comparisons. Aligning the small RNA reads to the genome and classifying each read against genomic features, we observed a substantial reduction in endo-siRNA reads corresponding to coding genes, transposons, pseudogenes, and intergenic space in the rnp-2(mg582) embryo library versus wild-type embryos; mog-2(ok1221) and rnp-3(ok1424) showed smaller changes (Fig. 1C). Examining size and first nucleotide distribution revealed that the 26G siRNAs were depleted dramatically (∼65%) in rnp-2(mg582) and to a lesser extent in the other two splicing mutants. This class of siRNAs was the most depleted species of endo-siRNA (Fig. 1D). 26G siRNAs in embryos bind to the ERGO-1 Argonaute protein and serve as primers for the generation by RNA-dependent RNA polymerases of the more abundant 22G siRNAs. Many of the 26G trigger siRNAs are generated from duplicated recently acquired C. elegans genes (Fischer et al. 2011). The eri-6/7 helicase gene is also required for the production during embryogenesis of 26G siRNAs (Fischer et al. 2011). Not all ERGO-1 target genes were depleted of 26G siRNAs in the splicing mutants that we tested; for example, the ERGO-1 target gene E01G4.5 was largely depleted of 26G siRNAs in the rnp-2 mutant, whereas another ERGO-1 target, T02G6.4, was unaffected (Fig. 1E; Supplemental Fig. S2).
We hypothesized two possible mechanisms by which a general mRNA splicing defect might cause a depletion of 26G siRNAs (or a combination of these mechanisms): (1) a defect in the mRNA splicing of a key factor in the generation of 26G siRNAs or (2) an accumulation of many poorly spliced mRNAs in a splicing mutant that depletes key factors for silencing, such as the ERGO-1 Argonaute or the ERI-6/7 helicase, to challenge the bandwidth of endo-siRNA pathways to generate 26G siRNAs. The peculiar trans-splicing of the key 26G siRNA biogenesis gene eri-6/7 was an excellent candidate for the first mechanism. eri-6/7 is assembled from divergently transcribed eri-6 and eri-7 primary transcripts that are trans-spliced into a single mRNA; this trans-splicing is sensitive to inactivation of the rnp-5/RNPS1 mRNA exon junction complex splicing cofactor gene (Fischer et al. 2008). Quantitative RT–PCR (qRT–PCR) analysis using primers specific for eri-6/7 pre-mRNA trans-splicing revealed that trans-spliced eri-6/7 was depleted fivefold to 10-fold in two different viable rnp-2 mutants (Fig. 2A). Consistent with the model that trans-splicing of eri-6 to eri-7 is hypersensitive to (and perhaps an evolved sensor of) splicing efficiency, RNAi inactivation of the tcer-1/TCERG1-splicing factor causes transgene silencing, like a mutation in eri-6/7 (Fischer et al. 2013). Also in support of this model, inactivation of the rnp-5-splicing factor gene disrupts eri-6/7 trans-splicing (Fischer et al. 2008). Our genetic epistasis analysis also supports this model: The Eri phenotype caused by inactivation of the splicing factor tcer-1 (Fischer et al. 2013) is strongly suppressed by expression of a prespliced eri-6/7 cDNA expression construct that is no longer trans-spliced (Supplemental Table S2). Thus, depletion of ERGO-1-specific 26G siRNAs in the rnp-2-splicing mutant can be explained by the defect in the trans-splicing of eri-6/7.
Figure 2.

rnp-2 mutations disrupt eri-6/7 trans-splicing and cause an Eri phenotype. (A) qRT–PCR for trans-splicing of the enhanced RNAi pathway gene eri-6//7 in wild type compared with two different rnp-2 mutants. (B) Eri phenotypes of splicing mutants; eri-6(mg379) was used as a positive control for a strong Eri phenotype.
Consistent with the disruption of eri-6/7 trans-splicing, the rnp-2 mutants have an Eri phenotype (Fig. 2B). The Eri phenotype of the eri-6/7 or ergo-1 mutants suggests that the silencing of poorly conserved target genes saturates the capacity of RNAi to silence using exogenous triggers such as feeding dsRNA so that when eri-6/7 or ergo-1 silencing is disabled, exogenous RNAi is enhanced (Fischer et al. 2011). One simple test for Eri uses dsRNA targeting the lir-1 gene, which has no phenotype in wild type, but, in many eri mutants, the siRNAs spread to then silence the second gene in this operon, lin-26, causing a lethal arrest (Pavelec et al. 2009). The rnp-2 mutant shows a potent lethal phenotype on lir-1 RNAi, like eri-1 and eri-6/7 mutants, suggesting that the rnp-2 mutants are Eri in this context of nuclear RNAi (Fig. 2B). In another test for Eri, feeding RNAi using dsRNAs from the genes hmr-1 or dpy-13 shows a more dramatic phenotype on strains carrying eri mutations, such as ergo-1 and eri-6/7 (Fischer et al. 2011). dpy-13 encodes one member of a highly duplicated collagen family; in eri mutants, the dpy-13 dsRNA trigger may promiscuously silence additional members of the collagen family, explaining the more lethal dpy-13 RNAi phenotype in an eri mutant compared with the viable but dumpy dpy-13-null allele phenotype. Feeding RNAi using hmr-1 or dpy-13 dsRNAs showed that the rnp-2 mutant is also enhanced for RNAi using these tester gene inactivations (Fig. 2B).
