

In this study, Ahmed et al. demonstrate that oxidative damage of telomeres inhibits telomerase activity at chromosome ends in cancer cells. Deletion of two antioxidant enzymes—PRDX1 and MTH1, needed for protecting telomeres from oxidative damage—results in loss of telomeric DNA in an oxygen concentration-dependent manner due to inhibition of telomerase, thus providing new insights into the role of antioxidant systems that are required to protect telomeres from oxidation.
Keywords: MTH1, PRDX1, aging, cellular senescence, oxidative stress, telomerase, telomeres
Abstract
Telomerase counteracts telomere shortening and cellular senescence in germ, stem, and cancer cells by adding repetitive DNA sequences to the ends of chromosomes. Telomeres are susceptible to damage by reactive oxygen species (ROS), but the consequences of oxidation of telomeres on telomere length and the mechanisms that protect from ROS-mediated telomere damage are not well understood. In particular, 8-oxoguanine nucleotides at 3′ ends of telomeric substrates inhibit telomerase in vitro, whereas, at internal positions, they suppress G-quadruplex formation and were therefore proposed to promote telomerase activity. Here, we disrupt the peroxiredoxin 1 (PRDX1) and 7,8-dihydro-8-oxoguanine triphosphatase (MTH1) genes in cancer cells and demonstrate that PRDX1 and MTH1 cooperate to prevent accumulation of oxidized guanine in the genome. Concomitant disruption of PRDX1 and MTH1 leads to ROS concentration-dependent continuous shortening of telomeres, which is due to efficient inhibition of telomere extension by telomerase. Our results identify antioxidant systems that are required to protect telomeres from oxidation and are necessary to allow telomere maintenance by telomerase conferring immortality to cancer cells.
Telomeres protect chromosome ends from degradation, DNA recombination, DNA end-joining, and the DNA damage response (DDR) (de Lange 2009; Denchi and Sfeir 2016). Telomere protection is mediated by the telomeric DNA repeats consisting of 5′-TTAGGG-3′/5′-CCCTAA-3′ sequences, which recruit the shelterin and other telomeric proteins that are required for telomere functions. Telomeres shorten due to the end replication problem and the nucleolytic processing of chromosome ends with every round of DNA replication (Soudet et al. 2014). Short telomeres activate a DDR, which induces cellular senescence or apoptosis (d'Adda di Fagagna et al. 2003; Denchi and de Lange 2007). Further erosion of telomeric repeats leads to loss of protection of chromosome ends from DNA end-joining repair activities, leading to chromosome end fusions and severe genome instability (Jones et al. 2014). The latter is referred to as telomere crisis (Maciejowski and de Lange 2017). Short telomere-mediated senescence and crisis occur during tumorigenesis. Short telomere-mediated senescence also becomes prevalent during aging.
Telomere shortening is counteracted by the cellular reverse transcriptase telomerase, which uses an internal RNA moiety as a template to add telomeric repeats to the ends of chromosome (Greider and Blackburn 1989; Lingner et al. 1997). In humans, telomerase expression is tightly regulated during development (Forsyth et al. 2002). It is expressed during early embryonic development in all cells but later in life only in germ and stem cells. Upon differentiation, expression of the catalytic subunit of telomerase hTERT is repressed, thus limiting the replicative potential of somatic cells. The suppression of telomerase limits the replicative potential of precancerous tissues and therefore provides a powerful tumor-suppressive mechanism. Cancer cells overcome this barrier through up-regulation of hTERT, which frequently involves mutations in the hTERT promoter (Horn et al. 2013; Huang et al. 2013).
Reactive oxygen species (ROS) are generated by UV light, ionizing radiation, and a wide range of chemicals (De Bont and van Larebeke 2004). In addition, mitochondria are a major endogenous source for ROS production, since, during oxidative phosphorylation at a low frequency, O2 is incompletely reduced to superoxide radical (O2−) instead of water. ROS are highly reactive and can damage nucleic acids, nucleotides, lipids, and proteins if not removed by the cell's armory of antioxidant enzymes, which includes peroxidases, catalase, glutathione peroxidases, and peroxiredoxins (Perkins et al. 2015). Of these, peroxiredoxin 1 (PRDX1) has been shown to associate with telomeric chromatin and protect telomeres from oxidative damage (Aeby et al. 2016). In addition, the nudix phosphohydrolase superfamily enzyme 7,8-dihydro-8-oxoguanine triphosphatase (MTH1), which is involved in nucleotide pool sanitization, has been shown to contribute to telomere intactness (Fouquerel et al. 2016).
Owing to its sequence and structure, telomeric DNA is thought to be particularly susceptible to ROS-mediated cleavage and base modifications (for reviews, see von Zglinicki 2002; Ahmed and Lingner 2018). In vitro experiments demonstrated that telomeric DNA is more reactive with ROS than nontelomeric sequences (Oikawa and Kawanishi 1999). Oxidized DNA bases are repaired by base excision repair (Jacobs and Schar 2012). This pathway requires an intact complementary DNA strand that is used as a template during the repair reaction upon excision of the oxidized nucleotides. However, telomeres are partially single-stranded. The telomeric 5′-TTAGGG-3′ repeats containing a 3′ overhang, which has a length of 50–100 nucleotides at human telomeres, may be particularly susceptible to persisting damage as long as it is not engaged in a t-loop configuration in which the 3′ overhang is tucked into the double-stranded part of the telomere (Doksani et al. 2013).
