

Abstract
Objective
Hypoxia-inducible factor 1 (HIF-1) and activator protein 1 (AP-1) are important transcription factors regulating expression of genes involved in cell survival. HIF-1α and c-Jun are key components of HIF-1 and AP-1, respectively, and are regulated by epidermal growth factor receptor (EGFR)-mediated cell signaling and tumor microenvironmental cues. The roles of HIF-1α and c-Jun in development of resistance to EGFR tyrosine kinase inhibitor (TKI) in non-small cell lung cancer (NSCLC) with activating mutation of EGFR have not been explored. In this study, we investigated the roles of HIF-1α and c-Jun in mediating primary and acquired resistance to gefitinib in NSCLC cells with activating mutation of EGFR.
Materials and Methods
Changes in HIF-1α protein and in total and phosphorylated c-Jun levels in relation to changes in total and phosphorylated EGFR levels before and after gefitinib treatment were measured using Western blot analysis in NSCLC cells sensitive or resistant to gefitinib. The impact of overexpression of a constitutively expressed HIF-1α (HIF-1α/ΔODD) or a constitutively active c-Jun upstream regulator (SEK1 S220E/T224D mutant) on cell response to gefitinib was also examined. The effect of pharmacological inhibition of SEK1-JNK-c-Jun pathway on cell response to gefitinib was evaluated.
Results
Downregulation of HIF-1α and total and phosphorylated c-Jun levels correlated with cell inhibitory response to gefitinib better than decrease in phosphorylated EGFR did in NSCLC cells with intrinsic or acquired resistance to gefitinib. Overexpression of HIF-1α/ΔODD or SEK1 S220E/T224D mutant conferred resistance to gefitinib. There exists a positive feed-forward regulation loop between HIF-1 and c-Jun. The JNK inhibitor SP600125 sensitized gefitinib-resistant NSCLC cells to gefitinib.
Conclusions
HIF-1α and c-Jun functionally cooperate in development of resistance to gefitinib in NSCLC cells. The translational value of inhibiting HIF-1α/c-Jun cooperation in overcoming resistance to EGFR TKI treatment of NSCLC cells with activating mutation of EGFR deserves further investigation.
Keywords: Non-small cell lung cancer, EGFR mutation, TKI, HIF-1α, c-Jun
1. Introduction
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have been used worldwide to treat patients with advanced non-small cell lung cancer (NSCLC) since the first-generation EGFR TKIs (gefitinib and erlotinib) were approved in early 2000s. In NSCLC patients with tumors harboring EGFR activating mutations in exons 19–21 of EGFR kinase domain, treatment with a first-generation EGFR TKI prolonged progression-free survival time by about 9.7 months [1].
However, not all NSCLC patients with the EGFR activating mutations in their tumors have such favorable outcomes in response to EGFR TKIs. About 20% to 30% of NSCLC patients with the EGFR activating mutations in their tumors have no objective tumor-regressive response to initial treatment with a first-generation EGFR TKI [1–4]. A number of mechanisms can cause intrinsic (primary) resistance of NSCLC to EGFR TKIs, including EGFR downstream pathway redundancy (activated by overlapping signal pathways), pathway reactivation (independent of EGFR due to oncogenic mutations or mutational inactivation of key signaling molecules, such as Ras [5,6] and PTEN [7,8]), and pathway alternation (escape from EGFR signaling regulation via recruiting an alternate signaling pathway) [9]. In addition, tumor microenvironmental cues and tumor heterogeneity can cause intrinsic resistance to EGFR TKI [10].
Moreover, even in patients who initially have a partial or complete response to an EGFR TKI, acquired resistance may ultimately occur. The mechanisms underlying development of acquired resistance of NSCLC to first-generation EGFR TKIs include EGFR T790M secondary mutation (present in ~60% cases of acquired resistance), MET amplification (5%–10%), PIK3CA mutation (~5%), BRAF mutation (~1%), and small-cell cancer transformation (~5%) [11]; in another approximately 20% to 25% of cases of acquired resistance, the underlying mechanisms remain unclear.
Many NSCLC patients, whose tumor initially responds to a first-generation EGFR TKI but develops resistance due to secondary EGFR T790M mutation, benefit from treatment with a third-generation EGFR TKI, such as osimertinib; however, a significant percentage of patients with acquired resistance due to EGFR T790M mutation do not respond to a third-generation EGFR TKI [12,13]. Novel insights into the mechanisms of resistance to EGFR TKIs are critical for developing new therapeutic strategies for improving the outcome of NSCLC patients with advanced disease.
Hypoxia-inducible factor-1 (HIF-1), a master regulator of response to tumor hypoxia, is a heterodimer consisting of an oxygen-sensitive alpha subunit (HIF-1α) and a constitutively expressed beta subunit (HIF-1β) [14–18]. The level of HIF-1α is increased dramatically in hypoxic tumor microenvironments because of decreased ubiquitination and degradation of HIF-1α protein associated with tumor hypoxia [19,20]. The level of HIF-1α is also upregulated by aberrant cell signaling through increased expression [21–25]. We previously showed that downregulation of HIF-1α through inhibiting EGFR downstream cell signaling is required for the antiproliferative effects of the anti-EGFR antibody cetuximab in head and neck cancer, colorectal cancer, and NSCLC models [26–36].