Because an mRNA splicing mutation is defective for the ERGO-1 pathway of duplicated gene silencing, we also surveyed the splicing mutants for other RNAi defects. Using feeding RNAi against GFP, the splicing mutant animals were competent to silence GFP in the first generation of feeding RNAi: Animals expressing a bright nuclear GFP transgene (sur-5-gfp) showed potent GFP silencing upon exposure to gfp RNAi in the parental generation. RNAi gene inactivation in wild-type C. elegans will typically persist into the first generation of progeny even if the feeding of Escherichia coli bearing gfp dsRNA is discontinued after the parental generation (a chase with feeding of E. coli OP50 that does not express any GFP dsRNA) (F1, Fig. 3A; Timmons et al. 2001). We also tested the splicing mutants for intergenerational inheritance of siRNAs, a phenotype disrupted by the nrde mutants, which are unable to propagate exogenous RNAi (exo-RNAi) silencing from the parental generation to the progeny F1 generation (Burton et al. 2011). The Nrde pathway resides downstream from siRNA generation by Mutator foci and includes an Argonaute (NRDE-3 in the soma; HRDE-1 in the germline), the highly conserved nuclear protein NRDE-2, and the nematode-specific proteins NRDE-1 and NRDE-4. In the rnp-3 and mog-2 mutants and a nrde-2 control mutant, RNAi of GFP failed to transmit to the F1 generation (Fig. 3B,C). rnp-2 mutant F1s still had potently silenced GFP. Thus, the splicing functions of rnp-3 and mog-2 are necessary for the production of or response to inherited siRNAs mediated by the nrde pathway.
Figure 3.

U2 snRNP-specific splicing genes exhibit a Nrde phenotype in a GFP transgene silencing assay. (A) Strategy for examining the parental (P0)-to-F1 transmission of RNAi phenotypes of wild type versus mutants. (B) Representative fluorescence microscopy images for GFP signal in the parental (P0)-to-F1 RNAi transmission test; the nrde-2(gg091) mutant was used as a positive control (Burton et al. 2011). (C) Quantitation of SUR-5-GFP transgene desilencing in the F1 generation of parents that were grown on E. coli expressing double-stranded gfp RNA.
In addition to the 26G siRNAs, 22G endo-siRNAs from coding genes are substantially changed in rnp-2 mutant embryos. 22G siRNAs are classified based on the Argonaute proteins to which they bind; the WAGO clade of C. elegans Argonautes binds to 22G siRNAs and, in turn, silences target genes bearing sequence complementarity; WAGO 22G siRNAs require the Mutator genes, including mut-16, and are synthesized in “Mutator foci” adjacent to P granules in the germline. The Argonaute CSR-1 complexes with 22G siRNAs and promotes proper chromosome segregation during mitosis in the germline (Billi et al. 2014). More than 200 gene loci were more than fivefold depleted of siRNA in rnp-2 mutants (vs. wild type); 119 of these depleted loci were characterized previously as WAGO targets, and five are CSR-1 targets (Zhang et al. 2011). Conversely, 70 genes exhibited greater than fivefold increased siRNA counts; three genes with increased siRNAs in rnp-2 mutants were WAGO targets, and none were CSR-1 targets (Fig. 4; Supplemental Table S3). Furthermore, 20 pseudogenes were more than fivefold depleted of siRNAs, four of which were identified previously as ERI-6/7 target pseudogenes (Supplemental Table S4). Genes that were strongly depleted for endo-siRNAs in rnp-2 mutants include K07A1.3, a poorly conserved WAGO Argonaute target gene (Fig. 4). The genes with increased siRNAs in rnp-2 mutants are well conserved and often possess more clearly defined functional annotations relative to WAGO target genes, suggesting that the WAGO pathway may be mistargeting these genes when splicing is aberrant (Fig. 4; Supplemental Table S3). Genes that were strongly enriched for endo-siRNA reads in rnp-2 mutants include Y51A2D.8, which encodes an ortholog of human cathepsin O (CTSO); it is strongly conserved in Caenorhabditis nematodes and moderately conserved in other animals (Fig. 4). Genes with the highest siRNA counts (reads per million [RPM]) were the most strongly depleted siRNAs in a mut-16 mutant (Phillips et al. 2012), but the 250 most abundant siRNA-generating coding gene loci in wild type are underrepresented in rnp-2-affected genes (Supplemental Fig. S3). Thus, disruptions in rnp-2 cause defects in both the ERI-6/7 pathway and siRNA formation across diverse WAGO pathway target genes.
Figure 4.
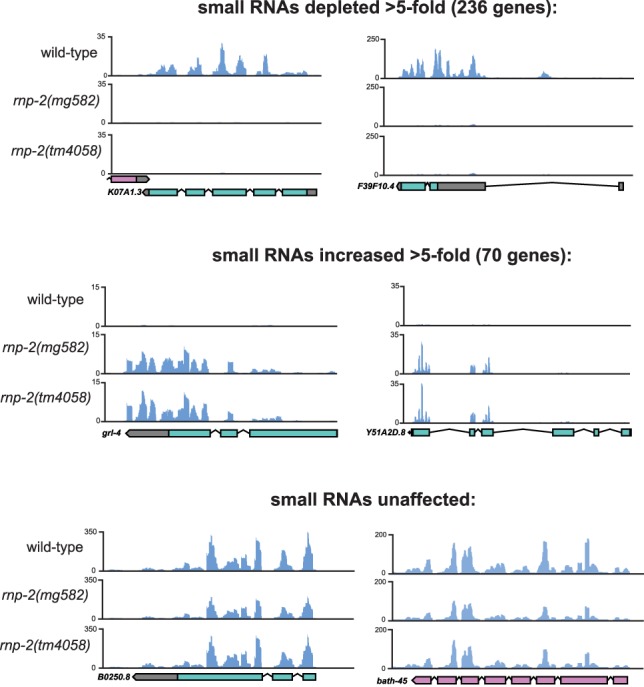
Genome browser images of genes bearing siRNAs that are depleted (top), enriched (middle), or unaffected (bottom) in two different rnp-2 mutants compared with wild type. The Y-axis in each graph indicates reads per million.