The effects of ROS on telomere length and telomerase are not well understood. In telomerase-negative cells, ROS have been shown to accelerate the onset of cellular senescence (Forsyth et al. 2003; Passos et al. 2007; Ahmed et al. 2008; Britt-Compton et al. 2009). However, enhanced telomere shortening was seen in only a subset of these studies, suggesting that ROS promote senescence by telomere-independent damage. Based on in vitro studies, ROS have been proposed to either promote (Fouquerel et al. 2016; Lee et al. 2017) or repress (Aeby et al. 2016; Fouquerel et al. 2016) telomerase activity. In vitro, certain oxidized DNA substrates containing 8-oxoguanosine as a terminal nucleotide efficiently prevented extension by telomerase (Aeby et al. 2016). Furthermore, the oxidized nucleotide 8-oxo-2′deoxyguanosine-5′-triphosphate (8-oxo dGTP) causes premature chain termination when incorporated by telomerase (Aeby et al. 2016; Fouquerel et al. 2016). On the other hand, it has been proposed that oxidative DNA damage promotes telomerase activity by destabilizing G-quadruplex DNA structures (Fouquerel et al. 2016; Lee et al. 2017). In addition, ROS damage of telomeric DNA can interfere with binding of the shelterin components telomeric repeat-binding factor 1 (TRF1) and TRF2 in vitro (Opresko et al. 2005). Partial depletion of TRF1, TRF2, or POT1 leads to telomere length increase (van Steensel and de Lange 1997; Smogorzewska et al. 2000; Ancelin et al. 2002; Loayza and de Lange 2003).
In this study, in order to study the impact of ROS on telomere maintenance, we disrupted the genes for PRDX1 and MTH1 in cancer cells and determined the consequences on telomere maintenance. We demonstrated that upon oxidative stress, PRDX1 and MTH1 contribute nonredundant roles to retain telomeres in a telomerase-extendible state. In the absence of both, telomeres shorten continuously due to the efficient inhibition of telomerase. Thus oxidative damage of telomeres efficiently inhibits telomerase function in cancer cells.
Results
PRDX1 and MTH1 cooperate to reduce oxidation of guanine in the genome
PRDX1 reduces the burden of ROS, reducing H2O2 to H2O (Fig. 1A). MTH1 hydrolyzes oxidized nucleotides to monophosphates, preventing their incorporation into DNA during DNA replication and by telomerase (Fig. 1A). To study the effects of oxidative stress and the roles of PRDX1 and MTH1 on telomere maintenance, we disrupted (using CRISPR/Cas9 technology) MTH1 in HCT116 colon cancer cells and in HCT116 PRDX1 knockout cells that have been described previously (Aeby et al. 2016). Cells were cloned and analyzed for loss of PRDX1 and MTH1 expression by Western blot analysis (Fig. 1B,C). Sequencing of the MTH1 loci in cell clones revealed the presence of short deletions in the vicinity of the guiding RNA target sites downstream from the ATG start codon, leading to frameshift mutations and premature stop codons (Supplemental Fig. S1A,B). In addition, the Western blot analysis revealed that PRDX1 knockout clones expressed increased amounts of MTH1 (Fig. 1B), and MTH1 knockout clones increased levels of PRDX1 (Fig. 1C). This suggests that cells respond to increased oxidative damage that may be elicited upon disruption of one antioxidant system by increasing the expression of others. In agreement with this notion, HCT116 wild-type cells showed oxygen concentration-dependent expression of MTH1, PRDX1, and the PRDX1 homolog PRDX2 (Fig. 1D).
Figure 1.

MTH1 and PRDX1 cooperate for safeguarding the genome from oxidation. (A) Schematic representation of the enzymatic functions of PRDX1 and MTH1 and the putative effects of oxidation of telomere substrates on telomere maintenance by telomerase. PRDX1 reduces ROS. MTH1 hydrolyzes 8-oxo dGTP to 8-oxo dGMP, preventing its usage by telomerase and other DNA polymerases. In vitro experiments suggested that oxidation of telomeric DNA can either promote or inhibit telomerase. (B) Western blot analysis of MTH1 levels in two HCT116 wild-type cell clones (WT1 and WT2), two PRDX1 knockout clones (P KO1 and P KO2), and three PRDX1/MTH1 double-knockout clones (PM DKO1–3). The relative MTH1 levels are indicated. Tubulin was used as a loading control. (C) Western blot analysis of PRDX1 and MTH1 levels in HCT116 wild-type (WT1 and WT2), MTH1 -knockout (M KO1 and M KO2), and PRDX1/MTH1 double-knockout (PM DKO1 and PM DKO2) cells. The relative PRDX1 levels are indicated. (D) Western blot analysis of MTH1, PRDX1, and PRDX2 in HCT116 cells grown in incubators with different O2 concentrations. (E) Visualization of 8-oxo dG in DNA of wild-type, PRDX1 knockout (P KO), MTH1 knockout (M KO), and PRDX1/MTH1 double-knockout (PM DKO) cells by immunostaining with anti-8-oxoguanine (8-oxo G) antibody.
To determine the roles of MTH1 and PRDX1 in suppressing oxidation of guanine in the genome, we stained pre-extracted and RNase A-treated nuclei of HCT116 wild-type, PRDX1 knockout, MTH1 knockout, and PRDX1/MTH1 double-knockout cells with antibodies recognizing 8-oxoguanine (8-oxo G). Analysis by indirect immunofluorescence showed oxygen concentration-dependent increased incorporation of 8-oxoguanine into the genome (Fig. 1E). The staining for 8-oxo G was enhanced in MTH1 knockout cells and further pronounced in the PRDX1/MTH1 double-knockout cells. Together, these results indicate that MTH1 and PRDX1 cooperate to reduce 8-oxo G in the genome.
PRDX1 and MTH1 prevent oxygen-induced continued telomere shortening
To determine the putative roles of PRDX1 and MTH1 in telomere maintenance, we analyzed telomere length changes by Southern blot analysis of wild-type, PRDX1 knockout, and MTH1 knockout HCT116 clones upon propagation in tissue culture chambers containing different oxygen concentrations (Fig. 2). The individual clones showed different starting telomere lengths, which is typically observed in cell cloning experiments (Colgin et al. 2003). In 20% oxygen, the wild-type and PRDX1 knockout clones showed no telomere shortening upon propagation. The MTH1 knockout clone lost 15 base pairs (bp) of telomeric DNA per population doubling (PD), and the PRDX1/MTH1 double-knockout lost 23 bp/PD (Fig. 2A). The telomere shortening in MTH1 knockout and PRDX1/MTH1 double-knockout cells was progressive and concerned the entire population of telomeres (Supplemental Fig. S2). In 40% oxygen, the PRDX1 knockout clones were not viable for extended periods of time. Telomere shortening was enhanced in the two MTH1 knockout clones to 35 and 43 bp/PD. In 5% oxygen, telomere lengths remained nearly stable in all clones upon propagation (Fig. 2C). One of the PRDX1/MTH1 double knockouts showed a slow shortening rate of 6 bp/PD. To further corroborate these results and exclude off-target effects, we cloned the MTH1 cDNA into a retroviral vector and infected wild-type and MTH1 knockout clones. The derived cells expressed similar amounts of transgenic MTH1, as assessed by Western blot analysis (Supplemental Fig. S3A). The analysis of telomere lengths in cell populations grown in 20% oxygen demonstrated that expression of MTH1 from the retroviral vector rescued the telomere shortening phenotype of the MTH1 knockout clones (Supplemental Fig. S3B,C). Together, the quantification of these and other Southern blots indicated that MTH1 and PRDX1 cooperate to prevent oxygen-induced telomere shortening (Fig. 2D).