Activator protein-1 (AP-1) is a transcription factor that regulates gene expression in response to a variety of extracellular stimuli, growth factors, cytokines, heat shock, UV irradiation, hypoxia, and so forth [37]. AP-1, like HIF-1, is a dimeric complex; AP-1 is assembled through heterodimerization between members containing a leucine zipper motif, including c-Jun, c-Fos, ATF (activating transcription factor), and MAF (musculoaponeurotic fibrosarcoma) [38,39]. c-Jun is regulated by a combination of enhanced expression and phosphorylation on specific serine residues (S63 and S73) of c-Jun by c-Jun N-terminal kinase (JNK) [40–42], which is also known as stress-activated MAP kinase (SAPK), a member of the mitogen-activated protein (MAP) kinase family [43]. JNK is activated by dual phosphorylation on threonine and tyrosine residues (T183 and T185) by a member of the MAPKK group of protein kinases [44,45], specifically, by SAPK/Erk kinase (SEK) [46].
AP-1 has been shown to functionally cooperate with HIF-1 in hypoxia-induced gene transcription [47]. The response of AP-1/c-Jun to chronic hypoxia was reported to be HIF-1α-dependent [48]. In this study, we tested the hypotheses that HIF-1α serves as a biomarker of NSCLC cell response to gefitinib and that HIF-1α and c-Jun functionally cooperate in mediating resistance to gefitinib in NSCLC cells with activating mutation of EGFR.
2. Materials and methods
2.1. Reagents
Gefitinib (ZD1839) was purchased from Sigma-Aldrich Corp. (St. Louis, MO). SP600125 (1,9-pyrazoloanthrones) was purchased from Calbiochem/EMD Chemicals Inc. (Gibbstown, NJ). Antibodies against total and S73-phosphorylated c-Jun, total and T183/Y185-phosphorylated JNK, total and Y1068-phosphorylated EGFR, PARP, and β-actin were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-HIF-1α antibody was purchased from BD Biosciences. cDNA constructs containing HIF-1α/ΔODD and constitutively active SEK1 S220E/T224D mutant were provided by Dr. L. Eric Huang (University of Utah School of Medicine, Salt Lake City, UT) and Dr. Jonathan Kurie (The University of Texas MD Anderson Cancer Center, Houston, TX), respectively. Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA).
2.2. Cell lines and cell cultures
Human lung adenocarcinoma cells H3255 (harboring EGFR L858R mutation in exon 21), HCC827 (harboring EGFR E746-A750 in-frame deletion in exon 19), H1650 (harboring EGFR E746-A750 in-framedeletion in exon 19 and PTEN mutation), and H1975 (harboring EGFR L858R mutation in exon 21 and T790M mutation in exon 20) were maintained in Dulbecco’s modified Eagle’s medium/F12 medium supplemented with 10% FBS, 100U/mL penicillin, and 100 μg/mL streptomycin under the condition of 5% CO2 at 37°C.
2.3. Western blot analysis
After desired treatments, cells were washed twice with cold PBS and lysed in a lysis buffer (50mM Tris, pH 7.4, 150 mM NaCl, 0.5% NP-40, 50mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 25μg/ml aprotinin, and 25 μg/ml leupeptin) for 15minutes on ice [36,49]. Insoluble cell debris was removed by centrifugation. Equal amounts of protein lysates, quantified by Pierce Coomassie Plus, were separated by SDS–PAGE, blotted onto nitrocellulose, and probed with various primary antibodies. The signals were visualized using an enhanced chemiluminescence detection kit (GE Healthcare, Piscataway, NJ), and the bands of interest were quantified using Image J.
2.4. MTT proliferation assay
Cells were seeded in 48-well plates with 0.2 mL/well of medium containing 0.5% FBS at 37°C. Following the indicated treatment, cells were incubated with 20 μL/well of 10mg/mL MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) for 2hours and then lysed with a lysis buffer (200 μL/well) containing 20% SDS in dimethyl formamide/H2O (1:1, v/v; pH 4) at 37°C overnight. The relative number of surviving cells in each group was determined by measuring the optical density (OD) of the cell lysates at an absorbance wavelength of 570 nm. The OD value of each treatment group was expressed as a percentage of the OD value of the untreated control cells [32,50]. IC50 value was determined using Excel FORECAST.
2.5. cDNA transfection
Cells were transfected with cDNA plasmids using Lipofectamine 2000. Briefly, cells were seeded in 60-mm plates at 80% to 90% confluency overnight. cDNA and Lipofectamine 2000 were mixed in 100 μL of minimal essential medium (Opti-MEM, Life Technologies, Carlsbad, CA) for 15 minutes, and then this mixture was added into the culture medium. Six hours later, the medium was replaced with regular medium, and the cells were cultured for an additional 48 hours before detection of expression of cDNA constructs by Western blotting.
2.6. Statistical analysis
Experimental data are presented as mean ± standard deviation. Student’s t-test was used in statistical analyses using SPSS 19.0 software. P value <0.01 was considered statistically significant.
