

This review by Levine and Holland reviews the sources of mitotic errors in human tumors and their effect on cell fitness and transformation. They discuss new findings that suggest that chromosome missegregation can produce a proinflammatory environment and impact tumor responsiveness to immunotherapy and survey the vulnerabilities exposed by cell division errors and how they can be exploited therapeutically.
Keywords: aneuploidy, chromosomal instability, mitosis
Abstract
Mitosis is a delicate event that must be executed with high fidelity to ensure genomic stability. Recent work has provided insight into how mitotic errors shape cancer genomes by driving both numerical and structural alterations in chromosomes that contribute to tumor initiation and progression. Here, we review the sources of mitotic errors in human tumors and their effect on cell fitness and transformation. We discuss new findings that suggest that chromosome missegregation can produce a proinflammatory environment and impact tumor responsiveness to immunotherapy. Finally, we survey the vulnerabilities exposed by cell division errors and how they can be exploited therapeutically.
To err is human
Each day, millions of cells in our bodies undergo division to support growth and replace lost or damaged cells in our tissues. Numerous safeguards have evolved to ensure that these divisions proceed only under ideal growth conditions and with high fidelity. Mistakes during mitosis lead to the production of daughter cells with too many or too few chromosomes, a feature known as aneuploidy. Nearly all aneuploidies that arise due to mistakes in meiosis or during early embryonic development are lethal, with the notable exception of trisomy 21 in humans. However, mitotic errors that give rise to aneuploidy later in life have been linked to aging and tumorigenesis (Naylor and van Deursen 2016).
Aneuploidy is a very common feature of cancer, arising in almost 70% of solid human tumors (Duijf et al. 2013). In addition to alterations in chromosome number, tumor cells show frequent structural alterations in chromosomes that include deletions, amplifications, and translocations. Errors in mitosis are the major source of numerical changes in chromosome number observed in cancer and also have been recognized recently to be a contributing factor in the generation of chromosomal rearrangements (Bakhoum et al. 2014; Leibowitz et al. 2015). In this review, we discuss possible sources of mitotic errors and the effect of these mistakes on cell physiology and tumorigenesis. We then describe recent findings suggesting that errors in cell division are recognized by the immune system and that tumor cells with complex karyotypes may evolve mechanisms to counteract this recognition. We conclude with a discussion of how mistakes in cell division or their associated consequences can be targeted therapeutically to benefit patients with cancer.
Sources of mitotic errors
Cancer genomes are fluid, shape-shifting entities owing to a variety of genetic instability phenotypes, each of which exhibits its own unique mutational signature. Chromosomal instability (CIN) refers to the ongoing acquisition of genomic alterations that involve high rates of chromosome gain and loss (Lengauer et al. 1997). CIN is recognized as a general property of most aneuploid cancer cell lines and drives intratumoral heterogeneity, which allows for adaptation to changing environmental conditions (van Jaarsveld and Kops 2016). It is important to recognize that CIN and aneuploidy are distinct traits that are likely to have different impacts on tumor evolution and clinical behavior. While aneuploidy is a genetic state, CIN refers to the rate at which karyotypes diverge. Therefore, while CIN invariably leads to aneuploidy, cells can be stably aneuploid without exhibiting CIN. Below, we discuss the causes of CIN and how cell division errors contribute to the evolution of malignant karyotypes in human cancers.
Spindle assembly checkpoint (SAC) defects
The objective of mitosis is to faithfully segregate the replicated chromosomes into two new daughter cells. This is achieved by the attachment of chromosomes to microtubules (MTs) of the mitotic spindle apparatus. Chromosomes attach to ends of MTs at specialized protein structures, known as kinetochores, that assemble onto centromeric chromatin. Replicated chromosomes have two kinetochores, and biorientation is achieved when each sister kinetochore binds MTs oriented toward opposite spindle poles. A surveillance mechanism known as the SAC delays the separation of the sister chromatids at anaphase until all of the kinetochores have made correct attachments to spindle MTs (Fig. 1A). Components of the SAC localize to unattached kinetochores and function in a biochemical signaling cascade to inhibit activation of the CDC20-bound anaphase-promoting complex/cyclosome (APC/CCdc20), an E3 ubiquitin ligase that targets Cyclin B and Securin for degradation by the proteasome (Fig. 1A). Securin destruction liberates Separase, which then cleaves and inactivates the cohesin complex that holds the sister chromatids together, thereby allowing sister chromatid separation and the onset of anaphase (Fig. 1B). Degradation of Cyclin B inactivates cyclin-dependent kinase 1 (Cdk1), allowing mitotic exit and the completion of cell division.
Figure 1.
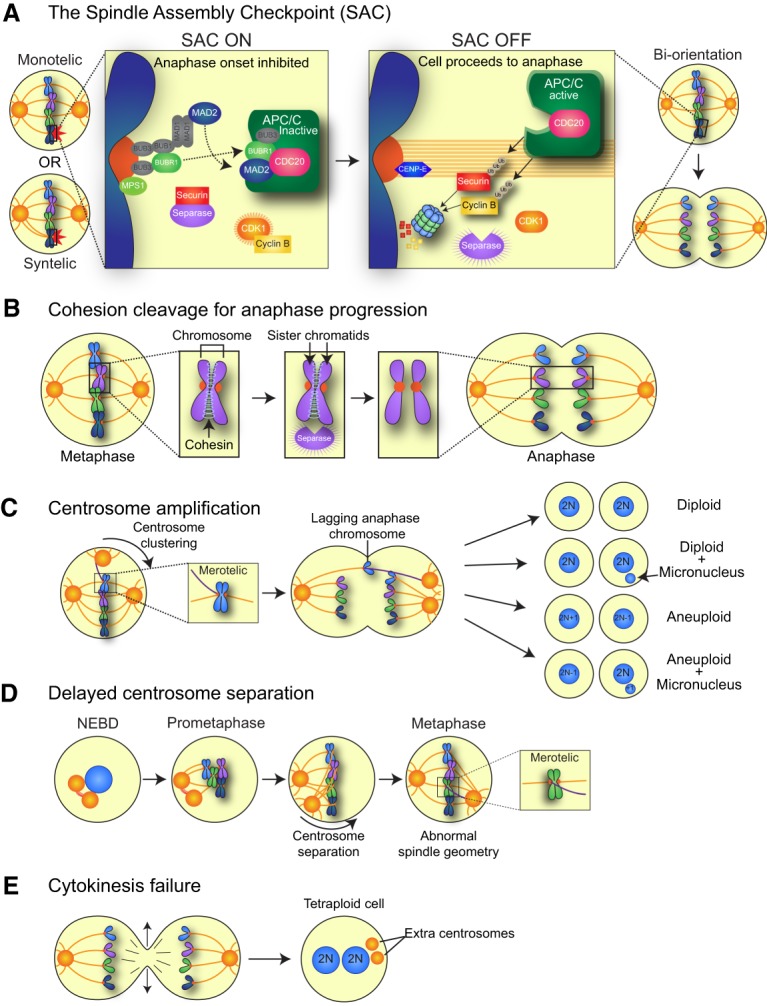
Chromosome segregation and sources of mitotic errors. (A) Unattached kinetochores activate an inhibitory SAC signal, which in turn blocks progression to anaphase. The target of the SAC is the APC/C, an E3 ubiquitin ligase that targets several proteins for degradation, including Cyclin B1 and Securin. When all kinetochores are correctly attached to MTs emerging from opposite poles of the cell (biorientation), the SAC is silenced, and APC/CCDC20 ubiquitinates and targets for degradation Cyclin B (to inactivate CDK1 and allow for mitotic exit) and Securin (to liberate the protease Separase and initiate the onset of anaphase). (B) Replicated sister chromatids are held together by the cohesin complex of proteins. Following silencing of the SAC, Securin is degraded, and the protease Separase is activated. Separase cleaves the cohesin complex to allow for sister chromatid separation and anaphase onset. (C) Extra centrosomes can generate a transient multipolar spindle, which, following centrosome clustering, leads to an increased rate of merotelic attachments, where one sister kinetochore is attached to MTs emerging from opposite poles. Merotelically attached chromosomes can lag in the middle of the spindle during anaphase and may subsequently be missegregated or incorporated into micronuclei. (D) After centrosome duplication, the two centrosomes are attached by a protein linker. This linker is disassembled prior to mitotic entry to allow the centrosomes to migrate apart and form opposite poles of the spindle. Delays in centrosome separation can lead to misattached chromosomes and/or abnormal spindle geometry that results in increased rates of chromosome missegregation. (E) Cleavage furrow regression leads to cytokinesis failure and the formation of a binucleate tetraploid cell with twice the normal centrosome content.
In mammals, inactivation of the SAC leads to dramatic chromosome segregation errors; thus, the SAC is essential for organismal development and the viability of most mammalian cells. However, while most cells require the SAC for continued growth, examples have emerged where the requirement for the SAC can be bypassed. For example, extending the time for chromosome alignment by lowering APC/C activity can render the SAC nonessential in human colorectal cancer cells (Wild et al. 2016; Sansregret et al. 2017), and some cells lacking the SAC component MAD2 can proliferate in vitro and in vivo if p53 is inactivated to allow tolerance to high levels of genome instability (Burds et al. 2005; Foijer et al. 2017).
The SAC is not an all-or-nothing response, but rather the strength of the signal depends on the number of unattached kinetochores (Collin et al. 2013). Thus, mutations that weaken the SAC can result in precocious anaphase onset before complete kinetochore attachment, which dramatically increases the probability of chromosome missegregation. Mouse models have shown that attenuating the SAC promotes aneuploidy and genome instability in vivo (Simon et al. 2015). Moreover, mutations in the SAC proteins TRIP13 and BUBR1 cause mosaic variegated aneuploidy (MVA), a rare disorder characterized by high levels of aneuploidy and an increased incidence of tumorigenesis (Hanks et al. 2004; Suijkerbuijk et al. 2010; Yost et al. 2017). Nevertheless, mutations in SAC genes are rare in human tumors, and cells with CIN do not generally enter anaphase precociously, indicating that SAC dysfunction is not a major contributor to the mitotic errors and karyotypic heterogeneity observed in human cancer cells (Holland and Cleveland 2012b).
Cohesion defects
The separation of the chromosomes at anaphase relies on the timely loss of sister cohesion (Fig. 1B). In principle, defects in the cohesin complex could result in precocious sister chromatid separation and chromosome segregation errors. Consistently, recent work has suggested that the deterioration of cohesion with advanced maternal age is a leading cause of meiotic defects and age-related aneuploidy in female oocytes (El Yakoubi and Wassmann 2017). Genes involved in sister chromatid cohesion have also been found to be mutated in colorectal cancers (Barber et al. 2008) and a wide range of myeloid neoplasms (The Cancer Genome Atlas Research Network 2013; Kon et al. 2013). However, the consequence of these mutations on chromosome segregation fidelity has not been tested. Since cohesin plays a major role in higher-order chromatin organization during interphase, cohesin defects could result in dysregulated gene expression that drives tumor development. Indeed, while recurrent mutations of the cohesin component STAG2 have been observed in diverse tumor types, many of these tumor-associated mutations do not adversely affect chromosome segregation (Solomon et al. 2011; Balbas-Martinez et al. 2013; Kim et al. 2016). This suggests that STAG2 mutations and possibly other tumor-associated mutations in the cohesin complex exert their tumorigenic effects outside of mitosis.
Merotelic attachments
While cells with CIN rarely display evidence of SAC or cohesion defects, they do exhibit an increase in lagging anaphase chromosomes as a consequence of increased merotelic kinetochore–MT (K–MT) attachments (Gascoigne and Taylor 2008; Thompson and Compton 2008). Initial capture of spindle MTs by kinetochores is asynchronous and stochastic. Consequently, erroneous K–MT attachments must be converted into bioriented attachments to permit faithful chromosome segregation. Merotelic attachments occur when a single kinetochore becomes bound to MTs anchored at both spindle poles (Fig. 1C). These types of attachments do not trigger a SAC-dependent mitotic delay, often resulting in anaphase onset without resolving the defect.
