

Abstract
Targeting somatically activated oncogenes has revolutionized the treatment of non-small cell lung cancer (NSCLC). Mutations in the gene mesenchymal-epithelial transition (MET) near the exon 14 splice sites are recurrent in lung adenocarcinoma and cause exon skipping (METΔ14). Here we analyzed 4,422 samples from 12 different malignancies to estimate the rate of said exon skipping. METΔ14 mutation and transcript were most common in lung adenocarcinoma. Endogenously expressed levels of METΔ14 transformed human epithelial lung cells in a Hepatocyte growth factor (HGF)-dependent manner. Additionally, overexpression of the orthologous mouse allele induced lung adenocarcinoma in a novel, immunocompetent mouse model. Met inhibition showed clinical benefit in this model. Additionally, we observed a clinical response to crizotinib in a patient with METΔ14-driven NSCLC, only to observe new missense mutations in the MET activation loop, critical for binding to crizotinib, upon clinical progression. These findings support genomically selected clinical trials directed towards METΔ14 in a fraction of NSCLC patients, confirm second-site mutations for further therapeutic targeting prior to and beyond acquired resistance, and provide an in vivo system for the study of METΔ14 in an immunocompetent host.
Keywords: Targeted therapy, Non-small cell lung cancer, acquired resistance
Introduction
The mesenchymal-epithelial transition (MET) proto-oncogene has been extensively studied in human cancer. DNA amplification of wild-type MET, fusion of MET from chromosomal rearrangement and activating kinase-domain point mutations have all been identified as independent mechanisms of MET/HGF axis activation in cancer (1). Somatic mutations at or around the splice junctions of MET exon 14 (METΔ14) are a recurrent mechanism of MET activation and lead to a METΔ14 protein that lacks the intracellular juxtamembrane domain (2). The METΔ14 mutation and transcript occurs in around 3–4% of lung adenocarcinoma (LUAD) (3,4). Exon 14 of MET encodes an ubiquitin ligase site (Y1003), which promotes MET degradation (5). As such, extended protein half-life has been proposed as a selective force for METΔ14 mutations in cancer (2). Case-reports have suggested varying degrees of responsiveness to experimental- and FDA-approved agents that inhibit MET in patients with tumors harboring METΔ14 alleles(6) but preclinical models and clinical trials are lacking to date. Finally, acquired mutations in the MET gene have been observed recently in NSCLCs; one (METY1230C) following treatment with chemotherapy, radiation and crizotinib (7), and another (METD1228V) with an experimental MET inhibitor in an EGFR-mutant lung adenocarcinoma without pretreatment MET mutations (8). These reports suggest that second-site, acquired resistance might be combated by next-generation MET inhibitors. Several unanswered questions remain. Malignancies other than lung cancer have not been screened for the METΔ14 mutation and transcript. Additionally, the role ligand (HGF) plays in signaling from the mutant receptor and the biochemical activity of this mutant protein have not been extensively characterized.
We describe the prevalence of METΔ14 in solid tumors and define the role of METΔ14 using both in vitro and in vivo models. We identify acquired resistance to crizotinib in a patient harboring a METΔ14 mutation. These findings qualify METΔ14 mutations as drivers of lung adenocarcinoma and identify a subpopulation of patients whom may benefit from further development of targeted MET/HGF therapies and suggest that combinations of type I and II MET inhibitors might be best deployed together, to prevent activation loop mutations that drive single agent resistance.
Materials and Methods
mRNA expression analysis and Genomic analysis
TCGA PanCancer exon level RNAseq data was obtained (9) and mRNA data for seven types of cancers (READ, COAD, LUSC, KIRC, HNSC, BLCA, LUAD) were downloaded from UCSC. Genomic coordinates for MET exon 13 (chr7: 116411552 -116411708), exon 14 (Chr7: 116411903 -116412043) and exon 15 (Chr7: 116414935 -116415165) were used to retrieve the expression of these three exons. We used a ratio of exon 14 to the average of neighboring exons 13 & 15 [r=exon 14*2/(exon13+exon15)] (3). Samples with rate of skipping R=(1-r)>0.6 were considered as exon 14 skipping (Δ14), including partial skipping 0.6<R<0.9 and near complete skipping R>0.9. In TCGA_LUAD, the samples were stratified into MET wt and METΔ14 based on above predicted METΔ14 mRNA status, HGF and MET mRNA expression in two groups was plotted. Samples with METΔ14 skipping identified in latest available TCGA LUAD RNAseq exon level data (n=571) were manually searched with corresponding MET genotyping in TCGA LUAD samples with known whole exome sequences (10). For specimens that had MET exon 14 skipping at the RNA-level, but lacked somatic mutation calls, exome sequencing BAMs were downloaded from the NCI Genomic Data Commons (GDC) (11). Soft-clipped reads near the genomic region of MET exon 14 were identified as potentially misaligned due to a larger deletion. The full sequence of the soft-clipped reads were realigned using BLAT (12). BLAT alignments supported larger deletions in these samples, with multiple read support. Normalized read counts were log2 transformed, scaled and centered in R program. Two-tailed student’s t-test was used for two group samples statistical analysis in R (Version 2.15).