Figure 2.

Oxidative stress-dependent telomere shortening in MTH1 knockout and MTH1/PRDX1 double-knockout cells. (A–C) TRF length determination by Southern blot analysis of genomic DNA. Genomic DNAs from HCT116 wild-type and knockout cells grown at 20% O2 (A), 40% O2 (B), or 5% O2 (C) for the indicated PDs were purified, restriction-digested, resolved on 0.7% agarose gels, and analyzed upon in-gel hybridization using a random radiolabeled telomeric probe. Averaged rates of telomere shortening (base pairs/PD), indicated below the gels, were deduced by quantifying the changes in telomere length between the indicated consecutive PDs. (D) Comparison of telomere shortening rates between MTH1 knockout (M KO) and PRDX1/MTH1 double-knockout (PM DKO) cells exposed to different O2 concentrations. The graph represents the median TRF lengths as a function of PDs. The graphs for MTH1 knockout (5% O2), MTH1 knockout (20% O2), and PRDX1/MTH1 double knockout (20% O2) were derived from Supplemental Figure S2B, while MTH1 knockout (40% O2) data points were derived from B.
PRDX1 and MTH1 are required for telomere elongation under oxidative stress
In order to test whether PRDX1 and MTH1 are required to allow telomerase-mediated telomere elongation under oxidative stress, we expressed a mutant POT1 allele, referred to as POT1-ΔOB (Loayza and de Lange 2003), in the wild-type and mutant HCT116 cells (Fig. 3A). POT1-ΔOB lacks the oligosaccharide/oligonucleotide-binding (OB) fold domain for binding single-stranded telomeric DNA and induces rapid telomerase-mediated telomere elongation (Loayza and de Lange 2003). All transduced cell lines showed similar amounts of active telomerase when measured by the RQ-TRAP assay, indicating that oxidative stress and the absence of MTH1 and PRDX1 did not affect the biogenesis of active telomerase (Fig. 3B). Telomere elongation upon propagation was first measured in cells grown in incubators containing an atmospheric concentration of 5% O2. Wild-type, MTH1 knockout, and PRDX1/MTH1 double-knockout cells elongated their telomeres at a similar rate of ∼100 bp/PD (Fig. 3C,D). However, when grown in incubators containing a 20% or 40% O2 atmosphere, telomere elongation was strongly inhibited in the mutant cell lines. The wild-type cells still elongated their telomeres at 105 and 82 bp/PD at 20% and 40% oxygen, respectively (Fig. 3E,F). On the contrary, disruption of MTH1 or MTH1 and PRDX1 together reduced the elongation rate in 20% O2 to 38 and 9–11 bp/PD, respectively. In 40% O2, telomeres shortened in the absence of MTH1, with faster shortening upon codisruption of PRDX1 (Fig. 3E,F). These results indicated that MTH1 and PRDX1 cooperate to allow telomerase-mediated telomere elongation under oxidative stress conditions.
Figure 3.

Inefficient elongation of telomeres in MTH1 and PRDX1 knockout cells at high O2 concentrations. (A) Western blot analysis of N-MycPOT1ΔOB in wild-type and knockout cells by immunoblotting using an anti-N-Myc antibody (9E11). Where indicated (+), wild-type and knockout HCT116 cells were stably infected with a retroviral construct expressing N-MycPOT1ΔOB. (B) Comparison of telomerase activity (RQ-TRAP assay) in the indicated knockout cells relative to wild type. (C) TRF analysis of cells expressing N-MycPOT1ΔOB grown at 5% O2. Upon completion of puromycin selection at PD10, cells were further propagated for the indicated PDs. (D) Distribution profile of telomere fragments from cells obtained in C. Cumulative fractions of telomere fragments with different sizes were plotted to generate the telomere fragment distribution profiles of wild-type and knockout cells. (E) TRF analysis of cells expressing N-MycPOT1ΔOB grown at 20% O2 and 40% O2. (F) Distribution profile of telomere fragments from cells grown at 20% O2.
PRDX1 and MTH1 promote telomerase activity at chromosome ends under oxidative stress
To further substantiate the supportive roles of PRDX1 and MTH1 for telomerase-mediated telomere elongation under oxidative stress conditions, we expressed a modified telomerase RNA moiety (hTR) containing a mutant template (referred to as TSQ1) from a retroviral vector in the wild and mutant cell lines. Instead of the canonical 5′-TTAGGG-3′ telomeric DNA repeats, TSQ1–hTR directs the addition of 5′-GTTGCG-3′ repeats to the 3′ ends of chromosomes (Fig. 4A; Diolaiti et al. 2013). The activity of TSQ1 telomerase was compared in an adapted RQ-TRAP assay that amplified the TSQ1 mutant but not wild-type telomeric repeats. All TSQ1–hTR transduced cell lines showed comparable levels of mutant telomerase activity (Fig. 4B). Incorporation of TSQ1 repeats into telomeric DNA was determined by Southern hybridization of total DNA with TSQ1-specific probes after growth for zero or 21 PDs in an incubator containing 5% or 20% O2 (Fig. 4C). Alu repeat DNA was detected as a loading control. Quantification of signals revealed that at 5% O2, wild-type and knockout cells exhibited comparable levels of TSQ1 repeat DNA incorporation after 21 PDs. However, when grown in the presence of 20% O2, TSQ1 repeat DNA incorporation was reduced in MTH1 knockout cells when compared with wild type, and a further reduction of the TSQ1 repeat DNA was obtained with the MTH1/PRDX1 double-knockout cells (Fig. 4D). These results confirm the notion that telomerase is inhibited at chromosome ends in the absence of MTH1 and PRDX1 under oxidative stress conditions.