siRNAs at spliced and unspliced junctions
Because invading foreign nucleic acids such as viruses are likely to carry splicing signals that can function in many hosts but are not as highly selected within that host lineage to splice with the RNA-splicing machinery of that species, we analyzed whether there is a relationship between splicing signals, gene conservation, and the distribution of endo-siRNAs across intron–exon and exon–exon splice boundaries in the genome. We devised a splice site score based on the most commonly occurring bases at or proximal to the splice donor (“GU”) and splice acceptor (“AG”) sequences of C. elegans introns (Fig. 5A). By this metric, the average splice site score [Σ − log(Prob)] for introns across the C. elegans genome is ∼6.2. A higher score indicates an intron with a splice donor and/or acceptor that is more divergent from those of introns that are usually harbored in well-conserved ancient genes. Next, we assessed the abundance of endo-siRNAs that span either spliced exon–exon junctions (EEJ siRNAs) or unspliced intron–exon junctions (IEJ siRNAs). A majority of C. elegans endo-siRNAs that target coding genes maps to exons rather than introns (Gent et al. 2010). However, the extent to which siRNAs map to unspliced transcripts has not been carefully examined. We examined siRNA reads directly spanning either 5′ exon–intron junctions or 3′ IEJs (IEJ siRNAs). We chose IEJ siRNAs as a representation of siRNAs that generally lie in intronic space; measuring the abundance of siRNAs that lie completely within intron space is complicated by the large number of repetitive elements, embedded coding genes, miRNAs, and other genomic features harbored within introns. Thus, EEJ siRNAs represent siRNAs produced from mRNAs after splicing, whereas IEJ siRNAs represent siRNAs most likely produced by mRNAs before they are spliced in the nucleus. A high IEJ/EEJ siRNA ratio could be an indication of poorly spliced mRNAs that are detected by the RNAi machinery for silencing. We removed all IEJ siRNAs from our analysis that did not overlap with an experimentally validated intron (derived from total RNA sequencing [RNA-seq] of wild-type adult C. elegans) (see Supplemental Table S5). The abundance of IEJ siRNAs varied widely among endo-RNAi targets; certain genes harbor particularly abundant IEJs, suggesting that the splicing signals at those corresponding introns might be particularly poor. Candidate IEJ siRNAs can be bona fide or could represent differentially spliced mRNAs, where the intron in one cell type or physiological condition is an exon in another cell type or physiological condition. We therefore curated all IEJ and EEJ siRNAs after removing introns that overlap alternatively spliced exons using total RNA-seq (which removed 3% IEJ reads) (Supplemental Table S5). An example of a gene with abundant IEJ siRNAs is the poorly conserved gene F52G3.3 (Fig. 5B), which has both a small number of EEJ siRNAs and abundant IEJ siRNAs spanning the splice acceptor site in the second intron of the gene (Fig. 5B). This intron also has a poor intron quality score of 11.2 due to the divergence from consensus sequences at both the splice donor and acceptor sites (Hollins et al. 2005).
Figure 5.

siRNA densities across spliced and unspliced junctions as a function of splice site quality, intron number, and gene conservation. (A) Logo diagram depicting the most frequently occurring bases at and proximal to the splice donor and acceptor sequences averaged across all C. elegans introns. Orange bars below the logo diagram represent the bases that were used to calculate divergence from both splice donor and acceptor sequences. (B) F52G3.3 is an example of a poorly conserved gene for which the second (highly divergent) intron generates abundant IEJ siRNAs. (C) Genome-wide analysis of splice site scores of all introns (Y-axis) versus the ratio of IEJ to EEJ siRNAs. Note that poor splice site quality scores (a high number) track with more siRNAs generated across splice sites (IEJ siRNAs). (D) The box/whisker plot is similar to that in C, but the Y-axis is the conservation of all genes between C. elegans and Caenorhabditis briggsae calculated using BLAST scores. Note that poorly conserved genes generate more siRNAs across splice sites (IEJ siRNAs). (E) IEJ/EEJ versus intron number genome-wide. (F) Small RNAs that map to the vicinity of the Tc1 transposon intron are displayed; the Tc1 gene structure is depicted above the small RNA reads.