3. Results
3.1. Downregulation of HIF-1α and total and phosphorylated c-Jun protein correlates with cell inhibitory response to gefitinib in NSCLC cells
As shown in Fig. 1, four human NSCLC cell lines harboring EGFR mutation in exon 19 or 21, which is generally considered a predictor of response to the first-generation EGFR TKIs [51–53], responded differently to treatment with gefitinib. HCC827 and H3255 cells were sensitive to gefitinib, with IC50 values of 8.26 nM and 22.75 nM, respectively (Fig. 1A, left). In contrast, H1650 and H1975 cells were resistant to gefitinib, with IC50 values greater than 2000 nM (Fig. 1A, right). As shown in Fig. 1B, after overnight exposure to gefitinib at various concentrations, HCC827 and H3255 cells but not H1650 and H1975 cells exhibited apoptosis as indicated by appearance of PARP cleavage.
Fig. 1. Downregulation of HIF-1α and c-Jun protein correlates with cell inhibitory response to gefitinib treatment in NSCLC cells.
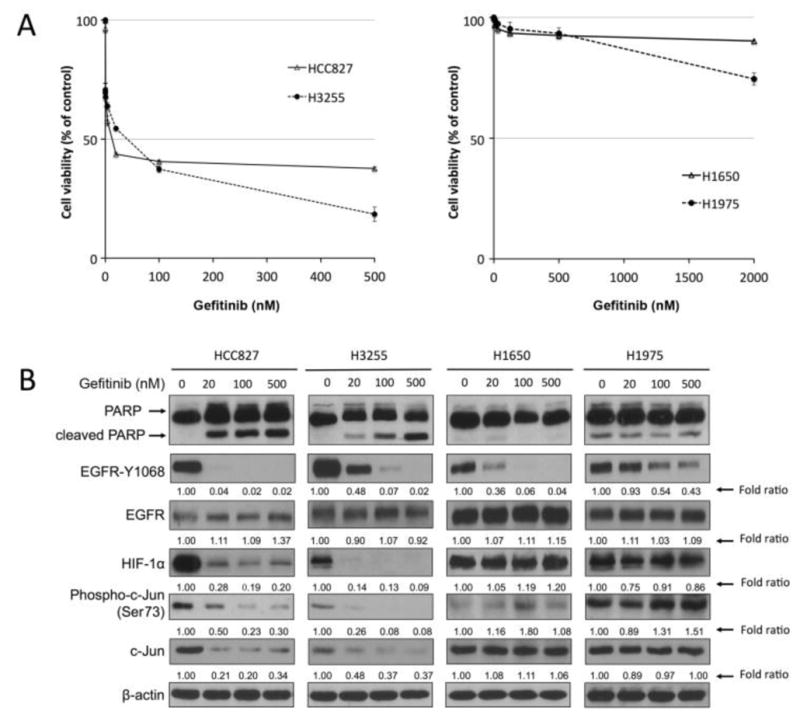
A. HCC827 and H3255 cells (left panel) and H1650 and H1975 cells (right panel) were treated with the indicated concentrations of gefitinib for 5 days. Cell survival was measured by MTT proliferation assay. B. HCC827, H3255, H1650, and H1975 cells were treated with the indicated concentrations of gefitinib for 24 hours. Cells were then harvested, and equal amounts of protein lysates were subjected to Western blot analysis with the indicated antibodies. The level of β-actin was used as a reference of lysate protein loading control of each cell line.
Of note, the tyrosine kinase activity of EGFR was inhibited in an EGFR TKI dose-dependent manner in all four cell lines, although there were differences in sensitivity: HCC827 (gefitinib-sensitive) was the most sensitive, H3255 (gefitinib-sensitive) and H1650 (gefitinib-resistant) had intermediate sensitivity, and H1975 (gefitinib-resistant) was relatively less sensitive. In contrast, levels of HIF-1α and c-Jun seemed to be more closely associated with the response to gefitinib treatment. Specifically, both HIF-1α and c-Jun showed remarkable decrease in protein level detected by Western blotting after gefitinib treatment in the gefitinib-sensitive cell lines (HCC827 and H3255) but not in the gefitinib-resistant cell lines (H1650 and H1975) at doses as high as 500 nM gefitinib. This interesting finding suggests that HIF-1α and c-Jun are important signaling molecules mediating NSCLC cell response to EGFR TKI.
3.2. Levels of HIF-1α and c-Jun are increased in NSCLC cells with acquired resistance to gefitinib
To further investigate whether decreased levels of HIF-1α and c-Jun correlate with inhibitory response to EGFR TKI treatment, we exposed H3255 and HCC827 cells (both gefitinib-sensitive) to gefitinib at increasing concentrations for an extended period of time. Through this process, we generated a subline of H3255 cells, H3255RR, that was relatively resistant to gefitinib compared to the parental H3255 cells, and a subline of HCC827 cells, HCC827R, that exhibited remarkable resistance to gefitinib. As shown in Fig. 2A, the IC50 value for H3255RR was 92 nM, approximately 11 times the IC50 value for the parental H3255 cells (8.26 nM, Fig. 1A), and the IC50 value for HCC827R was over 500 nM, dramatically higher than the IC50 value for the parental HCC827 cells (22.75 nM, Fig. 1A).
Fig. 2. The levels of HIF-1α and c-Jun are increased in NSCLC cells with acquired resistance to gefitinib.