Figure 4.

Reduced telomerase activity at chromosome ends in MTH1 knockout and MTH1/PRDX1 double-knockout cells. (A) Schematic representation of wild-type and TSQ1 mutant telomerase specifying synthesis of TSQ1 (5′-GTTGCG-3′)n telomeric repeats. Lentiviral constructs harboring the mutant form of hTR-encoding TSQ1 repeats were introduced in wild-type and knockout cells. (B) Comparison of telomerase activity of the TSQ1–hTR–telomerase complex in wild-type and knockout cells. Cell lysates of wild-type and knockout cells expressing TSQ1–hTR (indicated by +) were analyzed in a modified RQ-TRAP assay using a substrate and a primer that specifically detects incorporated GTTGCG repeats (Materials and Methods). (C) Dot blot analysis of genomic DNA digested with restriction enzymes. At PD0 and PD21, total GTTGCG signal and Alu signal were detected upon hybridization with specific radiolabeled probes. PD0 represents the day of completion of puromycin selection. (D) Quantification of the TSQ1 signal in C. The TSQ1 signal was normalized to the Alu signal and is expressed relative to wild type. Error bars correspond to SD obtained from three independent experiments. (**) P = 0.0016; (****) P < 0.0001; (ns) P > 0.05, unpaired t-test two-tailed P-value compared with the corresponding wild type.
Telomere shortening in PRDX1/MTH1 double-knockout cells culminates in DDR
We assessed the long-term effects of telomere shortening in MTH1 knockout and PRDX1/MTH1 double-knockout cells after 30 and 110 PDs of growth in 20% O2-containing incubators (Fig. 5). At the 110 PD time point, the two double-knockout cells had TRF lengths of 3.6–4.2 kb when assessed on Southern blots (Fig. 5A,B). Upon cell propagation, analysis of metaphase chromosomes stained with DAPI (in which telomeres were detected by fluorescence in situ hybridization [FISH] with a fluorescently labeled telomeric probe) revealed an increased presence of intrachromosomal telomere fusions and chromosome ends lacking a telomeric signal altogether (Fig. 5C–E; Supplemental Fig. S4). At the same time, the double-knockout cells showed increased accumulation of γ-H2AX at telomeres in late PDs (Supplemental Fig. S5). Western blot analysis showed increased cellular levels of the DDR markers γ-H2AX and, at Ser33, phosphorylated RPA32 (Fig. 5F). In addition, expression of the p53 target p21 was increased at the 110 PD time point. Altogether, the data support the conclusion that a DDR was activated due to telomere shortening in PRDX1/MTH1 double-knockout cells.
Figure 5.

MTH1–PRDX1 double knockouts accumulate extremely short telomeres and DNA damage markers. (A) TRF analysis at PD30 and PD110. The indicated clones were grown in incubators containing 20% O2. (B) Distribution profile of telomere fragments at the indicated PDs of A. (C) Analysis of telomere signals (red) in metaphase chromosomes (blue) of clones grown for 120 PDs. White arrows indicate telomere signal-free ends, and arrowheads indicate intrachromosomal telomere fusions. (Blue) DAPI-stained chromosomal DNA; (red) TeloC probe for telomeric DNA. (D) Quantification of intrachromosomal telomere fusions. (E) Quantification of telomere-free ends across different PDs. (F) Western blot analysis of DNA damage markers in PRDX1/MTH1 double-knockout (PM DKO) cells grown for 30 and 110 PDs. For D and E, >2700 telomeres were scored for each sample.
Discussion
Telomere maintenance by telomerase is crucial for highly proliferating cells, including germ cells, embryonic cells, stem cells, and cancer cells. Oxidative damage of telomeres has been proposed to either enhance or inhibit telomerase activity at chromosome ends (Aeby et al. 2016; Fouquerel et al. 2016; Lee et al. 2017). Oxidation of telomeric DNA was suggested to inhibit the formation of G-quadruplex DNA (which inhibits telomerase) (Fouquerel et al. 2016; Lee et al. 2017) and reduce shelterin binding, rendering telomerase more active at chromosome ends. On the other hand, oxidized guanine at the 3′ terminus of telomeric DNA inhibited telomerase, resulting in chain termination when tested in vitro in telomerase assays (Aeby et al. 2016; Fouquerel et al. 2016). Our results obtained in HCT116 cancer cells demonstrate that the telomerase inhibitory effects of ROS-damaged telomeres prevail. The analysis became possible by removing two antioxidant enzymes, MTH1 and PRDX1, both of which have been implicated previously in the protection of telomeres from oxidative damage. Thus, the effects of ROS on telomere length could be assessed under relatively mild oxidative stress conditions, avoiding growth-inhibiting pleiotropic cellular damage and massive DNA backbone cleavage events that have been observed upon acute oxidative stress in previous studies (von Zglinicki et al. 2000; Aeby et al. 2016). Notably, when grown in incubators containing a 20% O2 atmosphere, MTH1 knockout and PRDX1/MTH1 double-knockout cells exhibited growth rates comparable with those of wild-type cells without causing major DNA breakage events of genomic or telomeric DNA that could be detected in metaphase chromosomes (Supplemental Fig. S4). We demonstrate that concomitant deletion of PRDX1 and MTH1 leads to continuous telomere shortening in dependency of oxygen concentration. The observed telomere shortening was due to inhibition of telomerase based on the following observations. First, the telomere elongation kinetics in POT1-ΔOB-expressing cells, which unleashes telomerase activity in wild-type cells, showed reduced to complete inhibition of telomere extension kinetics in MTH1 knockout and MTH1/PRDX1 double-knockout cells in an oxygen concentration-dependent manner. Second, the TSQ1 mutant allele of telomerase showed reduced cellular activity at chromosome ends in the absence of MTH1 and PRDX1, giving lower amounts of TSQ1 telomeric repeats upon cell propagation.