Most merotelically attached chromosomes segregate correctly during anaphase (Cimini et al. 2004). However, a proportion of chromosomes with these attachments are delayed in their segregation and end up lagging in the middle of the spindle (Fig. 1C). Lagging anaphase chromosomes are frequently observed in chromosomally unstable cancer cells. These tardy chromosomes can be missegregated to produce aneuploid daughter cells (Cimini et al. 2001; 2003). More frequently, however, lagging chromosomes are segregated to the correct daughter cell but fail to reach the main chromosome mass prior to nuclear envelope reassembly and are partitioned into a micronucleus (Thompson and Compton 2011). As described below, DNA trapped within micronuclei undergoes extensive DNA damage that can lead to chromosome rearrangements (Zhang et al. 2015). Merotelic attachments are thus likely to be a major source of genetic instability in human tumors; three main sources of these attachment errors—hyperstabilized K–MT interactions, centrosome amplification, and altered timing of centrosome separation—are discussed below.
K–MT stability
Efficient correction of erroneous K–MT attachments requires the detachment of MTs from inappropriately attached kinetochores. Consequently, reducing the turnover of K–MT interactions allows for the persistence of erroneous attachments and increases the frequency of chromosome segregation errors. Chromosomally unstable tumors exhibit hyperstable K–MT interactions relative to euploid cells (Bakhoum et al. 2009a). Moreover, reducing K–MT attachment stability restores faithful chromosome segregation in cells with CIN (Bakhoum et al. 2009b). This suggests that elevated K–MT attachment stability is a major driver of chromosome segregation errors. Overexpression of MAD2 or loss of STAG2 has been shown to result in hyperstabilized K–MT attachments that can predispose cells to CIN, but the molecular defects that cause an increase in K–MT attachment stability in most cells with CIN remain unclear (Kabeche and Compton 2012; Kleyman et al. 2014).
Centrosome amplification
A further source of merotelic attachments arises from the acquisition of extra copies of the centrosome, known as centrosome amplification (Fig. 1C). Supernumerary centrosomes are a common feature of human cancers and can arise through several different pathways, including a cell division failure, cell fusion, and centrosome overduplication (Chan 2011; Nigg and Holland 2018). The presence of extra centrosomes leads to the formation of a multipolar mitotic spindle, which, if not corrected prior to anaphase, results in the segregation of chromosomes into more than two daughter cells. Live-cell imaging has revealed that the progeny of multipolar divisions are frequently inviable, since daughter cells are unlikely to inherit a full complement of chromosomes (Ganem et al. 2009). The best-characterized mechanism for dealing with this burden is the clustering of extra centrosomes to form a pseudobipolar spindle (Fig. 1C; Quintyne et al. 2005; Basto et al. 2008; Kwon et al. 2008; Leber et al. 2010). Efficient centrosome clustering is required for the survival of cancer cells with extra centrosomes and requires multiple factors, including the minus end-directed motor protein HSET/KIFC1 (Kwon et al. 2008). A recent study revealed that centrosome clustering in epithelial cells was inhibited by E-Cadherin, which increases cortical contractility and suppresses centrosome movement (Rhys et al. 2018). Loss of E-Cadherin is frequently observed in breast cancer cells with high levels of centrosome amplification, suggesting that cancer cells can select for genetic changes that enable efficient centrosome clustering. While the coalescence of centrosomes in a multipolar spindle provides a pathway to avoid lethal divisions, it also promotes the formation of merotelic K–MT attachments that lead to lagging anaphase chromosomes (Ganem et al. 2009; Silkworth et al. 2009). This provides an explanation for the association of centrosome amplification with CIN and aneuploidy.
Besides supernumerary centrosomes, additional mechanisms can contribute to multipolarity and/or aberrant spindle geometry in cancer cells. For example, multipolar spindles can form independently of centrosome amplification following a loss of spindle pole integrity (Maiato and Logarinho 2014). In addition, overexpression of Aurora A kinase or loss of its negative regulator, CHK2 kinase, increases MT assembly rates. This leads to transient alterations in spindle geometry that promote the generation of erroneous K–MT attachments and lagging anaphase chromosomes (Ertych et al. 2014). Since overexpression of Aurora A and loss of CHK2 occur frequently in human cancers, this may represent an important pathway influencing CIN in tumors.
Timing of centrosome separation
The improper timing of centrosome separation prior to cell division is emerging as an additional source of genetic instability (Nam et al. 2015). After centrosome duplication, the two centrosomes are connected by a protein linker, which is dissolved prior to entry into mitosis (Fig. 1D). Both delaying and accelerating centrosome separation elevate the frequency of chromosome misattachments to the mitotic spindle, leading to chromosome segregation errors (Silkworth et al. 2012; Zhang et al. 2012; Nam and van Deursen 2014; Kanakkanthara et al. 2016; van Ree et al. 2016). The deubiquitinase USP44 localizes to the centrosome, and loss of this protein results in incomplete centrosome separation and elevated frequencies of lagging chromosomes. Importantly, USP44 knockout mice are prone to aneuploidization and spontaneous tumor formation (Zhang et al. 2012). Defective centrosome separation can also occur as a result of misregulation of the EG5/KIF11 motor protein that drives centrosome separation. Overexpression of EG5 leads to chromosome missegregation and increased tumor incidence (Castillo et al. 2007). Moreover, in addition to negatively regulating PI(3)K signaling through its phosphatase activity, the tumor suppressor protein PTEN also functions to promote the centrosomal recruitment of EG5 and control timely centrosome separation (van Ree et al. 2016). Mice carrying a PTEN mutant that is defective in promoting EG5 loading onto the centrosomes but active in antagonizing PI(3)K signaling show increased aneuploidy and tumor susceptibility, suggesting that PTEN's centrosomal role contributes to its tumor-suppressive function (van Ree et al. 2016). Defects in centrosome dynamics may therefore constitute a source of erroneous kinetochore attachments, which in turn drive CIN in human tumors.
Tetraploidy
A final source of mitotic errors arises from the proliferation of tetraploid cells, which have twice the normal chromosome content. Tetraploidy generally occurs through one of three main mechanisms. The first is cytokinesis failure, where daughter cells fail to separate following cell division (Fig. 1E). Second, tetraploidization can occur as a result of cell fusion, which can occur spontaneously or as a result of viral infection (Duelli et al. 2005, 2007). Finally, tetraploid cells can arise by endoreduplication, in which two rounds of DNA replication occur without an intervening cell division. Of these three main pathways, cytokinesis failure or mitotic failure is likely to be the major mechanism contributing to the production of tetraploid cells in premalignant lesions. One mechanism by which this can occur is via retention of chromatin in the middle of the spindle, which can induce furrow regression (Steigemann et al. 2009). Cytokinesis failure has also been reported to occur following entosis, where viable cells that are internalized by neighboring cells block furrow ingression (Krajcovic et al. 2011).
Proliferating tetraploid cells are genomically unstable and able to promote tumorigenesis in mice (Fujiwara et al. 2005; Davoli and de Lange 2012). The instability of tetraploid cells arises because these cells have twice the normal number of centrosomes, which endows cells with CIN (Fig. 1E; Ganem et al. 2009; Silkworth et al. 2009). In addition, the extra chromosomes in tetraploid cells buffer against deleterious mutations in essential and haploinsufficient genes, allowing continued growth in the face of otherwise lethal genomic alterations. Computational analysis of ∼5000 cancer genome sequences suggested that ∼37% of human cancers undergo a genome doubling event at some point during their evolution (Zack et al. 2013). Correspondingly, tetraploidy may represent an early step in tumorigenesis and has been observed in premalignant lesions in the esophagus and cervix as well as in non-small cell lung cancer (Galipeau et al. 1996; Olaharski et al. 2006; Jamal-Hanjani et al. 2017). Genome doubling is thus likely to represent an important intermediate in the development of many genomically unstable human tumors.
Consequences of mitotic errors
Mitotic errors lead to DNA damage
Mitotic errors have long been recognized to be a major source of whole-chromosomal aneuploidy, but recent evidence has also linked chromosome segregation errors to the generation of DNA damage that promotes structural alterations in chromosomes. Structural rearrangements alter the linear organization of chromosomes and are an established driver of tumorigenesis. Emerging evidence has suggested that lagging anaphase chromosomes, in addition to having a high risk of missegregation, are uniquely susceptible to the acquisition of DNA damage.
Chromosomes that lag in the middle of the spindle can be damaged if they fail to clear the spindle midzone prior to completion of cytokinesis (Fig. 2A). These chromosomes become trapped in the cleavage furrow, generating DNA double-strand breaks that are erroneously repaired to produce unbalanced translocations (Janssen et al. 2011). In addition to suffering direct DNA damage during cytokinesis, lagging chromosomes are often partitioned into micronuclei, where they acquire DNA damage in the following cell cycle. This arises in part because the nuclear envelope of micronuclei is unusually fragile and prone to spontaneous rupture, exposing the micronuclear DNA to potentially damaging cytoplasmic components (Hatch et al. 2013). Collapse of the micronuclear envelope during S phase leads to stalled replication and associated DNA damage (Crasta et al. 2012; Zhang et al. 2015). Moreover, micronuclei show delayed DNA replication kinetics, resulting in cells that enter into mitosis while replication of the micronuclear DNA is ongoing (Crasta et al. 2012). This leads to premature condensation and the fragmentation of the micronuclear chromosome (Crasta et al. 2012; Ly et al. 2017).
Figure 2.
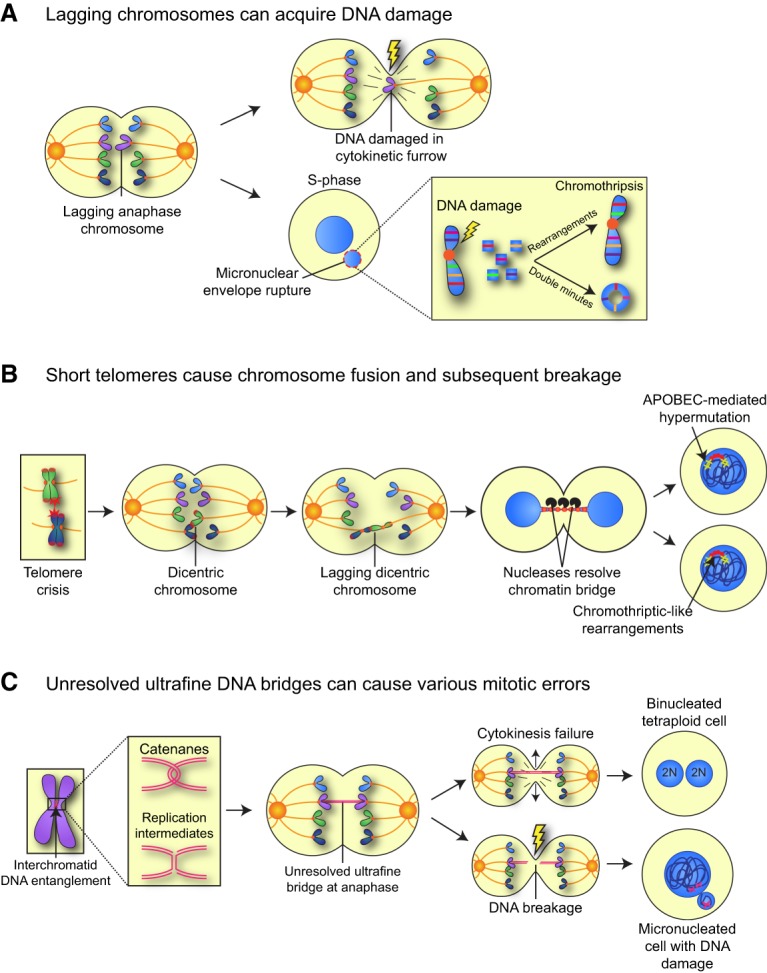
Mitotic errors can generate DNA damage. (A) Lagging chromosomes in anaphase can acquire DNA damage directly by being trapped in the spindle midzone during cytokinesis. In addition, lagging chromosomes that are partitioned into micronuclei can acquire DNA damage in interphase of the subsequent cell cycle. Extensive damage leads to chromosome shattering, a phenomenon known as chromothripsis, which results in the production of highly localized chromosome rearrangements and/or the production of double-minute chromosomes. (B) Extensive shortening of telomeres (telomere crisis) can result in the end-to-end fusion of two telomeres and the generation of a dicentric chromosome. Dicentric chromosomes can attach to opposite sides of the cell and be pulled apart during mitosis, resulting in a chromatin bridge that connects the two daughter nuclei. The nuclear membrane surrounding the bridging DNA ruptures in interphase, and the exposed DNA can be subject to cleavage by a cytoplasmic nuclease to resolve the bridge. The DNA exposed to the cytoplasm may undergo chromothriptic-like chromosome rearrangements and/or hypermutation generated by APOBEC cytidine deaminases. (C) DNA entanglements between sister chromatids can form at underreplicated regions or as a result of persistent DNA catenation. If these linkages are not resolved by topoisomerases and helicases, they can form ultrafine DNA bridges that connect the segregating sister chromatids in anaphase. Ultrafine bridges can lead to cytokinesis failure, resulting in a binucleated cell, or be broken during anaphase, creating DNA damage and micronuclei.