A. HCC827R and H3255RR cells were treated with the indicated concentrations of gefitinib for 5 days. Cell survival was measured by MTT proliferation assay. B. HCC827, HCC827R, H3255 and H3255RR cells were treated with 20 nM gefitinib or DMSO vehicle control for 24 hours. Cells were then harvested, and equal amounts of protein lysates were subjected to Western blot analysis with the indicated antibodies. The level of β-actin was used as a reference of lysate protein loading control of each cell line.
We found that, compared with their respective parental cells, both H3255RR and HCC827R cells exhibited much higher basal protein levels of HIF-1α and total and phosphorylated c-Jun (Fig. 2B). In the H3255RR cells, even though the resistance to gefitinib relative to parental H3255 cells was not as great as the resistance of HCC827R cells relative to parental HCC827 cells, the increase in baseline HIF-1α and c-Jun levels between parental and resistant cells was remarkable, as shown by the fold ratios (Fig. 2B). The levels of HIF-1α and total and phosphorylated c-Jun were not decreased by gefitinib in HCC827R cells. In H3255RR cells, although the levels of HIF-1α and total and phosphorylated c-Jun were decreased by gefitinib, the magnitude of the decrease was less than the magnitude of the decrease in the parental H3255 cells, as shown by the fold ratios (Fig. 2B). The decrease likely reflected the difficulty of developing H3255 sublines resistant to gefitinib. As shown in Fig. 2A, H3255RR cells retained considerable sensitivity to gefitinib even though they were relatively resistant compared to the parental H3255 cells.
3.3. c-Jun expression is induced under hypoxia and upregulated by HIF-1, and HIF-1α and c-Jun contribute to resistance to gefitinib in NSCLC cells
We next investigated the functional roles of HIF-1α and c-Jun and their interactions in mediating resistance to gefitinib. Tumor hypoxia is common in solid tumors, including NSCLC. HIF-1α rapidly accumulates in cells under hypoxia, and c-Jun is known to be activated by HIF-1 under hypoxia in certain experimental conditions. We compared HIF-1α and c-Jun levels between NSCLC cells cultured under normoxia and hypoxia. As shown in Fig. 3A, overnight incubation of the cells in a hypoxic chamber with 1% oxygen led to an increase in HIF-1α level in all four NSCLC cell lines, which had various degrees of sensitivity to gefitinib. The low-oxygen condition also led to increases in the levels of both total and phosphorylated c-Jun protein and increases in the levels of both total and phosphorylated JNK1 protein, with the degrees of increase differing by cell type. Since hypoxia is one of the characteristic features of solid tumors, these data suggested that the hypoxic microenvironment might play a role in development of resistance to EGFR TKI in NSCLC.
Fig. 3. c-Jun expression is induced under hypoxia and upregulated by HIF-1, and c-Jun and HIF-1α contribute to resistance to gefitinib in NSCLC cells.

A. HCC827, H3255, H1650, and H1975 cells were cultured in normoxia or in a hypoxia chamber (1% O2) for 24 hours. Cells were then harvested immediately, and equal amounts of protein lysates were subjected to Western blot analysis with the indicated antibodies. B. HCC827 and H3255 cells were transfected with a construct containing HIF-1α/ΔODD mutant or pcDNA3.1 control vector as described in Materials and Methods. Cell lysates were prepared for Western blotting with the indicated antibodies. C. HCC827 and H3255 cells expressing control vector or HIF-1α/ΔODD mutant were treated with 20 nM gefitinib or vehicle control for 24 hours. Cell lysates were prepared for Western blotting with the indicated antibodies. The level of β-actin was used as a reference of lysate protein loading control of each cell line.
To test our hypothesis that hypoxia contributes to development of resistance to gefitinib in part through upregulating c-Jun, we introduced a constitutively expressed HIF-1α construct containing a HIF-1α oxygen-dependent degradation domain deletion mutant (HIF-1α/ΔODD) in HCC827 and H3255 cells. We previously reported that HIF-1α/ΔODD mutant retains the majority of the transcriptional activity of full-length HIF-1α and can be stably overexpressed in a normoxic environment [35]. Compared to the control vector-transfected cells, HCC827 and H3255 cells expressing HIF-1α/ΔODD exhibited higher levels of total and phosphorylated c-Jun and JNK (Fig. 3B). This result was consistent with our observation of increase in total and phosphorylated c-Jun and JNK under hypoxia (Fig. 3A). To examine whether HIF-1α has a causal role in mediating resistance to gefitinib, we examined the induction of apoptosis after gefitinib treatment in NSCLC cells with and without overexpression of HIF-1α/ΔODD. As shown in Fig. 3C, there was a noticeable decrease in the level of PARP cleavage in HCC827 HIF-1α/ΔODD cells and H3255 HIF-1α/ΔODD cells compared to the results in the respective control vector-transfected cells. This finding indicated that HIF-1α overexpression plays a critical role in mediating resistance to EGFR TKI treatment in NSCLC.