We therefore propose the following model (Fig. 6). With oxidative stress, the concentration of 8-oxo dGTP increases. In wild-type cells, the bulk of 8-oxo dGTP is dephosphorylated by MTH1 to 8-oxo dGMP, preventing its incorporation by telomerase into telomeric repeats. In the absence of MTH1, 8-oxo dGTP concentration increases, leading to its frequent incorporation by telomerase into telomeric repeats, causing chain termination. The effects of MTH1 loss are further exacerbated in cells that lack PRDX1. PRDX1 reduces ROS and therefore will also contribute to the reduction of cellular 8-oxo dGTP. In addition, PRDX1 may reduce the direct ROS-mediated damage of telomeric DNA. These complementary roles of MTH1 and PRDX1 can explain the additive effects of MTH1 and PRDX1 loss. Both of these enzymes were shown previously to contribute to the protection of telomeres. PRDX1 is enriched with telomeric chromatin and protects telomeric DNA from acute oxidative damage (Aeby et al. 2016). MTH1 depletion in cells with short telomeres showed enhanced damage (Fouquerel et al. 2016). In the present study, for the first time, the effects on telomerase in live cells are assessed.
Figure 6.
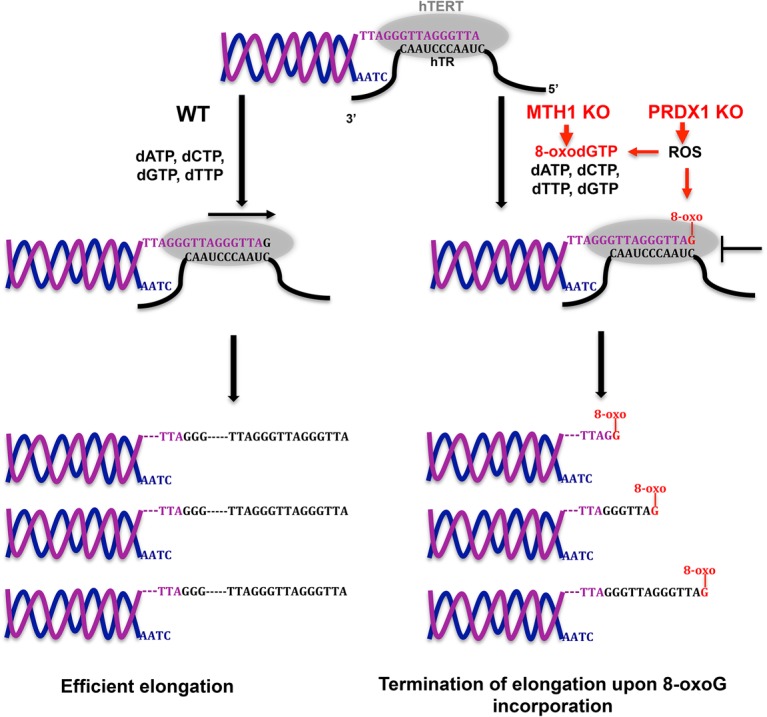
Model for telomerase inhibition in MTH1 knockout and PRDX1/MTH1 double-knockout cells. ROS increase the concentration of 8-oxo dGTP. In MTH1 knockout cells, 8-oxo dGTP is not hydrolyzed to 8-oxo dGMP. Telomerase incorporates 8-oxo G, leading to premature chain termination. Loss of PRDX1 further enhances telomerase inhibition. PRDX1 loss increases ROS, promoting oxidation of dGTP to 8-oxo dGTP. PRDX1 loss may also enhance direct ROS-mediated damage of telomeres at the chromosome 3′ end, leading to inhibition of telomerase.
The inhibitory effects of oxidative stress at telomeres on telomere maintenance by telomerase reported here may not be conserved in all eukaryotes. In Saccharomyces cerevisiae, deletion of the major peroxiredoxin TSA1 caused telomerase-dependent telomere elongation (Lu et al. 2013). This effect correlated with reduced binding of Rap1 to telomeres, which negatively regulates telomerase. This suggests that S. cerevisiae telomerase is more tolerant of 8-oxo G than human telomerase.
In human cells, telomerase activity at chromosome ends depends on the telomerase holoenzyme component, the telomerase recruiter TPP1, and checkpoint kinases (for review, see Schmidt and Cech 2015). Here, we uncover critical roles of antioxidant enzymes to allow telomere maintenance by telomerase. These enzymes function to keep the telomerase substrate dGTP and telomeric DNA in an unoxidized form that otherwise inhibits the enzyme. MTH1 and PRDX1 therefore appear most critical in cells that express telomerase during development and in stem cells to maintain telomeres in a telomerase-extendible state. Therefore, ROS damage of telomeres may be particularly detrimental in cells whose viability relies on telomerase. Oxidative damage of DNA increases with age (Fraga et al. 1990; Lu et al. 2004), which therefore may cause age-dependent telomerase inhibition in stem cells. Thus, our findings may explain telomere exhaustion in stem cells triggering age-related tissue exhaustion, such as bone marrow failure, muscle loss, and others (Aubert and Lansdorp 2008; Mourkioti et al. 2013). These conclusions highlight some disadvantages of having ssDNA at chromosome 3′ ends and using telomerase instead of semiconservative DNA replication to synthesize the very ends of chromosomes. In particular, telomerase is not supported by DNA repair systems that rely on a complementary DNA strand (David et al. 2007) and is not backed up by translesion DNA polymerases that can overcome unrepaired obstacles in dsDNA (Zahn et al. 2011; Sale et al. 2012).
Telomerase up-regulation in tumors is crucial for the immortal phenotype of cancer cells. Telomerase inhibition has been shown to induce telomere shortening, leading to death of tumor cells (Hahn et al. 1999). The shortening rates observed here in cells lacking MTH1 and PRDX1 are similar to the ones reported previously upon inhibition of telomerase (Hahn et al. 1999; Damm et al. 2001; Asai et al. 2003). Thus, this work also opens novel avenues to target telomeres and telomerase in cancer cells, for which efficient drug regimens remain to be described. Notably, recent data indicate that cancer cells are more vulnerable than noncancer cells to ROS (Sabharwal and Schumacker 2014). Thus, increasing ROS may preferentially target cancer cells, and several chemotherapeutic cancer drugs as well as ionizing radiation either induce ROS or reduce the cellular antioxidant capacity (Trachootham et al. 2009). It will be interesting to test whether these drugs may be used to target telomeres and whether they can be boosted for telomerase inhibition by combing them with inhibitors for MTH1 and PRDX1.