Cell lines and cell culture
The AALE parental cell line was established and maintained as described (13) (14). The AALE parental cell line and the following stable cell lines were received as gifts in 2015 from Dr. Choi and Dr. Berger in Dr. Meyerson’s lab at Dana-Farber Cancer Institute. No mycoplasma testing was performed. AALE MET sg pool and AALE GFP sg pool were infected with sgRNA encoded by pXPR001 and selected in puromycin (1.5 μg/mL). AALE MET sg 1F10 single clone and 2E9 single clone were derived from transient transfection with pXPR001-METsg, followed by isolation by limiting dilution. MET exon 14 deletion was confirmed by PCR and immunoblotting. AALE cell lines expressing ectopic MET Δ14, METwt, RFP, TPR-MET, TPR-MET ex14 wt or TPR-MET ex14 Y1003F were infected with corresponding pLX302 lentivirus and selected in 1.5 μg/ml puromycin.
Protein stability
The experiment was performed as described (2). AALE cells were treated with 75ug/ml cycloheximide (CHX) after 5 min. exposure to HGF (50ng/ml). Protein half-life was calculated from exponential regression of MET protein band intensity as assessed on a LiCor® machine.
Trypsin assay
The experiment is a modified version of that previously described (15), intracellular (internalized) MET is protected from exogenous trypsin in cell culture while cell surface-displayed MET is trypsin sensitive (15). As schematized in Supplementary Fig. 2B, AALE cells were grown to 50% confluence and stimulated with HGF for various time periods. Subsequently, cells were shifted to 4 °C, washed twice with PBS and for 10 minutes in ice-cold pH 3.7 medium (pH adjusted with HCl). After two washes with ice-cold PBS the cells were treated with 0.1% Trypsin in PBS on ice for 30 minutes followed by the addition of TNS (CC-5002, Lonza) and PBS washes. The cells were lysed and handled as described in the immunoblotting section in supplementary method.
Ras Binding Domain pull-down Assays
The experiment was performed as described (16) according to Ras activation assay kit instructions (BK008, Cytoskeleton).
Mouse model
All of the animal experiments were done under protocols reviewed and approved by UCSF animal care committee. Trp53f/f mouse has been described (17). We used 1×106 active viral particles/mL for pHAGE-METΔ14 and pHAGE-GFP and 1×107 for pHAGE-KRASG12D (n=3 mice each). Mice were euthanized at protocol-defined endpoint. See supplementary methods for Preclinical Therapeutic Studies with Micro X-ray Computed Tomography (μCT).
Clinical tumor and cell-free DNA sequencing
Pretreatment tumor sequencing at diagnosis was performed by Foundation Medicine (Cambridge, MA) (4). Cell-free DNA sequencing at the time of progression was performed by Guardant Health (Redwood City, CA) (18). GCP practice and institutional review board (IRB) regulation were followed in the study of patient material.
Results
MET Exon 14 Skipping Occurs in Lung Adenocarcinoma, but Rarely in Other Malignancies
To determine the frequency with which MET-encoded mRNA transcripts specifically exclude exon 14 (i.e. METΔ14) in various cancer types, we examined RNAseq from a total of 4,422 samples from the TCGA Pan-Cancer-12 set composed of cases from 12 different malignancies (9). First we examined total mRNA expression of MET and its ligand HGF and found only lung adenocarcinoma (LUAD) and clear cell kidney cancer (KIRC) had high levels of both transcripts (Supplementary Fig. S1A, S1B). Next we used exon-level analysis to calculate a ratio of MET exon 14 to the average of neighboring exons 13&15 [r=exon 14*2/(exon13+exon15)] (3) to identify 16 LUAD samples from 571 (2.8%) (10) with METΔ14 skipping (Fig. 1A, and Fig. S1C) while the other 6 cancer types have no or low (<1.2%) rates of MET exon 14 skipping. Fourteen of 16 METΔ14 samples (87%) showed somatically encoded DNA MET alterations around splice sites (4) (Fig. 1B and table S1). Fifteen out of 2,582 samples from cancers other than lung showed RNA evidence of exon 14 skipping by the above metric. No splice site mutations supporting a somatic origin for this skipping were seen in other cancer types. Total MET and HGF mRNA expression did not differ between METΔ14 and MET wt samples (Fig. S1D). In summary, the relatively high METΔ14 skipping rate and high total MET and HGF expression level suggest LUAD as the appropriate setting for a deeper, functional investigation of METΔ14 function.
Figure 1.