3.4. c-Jun phosphorylation upregulates HIF-1α expression and renders NSCLC cells resistant to gefitinib
To further understand the interaction between HIF-1α and c-Jun in mediating NSCLC resistance to gefitinib, we used both genetic and small-molecule-inhibitor approaches to investigate the role of c-Jun in the scenario. SAPK/Erk kinase (SEK1), also known as MKK4 or JNK kinase, can activate the MAP kinase (SAPK and JNK) and subsequently activates c-Jun by phosphorylating c-Jun on serine 63 and serine 73 within its transcriptional activation domain [40,41]. We transfected HCC827 and H3255 cells with a construct containing a constitutively active SEK1 S220E/T224 mutant. As shown in Fig. 4A, transfection with SEK1 S220E/T224D mutant successfully upregulated the levels of phosphorylated SAPK/JNK and phosphorylated c-Jun in HCC827 and H3255 cells. Overexpression of SEK1 S220E/T224D mutant reduced the induction of PARP cleavage by gefitinib treatment in both cell lines (Fig. 4B), suggesting that c-Jun activation can inhibit gefitinib-induced apoptosis in NSCLC cells. We also found that overexpression of SEK1 S220E/T224D mutant upregulated the level of HIF-1α (Fig. 4A). These data suggested that there is reciprocal regulation between HIF-1α and c-Jun that forms a positive feed-forward loop that facilitates development of cellular resistance to gefitinib in NSCLC cells.
Fig. 4. c-Jun phosphorylation upregulates HIF-1α expression and renders NSCLC cells resistant to gefitinib.

A. HCC827 and H3255 cells were transfected with a construct containing SEK1 S220E/T224D or pcDNA3.1 control vector as described in Materials and Methods. Cell lysates were prepared for Western blotting with the indicated antibodies. B. HCC827 and H3255 cells expressing SEK1 S220E/T224D or the control vector were treated with 20 nM gefitinib or vehicle control for 24 hours. Cell lysates were prepared for Western blotting with the indicated antibodies. The level of β-actin was used as a reference of lysate protein loading control of each cell line.
3.5. SP600125 inhibits c-Jun phosphorylation, downregulates HIF-1α level, and promotes induction of apoptosis by gefitinib in gefitinib-resistant NSCLC cells
To examine whether pharmacological inhibition of JNK may overcome resistance to gefitinib in NSCLC cells, we subjected gefitinib-resistant H1650 and H1975 cells to overnight treatment with gefitinib with and without SP600125, a potent JNK and c-Jun inhibitor [54,55]. As shown in Fig. 5A, the levels of phosphorylated SAPK/JNK and phosphorylated c-Jun were remarkably decreased after SP600125 treatment. Furthermore, HIF-1α protein level was downregulated in both cell lines. This result was consistent with our finding shown in Fig. 4A that phosphorylation of c-Jun led to upregulation of HIF-1α.
Fig. 5. SP600125 inhibits c-Jun phosphorylation, downregulates HIF-1α level, and promotes induction of apoptosis by gefitinib in gefitinib-resistant NSCLC cells.

A. H1650 and H1975 cells were treated with 10 μM SP600125 or vehicle control for 24 hours. Cell lysates were then prepared and subjected to Western blot analysis with the indicated antibodies. B. H1650 and H1975 cells were treated with vehicle control, 500 nM gefitinib, 10 μM SP600125, or 500 nM gefitinib and 10 μM SP600125. Cell lysates were then prepared for Western blotting with the indicated antibodies. C. H1650 and H1975 cells were treated with 500 nM gefitinib, 10 μM SP600125, or 500 nM gefitinib and 10 μM SP600125. Cell inhibition was measured by MTT proliferation assay. The data are presented as a percentage of cells treated with vehicle control (n=3; **P <0.005).
Fig. 5B shows that the combination of SP600125 and gefitinib sensitized H1650 and H1975 cells to treatment with gefitinib through induction of apoptosis, whereas gefitinib alone did not increase PARP cleavage beyond the level in untreated cells. Fig. 5C shows that gefitinib alone inhibited cell growth by only 2.25% in H1650 cells and 4.36% in H1975 cells. SP600125 treatment alone inhibited cell growth only moderately (by 12.47% in H1650 cells and 28.69% in H1975 cells). However, combination treatment with gefitinib and SP600125 inhibited cell growth by 45.33% in H1650 cells and 69.1% in H1975 cells.
Together, our findings indicate that inhibition of the c-Jun and HIF-1α regulatory loop is a novel approach to overcoming resistance to gefitinib in NSCLC.
4. Discussion
In the present study, we explored whether HIF-1α and c-Jun functionally cooperate in mediating resistance of NSCLC cells with activating mutation of EGFR to gefitinib, a prototype first-generation EGFR TKI. We found that downregulation of HIF-1α and c-Jun was more closely correlated with cell inhibitory response to the EGFR TKI gefitinib than was decrease in phosphorylated EGFR in NSCLC cells. NSCLC cells with intrinsic or acquired resistance to gefitinib exhibited higher basal levels of HIF-1α and total and phosphorylated c-Jun. Experimental overexpression of a constitutively expressed HIF-1α (HIF-1α/ΔODD) construct or a constitutively active SEK1 S220E/T224D mutant in NSCLC cells upregulated total and phosphorylated levels of c-Jun and conferred resistance to gefitinib. Pharmacological inhibition of JNK-c-Jun pathway sensitized gefitinib-resistant NSCLC cells to gefitinib treatment.