Materials and methods
Cell lines
Human HCT116 cell lines were used for all of the experiments and genetic manipulations. Retroviruses and lentiviruses harboring the desired DNA constructs were produced in HEK293T cells upon Lipofectamine 2000-mediated (Invitrogen) transfection of viral constructs. HCT116 cells stably expressing POT1-ΔOB or MTH1 from cDNAs were generated by infecting with retroviruses followed by 7 d of 1 µg/mL puromycin (InvivoGen) selection. HCT116 cells expressing TSQ1–hTR were generated by lentiviral transduction and puromycin selection for 7 d. All cells were cultured in DMEM supplemented with 10% FBS and maintained in a 5% CO2 incubator at 37°C and the indicated concentrations of O2.
Generation of MTH1 knockout cell lines
The CRISPR–Cas9 system was used to target the MTH1 gene locus (NC_000007.14; gene ID 4521). The plasmid pSpCas9(BB)-2A-puro was obtained from Addgene (catalog no. 62988). The oligonucleotide harboring the guide RNA (gRNA) target sequence against exon 3 (MTH1gRNA: CACCCATGAAAAAGCGAGGCTTCG) was designed using the Optimal CRISPR design tool (http://crispr.mit.edu). A gRNA-harboring oligonucleotide and its complementary oligonucleotide were annealed to make the dsDNA substrate for the ligation in BpiI linearized vector. The resulting construct was transfected in HCT116 wild-type or PRDX1 knockout cells using Lipofectamine 2000 (Invitrogen). After 3 d of 1 µg/mL puromycin (InvivoGen) selection, cells were diluted to generate single-cell colonies. Several single-cell colonies were screened for the absence of MTH1 using Western blotting with C-terminal-specific anti-MTH1 antibody (1:5000 dilution; Abcam, ab200832). PRDX1–MTH1 double-knockout cell lines were generated by transfecting the HCT116 PRDX1 knockout cells (Aeby et al. 2016) with the above-described construct. Single-cell MTH1 knockout clones were sequenced to identify the disrupted alleles.
Immunoblotting
Cell pellets were resuspended in Laemmli buffer and boiled for 5 min, and proteins were resolved on 4%–20% Mini Protean TGX (Bio-Rad). Proteins were transferred to nitrocellulose blotting membranes (Amersham), blocked with blocking solution (5% nonfat dry milk in PBS/0.1% Tween 20) for 30 min, and incubated with the appropriate primary antibody in blocking solution overnight at 4°C. The membranes were washed three times for 5 min with PBS/0.1% Tween 20 followed by incubation with HRP-conjugated secondary antibody (Promega) and chemiluminescence detection using a Western blotting detection kit (Western bright ECL, Advansta). Visualization of 8-oxo G was performed using the previously published protocol for nuclear 8-oxo G staining (Ohno et al. 2009).
Telomere length analysis
For in-gel analysis of telomere length, genomic DNA was purified from cells with the Wizard genomic DNA purification kit (Promega). Six micrograms of genomic DNA was subjected to restriction digestion with HinfI, RsaI, MnlI, HphI, and AluI (10 U of each) in a 50-µL reaction volume containing 1× CutSmart buffer (New England Biolabs). Two micrograms of digested DNA was mixed with 1× DNA loading buffer (10 mM Tris-HCl at pH 7.6, 0.03% bromophenol blue, 0.03% xylene cyanol FF, 60% glycerol, 60 mM EDTA), loaded on 0.7% agarose gel in 1× TBE, and fractionated by gel electrophoresis at 2 V/cm for 16 h. Dried gels were treated for 30 min each with denaturation buffer (0.5 M NaOH, 1.5 M NaCl) and neutralization buffer (0.5 M Tris-HCl at pH 7.5, 1.5 M NaCl) before prehybridization with Church mix for 2 h and overnight incubation at 50°C with randomly labeled TeloC probe (Grolimund et al. 2013). After hybridization, the gels were washed at 55°C (once with 4× SSC, once with 4× SSC + 0.1% SDS, and twice with 2× SSC + 0.1% SDS) and exposed to a phosphorimager screen overnight. After exposure, screens were scanned on a Typhoon phosphorimager (GE). The detection of telomere fragments was performed by densitometry-based AIDA software and subsequently analyzed by Graphpad Prism 7.
RQ-TRAP assay
The RQ-TRAP assay was performed as described (Wege et al. 2003). HCT116 cells were harvested, washed once in PBS, and resuspended in NP40 lysis buffer (10 mM Tris-HCl at pH 8.0, 1 mM MgCl2, 1 mM EDTA, 1% [v/v] NP-40, 0.25 mM sodium deoxycholate, 10% [v/v] glycerol, 150 mM NaCl, 5 mM β-mercaptoethanol, 0.1 mM PMSF [phenylmethylsulfonyl fluoride]). After 30 min of incubation on ice, extracts were centrifuged at 16,000g for 20 min at 4°C. Protein concentration of cell-free lysates was determined by BCA protein assay kit (Thermo Fischer Scientific). Lysate (0.5 µg) was added to 25-µL reactions in a 96-well plate containing 100 ng of TS (5′-AATCCGTCGAGCAGAGTT-3′) and 100 ng of ACX (5′-GCGCGGCTTACCCTTACCCTTACCCTAACC-3′) or 100 ng of TS-TSQ1 (5′-AATCCGTCGAGCAGAGGT-3′) and 100 ng of TSQ1-ACX (5′-GCGCGCCATCCGCATCCGCATCCGCAACCG-3′) primer sets and 1× Power SYBR Green PCR mix (Applied Biosystems). The reaction mix was incubated for 30 min at 30°C and 10 min at 95°C and amplified for 40 PCR cycles for 15 sec at 95°C and 60 sec at 60°C in a 7900 HT Fast real-time PCR system (Applied Biosystems). Baseline and threshold were set to be 10 standard deviations (SD) above the background (10 SD = one power of 10) to determine the threshold cycle (Ct). Telomerase activity was expressed relative to wild type. Samples were serially diluted to verify the linearity of the RQ-TRAP reaction. Heat-inactivated or RNase-treated extracts and lysis buffer control were used to verify telomerase activity-dependent generation of amplification products.