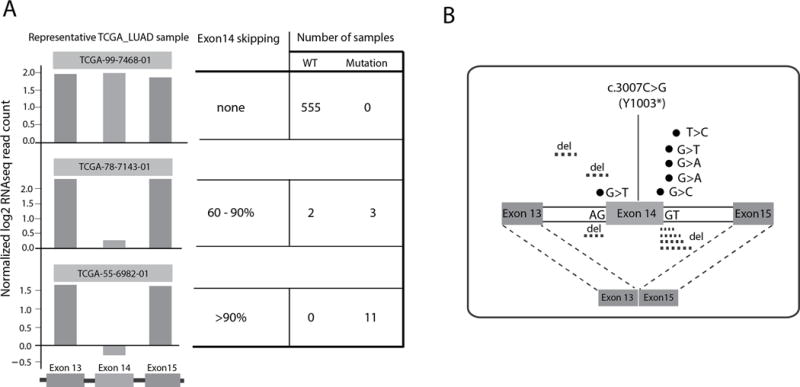
A) Lung adenocarcinoma samples with no, partial or near-complete MET exon 14 skipping and number of samples with wild type (wt) and mutant MET in each group. B) Single nucleotide variants and deletions seen near exon 14 in lung adenocarcinoma samples with mRNA evidence of exon 14 skipping.
MET Exon 14 Skipping Transforms Human Lung Epithelial Cells in an HGF-Dependent Manner
The majority of METΔ14 skipping occurs without MET amplification, at least in early-stage LUAD (table S1) (19). To model the effects of METΔ14 expression from the endogenous promoter in a lung cancer-relevant cell type, we used CRISPR to target the MET intron/exon 14 junctions in AALE cells, an immortalized, non-transformed, human tracheobronchial epithelial cell line (14). sgRNA/CAS9-editing led to MET exon14 skipping and METΔ14mRNA expression confirmed by RT-PCR (Fig. S2A) and immunoblotting (Fig. 2A). Importantly, this endogenously-transcribed system allowed us to decouple METΔ14’s biochemical function from overexpression, adding new knowledge to the existing ectopic expression cell line models which inevitably overexpress MET protein when introducing the mutation(4). Expression of METΔ14 from the endogenous locus (METsg) renders AALE cells anchorage independent in an HGF dose-dependent manner (Fig. 2A), despite equivalent or even lower total protein levels of the exon-skipped receptor compared to wild type MET in GFPsg control AALE cells. Similarly, ectopically expressed METΔ14 cDNA transformed AALE cells in an HGF-dependent manner more efficiently than equivalently expressed wt MET under the same HGF concentration (Fig. 2B). TPR-MET, a cytosolic, hyperactive, HGF-independent MET fusion mutant with strong transforming ability(20) was used as a control for both colony formation and HGF independence (Fig. 2B). These results suggest that METΔ14 is more active than wild type MET, but depends on HGF for full receptor activation and cellular transformation.
Figure 2.
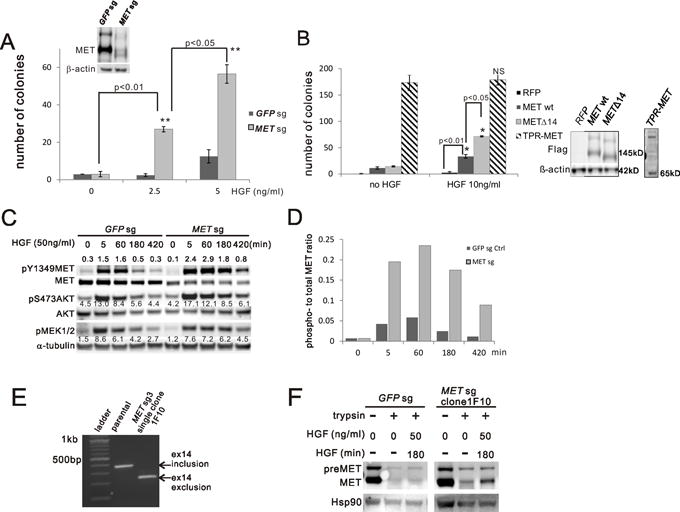
METΔ14 mutants transform AALE cells in a HGF-dependent manner and are hyperactive. A) Anchorage-independent growth was assessed by soft-agar assay comparing AALE cells targeted with GFP (GFPsg) or MET intron/exon 14 skipping sgRNA (METsg) under the indicated HGF concentrations. Expression of MET wt and METΔ14 was confirmed by immunoblotting. The sum of colonies from 5 random fields at week 3 is reported as the mean of duplicates (±SD). ** p<0.01 B) Anchorage-independent growth was assessed by soft-agar assay comparing AALE cells with stable ectopic expression of indicated constructs. Immunoblotting confirms expression of FLAG-tagged constructs. The sum of colonies from 5 random fields at week 3 is reported as the mean of duplicates (±SD). * p<0.05 C) AALE cell lysates were collected from GFPsg or METsg cells with HGF stimulation at the indicated concentration and time. Expression and phosphorylation of MET (pY1349), AKT (pS473) and MEK1/2 (pS217/221) were measured by immunoblotting. α-tubulin is a loading control. Values next to each band are quantified band intensity. D) Ratio of phospho- to total-MET band intensity from (C) at the indicated time points. E) MET exon 14 status (inclusion or exclusion) was determined by reverse transcription PCR from AALE parental cells and single clone(1F10) selected from MET sgRNA targeted cells (MET sg3). F) Trypsin assay: EGF starved AALE cells were induced by HGF (50ng/ml) for indicated time and then washed and cooled down on ice followed by a treatment with trypsin for 30min. Cell lysates were immunoblotted with MET and HSP90 as loading control.