Massive DNA damage occurring on chromosomes isolated within micronuclei can produce complex patterns of localized chromosome rearrangements that are highly reminiscent of those observed following a phenomenon known as “chromothripsis” (Fig. 2A; Zhang et al. 2015). Chromothripsis is characterized by the presence of extensive chromosomal rearrangements restricted to one or a few chromosomes (Stephens et al. 2011; Holland and Cleveland 2012a). These alterations have been observed in a broad array of tumor types and occur at a higher frequency in specific types of cancers, including those that arise from the blood and brain (Rode et al. 2016). Therefore, partitioning of chromosomes into micronuclei offers an attractive mechanistic explanation for how mitotic errors promote acquisition of highly localized DNA damage.
The shattering and subsequent reassembly of chromosome fragments during chromothripsis can also lead to the production of circular acentric chromosomes, known as double minutes (Fig. 2A; Stephens et al. 2011; Zhang et al. 2015). These circular chromosomes can be present at very high numbers and often harbor oncogenes that drive tumor development. Double minutes have been observed in nearly half of tumors, and their random segregation during cell division leads to heterogeneity in oncogene copy number that renders tumors more adaptable to changing environmental conditions (Turner et al. 2017).
A further source of DNA damage emerges following telomere crisis, where extensive telomere shortening leads to the end-to-end fusion of two telomeres and the generation of a dicentric chromosome with two independent MT attachment sites (Fig. 2B). Dicentric chromosomes produce chromatin bridges that connect the two daughter nuclei in early G1. During anaphase, the dicentric chromosome can undergo breakage, which, following repair, can lead to nonreciprocal translocations (Artandi et al. 2000). In some instances, the trapped DNA from dicentric chromosomes can promote cytokinesis failure and the generation of tetraploid cells (Davoli and de Lange 2012). More frequently, however, the daughter cells migrate apart from each other, and the nuclear membrane surrounding the bridged DNA ruptures, allowing the exposed chromatin to be attacked by a cytoplasmic nuclease, resulting in the resolution of the bridge (Maciejowski et al. 2015). Sequencing revealed clustered chromothriptic-like rearrangements arising from the chromatin that was presumably trapped within the bridge. Surprisingly, these same regions were also associated with hypermutation or “kataegis,” which may arise from editing of exposed ssDNA on chromatin bridges by APOBEC cytidine deaminases (Fig. 2B). Such a pathway could explain how focal rearrangements and regions of clustered hypermutation are acquired as a result of mitotic errors.
Finally, DNA damage can also arise from the inappropriate resolution of DNA ultrafine bridges (UFBs). UFBs are thin segments of naked DNA that connect the segregating sister chromatids at anaphase. They are formed as a result of topological links between sister chromatids that arise as a result of persistent DNA catenation or replication stress (Bizard and Hickson 2018). Replication stress has been proposed to contribute to genomic alterations and CIN as a result of attempts to segregate underreplicated regions of the genome (Burrell et al. 2013). These DNA entanglements often occur at defined chromosomal loci, such as common fragile sites or centromeric regions, and are usually resolved prior to mitosis. If left unresolved, UFBs can break during anaphase and form micronuclei or lead to a failure or abscission, resulting in the production of binucleated tetraploid cells (Fig. 2C). These events occur frequently in some cancer cells, indicating that aberrant resolution of UFBs can lead to mitotic DNA damage that contributes to numerical and structural alterations in the tumor karyotype (Chan et al. 2009; Naim and Rosselli 2009; Tiwari et al. 2018).
Mitotic errors can trigger activation of p53
In studying the immediate effects of cell division errors on cellular proliferation, a common theme has emerged: Mistakes in cell division frequently lead to activation of the tumor suppressor protein p53, which in turn induces a cell cycle arrest, senescence, or apoptosis. The triggers for p53 activation following mitotic errors are complex and multifactorial. Activation of p53 occurs following chromosome missegregation, and, correspondingly, loss of p53 is frequently associated with aneuploidy in human cancers (Burds et al. 2005; Li et al. 2010; Thompson and Compton 2010). However, whether aneuploidy itself directly triggers p53 activation has remained unclear. Recently, two studies shed some light on this lingering question by showing that p53 activation is a potential but not obligatory outcome of chromosome missegregation (Santaguida et al. 2017; Soto et al. 2017). Complex aneuploidies that involve structural alterations do trigger p53 activation, likely as a result of DNA damage that is acquired during or following some mitotic errors. On the other hand, simple aneuploidies that involve only a small fraction of the genome can be propagated in a p53-competent background (Santaguida et al. 2017; Soto et al. 2017). These data suggest that whole-chromosome imbalances per se are not sufficient to activate p53, but events associated with chromosome missegregation or aneuploidy may do so.
Additional features of an erroneous mitosis could also contribute to p53 activation after division. For example, aneuploidy was proposed to increase the levels of reactive oxygen species that lead to activation of the ataxia telangiectasia-mutated (ATM) DNA damage signaling kinase and p53 (Fig. 3A; Li et al. 2010). Moreover, p53 stabilization following chromosome missegregation has been linked with the entrapment of chromatin in the cytokinetic furrow or the damage that accumulates in underreplicated DNA contained within micronuclei (Fig. 3A; Janssen et al. 2011; Crasta et al. 2012). A prolonged mitosis even in the absence of overt cell division errors can also trigger a p53-dependent cell cycle arrest in nontransformed cells (Uetake and Sluder 2010). Eliminating cells that delay in mitosis could serve as a quality control to prevent the proliferation of cells that have experienced stresses during mitosis (Lambrus and Holland 2017). Finally, recent work has proposed that differential phosphorylation of histones on lagging anaphase chromosomes is required to initiate p53 stabilization in the subsequent G1 (Hinchcliffe et al. 2016). However, how specific missegregated chromosomes could be marked by histone phosphorylation and how this feeds into p53 activation remain unclear.
Figure 3.
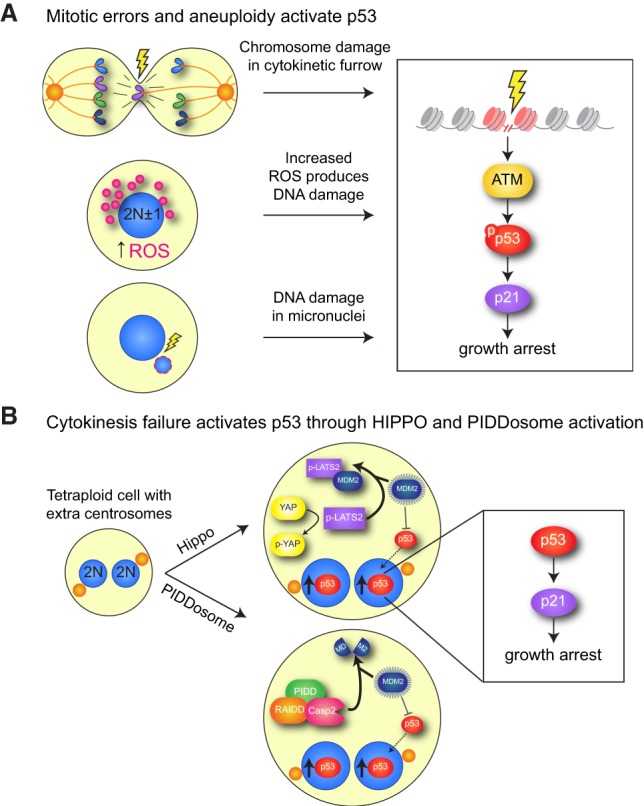
Mitotic errors can trigger activation of p53. (A) Lagging chromosomes that become damaged in the cleavage furrow or in micronuclei can elicit the canonical DNA damage repair pathway that activates p53. In addition, aneuploidy can also cause increased reactive oxidative species that lead to activation of DNA damage signaling. (B) Cytokinesis failure has been proposed to activate p53 through two distinct pathways. (1) Activation of the Hippo pathway. The Hippo pathway kinase LATS2 is activated in a tetraploid cell, leading to the phosphorylation and cytoplasmic sequestration of the transcription factor YAP. In addition, LATS2 binds and inactivates MDM2, a negative regulator of p53 stability. Inhibiting MDM2 allows for the increased accumulation of p53, which up-regulates p21 to elicit a growth arrest. (2) Activation of PIDDosome signaling. Tetraploid cells activate the PIDDosome, a multiprotein complex comprised of PIDD and RAIDD that in turn activates Caspase-2 (CASP2). Caspase 2 cleaves and inactivates MDM2, allowing p53 stabilization.
Given the potential harm that can arise as a result of the uncontrolled proliferation of tetraploid cells, it is perhaps unsurprising that mammalian cells have also evolved systems to limit the division of cells with increased ploidy. While some cells can proliferate following cytokinesis failure (Uetake and Sluder 2004; Wong and Stearns 2005), in many instances, tetraploid cells arrest in G1 due to the stabilization of p53 and up-regulation of the CDK inhibitor p21 (Andreassen et al. 2001; Kuffer et al. 2013). Two distinct pathways have been implicated in activating p53 to suppress the proliferation of tetraploid cells (Fig. 3B). In one pathway, cytokinesis failure activates the Hippo pathway kinase LATS2, which stabilizes p53 and inactivates the progrowth transcriptional regulator YAP (Ganem et al. 2014). In an alternative pathway, cytokinesis failure promotes the activation of the PIDDosome, a multiprotein complex that activates Caspase-2, leading to subsequent p53 stabilization (Fava et al. 2017). The presence of extra centrosomes in tetraploid cells has been suggested to be a key trigger for the activation of both LATS2 and the PIDDosome, but the extent to which these pathways act independently or collaborate to restrain the growth of tetraploid cells remains to be determined.
Since mitotic errors frequently trigger a cell cycle arrest in nontransformed cells, tumor cells with CIN are likely to acquire alterations that allow them to circumvent the anti-proliferative effects of p53 activation. Indeed, disruption of p53 is one of the most frequent events in tumorigenesis and allows cells to tolerate a broad range of insults, including CIN. Caspase 2 has been proposed to activate p53 following chromosome missegregation by cleaving MDM2, an E3 ubiquitin ligase that acts to target p53 for degradation by the proteasome. Consistently, loss of Caspase 2 is associated with an increased tolerance for karyotype imbalances (Dorstyn et al. 2012; Puccini et al. 2013; Dawar et al. 2017). In colorectal tumors, mutations in BCL9L confer tolerance to CIN by reducing Caspase 2 levels and preventing p53 stabilization (Lopez-Garcia et al. 2017). Finally, overexpression of cyclin D1 enables cells to circumvent a G1 arrest following genome doubling by sequestering p21 (Crockford et al. 2017). In summary, mitotic errors can directly or indirectly trigger activation of p53; thus, mechanisms that suppress or circumvent p53 activation are likely to be key contributors to the propagation of chromosomally unstable tumor cells.
Impact of mitotic errors on cell fitness
Given the detrimental effects of mitotic errors on genome stability, the question that naturally arises is how frequently these events occur in vivo. While mitotic errors are difficult to observe directly in tissues, several studies have measured the degree of aneuploidy in normal cells using fluorescence in situ hybridization (FISH), chromosome spreads, or spectral karyotyping. Surprisingly, initial estimates performed with FISH in healthy tissues suggested that 30%–50% of cells in the mammalian brain (Rehen et al. 2001; Pack et al. 2005; Yurov et al. 2007; Faggioli et al. 2012) and up to 50% of cells in the liver are aneuploid (Duncan et al. 2010, 2012). More recently, however, single-cell sequencing studies in these same tissues reported much lower levels of aneuploidy (<5% of cells), and similar low rates were observed in the skin (McConnell et al. 2013; Cai et al. 2014; Knouse et al. 2014; van den Bos et al. 2016). Since single-cell sequencing offers a more reliable technology for examining karyotypes at high resolution in an unbiased manner, these data indicate that cells with abnormal karyotypes are likely to be rare in healthy tissues (Bakker et al. 2015).