Dot blot analysis of DNA
DNA was purified and eluted using the PCR cleanup kit (Qiagen). DNA was loaded on Hybond N+ membrane using a dot blot apparatus (Bio-Rad), and telomeric sequences were detected using a randomly labeled TeloC probe (Grolimund et al. 2013). For the detection of Alu sequences, membranes were stripped and reprobed with a 5′ end 32P-labeled oligonucleotide probe specific for Alu DNA (5′-GTGATCCGCCCGCCTCGGCCTCCCAAAGTG-3′).
For the detection of TSQ1 sequences, restriction-digested genomic DNA was pipetted on a Hybond N+ membrane (Amersham), UV cross-linked, and sequentially treated with denaturation and neutralization buffers for 15 min each. Church mix prehybridized membrane was incubated with 5′ end 32P-labeled oligonucleotide TSQ1 probe (5′-CCGCAACCGCAACCGCAA-3′) for the detection of incorporated TSQ1 repeat sequences.
Metaphase telomere and TIF analysis
Cells were treated with 0.1 μg/mL demecolcine for 6 h, washed with 1× PBS, and harvested. Harvested cells were resuspended in hypotonic solution (0.056 M KCl) for 7 min and subsequently fixed in methanol:acetic acid (3:1) overnight. The treated cell suspensions were dropped onto slides to prepare metaphase spreads and dried for 16–24 h before FISH. For FISH, slides were rehydrated in 1× PBS for 5 min, treated with 4% formaldehyde in PBS for 5 min, and again washed three times with PBS followed by sequential dehydration with 70%, 95%, and 100% ethanol (5 min each). FISH hybridization solution (70% formamide, 10 mM Tris-HCl at pH 7.4, 1% [w/v] blocking reagent [Roche], 89 µM Cy3-TeloC PNA probe [PNA Bio, Cy3-CCCTAACCCTAACCCTAA-3′, catalog no. F1002]) was applied to air-dried slides and incubated for 3 min at 80°C on a heating block. After 3 h of hybridization at room temperature, cells were washed twice with wash buffer 1 (10 mM Tris-HCl at pH 7.4, 70% formamide) and once with wash buffer 2 (100 mM Tris-HCl at pH 7.4, 150 mM NaCl, 0.08% Tween-20). Subsequently, slides were incubated with 0.1 µg/mL DAPI in wash buffer 2 for 5 min and washed with wash buffer 2. After washing, slides were sequentially dehydrated in ethanol, air-dried, and mounted in VectaShield anti-fade medium (Vector Laboratories). For TIF analysis by immunofluorescence-FISH, cells were grown on coverslips and fixed in 4% formaldehyde. Immunofluorescence staining was performed by incubating the fixed cells with anti-γH2AX-specific (1:500 dilution; Millipore) antibody for 1 h at room temperature followed by 30 min of incubation with Alexa fluor 633-conjugated anti-mouse secondary antibody. Following immunofluorescence, FISH was performed as described above. The images were acquired using a Zeiss Axio plan 2 microscope system at 100× magnification.
Quantification and statistical analysis
Immunoblot, dot blot, and TRF gel analyses were performed using AIDA software. The mean intensity of the immunoblot and dot blot was measured within the defined area containing the gel band or dot in the blot. The intensity of telomere fragments was measured within the defined lane area harboring the signal of telomere fragments. For the calculation of telomere length, signals of TRFs were normalized with their molecular mass to correct for hybridization efficiency. Statistical parameters, statistical tests used, number of events scored, standard deviation, and statistical significance are included in each figure's legend. Statistical analysis was carried out using Graphpad Prism 7 software, and differences were considered statistically significant when P < 0.05 by two tailed Student's t-test.
Supplementary Material
Acknowledgments
We thank Bradley A. Stohr (University of California at San Francisco) for providing lentiviral constructs for the expression of TSQ1–hTR. Research in J.L.’s laboratory was supported by the Swiss National Science Foundation (SNSF), the SNSF-funded National Center of Competence in Research RNA and Disease Network, the Swiss Cancer League, and École Polytechnique Fédérale de Lausanne.
Author contributions: W.A. and J.L. designed the research, W.A. carried out experiments, and J.L. and W.A. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.313460.118.