METΔ14 Increases and Prolongs RAS/AKT & RAS/ERK pathway signaling
To study the mechanism(s) underlying the transforming ability of METΔ14, we measured signaling through the MET/HGF axis to downstream effector pathways. We observed that cells with expression of METΔ14 from the endogenous locus in AALE (METsg) resulted in higher and more durable levels of phospho-Y1349 MET after HGF-stimulation as compared to MET wt-expressing AALE cells (Fig. 2C and 2D). MET/HGF signaling is known to activate Ras and its downstream PI3’K-AKT and MEK-ERK pathways. AKT and MEK1/2 phosphorylation likewise increase more rapidly and persist longer after HGF stimulation (Fig. 2C), suggesting that the strength and/or duration of HGF-MET signaling is increased in cells expressing METΔ14. MET internalization/endocytosis after HGF binding plays a direct role in tumorigenesis in part because activated MET continues to signal to effectors after internalization (15). To study MET internalization, we deployed a sgRNA/CAS9-edited AALE single clone: 1F10, harboring complete, (presumably bi-allelic) editing of MET (Fig. 2E). The addition of trypsin removes extracellular proteins and spares internalized MET, rendering it detectable by immunoblotting after cell lysis (Fig. S2B diagram). After HGF stimulation, METΔ14 is rapidly internalized and persists in an intercellular, active state longer than does wt MET (Fig. 2F), possibly explaining the prolonged METΔ14 signaling observed in this system.
Loss of MET exon 14 Moderately Increases MET protein stability
Deletion of Exon 14 of MET prevents ubiquitination and therefore fosters stabilization of MET protein (2). We measured the half-lives of endogenously-expressed wt MET and METΔ14 protein after HGF stimulation in two individual informative sgRNA/CAS9-edited AALE clones. 1F10 has complete MET editing (Fig. 2D), while clone 2E9 has hemizygous editing of MET (Fig. S3A, right lane). Immunoblotting confirmed that 1F10 expresses exclusively METΔ14 while the 2E9 clone expresses equivalent amounts of both wt MET (upper band) and METΔ14 (lower band) (Fig. S3B). Immunoblotting of cells treated with cycloheximide and serially collected at different time points (Fig. 3A) shows that METΔ14 has a 15% longer protein half-life (t1/2) (Fig. S3C) in both homozygous clone 1F10 (7.4 hr) compared to wt parental (6.5 hr) and in hemizygous clone 2E9 (8.5hr vs. 7.6hr Fig. S3C). Pooled METsg cells also showed similar trends, with a METΔ14 half-life of 7.9 hr compared to the 6.2 hr for MET wt in the GFPsg pool (control) (Fig. S3D and S3E). This modest increase in t1/2 confirms the role of exon 14-encoded Cbl-binding-site in MET stability, but suggests additional, negative regulatory functions possibly attributable to the exon 14-encoded domains.
Figure 3.
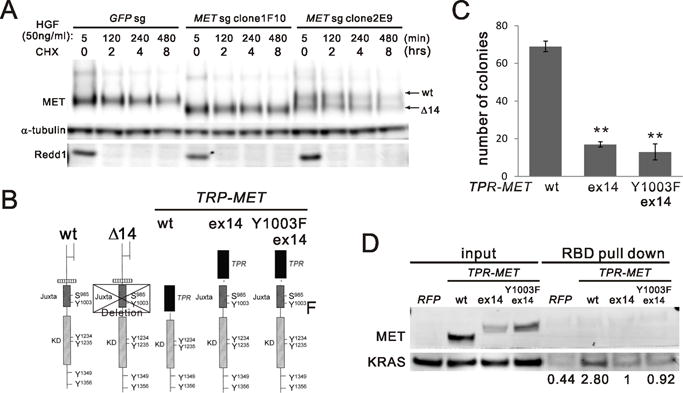
A) MET protein half-life. AALE GFPsg cells or METsg single clone 1F10 and 2E9 were treated with cycloheximide (CHX) for indicated time after 5 min of HGF stimulation and washout. Cell lysates were immunoblotted with MET antibody and α-tubulin antibody and Redd1 antibody for CHX control. B) a schematic panel of MET protein domains in wild type (wt) and mutants. Exon 14 skipping (Δ14), exon 14 (ex14). C) Comparing AALE cells with stable ectopic expression of V5 tagged TPR-MET wt (wt) and mutants including wt exon14 insertion (ex14) and Y1003F exon14 insertion (Y1003F ex14). Anchorage-independent growth was assessed as described in (Fig. 2A). **p<0.01. D) RAS-binding domain (RBD) pull down assay comparing RAS activation in AALE cells described in (C).