Examining the relationship of the IEJ/EEJ ratio across the whole genome as a function of splice site score revealed that highly divergent introns are more likely to have abundant IEJ siRNAs (relative to EEJ siRNAs) (Fig. 5C). Because we hypothesized that genes with poor intron scores might have been acquired by viral infection, we also plotted genome-wide IEJ/EEJ ratios as a function of gene conservation between C. elegans and the closely related species Caenorhabditis briggsae. Most C. elegans protein-coding genes have strong C. briggsae orthologs; these are very closely related species that can mate and cross-fertilize, but the hybrids are not viable. However, ∼10% of C. elegans genes are not found in C. briggsae (and vice versa) (Gupta and Sternberg 2003); these genes may have been acquired specifically in the C. elegans lineage (for example, by viral infection) or deleted in the C. briggsae lineage. Because viral genomes select for compact genes with few introns, we also assessed intron count per gene in our analysis. We observed that the ∼10% of C. elegans genes that are poorly conserved in C. briggsae have fewer introns and are more likely to have introns that generate relatively abundant IEJ siRNAs (Fig. 5D,E, respectively). This is reasonable if genes poorly conserved between C. elegans and closely related Caenorhabditis species have been acquired in C. elegans (for example, as integrated viruses) and if these horizontally transferred viruses have splicing signals that are nonoptimal for C. elegans. A full assessment of IEJ and EEJ siRNAs for all coding genes in wild-type and splicing mutant strains, including splice site scores, conservation score versus C. briggsae, and intron number per gene, is presented in Supplemental Table S6. We observed few differences in IEJ/EEJ siRNA ratios for individual genes between wild type and the splicing mutants. However, 26G siRNA reads corresponding to the EEJ were depleted in the rnp-2 mutants (Supplemental Table S7), suggesting that successful splicing of endo-RNAi transcripts might be critical for ERGO-1 26G siRNA formation.
In addition to poorly conserved coding genes, we also analyzed the Mariner/Tc1 family transposons of the C. elegans genome. Despite being robustly silenced by endo-RNAi, the multiple copies of Tc1 and Tc3 found throughout the genome are almost invariant at the nucleotide sequence level. Tc1 and Tc3 transposons have independently acquired a single small intron in or near their transposase coding sequence (Robertson 1995). Strikingly, while the Tc1 intron is rarely spliced (Sijen and Plasterk 2003), we detected only EEJ siRNAs at the Tc1 and Tc3 introns along the intron boundaries (Fig. 5F; data not shown), suggesting that the siRNA-generating machinery encounters only spliced Tc1 and Tc3 transcripts. This is consistent with the model that inefficient splicing is required for routing of transposon transcripts into the endo-RNAi pathway (Dumesic et al. 2013; Lee et al. 2013). Thus, the Tc1 and Tc3 transposons are routed to the siRNA-generating machinery as spliced substrates, while RNAi target-coding genes may not require splicing of their introns to be licensed as templates for siRNA formation. Finally, examining the small RNA size and first nucleotide distribution for both IEJ and EEJ siRNAs, there is a noticeable enrichment of reads representing 21U RNAs in the IEJ siRNA cohort, suggesting that 21U piRNAs may originate in the intronic space (which tends to be relatively A/T-rich) (Supplemental Fig. S4).
endo-RNAi target gene transcripts are selectively retained on the spliceosome
The yeast C. neoformans produces endo-siRNAs from transposon mRNAs that are selectively retained on spliceosomes due to suboptimal splicing signals, with more surveillance at the 3′ end of the intron (Dumesic et al. 2013). The observation that recently acquired C. elegans genes such as F52G3.3 harbor IEJ siRNAs and have suboptimal splicing signals prompted us to investigate whether a similar splicing surveillance occurs in metazoans. We carried out RNA immunoprecipitations from native C. elegans extracts using a tagged RNP-3/U2B′′ fusion protein in conjunction with qRT–PCR of both RNAi target and nontarget genes; we determined whether either cohort of genes was enriched in the RNP-3 splicing protein immunoprecipitations. We constructed a single-copy transgene encoding a C-terminal 3xFlag-tagged RNP-3 (Fig. 6A); we chose to use RNP-3 in this experiment because it was shown previously to be tightly associated with both precatalytically and catalytically active spliceosome complexes in human cells (while RNP-2/U1A was not) (Bessonov et al. 2008). The Flag-tagged RNP-3 transgene is functional; it rescued the slow growth/low brood size phenotypes of the rnp-3(ok1424)-null mutant as well as the sterility phenotype of rnp-3(ok1424) animals treated with rnp-2 RNAi (Supplemental Fig. S5). We queried whether RNP-3 immunoprecipitations could capture intact spliceosomes by monitoring the abundance of coimmunoprecipitated small nuclear RNAs (snRNAs) by qRT–PCR. While RNP-3::3xFlag predominantly retrieved the U2 snRNA (∼2000× enrichment), other snRNAs, including U4, U5, and U6, were enriched ∼30-fold to 130-fold over control immunoprecipitations (Fig. 6B), showing that RNP-3 immunoprecipitations capture precatalytic intact spliceosomes (Will and Luhrmann 2011). Other abundant nonspliceosomal RNAs (5S and 5.8S rRNA) were not enriched in RNP-3 immunoprecipitations (Fig. 4B). We monitored transcript abundance of abundant genes and select genes targeted by RNAi or Eri pathways, including transposons, eri-6/7, NRDE-3 targets, and WAGO targets (i.e., genes with corresponding siRNAs enriched in WAGO-1 immunoprecipitations) (Gu et al. 2009). We used droplet digital PCR (ddPCR) to detect RNAi target gene transcripts that are generally expressed at very low levels. In counting the transcript amplicon concentrations from two independent RNA immunoprecipitations, transcripts corresponding to highly expressed genes, including act-1/actin and glp-1/Notch, were not enriched or were relatively depleted in the RNP-3::3xFlag immunoprecipitation compared with total RNA derived from input lysate (suggesting that they are not tightly associated with the spliceosome) (Fig. 4C). Transcripts of WAGO target genes, including the poorly conserved ERI-6/7 target gene K02E2.11, were selectively retained in RNP-3 immunoprecipitations (Fig. 6C; Fischer et al. 2011). Because WAGO target transcript RNAs are often expressed at very low levels, it is likely that these transcripts are preferentially retained on intact spliceosomes in vivo. Tc1 and Tc3 transposon transcripts were also enriched in the RNP-3::3xFlag immunoprecipitations.