Despite being predicted to respond to first-line treatment with an EGFR TKI, 20% to 30% of NSCLC patients with activating EGFR mutations do not benefit from the treatment [12,13]. Even osimertinib, a potent and irreversible third-generation EGFR TKI, produced an overall objective response rate of only about 67% in the 80-mg cohort when the drug was used as first-line treatment for advanced NSCLC with activating mutation of EGFR [56,57]. Novel mechanisms contributing to resistance of advanced NSCLC to EGFR TKI thus need to be identified. Identification of novel molecular biomarkers is clinically important to further improve patient responses to EGFR TKI.
An important finding of our current study was that resistance to gefitinib in NSCLC cells with either intrinsic or acquired resistance did not entirely correlate with the level of EGFR tyrosine kinase inhibition shown by decrease in the level of EGFR activation-specific phosphorylation on Y1068. In other words, EGFR tyrosine kinase activity was noticeably inhibited by gefitinib in the gefitinib-resistant cell lines, although the level of EGFR tyrosine kinase inhibition was not as great as the level of inhibition seen in the gefitinib-sensitive cells. Current efforts to develop newer-generation EGFR TKIs remain focused on effective inhibition of EGFR tyrosine kinase when EGFR TKI induces a secondary T790M mutation in the tyrosine kinase domains, but our findings suggest that aberrations in cell signaling downstream of EGFR is an area that deserves more attention.
We recently reported that cell signaling downstream of EGFR plays important roles in mediating cancer cell response to the EGFR-blocking antibody cetuximab [27,31,34,35]. In particular, we found that downregulation of HIF-1α appears to correlate better than EGFR tyrosine kinase inhibition with cellular response to cetuximab treatment in head and neck cancer cells [35]. We also found that the role of HIF-1α in mediating resistance to cetuximab is linked to the key role of HIF-1 in regulating cancer metabolism [32,35].
In the present study, we found that basal levels of HIF-1α and total and phosphorylated c-Jun protein were higher in NSCLC cells that acquired resistance to EGFR TKI through long-term adaptation. These data indicated that the elevation of HIF-1α and c-Jun might play a role in development of EGFR TKI resistance in NSCLC. Results of our experiments involving overexpression of a constitutively expressed HIF-1α/ΔODD and a constitutively active SEK1 S220E/T224D mutant led us to propose the model depicted in Fig. 6. In this model, intrinsic genetic aberration, in other words, EGFR activating mutation, and extrinsic environmental cues, such as tumor hypoxia and stress, lead to upregulation of HIF-1α and c-Jun phosphorylation by upstream respective signaling and subsequent activation of an array of targeted genes through HIF-1 and AP-1. It was reported that a positive feed-forward loop exists between AP-1 and c-Jun, in which AP-1 could activate c-Jun promoter, thus enhancing c-Jun expression, which can further enhance the activity of AP-1 [58–60]. Our current data suggest that a positive feed-forward loop also exists between HIF-1α and c-Jun, in which constitutive activation of the SEK1-JNK-c-Jun pathway can upregulate HIF-1α protein level. Our data indicate that pharmacological interruption of these positive feed-forward loops using a small molecule inhibitor may offer a new approach for sensitizing EGFR TKI-resistant NSCLC cells to EGFR TKI. In vivo studies and more delicate mechanistic studies are needed to confirm our findings and further elucidate the important roles of HIF-1α and c-Jun in resistance to EGFR TKI. c-Jun and HIF-1α can bind to the transcriptional co-activators CBP/p300 and SRC-1 to enhance transcriptional activity [47,61]. c-Jun upregulation under hypoxia masks the ODD domain of HIF-1α, thus preventing HIF-1α from being ubiquitinated and degraded [62]. Whether these mechanisms play a role in the development of NSCLC cell resistance to EGFR TKI treatment needs to be further investigated.
Fig. 6. Proposed model of functional cooperation between HIF-1α and c-Jun in regulating primary and acquired resistance to gefitinib in NSCLC cells with activating mutation of EGFR.

HIF-1α and c-Jun are upregulated or phosphorylated by cell signal pathways downstream of EGFR and by environmental cues. Both HIF-1 and AP-1 activate transcription of target genes. AP-1 enhances c-Jun expression through a positive feed-forward loop. c-Jun upregulation prevents HIF-1α ubiquitination and degradation. HIF-1 may upregulate c-Jun expression through another positive feed-forward loop. *EGFR activating mutation.
In summary, our data reveal novel insights into potential roles of HIF-1α and c-Jun and their functional cooperation in mediating development of resistance to EGFR TKI in NSCLC cells with activating mutation of EGFR. Our findings expand current knowledge on the mechanisms of resistance to EGFR TKI. Further study is warranted of co-targeting HIF-1α and c-Jun as a novel approach to overcoming NSCLC resistance to EGFR TKI.
Highlights.
HIF-1α and c-Jun downregulation correlates with NSCLC cell response to gefitinib
Gefitinib-resistant NSCLC cells exhibit high basal levels of HIF-1α and c-Jun
Constitutively expressed HIF-1α upregulates c-Jun and makes NSCLC gefitinib resistant
SEK-JNK-c-Jun inhibition downregulates HIF-1α and sensitizes NSCLC to gefitinib
Acknowledgments
This work was supported by a China Shanghai Municipal Health Bureau project award (201540097) to SM and by a US National Institutes of Health R01 award (CA179015) to ZF.