MET exon 14-encoded Domains Regulate MET Kinase Activity beyond CBL Engagement
To dissect the effects of METΔ14 on protein activity from those related to protein turnover, we generated multiple AALE cell lines with stable ectopic expression of mutant TRP-MET alleles. Since TRP-MET lacks the MET exon 14 sequence (20), it has been used as a template upon which to study the independent effect(s) of exon 14 vs. those of the Y1003 CBL binding site on MET activity (21). We inserted either the wt or Y1003F exon 14 sequence into the TPR-MET fusion junction in frame with the c-terminus of MET (Fig. 3B diagram). We generated AALE cells with stable expression of these constructs respectively confirmed by immunoblotting (Fig. S3F). Insertion of wt exon 14 inhibits 80% of the transforming activity of TPR-MET (Fig. 3C and Fig. S3G). Introducing Y1003F, known to prevent Cbl binding (2) in exon 14 failed to reverse the inhibition of the transforming activity of TPR-MET by wt exon 14 sequence (Fig. 3C), consistent with previous reports in mouse fibroblasts (21). Similar effects are seen on RAS-GTP loading, a marker of RAS activation (Fig. 3D). Co-immunoprecipitation of alternatively-tagged proteins demonstrated that exon 14 insertion does not disrupt TPR-MET dimerization (Fig. S3H). These results confirm the previously described inhibitory role of exon 14(2) and suggest additional negative regulatory function(s) of the exon 14-encoded juxtamembrane domain of MET beyond prevention of CBL engagement.
MetΔ15 Induces Lung Adenocarcinoma in a Novel in vivo Mouse Model
To explore the role of METΔ14 in vivo we utilized a mouse lung cancer model (Fig. 4A) in which lentivirus encoding a cDNA in cis with Cre recombinase is delivered intranasal, leading to expression of the cDNA and ablation of floxed germ line elements in the same lung cancer precursor cell. Since amplification of MDM2 or TP53 mutation co-occur with the vast majority of patients with MET exon 14 mutations (4,6), we infected immunocompetent Trp53flox mice (22) with a lentivirus encoding mouse Met lacking exon15 (MetΔ15, the mouse equivalent of human METΔ14) in cis with Cre recombinase. Lentivirus encoding KrasG12D or GFP in cis with Cre served as controls. Mice receiving KrasG12D or MetΔ15 lentivirus became tachypneic and were euthanized at 3 months post infection. These animals all harbored high-grade lung adenocarcinoma while those receiving GFP + Cre virus did not (Fig. 4B and Fig. S4A). Immunohistochemistry staining showed that all tumors expressed the lung marker Ttf-1 and proliferation marker Ki67 (Fig. 4C) while the MetΔ15-driven tumors specifically showed phospho-Met. Delivery of a distinct lentivirus encoding sgRNA targeting the endogenous mouse Met exon15 exon/intron junction in cis with Cre to Trp53flox mice resulted in adenoma but not adenocarcinoma formation at three months, indicating likely additive effects of MetΔ15 activity and Met overexpression or inherent differences in cDNA expression vs. sgRNA editing, or both (Fig. 4C right panel). In summary, the lentivirus delivered MetΔ15 expression in combination with genetic mouse recapitulates some of the lung adenoma cases seen in human patients and is a model to study MetΔ15 inhibition in vivo.
Figure 4.
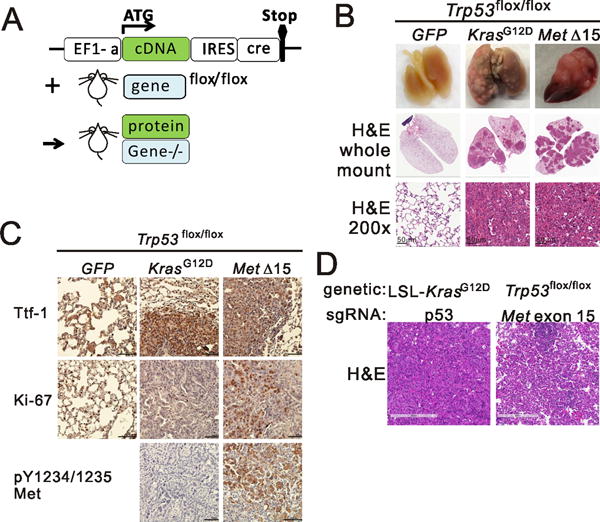
A new mouse model of MetΔ15-driven Lung Cancer. A) Schematic diagram of the combination mouse model. B) Whole mount lungs from Trp53flox/flox mice intranasal infected with indicated lentivirus. Top, macroscopic appearance of representative lungs; middle, Haematoxylin–eosin (H&E) staining of lungs, whole mount, bottom, H&E staining of lungs at high magnification. C) Representative immunohistochemistry of lungs from (B) stained with antibodies against the indicated proteins. D) Hematoxylin–eosin (H&E) staining of tumors from mice from indicated genetic background infected with the indicated sgRNA and cre encoding lentivirus.