Figure 6.

RNAi target genes are specifically enriched in spliceosome immunoprecipitations. (A, top) Anti-Flag Western blot showing that Flag-RNP-3 is efficiently immunoprecipitated from transgenic C. elegans native extracts. (Bottom) Total RNA electrophoresed on a 1% agarose/TBE gel. (B) qRT–PCR from an RNP-3::3xFlag RNA immunoprecipitation to monitor which subunits of the spliceosome are coimmunoprecipitated with RNP-3 (as indicated by the structural RNA components of each subunit: U1, U2, U4, U5, and U6 snRNAs). Nonspliceosomal structural RNAs 5S and 5.8S rRNAs were examined as negative controls. (C) ddPCR analysis monitoring the enrichment of either non-RNAi target (“conserved”) or RNAi target genes in RNP-3::3xFlag immunoprecipitations. Two experimental replicates are plotted together to show general trends of either enrichment or depletion of each transcript in RNP-3 immunoprecipitations.
To verify our ddPCR results, we carried out qRT–PCRs of K02E2.11 and observed enrichment of PCR amplicons in the RNP-3 immunoprecipitation indicative of unspliced transcripts (Supplemental Fig. S6). Many RNAi target transcripts that produce the most abundant siRNAs were not enriched in RNP-3 immunoprecipitations (Supplemental Fig. S7). As a control for transcript abundance, we quantitated several essential genes expressed at very low levels and did not observe enrichment of these transcripts in RNP-3 immunoprecipitations (Supplemental Fig. S7).
The retention of RNAi surveilled transcripts could occur at multiple points in C. elegans RNAi: (1) before the transcript is exported out of the nucleus into the P granule, (2) downstream from secondary siRNA synthesis by Mutator foci (Phillips et al. 2012), and (3) after recruitment of the nuclear RNAi (NRDE pathway) machinery to the endo-RNAi targeted gene locus. If WAGO target transcripts are retained on the spliceosome before being routed to P granules and Mutator foci as templates for 22G siRNA synthesis (Phillips et al. 2012), these transcripts should remain tightly associated to the RNP-3-3xFlag immunoprecipitations even when Mutator foci are genetically ablated. To test this hypothesis, we constructed (using CRISPR) a null mutation of mut-16 in the transgenic animals containing the single-copy RNP-3-3xFlag transgene in the rnp-3(ok1424) mutant. The CRISPR edit resulted in a stop and downstream frameshift in the first exon of mut-16: The mut-16(mg586) mutants were defective for RNAi (Rde) and, as expected for mut-16, were almost sterile at 25°C (Supplemental Fig. S8; data not shown). If spliceosome retention occurs upstream of siRNA amplification in the Mutator foci, as might be expected if the aberrant splicing of foreign genes was the trigger for RNAi destruction of a viral invader, then we would expect that the transposon transcripts retained in the RNP-3-3xFlag immunoprecipitation in the mut-16 mutant would not be affected dramatically. This was the case: Tc1 and Tc3 transcripts are still enriched in the RNP-3-3xFlag immunoprecipitation from mut-16(0) animals (Fig. 6C). Taken together, these results indicate that transcripts of certain genes targeted by endo-RNAi selectively accumulate on intact spliceosomes upstream of routing to cytoplasmic P granules and Mutator foci.
Discussion
Our results reveal a probable nuclear axis of RNAi upstream of endo-siRNA-generating pathways. We show that splicing in C. elegans is surveilled by small RNAs, with the levels of IEJ siRNAs elevated specifically in mRNAs with poor splice sites. Poor splicing due to defects in either cis-splicing signals in horizontally transferred genes or splicing caused by mutations in key splicing components focuses the RNAi pathway onto mRNAs that are spliced less efficiently than normal, with IEJs that are recognized as suboptimal. Those transcripts bearing the most IEJ siRNAs might be actively exported from the nucleus with retained introns; indeed, genetic disruption of the exon junction complex components induces the escape of unspliced transcripts from the nucleus in the germline (Shiimori et al. 2013). Thus, foreign genes may not properly engage the exon junction complex machinery due to poor splicing. In animals lacking rnp-2/U1A, splicing is not lethally disrupted but is sufficiently disrupted to cause a reorganization of the small RNA machinery onto transcripts that are not normally recognized as foreign. Interestingly, robust siRNA formation on “nonforeign” genes in rnp-2 mutants does not coincide with increased IEJ siRNAs toward these transcripts (i.e., the siRNAs appear to be mainly exonic) (Fig. 4), suggesting that both slow/inefficient splicing and splicing failure are signals for routing a transcript into the RNAi pathway. Our results indicate that the U1 snRNP-specific factor RNP-2 functions at early steps in endo-RNAi, while the U2-specific components RNP-3 and MOG-2 function in nuclear RNAi, which is generally viewed to occur later in the endo-RNAi pathway. How U1 and U2 snRNP-specific factors function so differently in endogenous RNAi pathways is not yet clear. The trans-splicing of the key silencing factor ERI-6/7 is also inhibited in rnp-2 mutants (Fig. 2) to disable the ERI pathway that competes with exogenous RNAi (Fischer et al. 2008). It is possible that the trans-splicing of ERI-6/7, which is unique to C. elegans in the Caenorhabditis genus (Fischer et al. 2008), may act as a sensor—a compensatory mechanism—to increase RNAi capacity when splicing defects titrate away exogenous RNAi capacity.