Low levels of aneuploidy in somatic tissues suggests that either the rates of mitotic errors in vivo are correspondingly low or that aneuploid cells are selected against/eliminated. While both assertions are likely correct, recent work has provided support for the idea that aneuploid cells are selected against in vivo. Hematopoietic stem cells (HSCs) with defined chromosome trisomies show a reduced fitness compared with euploid controls when transplanted into irradiated mice (Pfau et al. 2016). Similar experiments performed with chromosomally unstable BubR1H/H HSCs revealed that aneuploid cells were depleted from the peripheral blood over time. Importantly, nonproliferating tissues from BubR1H/H mice showed high levels of aneuploidy, while other regenerative tissues were largely euploid (Pfau et al. 2016). This suggests that in self-renewing adult tissues, aneuploid cells are under purifying selection and outcompeted by the relatively fitter euploid cells. In accord with these data, MVA patients that carry mutations in BUBR1 exhibit growth retardation and reduced brain size (Garcia-Castillo et al. 2008).
Similar to the observations made in vivo, aneuploidy is generally detrimental to cell proliferation in vitro (Gordon et al. 2012; Santaguida and Amon 2015). This fitness defect arises as a result of changes in the copy number of genes located on the aneuploid chromosomes (Torres et al. 2007, 2010; Pavelka et al. 2010; Stingele et al. 2012; Dephoure et al. 2014). The loss or gain of an entire chromosome alters the production of hundreds, if not thousands, of proteins. While altering the copy number of specific genes can bring about strong phenotypic changes, most phenotypes associated with aneuploidy arise from the simultaneous alteration of many gene products that have little effect when modified individually (Torres et al. 2007; Pavelka et al. 2010; Oromendia et al. 2012; Bonney et al. 2015). Analysis of yeast or human cells with extra copies of an individual chromosome revealed that while the abundance of most proteins correlated with increased gene dosage, ∼20%–25% of the proteins encoded on the additional chromosomes were expressed at close to diploid levels (Stingele et al. 2012; Dephoure et al. 2014). Importantly, the majority of these proteins is components of macromolecular complexes. These data suggest that aneuploid cells counteract the production of partially assembled multisubunit complexes by degrading uncomplexed subunits.
The degradation of protein subunits produces an increased load on protein folding and degradation pathways of aneuploid cells, explaining why these cells exhibit traits indicative of protetoxic stress (Torres et al. 2007; Oromendia et al. 2012; Sheltzer et al. 2012; Stingele et al. 2012). Aneuploid cells are also prone to protein aggregation and up-regulate autophagy-mediated protein degradation (Santaguida et al. 2015). The stress produced from aneuploidy-induced protein imbalances results in an increased sensitivity to compounds that inhibit autophagy or interfere with protein folding or degradation (Tang et al. 2011). In addition, mutations that compromise the ubiquitin–proteasome pathway produce synthetic fitness defects in aneuploid cells (Dodgson et al. 2016a,b). These data suggest that, along with chromosome-specific effects elicited by dosage alteration of certain genes, aneuploid cells also share a set of associated stress phenotypes that is largely independent of the specific karyotypic alteration.
The association of aneuploidy with decreased cellular fitness is seemingly counterintuitive with the observation that aneuploidy is a nearly universal feature of human tumors. This “aneuploidy paradox” remains to be fully resolved, but several possible explanations have emerged. First, the effects of chromosome segregation errors may be revealed only under the appropriate evolutionary constraints. Under the stringent selective pressure in the tumor microenvironment, most karyotypic alterations are expected to reduce cell fitness and be selected against. However, in rare instances, karyotypes may emerge that provide a selective advantage in a specific environmental setting. Indeed, aneuploidy has been shown to offer a selective advantage to yeast and human cells under conditions of environmental stress (Pavelka et al. 2010; Chen et al. 2012; Rutledge et al. 2016). Moreover, some nontumor cell lines acquire specific aneuploidies in culture that increase cellular proliferation, showing that certain aneuploid karyotypes can be beneficial even in the absence of cellular transformation (Ben-David et al. 2014). An alternative hypothesis for the prevalence of aneuploidy in human tumors is that cancer cells acquire alterations that allow them to tolerate the adverse stresses associated with karyotype imbalances. For example, loss of the deubiquitinating enzyme UBP6 improves the proliferation rate of several aneuploid yeast strains (Torres et al. 2010). Together, these studies show that aneuploidy does not inevitably suppress cellular proliferation but can in fact be selected for under specific environmental conditions.
Aneuploidy can promote further genome instability
Mitosis is a dynamic and finely tuned event that is particularly sensitive to perturbations in gene expression arising from karyotype alterations. Consistently, analysis in yeast has shown that aneuploid cells are less genomically stable and show increased rates of chromosome missegregation, mitotic recombination, and defective DNA damage repair (Sheltzer et al. 2011; Zhu et al. 2012). In human cells, specific aneuploidies have also been shown to increase mitotic error frequency (Nicholson et al. 2015; Passerini et al. 2016). In addition, while human cell lines with defined trisomies show reduced cell growth in vitro and in xenograft tumor assays, spontaneous karyotype evolution occurs during prolonged growth and improves cellular fitness (Sheltzer et al. 2017). These data suggest that aneuploidy can promote further karyotype instability and facilitate the acquisition of growth-promoting alterations.
Recent work has begun to elucidate how karyotype alterations impair mitotic fidelity. Even the presence of a single extra chromosome can trigger genomic instability by reducing the abundance of key replication proteins and impairing DNA replication (Passerini et al. 2016). These replication defects lead to the acquisition of DNA damage that promotes chromosome rearrangements and an increased frequency of mitotic errors (Santaguida et al. 2017). In addition, aneuploidy can also generate karyotype-specific phenotypic changes that lead to mitotic defects. For example, trisomy of chromosome 13 results in a cytokinesis defect because of increased expression of a gene encoded on the aneuploid chromosome (Nicholson et al. 2015). However, CIN is clearly not a necessary outcome of aneuploidy, as cells from individuals with specific trisomies exhibit rates of chromosome missegregation similar to those of euploid cells in vitro, and single-cell sequencing of neurons from an individual with Down syndrome failed to reveal additional aneuploidies (Valind et al. 2013; van den Bos et al. 2016).
Mitotic errors and tumorigenesis
The notion that mitotic errors could contribute to tumorigenesis was first postulated >100 years ago by Boveri (1914). However, whether cell division errors promote tumorigenesis or arise as a byproduct of transformation has remained an area of active debate. The fact that genes that control chromosome segregation are rarely mutated in human cancers raises the possibility that inducing CIN is a passenger event in tumor development. Indeed, inactivation of several tumor suppressor genes has been shown to promote CIN and aneuploidy (Manning et al. 2010; van Ree et al. 2016). Nevertheless, the weight of evidence suggests that, at least under some circumstances, mitotic errors do contribute to tumorigenesis. First, patients with MVA exhibit high levels of aneuploidy and an increased predisposition to certain types of cancers (Garcia-Castillo et al. 2008). Second, mitotic errors and aneuploidization can be found early during tumor evolution, and the extent of chromosomal aberrations correlates with tumor grade and poor prognosis (Mugneret et al. 2003; van de Wetering et al. 2007; Walther et al. 2008; M'Kacher et al. 2010; Bakhoum et al. 2011). Finally, perhaps the most persuasive evidence to support a causative link between CIN and tumor development comes from the study of mouse models with increased rates of mitotic errors caused by reduced or elevated levels of SAC components. Many of these models with CIN exhibit an increased incidence of spontaneous tumors and/or elevation of chemically or genetically induced tumor formation (Dobles et al. 2000; Michel et al. 2001; Babu et al. 2003; Dai et al. 2004; Jeganathan et al. 2006, 2007; Iwanaga et al. 2007; Sotillo et al. 2007; Weaver et al. 2007; Baker et al. 2009; Li et al. 2009; van Ree et al. 2010; Foijer et al. 2014). For an extensive discussion of the spectrum of tumors formed in these animals, see the following reviews: Holland and Cleveland (2009), Simon et al. (2015), and Naylor and van Deursen (2016).
How mitotic defects can act to promote tumor development remains an area of intense study. As a first path to facilitate tumorigenesis, CIN drives a continually evolving karyotype that produces genetic diversity in the tumor cell population. In addition to population-level genetic variation, the dosage imbalances produced by aneuploidy have been shown to reduce the robustness of biological networks and increase cellular variability (Beach et al. 2017). Together, this genetic and nongenetic heterogeneity creates phenotypic diversity. While the vast majority of alterations to the karyotype is expected to be detrimental, a small fraction of those changes could be advantageous and selected for during tumor evolution. The chromosomal location and relative density of tumor suppressor genes and oncogenes have been proposed to play an important role in shaping the tissue-specific patterns of aneuploidy observed in different types of cancer (Davoli et al. 2013; Sack et al. 2018). This may explain why certain chromosomal alterations are observed recurrently in some tumor types, such as gain of chromosome 8, which is observed in 25% of chronic myeloid leukemia cases and 10%–15% of cases of acute myeloid leukemia (Paulsson and Johansson 2007).
A widely proposed mechanism by which mitotic errors facilitate tumor development is through the loss of a chromosome that contains the remaining wild-type copy of a tumor suppressor gene. Indeed, this has been shown to occur in chromosomally unstable mice that were heterozygous for p53 or carried a mutated APC allele (Baker et al. 2009). In addition to promoting primary tumor growth, recent work functionally linked CIN with metastasis by showing that chromosomally unstable tumor cell lines are more likely to spread and form new tumors when compared with the same cells in which CIN was suppressed (Bakhoum et al. 2018). Finally, the genetic instability produced by CIN could contribute to the evolution of resistance in response to targeted anti-cancer therapies. In mice, CIN driven by overexpression of the SAC protein MAD2 provides the evolutionary fuel to facilitate tumor recurrence following withdrawal of the KRAS oncogene (Sotillo et al. 2010). Genetically engineered mice that recapitulate the ongoing karyotype changes observed in the majority of human tumors are thus likely to represent powerful models for testing the efficacy of emerging clinical drug candidates.
The most extensive characterization of the role of mitotic errors in tumorigenesis has emerged from the development of mouse models that possess elevated or reduced levels of SAC proteins. These animals display ongoing CIN and increased aneuploidy in cells and tissues. While many of these models are tumor-prone, some exhibit high levels of aneuploidy without an increase in tumor predisposition, demonstrating that the degree of aneuploidy is not an accurate predictor of tumor susceptibility (Baker et al. 2006). One possible explanation for this observation is that some of the proteins that are manipulated have functions outside of mitosis that confound interpretations of the tumor phenotypes (Funk et al. 2016). For example, SAC proteins have been proposed to play roles in insulin signaling (Choi et al. 2016), transcriptional repression (Yoon et al. 2004), DNA replication and repair (Sugimoto et al. 2004; Dotiwala et al. 2010), and membrane trafficking (Wan et al. 2014). An alternative possibility is that the genes manipulated to induce chromosome missegregation lead to distinct types of mitotic errors that change the karyotype in different ways. For example, reduction in the levels/activity of the MT motor protein CENP-E leads to the initiation of anaphase with polar chromosomes, resulting in whole-chromosome gain and loss events (Weaver et al. 2003). On the other hand, overexpression of MAD2 produces lagging anaphase chromosomes that can be subjected to DNA double-strand breaks and serve as a source of chromosomal rearrangements (Sotillo et al. 2007). It will be interesting to determine to what extent the frequency of DNA breaks that result from mitotic errors correlates with the propensity for tumor development.
Although considerable effort has been focused on modeling mitotic errors using mice with altered levels of SAC components, SAC dysfunction does not appear to be a major driver of CIN in human tumors. Given the established role of centrosome amplification in promoting CIN and its widespread presence in aneuploid human tumors, recent attention has turned to generating mice in which extra centrosomes could be generated by overexpressing PLK4, the master regulator of centrosome biogenesis (Marthiens et al. 2013; Coelho et al. 2015; Kulukian et al. 2015; Vitre et al. 2015; Sercin et al. 2016; Levine et al. 2017). Modest overexpression of Plk4 produced chronic centrosome amplification and aneuploidy in multiple tissues and was sufficient to drive the formation of lymphomas and squamous cell carcinomas (Levine et al. 2017). Strikingly, these tumors exhibited high levels of aneuploidy, ongoing chromosome segregation errors, and defective p53 signaling. Furthermore, tumors that formed as a result of centrosome amplification exhibited complex karyotypes that mimicked those frequently found in human tumors. It will now be valuable to develop additional animal models that mimic other mitotic aberrations frequently observed in human tumor cells, such as hyperstabilized K–MT interactions.