Therapeutic Benefit of Met inhibition in vivo
Crizotinib is FDA-approved for the treatment of ALK-fused NSCLC but also inhibits MET (23). Several groups have reported off-label use of crizotinib in NSCLC harboring MET exon 14 mutations (4),(19),(24). We deployed the MetΔ15/Trp53flox model to study crizotinib response in vivo on MetΔ15-driven tumors. Tumor-bearing mice were randomized by tumor volume (as assessed by micro-computed tomography performed at 8 weeks after lentiviral induction, Fig. 5A, D0 panel) to receive either crizotinib (50mg/kg/d) or vehicle. Crizotinib-treated mice had, on average, stable disease while control animals continued to progress (Fig. 5B and Fig. S4B). Immunostaining for phospho-MET showed that crizotinib treatment inhibited Met auto-phosphorylation and activation (Fig. 5C). Ki-67 reactivity (Fig. 5C and 5D) was also significantly lower in the crizotinib-treated group. These results suggest that crizotinib has a favorable, but suboptimal, therapeutic effect in this novel Met exon skipping driven mouse model of this important NSCLC subtype.
Figure 5.
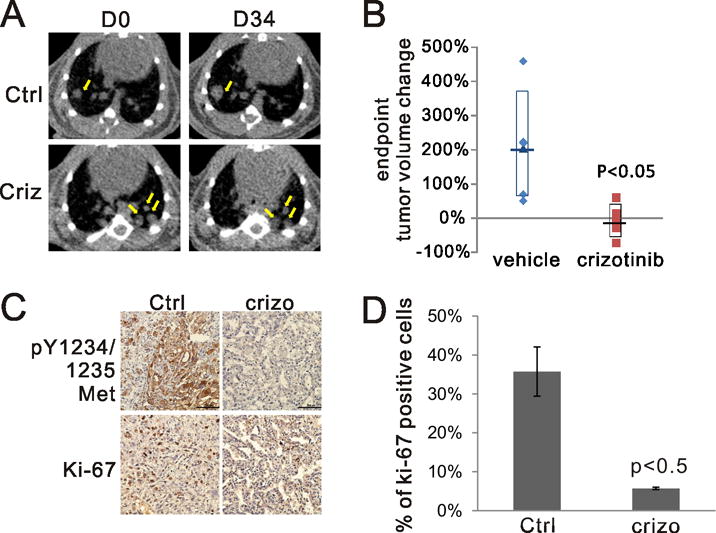
A) Representative microCT scans of mice receiving vehicle or crizotinib at the beginning (D0) and the end of treatment (D34). Yellow arrows point to representative tumor areas. B) The change of tumor volume reconstructed from microCT. % volume change= (D34 –D0)/D0 × 100%. C) Representative immunohistochemistry of lungs from vehicle or crizotinib treated groups stained with antibodies against the indicated proteins. Ctrl: vehicle treated, crizo: crizotinib treated. D) Quantification of Ki-67 positive cells from (C). Tumors from two mice for each group were evaluated.
Response to crizotinib and acquired resistance in a patient with METΔ14-Mutated NSCLC
We found coincident METΔ14 mutation with ~15× amplification, among other aberrations (Fig. 5A inserted table) in a 63-year-old, otherwise healthy, never-smoking female diagnosed with lung adenocarcinoma and pain. Systemic therapy with crizotinib was initiated after detection of METΔ14 mutation with ~15× amplification in tumor sequencing results. Restaging PET/CT demonstrated a dramatic improvement of disseminated disease involving the liver, adrenals, multiple osseous lesions and subcutaneous disease (Fig. 6A and Fig. S5A) and a near-complete radiographic response at three months (Fig.6B). Subsequent imaging at nine months showed progression in the liver (Fig. 6C). Cell-free DNA sequencing(18) was performed in the setting of clinical progression to explore mechanisms of acquired resistance without rebiopsy. Pretreatment MET exon 14 skipping mutation and MET amplification were identified along with four new, subclonal, missense mutations in MET: D1228N, Y1230H, Y1230S and G1163R (amino acid numbering according to UniProtKB P08581, Fig. 6C inserted table), all of which are located in the ATP docking pocket (25) (26) and have been previously and independently reported to activate the kinase, impair crizotinib binding (23) and to lead to resistance to other MET inhibitors in vitro (26). These mutations occur in trans from one another and in quantitatively lower allele fractions than the initial MET exon 14 mutations (Fig. S5B), indicating convergent, acquired resistance through activation loop interactions with crizotinib. This is the first clinical documentation of multiple secondary mutations in a crizotinib-treated case of METΔ14 driven lung adenocarcinoma, definitively proving that METΔ14 is the target of crizotinib and that METΔ14 is a therapeutic target in lung adenocarcinoma.