Footnotes
Conflict of interest
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 2.Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98:1817–1824. doi: 10.1111/j.1349-7006.2007.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 4.Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S, Shimizu E, Saito H, Toyooka S, Nakagawa K, Fukuoka M. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 5.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 6.Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol. 2013;31:1112–1121. doi: 10.1200/JCO.2012.43.0454. [DOI] [PubMed] [Google Scholar]
- 7.Endoh H, Yatabe Y, Kosaka T, Kuwano H, Mitsudomi T. PTEN and PIK3CA expression is associated with prolonged survival after gefitinib treatment in EGFR-mutated lung cancer patients. J Thorac Oncol. 2006;1:629–634. [PubMed] [Google Scholar]
- 8.VanderLaan PA, Rangachari D, Mockus SM, Spotlow V, Reddi HV, Malcolm J, Huberman MS, Joseph LJ, Kobayashi SS, Costa DB. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: Correlation with clinical outcomes. Lung Cancer. 2017;106:17–21. doi: 10.1016/j.lungcan.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgillo F, Della Corte CM, Fasano M, Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open. 2016;1:e000060. doi: 10.1136/esmoopen-2016-000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soria JC, Ramalingam SS. Osimertinib in EGFR mutation-positive advanced NSCLC. N Engl J Med. 2018;378:1262–1263. doi: 10.1056/NEJMc1801669. [DOI] [PubMed] [Google Scholar]
- 13.Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, Nogami N, Ohe Y, Mann H, Rukazenkov Y, Ghiorghiu S, Stetson D, Markovets A, Barrett JC, Thress KS, Janne PA. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36:841–849. doi: 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- 14.Maxwell PH, Pugh CW, Ratcliffe PJ. Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci U S A. 1993;90:2423–2427. doi: 10.1073/pnas.90.6.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3′ enhancer. Proc Natl Acad Sci U S A. 1994;91:6496–6500. doi: 10.1073/pnas.91.14.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 17.Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35:71–103. doi: 10.1080/10409230091169186. [DOI] [PubMed] [Google Scholar]
- 18.Semenza GL. Intratumoral hypoxia, radiation resistance, and HIF-1. Cancer Cell. 2004;5:405–406. doi: 10.1016/s1535-6108(04)00118-7. [DOI] [PubMed] [Google Scholar]
- 19.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 20.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 21.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- 22.Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- 23.Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–27981. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda R, Hirota K, Fan F, Jung YD, Ellis LM, Semenza GL. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem. 2002;277:38205–38211. doi: 10.1074/jbc.M203781200. [DOI] [PubMed] [Google Scholar]
- 25.Laughner E, Taghavi P, Chiles K, Mahon PC, Semenza GL. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1alpha) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol Cell Biol. 2001;21:3995–4004. doi: 10.1128/MCB.21.12.3995-4004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luwor RB, Lu Y, Li X, Liang K, Fan Z. Constitutively active Harvey Ras confers resistance to epidermal growth factor receptor-targeted therapy with cetuximab and gefitinib. Cancer Lett. 2011;306:85–91. doi: 10.1016/j.canlet.2011.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z. Requirement of hypoxia-inducible factor-1alpha down-regulation in mediating the antitumor activity of the anti-epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther. 2008;7:1207–1217. doi: 10.1158/1535-7163.MCT-07-2187. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Liang K, Li X, Fan Z. Responses of cancer cells with wild-type or tyrosine kinase domain-mutated epidermal growth factor receptor (EGFR) to EGFR-targeted therapy are linked to downregulation of hypoxia-inducible factor-1alpha. Mol Cancer. 2007;6:63. doi: 10.1186/1476-4598-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Li X, Lu H, Fan Z. 1,9-Pyrazoloanthrones downregulate HIF-1alpha and sensitize cancer cells to cetuximab-mediated anti-EGFR therapy. PLoS One. 2010;5:e15823. doi: 10.1371/journal.pone.0015823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Fan Z. The epidermal growth factor receptor antibody cetuximab induces autophagy in cancer cells by downregulating HIF-1α and Bcl-2 and activating the Beclin 1/hVps34 complex. Cancer Res. 2010;70:5942–5952. doi: 10.1158/0008-5472.CAN-10-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu H, Liang K, Lu Y, Fan Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1alpha. Cancer Lett. 2012;322:78–85. doi: 10.1016/j.canlet.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu H, Li X, Luo Z, Liu J, Fan Z. Cetuximab reverses the Warburg effect by inhibiting HIF-1-regulated LDH-A. Mol Cancer Ther. 2013;12:2187–2199. doi: 10.1158/1535-7163.MCT-12-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu H, Li X, Lu Y, Qiu S, Fan Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016;381:23–30. doi: 10.1016/j.canlet.