Materials and methods
Genetics, genome engineering, and generation of transgenic strains
All C. elegans strains were grown at 20°C unless indicated otherwise (Brenner 1974). Strains are listed in Supplemental Table S9. Mutants were backcrossed to wild type at least five times except for rnp-2(mg582), which was backcrossed twice after generation by CRISPR. rnp-2(mg582) was generated by nonhomologous end-joining-based CRISPR originally described in Friedland et al. (2013). The rnp-3::3xFlag single-copy transgenic strain (mgSi51) was generated by mos1-mediated single-copy insertion (mosSCI) (Frokjaer-Jensen et al. 2008). The promoter region (including the rnp-2 and rnp-3 ORFs) and rnp-3 3′ UTR fragments were amplified from N2 genomic DNA using Phusion high-fidelity polymerase (New England Biolabs/Finnzymes). The 3x-Flag sequence was introduced by primer overhangs on both 5′ and 3′ amplicon fragments along with a secondary “3x-Flag push” PCR step for the 3′ amplicon to complete the 3xFlag sequence. The fragments were assembled into pCFJ151 (previously linearized with BglII) by Gibson assembly (Gibson et al. 2009). mut-16(mg586) was engineered directly into mgSi51 by CRISPR using the dpy-10(cn64)-dominant “Roller” strategy (Arribere et al. 2014).
RNAi assays
Synchronized L1 larvae were fed E. coli expressing dsRNA targeting gfp, dpy-13, hmr-1, or lir-1. After ∼3 d at 20°C, animals were scored for GFP silencing (gfp), dumpy (dpy-13), sterile (hmr-1), or lethal (lir-1) phenotypes, respectively. Because the rnp-2 mutants are slightly dumpy, this complicated the interpretation of their phenotypes on dpy-13 RNAi as being a truly Eri phenotype (vs. simply exacerbating a subtle sick phenotype). For the synthetic enhancement screen with splicing factor knockdown, synchronized wild-type N2, rnp-2(mg582), or rnp-3(ok1424) L1s were fed E. coli expressing dsRNA corresponding to genes encoding core splicing factors (Fischer et al. 2008). After ∼3–4 d at 20°C, parental animals were scored for lethality and/or sterility phenotypes.
C. elegans fluorescence imaging and quantitation
Imaging of animals expressing SUR-5::GFP was carried out on a Zeiss Axio Zoom V16 microscope. Quantitation of populations of worms expressing SUR-5::GFP was carried out using a Copas Biosort (Union Biometrica). Imaging of worms expressing the mCherry-siR-1 siRNA reporter transgene (kindly provided by Taiowa Montgomery) was carried out using a Zeiss Axio Imager Z1 microscope.
Small RNA isolation and high-throughput sequencing
Two micrograms of total RNA isolated from C. elegans embryos was resolved by 15% urea/TBE PAGE, and 18- to 28-nt RNA was gel-purified. Purified small RNAs were treated with 5′ polyphosphatase (Epicentre) to remove 5′ diphosphate and triphosphate moieties, ligated to Ilumina HiSeq small RNA 5′ and 3′ adapters, and subjected to RT–PCR as described previously (Gu et al. 2009). Gel-purified PCR amplicons were sequenced on an Illumina HiSeq machine. Small RNAs were sequenced from both embryos and synchronized the L4 larval stage animals from rnp-2, rnp-3, and mog-2 animals. The developmental delay phenotypes of the rnp-2- and mog-2-splicing mutants made it difficult to properly synchronize wild-type and mutant animals at the L4 stage using time only; mRNA-seq analysis indicated that rnp-2 and rnp-3 mutants had a gene expression signature of late L3/early L4 (vs. mid-L4 for wild type) (see Supplemental Table S10).
Data processing and statistical analysis
Raw small RNA FASTQ sequencing files were processed as described previously (Shi et al. 2013). Small RNA sequence reads were extracted and aligned to the C. elegans genome (Wormbase genome release WS230 or WS235). Reads from wild-type and mutant small RNA libraries were normalized to the total number of million mapped reads. For scoring intron quality, we devised a quantitative estimator of sequence divergence between a given genomic position and the splice site motif derived from the bases at or proximal to the splice donor (“GU”) and splice acceptor (“AG”) sequences of C. elegans introns (Fig. 3A; Supplemental Fig. S2). Our initial analysis indicated that a poorly conserved ERI-6/7 target gene, ZK402.3, contained very abundant splice acceptor (3′) IEJ siRNAs at an intron whose intron quality score was 15.2 (i.e., very divergent from C. elegans introns harbored in conserved genes); however, this intron overlaps with an alternatively spliced exon that has not been annotated previously (data not shown). The donor and acceptor sequences that factor into the algorithm are directly above the orange boxes in the logo diagram in Figure 3A; the sequence motifs of donor and acceptor sequences are shown as logos of position weight matrices directly above the orange boxes. Using the simple assumption of independence between individual positions of the motif, we estimated the likelihood of a splice site to occur within a given genomic sequence as the product of probabilities for each of the nucleotides in the sequence to occur at a corresponding position within the splice site motif. We then calculated a score for the divergence of the sequence from the motif as the negative logarithm of this likelihood: S = −Σ log(pi). The average value of this divergence score for introns across the C. elegans genome is ∼6.2.