Although mitotic errors have long been implicated in driving cancer, it is becoming clear that in some contexts, increasing chromosome segregation errors can act to suppress tumorigenesis. In several examples where tumors develop with low rates of CIN, further increasing CIN suppressed tumor incidence (Weaver et al. 2007; Baker et al. 2009; Silk et al. 2013). Indeed, combining low rates of chromosome missegregation from expression of a mutant APC allele with additional CIN from reduced levels of CENP-E resulted in increased cell death that suppressed tumor progression but not initiation (Zasadil et al. 2016). This suggests that low rates of chromosome missegregation can promote tumor development, while high levels of CIN lead to the loss of essential chromosomes and tumor suppression (Funk et al. 2016). This explains the seemingly paradoxical observation that low levels of CIN are associated with a poor outcome in estrogen receptor-negative breast cancer, while high levels of CIN correlate with improved long-term survival (Birkbak et al. 2011; Roylance et al. 2011; Jamal-Hanjani et al. 2015). Since excessive chromosome segregation errors are lethal, tumors may select for alterations that antagonize the effects of excessive CIN. Subtly increasing the duration of mitosis by reducing APC/C activity reduces chromosome segregation errors in cells with CIN (Sansregret et al. 2017). Therefore, increasing the duration of mitosis could be a strategy used by cancer cells to tune the level of CIN and counteract the long-term fitness defects caused by excessive chromosome segregation errors. Consistently, single-cell genome sequencing of human breast tumors revealed that aneuploidy occurs early during tumor evolution but remains relatively stable during tumor outgrowth (Wang et al. 2014). This suggests that once a critical point has been reached, increased genome stability can be selected for to aid tumor growth.
Taken together, the available data suggest that mitotic errors can have distinct impacts at different points during tumor development. Low rates of mitotic errors can be tumor-promoting, particularly in the context of inactivating pathways that suppress the growth of aneuploid or polyploid cells. Nevertheless, higher rates of chromosome segregation errors lead to loss of essential chromosomes and tumor suppression. Identifying genetic alterations that cooperate to facilitate the transformation of chromosomally unstable cells is an important area of future work.
Mitotic errors trigger activation of the innate immune response
It has been appreciated since the 1960s that the immune system can recognize tumor antigens and eliminate nascent tumor cells (Bose 2017). These findings have spurred the development of immunotherapies that act to stimulate the immune system's ability to attack cancer cells that evolve to evade immune recognition. A compelling body of new experimental data supports the notion that the immune system can recognize and eliminate cells with complex karyotypes, raising the question of how these alterations are detected and how they contribute to shaping tumor evolution and therapeutic responses.
Cancer cells with abnormal karyotypes can emit signals that serve to increase their immunogenicity. One major signal is driven by the constitutive endoplasmic reticulum stress in aneuploid cells that leads to an increased exposure of immunogenic cell surface molecules and subsequent clearance by innate and adaptive immune cells (Fig. 4A; Senovilla et al. 2012; Acebes-Huerta et al. 2016). More recent work has implicated the innate immune receptor cGAS (cyclic GMP–AMP [cGAMP] synthase) as a key mediator of immune signaling in cells that undergo mitotic errors (Harding et al. 2017; Mackenzie et al. 2017). cGAS is a cytosolic DNA sensor that is activated upon binding to dsDNA to catalyze the production of the signaling molecule cGAMP (Sun et al. 2013; Wu et al. 2013). The second messenger cGAMP binds and activates STING (stimulator of interferon genes), leading to the production of type I interferon and other proinflammatory cytokines that trigger the immune response (Fig. 4B). Intracellular cGAMP can spread to neighboring cells through gap junctions to rapidly produce a paracrine proinflammatory signaling program (Ablasser et al. 2013).
Figure 4.
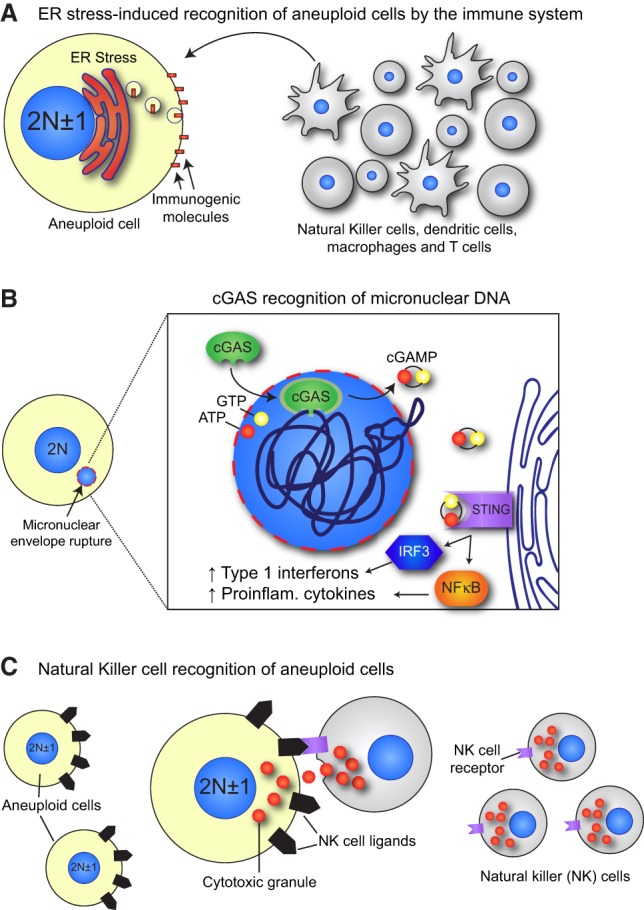
Mitotic errors activate the immune system. (A) Aneuploid cells exhibit a constitutive endoplasmic reticulum (ER) stress that leads to the increased surface exposure of immunogenic cell surface molecules, such as calreticulin. These are recognized by immune cells such as natural killer (NK) cells, dendritic cells, macrophages, and T cells that engulf or kill the aneuploid cell. (B) The micronuclear envelope is prone to rupture, leading to the exposure of the entrapped chromatin to cytoplasmic DNA-sensing molecules, such as cGAS. cGAS is activated by the exposed micronuclear DNA, allowing for conversion of ATP and GTP to the second messenger cGAMP. cGAMP activates STING, which causes IRF3- and NFκB-mediated expression of type 1 interferons and proinflammatory cytokines, respectively. (C) Aneuploid cells exhibit increased expression of NK cell-activating ligands, which allow recognition and killing of aneuploid cells by NK cells through their NKG2D and DNAM1 receptors.
During interphase, DNA is compartmentalized in the nucleus and inaccessible to cytosolic cGAS. Mitotic errors can cause whole chromosomes or parts of chromosomes to be partitioned into micronuclei. As discussed previously, chromosomes contained within micronuclei are subject to massive chromosome fragmentation following the spontaneous rupture of the micronuclear membrane (Crasta et al. 2012; Hatch et al. 2013; Zhang et al. 2015). This allows cGAS access to the damaged micronuclear DNA, triggering proinflammatory cytokine production (Fig. 4B). Micronuclear rupture may also release DNA fragments into the cytosol, explaining why chromosomally unstable cells have high levels of cytosolic DNA (Bakhoum and Landau 2017). Accordingly, inflammatory signals are up-regulated in cells with micronuclei generated by ionizing radiation (Mackenzie et al. 2017). Damaged DNA inside of micronuclei is therefore likely to act as an important source of immunostimulatory DNA.
The formation of micronuclei has also been identified as a critical mediator of cGAS–STING activation following DNA damage (Harding et al. 2017). DNA breaks that persist in mitosis lead to the generation of chromosome fragments that lack centromeres and cannot be segregated. These chromatin fragments escape from the nucleus and result in the formation of micronuclei in the next cell cycle. Recent work has shown that passage through mitosis and the formation of micronuclei are required for activation of cGAS–STING in response to DNA damage (Harding et al. 2017). Taken together, these data suggest that the cGAS–STING pathway can act as a cell-intrinsic surveillance pathway to detect the presence of cytosolic DNA resulting from DNA damage and chromosome segregation defects.
A final pathway to activate cGAS–STING after errors in mitosis is through the induction of cellular senescence (Dou et al. 2017; Gluck et al. 2017; Yang et al. 2017). Senescence is an irreversible growth arrest brought about by stresses such as ionizing radiation, oncogene activation, or persistent DNA damage (Tchkonia et al. 2013). cGAS has been shown to recognize cytosolic chromatin fragments released from the nucleus of senescent cells and in turn triggers the production of a variety of inflammatory mediators that activate the immune system, collectively known as the senescence-associated secretory phenotype (SASP) (Dou et al. 2017; Gluck et al. 2017; Yang et al. 2017). Arrested aneuploid cells have been shown to exhibit senescent characteristics and produce a SASP-like gene expression signature in vitro (Santaguida et al. 2017). These aneuploid cells also display increased expression of natural killer (NK) cell-activating ligands and are efficiently eliminated when cocultured with NK cells in vitro (Fig. 4C; Santaguida et al. 2017). Future work will focus on determining the importance of micronucleus- or senescence-induced cytosolic chromatin for triggering activation of cGAS–STING in aneuploid cells. Moreover, it remains to be established how effectively these pathways act to promote the immune clearance of aneuploid cells in vivo. Finally, it should be noted that activation of the cGAS–STING pathway in aneuploid tumors can act as a “double-edged sword” that both promotes clearance of cells with abnormal karyotypes and contributes to inflammation that drives tumorigenesis (Bose 2017).
Taken together, the evidence suggests that recognition of cancer cells by the immune system can be mediated by an aneuploid state or alterations associated with it. Tumor cells with complex karyotypes must thus evolve mechanisms to suppress recognition by the immune system. Accordingly, chromosomally unstable tumor cell lines avoid induction of proinflammatory signaling despite high levels of cytosolic DNA (Bakhoum and Landau 2017). Moreover, analysis of tumors from The Cancer Genome Atlas data sets revealed that cancers with highly aneuploid karyotypes exhibited reduced expression of genes related to cytotoxic immune functions and the production of proinflammatory cytokines (Buccitelli et al. 2017; Davoli et al. 2017; Taylor et al. 2018). A retrospective study of melanoma patients treated with immune checkpoint blockade anti-CTLA4 therapy showed that those with low levels of aneuploidy responded better. These data support the idea that the microenvironment of karyotypically deranged tumors is immunosuppressive and suggest that evaluating the levels of tumor aneuploidy could be used to predict responsiveness to immunotherapy.
Genetic alterations that enhance resistance to the host immune system have been identified in cancers (Khong and Restifo 2002). For example, loss of heterozygosity of human leukocyte antigen (HLA) occurs in 40% of non-small cell lung cancers and acts to suppress immune recognition by reducing neoantigen presentation (McGranahan et al. 2017). Therefore, identifying whether this or other mechanisms are responsible for the reduced immunogenicity of highly aneuploid tumors is an important area of future work that could offer insights into how to improve patient responsiveness to immunotherapy.
Exploiting mitotic errors for cancer therapy
Given the widespread prevalence of mitotic errors in human cancers, several approaches have been explored to target the mitotic apparatus or exploit weaknesses associated with the aneuploid state. With a long history of clinical efficacy, MT targeting agents are the most widely used anti-cancer drugs that target cell division. One of the most successful drugs in this class is paclitaxel, which has been used for decades to treat breast, ovarian, and lung cancer. Paclitaxel binds and stabilizes the MT lattice and, at high concentrations, arrests dividing cells in mitosis by preventing silencing of the SAC, leading to either cell death or senescence. However, clinically relevant doses of paclitaxel do not generate a mitotic arrest but rather lead to the formation of multipolar spindles that induce massive chromosome missegregation and cell death (Symmans et al. 2000; Orth et al. 2011; Zasadil et al. 2014). While killing of dividing cells is an attractive model for the anti-tumor actions of paclitaxel, this mechanism is difficult to reconcile with the slow proliferation rate of many solid tumors, which predicts that too few cells progress through mitosis in the presence of the drug to account for broad tumor killing (Mitchison 2012). Recently, an alternative “inflammatory micronucleation” model has been put forward that relies on the unusual ability of paclitaxel to induce the formation of many micronuclei in cells following exit from mitosis (Mitchison et al. 2017). In this model, micronucleation in a subset of cells that passes through mitosis in the presence of paclitaxel produces a proinflammatory signal that leads to en masse killing of tumor cells. While this proposal remains to be tested, it has the attractive feature of explaining why paclitaxel is more effective at killing solid tumors than other drugs that target the mitotic apparatus but do not induce micronucleation.