References
- Aeby E, Ahmed W, Redon S, Simanis V, Lingner J. 2016. Peroxiredoxin 1 protects telomeres from oxidative damage and preserves telomeric DNA for extension by telomerase. Cell Rep 17: 3107–3114. [DOI] [PubMed] [Google Scholar]
- Ahmed W, Lingner J. 2018. Impact of oxidative stress on telomere biology. Differentiation 99: 21–27. [DOI] [PubMed] [Google Scholar]
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G. 2008. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci 121: 1046–1053. [DOI] [PubMed] [Google Scholar]
- Ancelin K, Brunori M, Bauwens S, Koering CE, Brun C, Ricoul M, Pommier JP, Sabatier L, Gilson E. 2002. Targeting assay to study the cis functions of human telomeric proteins: evidence for inhibition of telomerase by TRF1 and for activation of telomere degradation by TRF2. Mol Cell Biol 22: 3474–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan R, Wunder E, et al. 2003. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res 63: 3931–3939. [PubMed] [Google Scholar]
- Aubert G, Lansdorp PM. 2008. Telomeres and aging. Physiol Rev 88: 557–579. [DOI] [PubMed] [Google Scholar]
- Britt-Compton B, Wyllie F, Rowson J, Capper R, Jones RE, Baird DM. 2009. Telomere dynamics during replicative senescence are not directly modulated by conditions of oxidative stress in IMR90 fibroblast cells. Biogerontology 10: 683–693. [DOI] [PubMed] [Google Scholar]
- Colgin LM, Baran K, Baumann P, Cech TR, Reddel RR. 2003. Human POT1 facilitates telomere elongation by telomerase. Curr Biol 13: 942–946. [DOI] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198. [DOI] [PubMed] [Google Scholar]
- Damm K, Hemmann U, Garin-Chesa P, Hauel N, Kauffmann I, Priepke H, Niestroj C, Daiber C, Enenkel B, Guilliard B, et al. 2001. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J 20: 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David SS, O'Shea VL, Kundu S. 2007. Base-excision repair of oxidative DNA damage. Nature 447: 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N. 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19: 169–185. [DOI] [PubMed] [Google Scholar]
- de Lange T. 2009. How telomeres solve the end-protection problem. Science 326: 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. 2007. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448: 1068–1071. [DOI] [PubMed] [Google Scholar]
- Denchi EL, Sfeir A. 2016. Stop pulling my strings—what telomeres taught us about the DNA damage response. Nat Rev Mol Cell Biol 17: 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diolaiti ME, Cimini BA, Kageyama R, Charles FA, Stohr BA. 2013. In situ visualization of telomere elongation patterns in human cells. Nucleic Acids Res 41: e176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doksani Y, Wu JY, de Lange T, Zhuang X. 2013. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155: 345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth NR, Wright WE, Shay JW. 2002. Telomerase and differentiation in multicellular organisms: turn it off, turn it on, and turn it off again. Differentiation 69: 188–197. [DOI] [PubMed] [Google Scholar]
- Forsyth NR, Evans AP, Shay JW, Wright WE. 2003. Developmental differences in the immortalization of lung fibroblasts by telomerase. Aging Cell 2: 235–243. [DOI] [PubMed] [Google Scholar]
- Fouquerel E, Lormand J, Bose A, Lee HT, Kim GS, Li J, Sobol RW, Freudenthal BD, Myong S, Opresko PL. 2016. Oxidative guanine base damage regulates human telomerase activity. Nat Struct Mol Biol 23: 1092–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. 1990. Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci 87: 4533–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. 1989. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337: 331–337. [DOI] [PubMed] [Google Scholar]
- Grolimund L, Aeby E, Hamelin R, Armand F, Chiappe D, Moniatte M, Lingner J. 2013. A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat Commun 4: 2848. [DOI] [PubMed] [Google Scholar]
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. 1999. Inhibition of telomerase limits the growth of human cancer cells. Nat Med 5: 1164–1170. [DOI] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. 2013. TERT promoter mutations in familial and sporadic melanoma. Science 339: 959–961. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. 2013. Highly recurrent TERT promoter mutations in human melanoma. Science 339: 957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs AL, Schar P. 2012. DNA glycosylases: in DNA repair and beyond. Chromosoma 121: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RE, Oh S, Grimstead JW, Zimbric J, Roger L, Heppel NH, Ashelford KE, Liddiard K, Hendrickson EA, Baird DM. 2014. Escape from telomere-driven crisis is DNA ligase III dependent. Cell Rep 8: 1063–1076. [DOI] [PubMed] [Google Scholar]
- Lee HT, Bose A, Lee CY, Opresko PL, Myong S. 2017. Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res 45: 11752–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Hughes TR, Shevchenko A, Mann M, Lundblad V, Cech TR. 1997. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276: 561–567. [DOI] [PubMed] [Google Scholar]
- Loayza D, de Lange T. 2003. POT1 as a terminal transducer of TRF1 telomere length control. Nature 424: 1013–1018. [DOI] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. 2004. Gene regulation and DNA damage in the ageing human brain. Nature 429: 883–891. [DOI] [PubMed] [Google Scholar]
- Lu J, Vallabhaneni H, Yin J, Liu Y. 2013. Deletion of the major peroxiredoxin Tsa1 alters telomere length homeostasis. Aging Cell 12: 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, de Lange T. 2017. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourkioti F, Kustan J, Kraft P, Day JW, Zhao MM, Kost-Alimova M, Protopopov A, DePinho RA, Bernstein D, Meeker AK, et al. 2013. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat Cell Biol 15: 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Oka S, Nakabeppu Y. 2009. Quantitative analysis of oxidized guanine, 8-oxoguanine, in mitochondrial DNA by immunofluorescence method. Methods Mol Biol 554: 199–212. [DOI] [PubMed] [Google Scholar]
- Oikawa S, Kawanishi S. 1999. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett 453: 365–368. [DOI] [PubMed] [Google Scholar]
- Opresko PL, Mason PA, Podell ER, Lei M, Hickson ID, Cech TR, Bohr VA. 2005. POT1 stimulates RecQ helicases WRN and BLM to unwind telomeric DNA substrates. J Biol Chem 280: 32069–32080. [DOI] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. 2007. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A, Nelson KJ, Parsonage D, Poole LB, Karplus PA. 2015. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem Sci 40: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabharwal SS, Schumacker PT. 2014. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14: 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE, Lehmann AR, Woodgate R. 2012. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 13: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Cech TR. 2015. Human telomerase: biogenesis, trafficking, recruitment, and activation. Genes Dev 29: 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, van Steensel B, Bianchi A, Oelmann S, Schaefer MR, Schnapp G, de Lange T. 2000. Control of human telomere length by TRF1 and TRF2. Mol Cell Biol 20: 1659–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudet J, Jolivet P, Teixeira MT. 2014. Elucidation of the DNA end-replication problem in Saccharomyces cerevisiae. Mol Cell 53: 954–964. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. 2009. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8: 579–591. [DOI] [PubMed] [Google Scholar]
- van Steensel B, de Lange T. 1997. Control of telomere length by the human telomeric protein TRF1. Nature 385: 740–743. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T. 2002. Oxidative stress shortens telomeres. Trends Biochem Sci 27: 339–344. [DOI] [PubMed] [Google Scholar]
- von Zglinicki T, Pilger R, Sitte N. 2000. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med 28: 64–74. [DOI] [PubMed] [Google Scholar]
- Wege H, Chui MS, Le HT, Tran JM, Zern MA. 2003. SYBR Green real-time telomeric repeat amplification protocol for the rapid quantification of telomerase activity. Nucleic Acids Res 31: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn KE, Wallace SS, Doublie S. 2011. DNA polymerases provide a canon of strategies for translesion synthesis past oxidatively generated lesions. Curr Opin Struct Biol 21: 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.