Immunoprecipitation
For each immunoprecipitation sample, worms were cultivated on ∼12 enriched NGM plates (15 cm) seeded with 2 mL of OP50 E. coli bacteria. Approximately 750,000–1,000,000 synchronized adult C. elegans were harvested after cultivation for 70 h at 20°C from starved L1 stage. Worms were washed off the plates using M9 buffer; packed worm pellets (1–1.5 mL) were washed in 50-mL Falcon tubes twice with 50 mL of M9 buffer followed by one 50-mL wash with cold distilled H2O and one 25-mL wash with cold immunoprecipitation buffer (20 mM HEPES at pH 7.5, 2 mM MgCl2, 150 mM NaCl, 0.5% NP-40). Packed worm pellets were frozen in liquid nitrogen and stored at −80°C until extract preparation. Worm pellets were ground to a fine powder in liquid nitrogen and dissolved in 12 mL of ice-cold immunoprecipitation buffer (supplemented with 1 mM DTT, 1× PMSF, 0.02 U/µL RNase inhibitor [RiboLock; Thermo Fisher], complete protease inhibitors [EDTA-free; Roche]). Crude lysates were clarified by centrifugation at 37,000g for 30 min at 4°C (Beckman, JA-25.50 rotor), and 12 mL of clarified lysate was precleared by rotating with 100 µL of Pierce control agarose resin (pre-equilibrated in immunoprecipitation buffer) for 1 h at 4°C. Immunoprecipitations were carried out by incubating 10 mL of precleared lysate with 50 µL of M2-Flag agarose (pre-equilibrated in immunoprecipitation buffer; Sigma-Aldrich) for 2 h (RNA immunoprecipitation #1) or overnight (RNA immunoprecipitation #2) at 4°C. Immunoprecipitation beads were settled completely by gravity, the supernatant was removed, and beads were washed five times with 1 mL of immunoprecipitation buffer in 1.5-mL Eppendorf tubes. Washed beads were divided into two volumes of 25 µL; protein and RNA were eluted off the beads using 50 µL of Laemelli buffer and 1 mL of Trizol, respectively.
RNA isolation, qRT–PCR, and ddPCR
RNA was isolated with Trizol (Life Technologies) using the manufacturer's guidelines. For cDNA synthesis, 2–5 µg of total input RNA was treated with Turbo DNase (Thermo Fisher Scientific) per the manufacturer's guidelines, RNA was extracted using phenol:chloroform (pH 8.0), washed with an equal volume of chloroform, and precipitated with ethanol and sodium acetate (pH 5.5). All of the RNA from each immunoprecipitation sample was reprecipitated in parallel. RNA was reverse-transcribed using 250 ng of random hexamers and SuperScript III (Thermo Fisher). Input cDNA was adjusted to a final concentration of 5 ng/µL, and immunoprecipitation cDNA was brought to a final volume of 500 µL in TE buffer. qRT–PCR was carried out using IQ SYBR Green supermix (Bio-Rad) according to manufacturer's guidelines. ddPCR was carried out using the QX200 AutoDG ddPCR system (Bio-Rad). All primer sequences used in this study are listed in Supplemental Table S8. We were unable to find a unique set of PCR primers to specifically query whether ZK402.3 transcripts are retained in the same manner (ZK402.3 has extensive nucleotide identity with several other ERI-6/7 target genes) (Fischer et al. 2011).
Accession numbers
Small RNA Illumina high-throughput sequencing data are available at the Gene Expression Omnibus (accession no. GSE113301).
Supplementary Material
Acknowledgments
We thank Shohei Mitani and the National Bioresource Project (Japan) for deletion strains, the Caenrohabditis Genetics Center for strains, and Tai Montgomery for reagents. This work was supported by National Institutes of Health (NIH) grant GM44619 (to G.R.) and NIH Ruth L. Kirschstein National Research Service Award F32 GM101802 (to M.A.N.).
Author contributions: M.A.N. and G.R. conceived and designed the experiments. M.A.N. and S.E.J.F. performed the experiments. M.A.N., S.E.J.F., F.J., A.A., and R.I.S. analyzed the data. M.A.N. and G.R. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311514.118.