The clinical success of paclitaxel spurred the development of multiple mitotic-specific drugs that target enzymes required for cell division (Dominguez-Brauer et al. 2015). For example, several groups have developed inhibitors of the SAC kinase MPS1 (Colombo et al. 2010; Jemaa et al. 2013; Tannous et al. 2013; Maachani et al. 2015; Wengner et al. 2016). These drugs override the mitotic checkpoint and increase the frequency of chromosome segregation errors, leading to the generation of inviable karyotypes. Excitingly, MPS1 inhibitors have been found to sensitize xenograft tumors to paclitaxel-induced killing by elevating the chromosome segregation errors above a threshold required for viability (Jemaa et al. 2013; Tannous et al. 2013; Maia et al. 2015). However, despite promising preclinical results, mitosis-specific drugs have shown limited efficacy in clinical trials and in most cases are outperformed by classic MT targeting agents such as paclitaxel (Komlodi-Pasztor et al. 2012). One reason for this discrepancy is likely to be the slow proliferation rate of tumors in vivo compared with cancer cell lines and xenograft tumors on which the drugs were tested (Komlodi-Pasztor et al. 2011). An additional barrier limiting the success of mitotic-specific drugs is their inability to discriminate between the divisions of normal cells and tumor cells, resulting in bone marrow toxicity that limits the dose and duration of treatment.
Next-generation mitosis-specific drugs are likely to be more successful if they exploit tumor-specific vulnerabilities. One example of such an approach is to target the divisions of cancer cells with extra centrosomes. Since cancer cells efficiently cluster extra centrosomes to avoid lethal multipolar divisions, inhibiting the pathways required for centrosome clustering will selectively destroy cells with extra centrosomes without affecting the growth of normal cells. The minus-end-directed motor protein HSET/KIFC1 has been identified as a major component required for centrosome clustering (Kwon et al. 2008). HSET is not required for the growth of cells with the normal number of centrosomes but is required for the viability of tumor cells with extra centrosomes. This knowledge has sparked the development of a suite of new drugs that aim to destroy tumor cells with extra centrosomes by suppressing centrosome clustering (Rebacz et al. 2007; Castiel et al. 2011; Raab et al. 2012; Shiheido et al. 2012; Kawamura et al. 2013; Pannu et al. 2014; Bhakta-Guha et al. 2015; Johannes et al. 2015; Li et al. 2015; Tan et al. 2015; Zhang et al. 2016). While these compounds have been shown to decluster centrosomes and induce lethal multipolar divisions in cell culture, whether these tumor-specific drugs achieve an enhanced therapeutic index and improved clinical efficacy remains to be determined.
In addition to targeting the cell division machinery, it may also be possible to expose the consequence of cell division errors by exploiting vulnerabilities associated with the aneuploid state itself. Aneuploid cells are more sensitive than euploid cells to compounds that exacerbate proteotoxic stress and metabolic stress (Tang et al. 2011). Moreover, pharmacological activation of both of these stress pathways acts synergistically to suppress the growth of chromosomally unstable xenograft tumors. Aneuploid cells also exhibit dysregulated sphingolipid metabolism that leads to increased levels of the proapoptotic lipid ceramide (Hwang et al. 2017; Tang et al. 2017). Correspondingly, pharmacological agents that increase ceramide levels are more toxic to aneuploid cells than diploid cells (Tang et al. 2017). Together, these studies offer a proof of principle that the aneuploid state can be exploited therapeutically and open the door to the possibility of generating broad-spectrum anti-cancer drugs that aim to exacerbate stresses inherent to aneuploid cells.
Perspective
Research into the basic mechanisms underlying faithful chromosome segregation has revealed insights into how mitotic errors contribute to intratumor heterogeneity, tumor progression, metastasis, and adaptive evolution in response to therapy. However, we still lack an in-depth understanding of the long-term impact of cell division errors in vivo. In the future, animal models that recapitulate the molecular defects responsible for promoting mitotic errors in human cancers will be instrumental in elucidating the physiological consequences of cell division errors and their impact on tumorigenesis. Such animal models will also aid in understanding the emerging link between cell division errors and the activation of the immune system. In particular, it remains unclear how effectively aneuploid cells are recognized and cleared by the immune system in vivo. Moreover, whether the proinflammatory consequences of cell division errors drive the evolution of an immunosuppressive tumor microenvironment remains to be tested.
The development of methodologies for evaluating the rates of cell division errors in vivo will be critical if they are to be leveraged for therapeutic intervention. It will therefore be important to move away from using FISH and metaphase spreads to measure aneuploidy and instead turn to more reliable and higher-resolution single-cell sequencing to evaluate karyotype changes. We will also benefit from further insight into how p53 is activated following cell division errors and how tumor cells evolve mutations to tune the rate of CIN. Understanding the impact of cell division errors on cellular physiology holds great promise for our understanding and treatment of cancer. In this regard, exploiting molecular differences in the ways that normal and tumor cells divide to selectively target the division of cancer cells is a particularly promising therapeutic avenue.
Acknowledgments
We thank our laboratory members for helpful discussions and apologize to colleagues whose work could not be cited due to space limitations. Work in the Holland laboratory was supported by a R01 research grant from the National Institutes of Health (NIH; GM 114119), an American Cancer Society Scholar grant (129742-RSG-16-156-01-CCG), and a March of Dimes Research grant (1-FY17-698) to A.J.H., and a National Research Service Award F31 grant from the NIH (GM122288) to M.S.L.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.314351.118.
References
- Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, Hornung V. 2013. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503: 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebes-Huerta A, Lorenzo-Herrero S, Folgueras AR, Huergo-Zapico L, Lopez-Larrea C, Lopez-Soto A, Gonzalez S. 2016. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. Oncoimmunology 5: e1074378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. 2001. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell 12: 1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. 2000. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406: 641–645. [DOI] [PubMed] [Google Scholar]
- Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, van Deursen JM. 2003. Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 160: 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, van Deursen JM. 2006. Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol 172: 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Jin F, Jeganathan KB, van Deursen JM. 2009. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 16: 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Landau DA. 2017. Chromosomal instability as a driver of tumor heterogeneity and evolution. Cold Spring Harb Perspect Med 7: a029611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Genovese G, Compton DA. 2009a. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr Biol 19: 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, Compton DA. 2009b. Genome stability is ensured by temporal control of kinetochore–microtubule dynamics. Nat Cell Biol 11: 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Danilova OV, Kaur P, Levy NB, Compton DA. 2011. Chromosomal instability substantiates poor prognosis in patients with diffuse large B-cell lymphoma. Clin Cancer Res 17: 7704–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Silkworth WT, Nardi IK, Nicholson JM, Compton DA, Cimini D. 2014. The mitotic origin of chromosomal instability. Curr Biol 24: R148–R149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. 2018. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553: 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker B, van den Bos H, Lansdorp PM, Foijer F. 2015. How to count chromosomes in a cell: an overview of current and novel technologies. Bioessays 37: 570–577. [DOI] [PubMed] [Google Scholar]
- Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, et al. 2013. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet 45: 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, et al. 2008. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci 105: 3443–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. 2008. Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach RR, Ricci-Tam C, Brennan CM, Moomau CA, Hsu PH, Hua B, Silberman RE, Springer M, Amon A. 2017. Aneuploidy causes non-genetic individuality. Cell 169: 229–242.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Arad G, Weissbein U, Mandefro B, Maimon A, Golan-Lev T, Narwani K, Clark AT, Andrews PW, Benvenisty N, et al. 2014. Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat Commun 5: 4825. [DOI] [PubMed] [Google Scholar]
- Bhakta-Guha D, Saeed ME, Greten HJ, Efferth T. 2015. Dis-organizing centrosomal clusters: specific cancer therapy for a generic spread? Curr Med Chem 22: 685–694. [DOI] [PubMed] [Google Scholar]
- Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, Tan IB, Richardson AL, Szallasi Z, Swanton C. 2011. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res 71: 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizard AH, Hickson ID. 2018. Anaphase: a fortune-teller of genomic instability. Curr Opin Cell Biol 52: 112–119. [DOI] [PubMed] [Google Scholar]
- Bonney ME, Moriya H, Amon A. 2015. Aneuploid proliferation defects in yeast are not driven by copy number changes of a few dosage-sensitive genes. Genes Dev 29: 898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose D. 2017. cGAS/STING pathway in cancer: Jekyll and Hyde story of cancer immune response. Int J Mol Sci 18: E2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T. 1914. Zur frage der entstenhung maligner tumoren. Gustav Fischer Verlag, Jena, Germany. [Google Scholar]
- Buccitelli C, Salgueiro L, Rowald K, Sotillo R, Mardin BR, Korbel JO. 2017. Pan-cancer analysis distinguishes transcriptional changes of aneuploidy from proliferation. Genome Res 27: 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burds AA, Lutum AS, Sorger PK. 2005. Generating chromosome instability through the simultaneous deletion of Mad2 and p53. Proc Natl Acad Sci 102: 11296–11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. 2013. Replication stress links structural and numerical cancer chromosomal instability. Nature 494: 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, Walsh CA. 2014. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep 8: 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiel A, Visochek L, Mittelman L, Dantzer F, Izraeli S, Cohen-Armon M. 2011. A phenanthrene derived PARP inhibitor is an extra-centrosomes de-clustering agent exclusively eradicating human cancer cells. BMC Cancer 11: 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo A, Morse HC III, Godfrey VL, Naeem R, Justice MJ. 2007. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res 67: 10138–10147. [DOI] [PubMed] [Google Scholar]
- Chan JY. 2011. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7: 1122–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, Hickson ID. 2009. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11: 753–760. [DOI] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R. 2012. Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E, Zhang X, Xing C, Yu H. 2016. Mitotic checkpoint regulators control insulin signaling and metabolic homeostasis. Cell 166: 567–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Howell B, Maddox P, Khodjakov A, Degrassi F, Salmon ED. 2001. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 153: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Moree B, Canman JC, Salmon ED. 2003. Merotelic kinetochore orientation occurs frequently during early mitosis in mammalian tissue cells and error correction is achieved by two different mechanisms. J Cell Sci 116: 4213–4225. [DOI] [PubMed] [Google Scholar]
- Cimini D, Cameron LA, Salmon ED. 2004. Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr Biol 14: 2149–2155. [DOI] [PubMed] [Google Scholar]
- Coelho PA, Bury L, Shahbazi MN, Liakath-Ali K, Tate PH, Wormald S, Hindley CJ, Huch M, Archer J, Skarnes WC, et al. 2015. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol 5: 150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J. 2013. The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol 15: 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo R, Caldarelli M, Mennecozzi M, Giorgini ML, Sola F, Cappella P, Perrera C, Depaolini SR, Rusconi L, Cucchi U, et al. 2010. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res 70: 10255–10264. [DOI] [PubMed] [Google Scholar]
- Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. 2012. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crockford A, Zalmas LP, Gronroos E, Dewhurst SM, McGranahan N, Cuomo ME, Encheva V, Snijders AP, Begum J, Purewal S, et al. 2017. Cyclin D mediates tolerance of genome-doubling in cancers with functional p53. Ann Oncol 28: 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, Mahmood R, Yang YM, Xu M, Rao CV. 2004. Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 64: 440–445. [DOI] [PubMed] [Google Scholar]
- Davoli T, de Lange T. 2012. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 21: 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. 2013. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155: 948–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Uno H, Wooten EC, Elledge SJ. 2017. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355: eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawar S, Lim Y, Puccini J, White M, Thomas P, Bouchier-Hayes L, Green DR, Dorstyn L, Kumar S. 2017. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 36: 2704–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O'Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM. 2014. Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. Elife 3: e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK. 2000. Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 101: 635–645. [DOI] [PubMed] [Google Scholar]
- Dodgson SE, Kim S, Costanzo M, Baryshnikova A, Morse DL, Kaiser CA, Boone C, Amon A. 2016a. Chromosome-specific and global effects of aneuploidy in Saccharomyces cerevisiae. Genetics 202: 1395–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodgson SE, Santaguida S, Kim S, Sheltzer J, Amon A. 2016b. The pleiotropic deubiquitinase Ubp3 confers aneuploidy tolerance. Genes Dev 30: 2259–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. 2015. Targeting mitosis in cancer: emerging strategies. Mol Cell 60: 524–536. [DOI] [PubMed] [Google Scholar]