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li X, Lu Y, Lu H, Luo J, Hong Y, Fan Z. AMPK-mediated energy homeostasis and associated metabolic effects on cancer cell response and resistance to cetuximab. Oncotarget. 2015;6:11507–11518. doi: 10.18632/oncotarget.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo J, Hong Y, Lu Y, Qiu S, Chaganty BK, Zhang L, Wang X, Li Q, Fan Z. Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett. 2017;384:39–49. doi: 10.1016/j.canlet.2016.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tao X, Lu Y, Qiu S, Wang Y, Qin J, Fan Z. AP1G1 is involved in cetuximab-mediated downregulation of ASCT2-EGFR complex and sensitization of human head and neck squamous cell carcinoma cells to ROS-induced apoptosis. Cancer Lett. 2017;408:33–42. doi: 10.1016/j.canlet.2017.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 38.Kerppola TK, Curran T. Maf and Nrl can bind to AP-1 sites and form heterodimers with Fos and Jun. Oncogene. 1994;9:675–684. [PubMed] [Google Scholar]
- 39.Morgan IM, Birnie GD. The serum response element and an AP-1/ATF sequence immediately downstream co-operate in the regulation of c-fos transcription. Cell Prolif. 1992;25:205–215. doi: 10.1111/j.1365-2184.1992.tb01395.x. [DOI] [PubMed] [Google Scholar]
- 40.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by MAP kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 41.Smeal T, Binetruy B, Mercola DA, Birrer M, Karin M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature. 1991;354:494–496. doi: 10.1038/354494a0. [DOI] [PubMed] [Google Scholar]
- 42.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 44.Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18:6087–6093. doi: 10.1038/sj.onc.1203129. [DOI] [PubMed] [Google Scholar]
- 45.Davis RJ. Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp. 1999;64:1–12. doi: 10.1515/9781400865048.1. [DOI] [PubMed] [Google Scholar]
- 46.Yan M, Dai T, Deak JC, Kyriakis JM, Zon LI, Woodgett JR, Templeton DJ. Activation of stress-activated protein kinase by MEKK1 phosphorylation of its activator SEK1. Nature. 1994;372:798–800. doi: 10.1038/372798a0. [DOI] [PubMed] [Google Scholar]
- 47.Alfranca A, Gutierrez MD, Vara A, Aragones J, Vidal F, Landazuri MO. c-Jun and hypoxia-inducible factor 1 functionally cooperate in hypoxia-induced gene transcription. Mol Cell Biol. 2002;22:12–22. doi: 10.1128/MCB.22.1.12-22.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laderoute KR, Calaoagan JM, Gustafson-Brown C, Knapp AM, Li GC, Mendonca HL, Ryan HE, Wang Z, Johnson RS. The response of c-jun/AP-1 to chronic hypoxia is hypoxia-inducible factor 1 alpha dependent. Mol Cell Biol. 2002;22:2515–2523. doi: 10.1128/MCB.22.8.2515-2523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li X, Lu Y, Liang K, Hsu JM, Albarracin C, Mills GB, Hung MC, Fan Z. Brk/PTK6 sustains activated EGFR signaling through inhibiting EGFR degradation and transactivating EGFR. Oncogene. 2012;31:4372–4383. doi: 10.1038/onc.2011.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 52.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 53.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 54.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS. c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. J Clin Invest. 2001;108:73–81. doi: 10.1172/JCI12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramalingam S, Yang JC, Lee CK, Kurata T, Kim DW, John T, Nogami N, Ohe Y, Janne PA. LBA1_PR: Osimertinib as first-line treatment for EGFR mutation-positive advanced NSCLC: updated efficacy and safety results from two Phase I expansion cohorts. J Thorac Oncol. 2016;11:S152. [Google Scholar]
- 57.Yang J, Ramalingam SS, Janne PA, Cantarini M, Mitsudomi T. LBA2_PR: Osimertinib (AZD9291) in pre-treated pts with T790M-positive advanced NSCLC: updated Phase 1 (P1) and pooled Phase 2 (P2) results. J Thorac Oncol. 2016;11:S152–S153. [Google Scholar]
- 58.Michiels C, Minet E, Michel G, Mottet D, Piret JP, Raes M. HIF-1 and AP-1 cooperate to increase gene expression in hypoxia: role of MAP kinases. IUBMB Life. 2001;52:49–53. doi: 10.1080/15216540252774766. [DOI] [PubMed] [Google Scholar]
- 59.Minet E, Michel G, Mottet D, Piret JP, Barbieux A, Raes M, Michiels C. c-JUN gene induction and AP-1 activity is regulated by a JNK-dependent pathway in hypoxic HepG2 cells. Exp Cell Res. 2001;265:114–124. doi: 10.1006/excr.2001.5180. [DOI] [PubMed] [Google Scholar]
- 60.Bandyopadhyay RS, Phelan M, Faller DV. Hypoxia induces AP-1-regulated genes and AP-1 transcription factor binding in human endothelial and other cell types. Biochim Biophys Acta. 1995;1264:72–78. doi: 10.1016/0167-4781(95)00116-x. [DOI] [PubMed] [Google Scholar]
- 61.Laderoute KR. The interaction between HIF-1 and AP-1 transcription factors in response to low oxygen. Semin Cell Dev Biol. 2005;16:502–513. doi: 10.1016/j.semcdb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Yu B, Miao ZH, Jiang Y, Li MH, Yang N, Li T, Ding J. c-Jun protects hypoxia-inducible factor-1alpha from degradation via its oxygen-dependent degradation domain in a nontranscriptional manner. Cancer Res. 2009;69:7704–7712. doi: 10.1158/0008-5472.CAN-09-0808. [DOI] [PubMed] [Google Scholar]