References
- Arribere JA, Bell RT, Fu BXH, Artiles KL, Hartman PS, Fire AZ. 2014. Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198: 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessonov S, Anokhina M, Will CL, Urlaub H, Luhrmann R. 2008. Isolation of an active step I spliceosome and composition of its RNP core. Nature 452: 846–850. [DOI] [PubMed] [Google Scholar]
- Billi AC, Fischer SE, Kim JK. 2014. Endogenous RNAi pathways in C. elegans. WormBook (ed. The C. elegans Research Community), WormBook, 10.1895/wormbook.1.170.1, http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NO, Burkhart KB, Kennedy S. 2011. Nuclear RNAi maintains heritable gene silencing in Caenorhabditis elegans. Proc Natl Acad Sci 108: 19683–19688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Albuquerque BF, Placentino M, Ketting RF. 2015. Maternal piRNAs are essential for germline development following de novo establishment of endo-siRNAs in Caenorhabditis elegans. Dev Cell 34: 448–456. [DOI] [PubMed] [Google Scholar]
- Drinnenberg IA, Fink GR, Bartel DP. 2011. Compatibility with killer explains the rise of RNAi-deficient fungi. Science 333: 1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR III, Bartel DP, Madhani HD. 2013. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 152: 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA, Ashe A, Piffaretti J, Wu G, Nuez I, Bélicard T, Jiang Y, Zhao G, Franz CJ, Goldstein LD, et al. 2011. Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol 9: e1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SEJ, Butler MD, Pan Q, Ruvkun G. 2008. Trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature 455: 491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Montgomery TA, Zhang C, Fahlgren N, Breen PC, Hwang A, Sullivan CM, Carrington JC, Ruvkun G. 2011. The ERI-6/7 helicase acts at the first stage of an siRNA amplification pathway that targets recent gene duplications. PLoS Genet 7: e1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Pan Q, Breen PC, Qi Y, Shi Z, Zhang C, Ruvkun G. 2013. Multiple small RNA pathways regulate the silencing of repeated and foreign genes in C. elegans. Genes Dev 27: 2678–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. 2013. Heritable genome editing in C. elegans via a CRISPR–Cas9 system. Nat Methods 10: 741–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. 2008. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat Genet 40: 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Lamm AT, Pavelec DM, Maniar JM, Parameswaran P, Tao L, Kennedy S, Fire AZ. 2010. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans soma. Mol Cell 37: 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. 2009. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol Cell 36: 231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta BP, Sternberg PW. 2003. The draft genome sequence of the nematode Caenorhabditis briggsae, a companion to C. elegans. Genome Biol 4: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollins C, Zorio DA, MacMorris M, Blumenthal T. 2005. U2AF binding selects for the high conservation of the C. elegans 3′ splice site. RNA 11: 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG, Lisch D. 1997. Transposable elements as sources of variation in animals and plants. Proc Natl Acad Sci 94: 7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Gabel HW, Kamath RS, Tewari M, Pasquinelli A, Rual JF, Kennedy S, Dybbs M, Bertin N, Kaplan JM, et al. 2005. Functional genomic analysis of RNA interference in C. elegans. Science 308: 1164–1167. [DOI] [PubMed] [Google Scholar]
- Law MJ, Chambers EJ, Katsamba PS, Haworth IS, Laird-Offringa IA. 2005. Kinetic analysis of the role of the tyrosine 13, phenylalanine 56 and glutamine 54 network in the U1A/U1 hairpin II interaction. Nucleic Acids Res 33: 2917–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, et al. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Maduro M, Li F, Li HW, Broitman-Maduro G, Li WX, Ding SW. 2005. Animal virus replication and RNAi-mediated antiviral silencing in Caenorhabditis elegans. Nature 436: 1040–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ. 2009. Small RNAs as guardians of the genome. Cell 136: 656–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery TA, Rim Y-S, Zhang C, Dowen RH, Phillips CM, Fischer SE, Ruvkun G. 2012. PIWI associated siRNAs and piRNAs specifically require the Caenorhabditis elegans HEN1 ortholog henn-1. PLoS Genet 8: e1002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelec DM, Lachowiec J, Duchaine TF, Smith HE, Kennedy S. 2009. Requirement for the ERI/DICER complex in endogenous RNA interference and sperm development in Caenorhabditis elegans. Genetics 183: 1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Montgomery TA, Breen PC, Ruvkun G. 2012. MUT-16 promotes formation of perinuclear Mutator foci required for RNA silencing in the C. elegans germline. Genes Dev 26: 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Brown KC, Montgomery BE, Ruvkun G, Montgomery TA. 2015. piRNAs and piRNA-dependent siRNAs protect conserved and essential C. elegans genes from misrouting into the RNAi pathway. Dev Cell 34: 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM. 1995. The Tc1-mariner superfamily of transposons in animals. J Insect Physiol 41: 99–105. [Google Scholar]
- Saldi T, Wilusz C, MacMorris M, Blumenthal T. 2007. Functional redundancy of worm spliceosomal proteins U1A and U2B′′. Proc Natl Acad Sci 104: 9753–9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Montgomery TA, Qi Y, Ruvkun G. 2013. High-throughput sequencing reveals extraordinary fluidity of miRNA, piRNA, and siRNA pathways in nematodes. Genome Res 23: 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiimori M, Inoue K, Sakamoto H. 2013. A specific set of exon junction complex subunits is required for the nuclear retention of unspliced RNAs in Caenorhabditis elegans. Mol Cell Biol 33: 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijen T, Plasterk RH. 2003. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature 426: 310–314. [DOI] [PubMed] [Google Scholar]
- Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. 2004. Crystal structure of Argonaute and its implications for RISC slicer activity. Science 305: 1434–1437. [DOI] [PubMed] [Google Scholar]
- Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, et al. 2013. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature 493: 694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, Fire A. 2001. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263: 103–112. [DOI] [PubMed] [Google Scholar]
- Will CL, Luhrmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Montgomery TA, Gabel HW, Fischer SE, Phillips CM, Fahlgren N, Sullivan CM, Carrington JC, Ruvkun G. 2011. mut-16 and other mutator class genes modulate 22G and 26G siRNA pathways in Caenorhabditis elegans. Proc Natl Acad Sci 108: 1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang J, Xu J, Zhang Z, Koppetsch BS, Schultz N, Vreven T, Meignin C, Davis I, Zamore PD, et al. 2012. UAP56 couples piRNA clusters to the perinuclear transposon silencing machinery. Cell 151: 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wang J, Schultz N, Zhang F, Parhad SS, Tu S, Vreven T, Zamore PD, Weng Z, Theurkauf WE. 2014. The HP1 homolog rhino anchors a nuclear complex that suppresses piRNA precursor splicing. Cell 157: 1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.