- Dorstyn L, Puccini J, Wilson CH, Shalini S, Nicola M, Moore S, Kumar S. 2012. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ 19: 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotiwala F, Harrison JC, Jain S, Sugawara N, Haber JE. 2010. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr Biol 20: 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli DM, Hearn S, Myers MP, Lazebnik Y. 2005. A primate virus generates transformed human cells by fusion. J Cell Biol 171: 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli DM, Padilla-Nash HM, Berman D, Murphy KM, Ried T, Lazebnik Y. 2007. A virus causes cancer by inducing massive chromosomal instability through cell fusion. Curr Biol 17: 431–437. [DOI] [PubMed] [Google Scholar]
- Duijf PH, Schultz N, Benezra R. 2013. Cancer cells preferentially lose small chromosomes. Int J Cancer 132: 2316–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB, Finegold MJ, Grompe M. 2010. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 467: 707–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ, Strom SC, Grompe M. 2012. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 142: 25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Yakoubi W, Wassmann K. 2017. Meiotic divisions: no place for gender equality. Adv Exp Med Biol 1002: 1–17. [DOI] [PubMed] [Google Scholar]
- Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuss S, Burfeind P, Aigner A, Wordeman L, Bastians H. 2014. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol 16: 779–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggioli F, Wang T, Vijg J, Montagna C. 2012. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum Mol Genet 21: 5246–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, Eiterer L, Demetz E, Weiss G, Geley S, Nigg EA, et al. 2017. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev 31: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F, Xie SZ, Simon JE, Bakker PL, Conte N, Davis SH, Kregel E, Jonkers J, Bradley A, Sorger PK. 2014. Chromosome instability induced by Mps1 and p53 mutation generates aggressive lymphomas exhibiting aneuploidy-induced stress. Proc Natl Acad Sci 111: 13427–13432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foijer F, Albacker LA, Bakker B, Spierings DC, Yue Y, Xie SZ, Davis S, Lutum-Jehle A, Takemoto D, Hare B, et al. 2017. Deletion of the MAD2L1 spindle assembly checkpoint gene is tolerated in mouse models of acute T-cell lymphoma and hepatocellular carcinoma. Elife 6: e20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. 2005. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437: 1043–1047. [DOI] [PubMed] [Google Scholar]
- Funk LC, Zasadil LM, Weaver BA. 2016. Living in CIN: mitotic infidelity and its consequences for tumor promotion and suppression. Dev Cell 39: 638–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ. 1996. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci 93: 7081–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D. 2009. A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Thery M, Camargo FD, Pellman D. 2014. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158: 833–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Castillo H, Vasquez-Velasquez AI, Rivera H, Barros-Nunez P. 2008. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am J Med Genet A 146A: 1687–1695. [DOI] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. 2008. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14: 111–122. [DOI] [PubMed] [Google Scholar]
- Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D. 2012. Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13: 189–203. [DOI] [PubMed] [Google Scholar]
- Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, et al. 2004. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 36: 1159–1161. [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. 2013. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154: 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe EH, Day CA, Karanjeet KB, Fadness S, Langfald A, Vaughan KT, Dong Z. 2016. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat Cell Biol 18: 668–675. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. 2009. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 10: 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. 2012a. Chromoanagenesis and cancer: mechanisms and consequences of localized, complex chromosomal rearrangements. Nat Med 18: 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. 2012b. Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 13: 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Gustafsson HT, O'Sullivan C, Bisceglia G, Huang X, Klose C, Schevchenko A, Dickson RC, Cavaliere P, Dephoure N, et al. 2017. Serine-dependent sphingolipid synthesis is a metabolic liability of aneuploid cells. Cell Rep 21: 3807–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga Y, Chi YH, Miyazato A, Sheleg S, Haller K, Peloponese JM Jr, Li Y, Ward JM, Benezra R, Jeang KT. 2007. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res 67: 160–166. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani M, A'Hern R, Birkbak NJ, Gorman P, Gronroos E, Ngang S, Nicola P, Rahman L, Thanopoulou E, Kelly G, et al. 2015. Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann Oncol 26: 1340–1346. [DOI] [PubMed] [Google Scholar]
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, et al. 2017. Tracking the evolution of non-small-cell lung cancer. N Engl J Med 376: 2109–2121. [DOI] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. 2011. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333: 1895–1898. [DOI] [PubMed] [Google Scholar]
- Jeganathan KB, Baker DJ, van Deursen JM. 2006. Securin associates with APCCdh1 in prometaphase but its destruction is delayed by Rae1 and Nup98 until the metaphase/anaphase transition. Cell Cycle 5: 366–370. [DOI] [PubMed] [Google Scholar]
- Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. 2007. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol 179: 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemaa M, Galluzzi L, Kepp O, Senovilla L, Brands M, Boemer U, Koppitz M, Lienau P, Prechtl S, Schulze V, et al. 2013. Characterization of novel MPS1 inhibitors with preclinical anticancer activity. Cell Death Differ 20: 1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes JW, Almeida L, Daly K, Ferguson AD, Grosskurth SE, Guan H, Howard T, Ioannidis S, Kazmirski S, Lamb ML, et al. 2015. Discovery of AZ0108, an orally bioavailable phthalazinone PARP inhibitor that blocks centrosome clustering. Bioorg Med Chem Lett 25: 5743–5747. [DOI] [PubMed] [Google Scholar]
- Kabeche L, Compton DA. 2012. Checkpoint-independent stabilization of kinetochore–microtubule attachments by Mad2 in human cells. Curr Biol 22: 638–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakkanthara A, Jeganathan KB, Limzerwala JF, Baker DJ, Hamada M, Nam HJ, van Deursen WH, Hamada N, Naylor RM, Becker NA, et al. 2016. Cyclin A2 is an RNA binding protein that controls Mre11 mRNA translation. Science 353: 1549–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura E, Fielding AB, Kannan N, Balgi A, Eaves CJ, Roberge M, Dedhar S. 2013. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 4: 1763–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong HT, Restifo NP. 2002. Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nat Immunol 3: 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, He X, Orr B, Wutz G, Hill V, Peters JM, Compton DA, Waldman T. 2016. Intact cohesion, anaphase, and chromosome segregation in human cells harboring tumor-derived mutations in STAG2. PLoS Genet 12: e1005865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleyman M, Kabeche L, Compton DA. 2014. STAG2 promotes error correction in mitosis by regulating kinetochore–microtubule attachments. J Cell Sci 127: 4225–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Wu J, Whittaker CA, Amon A. 2014. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci 111: 13409–13414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T. 2011. Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 8: 244–250. [DOI] [PubMed] [Google Scholar]
- Komlodi-Pasztor E, Sackett DL, Fojo AT. 2012. Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 18: 51–63. [DOI] [PubMed] [Google Scholar]
- Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. 2013. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet 45: 1232–1237. [DOI] [PubMed] [Google Scholar]
- Krajcovic M, Johnson NB, Sun Q, Normand G, Hoover N, Yao E, Richardson AL, King RW, Cibas ES, Schnitt SJ, et al. 2011. A non-genetic route to aneuploidy in human cancers. Nat Cell Biol 13: 324–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffer C, Kuznetsova AY, Storchova Z. 2013. Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 122: 305–318. [DOI] [PubMed] [Google Scholar]
- Kulukian A, Holland AJ, Vitre B, Naik S, Cleveland DW, Fuchs E. 2015. Epidermal development, growth control, and homeostasis in the face of centrosome amplification. Proc Natl Acad Sci 112: E6311–E6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. 2008. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 22: 2189–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Holland AJ. 2017. A new mode of mitotic surveillance. Trends Cell Biol 27: 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M, et al. 2010. Proteins required for centrosome clustering in cancer cells. Sci Transl Med 2: 33ra38. [DOI] [PubMed] [Google Scholar]
- Leibowitz ML, Zhang CZ, Pellman D. 2015. Chromothripsis: a new mechanism for rapid karyotype evolution. Annu Rev Genet 49: 183–211. [DOI] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B. 1997. Genetic instability in colorectal cancers. Nature 386: 623–627. [DOI] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D, et al. 2017. Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322 e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Wei Z, York JP, Zhang P. 2009. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol 185: 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. 2010. The ATM–p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci 107: 14188–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu W, Chen D, Boohaker RJ, Zhai L, Padmalayam I, Wennerberg K, Xu B, Zhang W. 2015. KIFC1 is a novel potential therapeutic target for breast cancer. Cancer Biol Ther 16: 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia C, Sansregret L, Domingo E, McGranahan N, Hobor S, Birkbak NJ, Horswell S, Gronroos E, Favero F, Rowan AJ, et al. 2017. BCL9L dysfunction impairs caspase-2 expression permitting aneuploidy tolerance in colorectal cancer. Cancer Cell 31: 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW. 2017. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19: 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maachani UB, Kramp T, Hanson R, Zhao S, Celiku O, Shankavaram U, Colombo R, Caplen NJ, Camphausen K, Tandle A. 2015. Targeting MPS1 enhances radiosensitization of human glioblastoma by modulating DNA repair proteins. Mol Cancer Res 13: 852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. 2015. Chromothripsis and kataegis induced by telomere crisis. Cell 163: 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia AR, de Man J, Boon U, Janssen A, Song JY, Omerzu M, Sterrenburg JG, Prinsen MB, Willemsen-Seegers N, de Roos JA, et al. 2015. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann Oncol 26: 2180–2192. [DOI] [PubMed] [Google Scholar]
- Maiato H, Logarinho E. 2014. Mitotic spindle multipolarity without centrosome amplification. Nat Cell Biol 16: 386–394. [DOI] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, Dyson NJ. 2010. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev 24: 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, Basto R. 2013. Centrosome amplification causes microcephaly. Nat Cell Biol 15: 731–740. [DOI] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, et al. 2013. Mosaic copy number variation in human neurons. Science 342: 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. 2017. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell 171: 1259–1271 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. 2001. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409: 355–359. [DOI] [PubMed] [Google Scholar]
- Mitchison TJ. 2012. The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell 23: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Pineda J, Shi J, Florian S. 2017. Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol 7: 170182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- M'Kacher R, Andreoletti L, Flamant S, Milliat F, Girinsky T, Dossou J, Violot D, Assaf E, Clausse B, Koscielny S, et al. 2010. JC human polyomavirus is associated to chromosomal instability in peripheral blood lymphocytes of Hodgkin's lymphoma patients and poor clinical outcome. Ann Oncol 21: 826–832. [DOI] [PubMed] [Google Scholar]
- Mugneret F, Callier P, Favre-Audry B. 2003. [Chromosomal abnormalities in acute myeloid leukaemias]. Pathol Biol 51: 314–328. [DOI] [PubMed] [Google Scholar]
- Naim V, Rosselli F. 2009. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol 11: 761–768. [DOI] [PubMed] [Google Scholar]
- Nam HJ, van Deursen JM. 2014. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol 16: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Naylor RM, van Deursen JM. 2015. Centrosome dynamics as a source of chromosomal instability. Trends Cell Biol 25: 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor RM, van Deursen JM. 2016. Aneuploidy in cancer and aging. Annu Rev Genet 50: 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JM, Macedo JC, Mattingly AJ, Wangsa D, Camps J, Lima V, Gomes AM, Doria S, Ried T, Logarinho E, et al. 2015. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. Elife 4: e05068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Holland AJ. 2018. Once and only once: mechanisms of centriole duplication and their deregulation in disease. Nat Rev Mol Cell Biol 19: 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaharski AJ, Sotelo R, Solorza-Luna G, Gonsebatt ME, Guzman P, Mohar A, Eastmond DA. 2006. Tetraploidy and chromosomal instability are early events during cervical carcinogenesis. Carcinogenesis 27: 337–343. [DOI] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. 2012. Aneuploidy causes proteotoxic stress in yeast. Genes Dev 26: 2696–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R, Mitchison TJ. 2011. Analysis of mitosis and antimitotic drug responses in tumors by in vivo microscopy and single-cell pharmacodynamics. Cancer Res 71: 4608–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack SD, Weil RJ, Vortmeyer AO, Zeng W, Li J, Okamoto H, Furuta M, Pak E, Lubensky IA, Oldfield EH, et al. 2005. Individual adult human neurons display aneuploidy: detection by fluorescence in situ hybridization and single neuron PCR. Cell Cycle 4: 1758–1760. [DOI] [PubMed] [Google Scholar]