Figure 6.
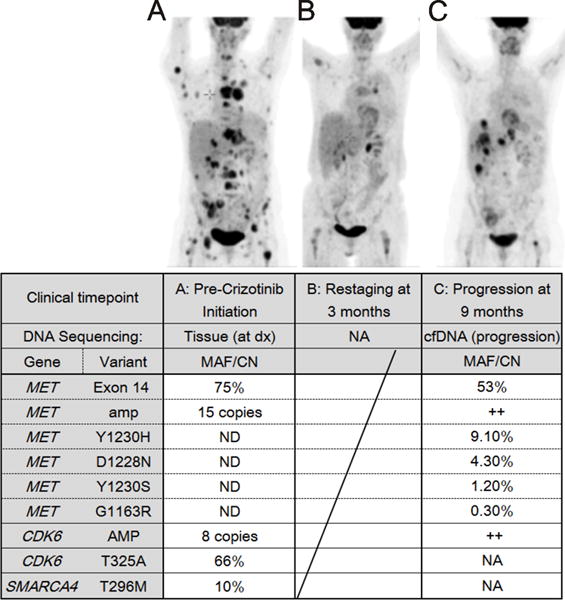
Acquired resistance to crizotinib in a 63-year-old NSCLC patient with METΔ14 mutation. Positron emission tomography of metastatic NSCLC in a 63-year-old female patient. A) Before treatment. B) After one month of crizotinib. C) Upon clinical progression. Inserted Table: Amp=amplification, Dx= diagnosis, cfDNA= cell free DNA, ND = Tested and Not Detected, NA= not assessed. MAF: Mutant Allele Fraction, CN: Copy Number, ++: amplified.
Discussion
The need to extend targeted approaches to larger fractions of patients with cancer is clear and compelling. We find 2.8% of lung adenocarcinomas show evidence of MET mRNA skipping, the vast majority of which result from acquired, somatic mutations and/or deletions in or around the exon 14-coding region of the gene, detectable with genomic profiling of MET. The two of 16 samples in which we observed evidence of mRNA skipping but could not detect DNA aberrations in MET could be either false positive mRNA predictions or harbor yet unexplained mechanisms for exon 14 exclusion in the mRNA transcript of MET. The clinical relevance of this population of NSCLC is large, as METΔ14 was at least as frequent as ALK and ROS1 fusions combined in one large dataset where both fusions and skipping were evaluable (10). Our integrative genomic, biochemical, animal and clinical analyses firmly establish METΔ14 as a driver event in NSCLC. We show that METΔ14 expression from the endogenous, non-amplified allele is transforming in an HGF-dependent manner, suggesting HGF levels as a possible independent determinant of activity, response and resistance to therapy. Our experiments here were designed to decouple METΔ14 catalytic activity from the effects of overexpression known to be transforming (27), a distinction our prior ectopic expression studies could not make(4). Our results with endogenous METΔ14 expression levels achieved through genomic editing of a relevant human cell type argue that the exon 14 encoded portion of MET negatively regulates the kinase activity as well as the half-life of the receptor.
Case series have demonstrated variable responses to crizotinib treatment in patients with genomic MET alterations including but not limited to METΔ14. We show for the first time that expression of the analogous MetΔ15 is oncogenic in adult mice and that its inhibition leads to demonstrable therapeutic benefit in vivo, proving MET itself (rather than an off-target kinase) as the most likely therapeutic target in the aforementioned clinical responses seen in patients. The use of this novel, preclinical model clarifies the likely target of a drug in routine use through directed interrogation of aberrations seen in actual patients. Variations of this flexible system could be deployed for evaluating numerous additional candidate driver oncogenes in a reliable, economically efficient and clinically relevant timeline.
Finally, we report the first case of acquired resistance to MET inhibition through multiple secondary missense mutations clustered in the activation loop of MET. This case emphasizes several important concepts in the targeted therapy of solid tumors. Most importantly, this is the first clinical documentation of multiple secondary mutations in the METΔ14 allele acquired upon clinical progression, definitively proving that METΔ14 is a therapeutic target. Secondly, it underscores the clinical relevance of tumor heterogeneity, as evidenced by multiple missense mutations in trans to one another, converging on residues critical for crizotinib binding. Finally, this case stresses the importance of unbiased reassessment of tumor genotype at progression, which in this case nominates available MET inhibitors with distinct modes of binding for treatment of progressive disease.