- Pannu V, Rida PC, Celik B, Turaga RC, Ogden A, Cantuaria G, Gopalakrishnan J, Aneja R. 2014. Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis 5: e1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passerini V, Ozeri-Galai E, de Pagter MS, Donnelly N, Schmalbrock S, Kloosterman WP, Kerem B, Storchova Z. 2016. The presence of extra chromosomes leads to genomic instability. Nat Commun 7: 10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson K, Johansson B. 2007. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol 55: 37–48. [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. 2010. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau SJ, Silberman RE, Knouse KA, Amon A. 2016. Aneuploidy impairs hematopoietic stem cell fitness and is selected against in regenerating tissues in vivo. Genes Dev 30: 1395–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puccini J, Shalini S, Voss AK, Gatei M, Wilson CH, Hiwase DK, Lavin MF, Dorstyn L, Kumar S. 2013. Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm-deficient mice. Proc Natl Acad Sci 110: 19920–19925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. 2005. Spindle multipolarity is prevented by centrosomal clustering. Science 307: 127–129. [DOI] [PubMed] [Google Scholar]
- Raab MS, Breitkreutz I, Anderhub S, Ronnest MH, Leber B, Larsen TO, Weiz L, Konotop G, Hayden PJ, Podar K, et al. 2012. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res 72: 5374–5385. [DOI] [PubMed] [Google Scholar]
- Rebacz B, Larsen TO, Clausen MH, Ronnest MH, Loffler H, Ho AD, Kramer A. 2007. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res 67: 6342–6350. [DOI] [PubMed] [Google Scholar]
- Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. 2001. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci 98: 13361–13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhys AD, Monteiro P, Smith C, Vaghela M, Arnandis T, Kato T, Leitinger B, Sahai E, McAinsh A, Charras G, et al. 2018. Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J Cell Biol 217: 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rode A, Maass KK, Willmund KV, Lichter P, Ernst A. 2016. Chromothripsis in cancer cells: an update. Int J Cancer 138: 2322–2333. [DOI] [PubMed] [Google Scholar]
- Roylance R, Endesfelder D, Gorman P, Burrell RA, Sander J, Tomlinson I, Hanby AM, Speirs V, Richardson AL, Birkbak NJ, et al. 2011. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol Biomarkers Prev 20: 2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge SD, Douglas TA, Nicholson JM, Vila-Casadesus M, Kantzler CL, Wangsa D, Barroso-Vilares M, Kale SD, Logarinho E, Cimini D. 2016. Selective advantage of trisomic human cells cultured in non-standard conditions. Sci Rep 6: 22828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. 2018. Profound tissue specificity in proliferation control underlies cancer drivers and aneuploidy patterns. Cell 173: 499–514.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansregret L, Patterson JO, Dewhurst S, Lopez-Garcia C, Koch A, McGranahan N, Chao WCH, Barry DJ, Rowan A, Instrell R, et al. 2017. APC/C dysfunction limits excessive cancer chromosomal instability. Cancer Discov 7: 218–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Amon A. 2015. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol 16: 473–485. [DOI] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, Amon A. 2015. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev 29: 2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Richardson A, Iyer DR, M'Saad O, Zasadil L, Knouse KA, Wong YL, Rhind N, Desai A, Amon A. 2017. Chromosome mis-segregation generates cell-cycle-arrested cells with complex karyotypes that are eliminated by the immune system. Dev Cell 41: 638–651.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. 2012. An immunosurveillance mechanism controls cancer cell ploidy. Science 337: 1678–1684. [DOI] [PubMed] [Google Scholar]
- Sercin O, Larsimont JC, Karambelas AE, Marthiens V, Moers V, Boeckx B, Le Mercier M, Lambrechts D, Basto R, Blanpain C. 2016. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat Cell Biol 18: 100–110. [DOI] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. 2011. Aneuploidy drives genomic instability in yeast. Science 333: 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A. 2012. Transcriptional consequences of aneuploidy. Proc Natl Acad Sci 109: 12644–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A. 2017. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell 31: 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiheido H, Terada F, Tabata N, Hayakawa I, Matsumura N, Takashima H, Ogawa Y, Du W, Yamada T, Shoji M, et al. 2012. A phthalimide derivative that inhibits centrosomal clustering is effective on multiple myeloma. PLoS One 7: e38878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. 2013. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci 110: E4134–E4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Scholl LM, Cimini D. 2009. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 4: e6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silkworth WT, Nardi IK, Paul R, Mogilner A, Cimini D. 2012. Timing of centrosome separation is important for accurate chromosome segregation. Mol Biol Cell 23: 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JE, Bakker B, Foijer F. 2015. CINcere modelling: what have mouse models for chromosome instability taught us? Recent Results Cancer Res 200: 39–60. [DOI] [PubMed] [Google Scholar]
- Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. 2011. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333: 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. 2007. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell 11: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. 2010. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature 464: 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M, Raaijmakers JA, Bakker B, Spierings DCJ, Lansdorp PM, Foijer F, Medema RH. 2017. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep 19: 2423–2431. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. 2009. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136: 473–484. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. 2011. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. 2012. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 8: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto I, Murakami H, Tonami Y, Moriyama A, Nakanishi M. 2004. DNA replication checkpoint control mediated by the spindle checkpoint protein Mad2p in fission yeast. J Biol Chem 279: 47372–47378. [DOI] [PubMed] [Google Scholar]
- Suijkerbuijk SJ, van Osch MH, Bos FL, Hanks S, Rahman N, Kops GJ. 2010. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res 70: 4891–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. 2013. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symmans WF, Volm MD, Shapiro RL, Perkins AB, Kim AY, Demaria S, Yee HT, McMullen H, Oratz R, Klein P, et al. 2000. Paclitaxel-induced apoptosis and mitotic arrest assessed by serial fine-needle aspiration: implications for early prediction of breast cancer response to neoadjuvant treatment. Clin Cancer Res 6: 4610–4617. [PubMed] [Google Scholar]
- Tan S, Guan X, Grun C, Zhou Z, Schepers U, Nick P. 2015. Gallic acid induces mitotic catastrophe and inhibits centrosomal clustering in HeLa cells. Toxicol In Vitro 30: 506–513. [DOI] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A. 2011. Identification of aneuploidy-selective antiproliferation compounds. Cell 144: 499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Yuwen H, Wang K, Bruno PM, Bullock K, Deik A, Santaguida S, Trakala M, Pfau SJ, Zhong N, et al. 2017. Aneuploid cell survival relies upon sphingolipid homeostasis. Cancer Res 77: 5272–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous BA, Kerami M, Van der Stoop PM, Kwiatkowski N, Wang J, Zhou W, Kessler AF, Lewandrowski G, Hiddingh L, Sol N, et al. 2013. Effects of the selective MPS1 inhibitor MPS1-IN-3 on glioblastoma sensitivity to antimitotic drugs. J Natl Cancer Inst 105: 1322–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, et al. 2018. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33: 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. 2013. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123: 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. 2008. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol 180: 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. 2010. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 188: 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. 2011. Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc Natl Acad Sci 108: 17974–17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Addis Jones O, Chan KL. 2018. 53BP1 can limit sister-chromatid rupture and rearrangements driven by a distinct ultrafine DNA bridging-breakage process. Nat Commun 9: 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. 2007. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924. [DOI] [PubMed] [Google Scholar]
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. 2010. Identification of aneuploidy-tolerating mutations. Cell 143: 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. 2017. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543: 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. 2004. Cell cycle progression after cleavage failure: mammalian somatic cells do not possess a ‘tetraploidy checkpoint’. J Cell Biol 165: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G. 2010. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 20: 1666–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valind A, Jin Y, Baldetorp B, Gisselsson D. 2013. Whole chromosome gain does not in itself confer cancer-like chromosomal instability. Proc Natl Acad Sci 110: 21119–21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bos H, Spierings DC, Taudt AS, Bakker B, Porubsky D, Falconer E, Novoa C, Halsema N, Kazemier HG, Hoekstra-Wakker K, et al. 2016. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer's disease neurons. Genome Biol 17: 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering CI, Horne MC, Knudson CM. 2007. Chromosomal instability and supernumerary centrosomes represent precursor defects in a mouse model of T-cell lymphoma. Cancer Res 67: 8081–8088. [DOI] [PubMed] [Google Scholar]
- van Jaarsveld RH, Kops G. 2016. Difference makers: chromosomal instability versus aneuploidy in cancer. Trends Cancer 2: 561–571. [DOI] [PubMed] [Google Scholar]
- van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM. 2010. Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol 188: 83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ree JH, Nam HJ, Jeganathan KB, Kanakkanthara A, van Deursen JM. 2016. Pten regulates spindle pole movement through Dlg1-mediated recruitment of Eg5 to centrosomes. Nat Cell Biol 18: 814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitre B, Holland AJ, Kulukian A, Shoshani O, Hirai M, Wang Y, Maldonado M, Cho T, Boubaker J, Swing DA, et al. 2015. Chronic centrosome amplification without tumorigenesis. Proc Natl Acad Sci 112: E6321–E6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther A, Houlston R, Tomlinson I. 2008. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut 57: 941–950. [DOI] [PubMed] [Google Scholar]
- Wan J, Zhu F, Zasadil LM, Yu J, Wang L, Johnson A, Berthier E, Beebe DJ, Audhya A, Weaver BA. 2014. A Golgi-localized pool of the mitotic checkpoint component Mad1 controls integrin secretion and cell migration. Curr Biol 24: 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, Chen K, Scheet P, Vattathil S, Liang H, et al. 2014. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512: 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Bonday ZQ, Putkey FR, Kops GJ, Silk AD, Cleveland DW. 2003. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol 162: 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. 2007. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11: 25–36. [DOI] [PubMed] [Google Scholar]
- Wengner AM, Siemeister G, Koppitz M, Schulze V, Kosemund D, Klar U, Stoeckigt D, Neuhaus R, Lienau P, Bader B, et al. 2016. Novel Mps1 kinase inhibitors with potent antitumor activity. Mol Cancer Ther 15: 583–592. [DOI] [PubMed] [Google Scholar]
- Wild T, Larsen MS, Narita T, Schou J, Nilsson J, Choudhary C. 2016. The spindle assembly checkpoint is not essential for viability of human cells with genetically lowered APC/C activity. Cell Rep 14: 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C, Stearns T. 2005. Mammalian cells lack checkpoints for tetraploidy, aberrant centrosome number, and cytokinesis failure. BMC Cell Biol 6: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ. 2013. Cyclic GMP–AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339: 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017. cGAS is essential for cellular senescence. Proc Natl Acad Sci 114: E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YM, Baek KH, Jeong SJ, Shin HJ, Ha GH, Jeon AH, Hwang SG, Chun JS, Lee CW. 2004. WD repeat-containing mitotic checkpoint proteins act as transcriptional repressors during interphase. FEBS Lett 575: 23–29. [DOI] [PubMed] [Google Scholar]
- Yost S, de Wolf B, Hanks S, Zachariou A, Marcozzi C, Clarke M, de Voer R, Etemad B, Uijttewaal E, Ramsay E, et al. 2017. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet 49: 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurov YB, Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Kutsev SI, Pellestor F, Beresheva AK, Demidova IA, Kravets VS, et al. 2007. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One 2: e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhsng CZ, Wala J, Mermel CH, et al. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. 2014. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med 6: 229ra243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil LM, Britigan EM, Ryan SD, Kaur C, Guckenberger DJ, Beebe DJ, Moser AR, Weaver BA. 2016. High rates of chromosome missegregation suppress tumor progression but do not inhibit tumor initiation. Mol Biol Cell 27: 1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Foreman O, Wigle DA, Kosari F, Vasmatzis G, Salisbury JL, van Deursen J, Galardy PJ. 2012. USP44 regulates centrosome positioning to prevent aneuploidy and suppress tumorigenesis. J Clin Invest 122: 4362–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D. 2015. Chromothripsis from DNA damage in micronuclei. Nature 522: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhai L, Wang Y, Boohaker RJ, Lu W, Gupta VV, Padmalayam I, Bostwick RJ, White EL, Ross LJ, et al. 2016. Discovery of a novel inhibitor of kinesin-like protein KIFC1. Biochem J 473: 1027–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Pavelka N, Bradford WD, Rancati G, Li R. 2012. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet 8: e1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]