Supplementary Material
Acknowledgments
We are grateful to the members of the Jura, Meyerson and Collisson labs for discussion and Yan Guo for bioinformatics consulting. We acknowledge the Preclinical Therapeutics Core and Small Animal Imaging Core at U.C.S.F. and the center for comparative medicine at U.C. Davis. We also would like to thank the Bandyopadhyay, Bivona, Scott, Jacks, Ruggero and McMahon labs for providing reagents.
Financial support:
M. Meyerson is supported by an American Cancer Society Research Professorship and by National Cancer Institute grant R35-CA197568. E. Collisson is supported by the Doris Duke Charitable Foundation, American Lung Association, Lung Cancer Research Foundation and NCI/NIH U24-CA21097 and R01-CA178015.
Abbreviations list
- MET
mesenchymal-epithelial transition
- HGF
Hepatocyte growth factor
References
- 1.Ma PC, Kijima T, Maulik G, Fox EA, Sattler M, Griffin JD, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63(19):6272–81. [PubMed] [Google Scholar]
- 2.Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006;66(1):283–9. doi: 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 3.Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9. doi: 10.1158/2159-8290.CD-15-0285. [DOI] [PubMed] [Google Scholar]
- 5.Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbe S, et al. Met/Hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol. 2005;25(21):9632–45. doi: 10.1128/MCB.25.21.9632-9645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 7.Ou SI, Young L, Schrock AB, Johnson A, Klempner SJ, Zhu VW, et al. Emergence of Preexisting MET Y1230C Mutation as a Resistance Mechanism to Crizotinib in NSCLC with MET Exon 14 Skipping. J Thorac Oncol. 2016 doi: 10.1016/j.jtho.2016.09.119. [DOI] [PubMed] [Google Scholar]
- 8.Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, et al. Acquired METD1228V Mutation and Resistance to MET Inhibition in Lung Cancer. Cancer Discov. 2016;6(12):1334–41. doi: 10.1158/2159-8290.CD-16-0686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–44. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607–16. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med. 2016;375(12):1109–12. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundberg AS, Randell SH, Stewart SA, Elenbaas B, Hartwell KA, Brooks MW, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene. 2002;21(29):4577–86. doi: 10.1038/sj.onc.1205550. [DOI] [PubMed] [Google Scholar]
- 14.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2(11):e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joffre C, Barrow R, Menard L, Calleja V, Hart IR, Kermorgant S. A direct role for Met endocytosis in tumorigenesis. Nat Cell Biol. 2011;13(7):827–37. doi: 10.1038/ncb2257. [DOI] [PubMed] [Google Scholar]
- 16.Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med. 2015;21(9):1038–47. doi: 10.1038/nm.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994–1004. [PMC free article] [PubMed] [Google Scholar]
- 18.Lanman RB, Mortimer SA, Zill OA, Sebisanovic D, Lopez R, Blau S, et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One. 2015;10(10):e0140712. doi: 10.1371/journal.pone.0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paik PK, Drilon A, Fan PD, Yu H, Rekhtman N, Ginsberg MS, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5(8):842–9. doi: 10.1158/2159-8290.CD-14-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wicker R, Bounacer A, Gascon A, Brison O, Sarasin A, Suarez HG. Cloning and sequencing of a tpr-met oncogene cDNA isolated from MNNG-transformed human XP fibroblasts. Biochim Biophys Acta. 1995;1264(3):254–6. doi: 10.1016/0167-4781(95)00174-3. [DOI] [PubMed] [Google Scholar]
- 21.Vigna E, Gramaglia D, Longati P, Bardelli A, Comoglio PM. Loss of the exon encoding the juxtamembrane domain is essential for the oncogenic activation of TPR-MET. Oncogene. 1999;18(29):4275–81. doi: 10.1038/sj.onc.1202791. [DOI] [PubMed] [Google Scholar]
- 22.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 23.Cui JJ, Tran-Dube M, Shen H, Nambu M, Kung PP, Pairish M, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54(18):6342–63. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 24.Awad MM. Impaired c-Met Receptor Degradation Mediated by MET Exon 14 Mutations in Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34(8):879–81. doi: 10.1200/JCO.2015.64.2777. [DOI] [PubMed] [Google Scholar]
- 25.Peach ML, Tan N, Choyke SJ, Giubellino A, Athauda G, Burke TR, Jr, et al. Directed discovery of agents targeting the Met tyrosine kinase domain by virtual screening. J Med Chem. 2009;52(4):943–51. doi: 10.1021/jm800791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiedt R, Degenkolbe E, Furet P, Appleton BA, Wagner S, Schoepfer J, et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011;71(15):5255–64. doi: 10.1158/0008-5472.CAN-10-4433. [DOI] [PubMed] [Google Scholar]
- 27.Toschi L, Janne PA. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin Cancer Res. 2008;14(19):5941–6. doi: 10.1158/1078-0432.CCR-08-0071. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.