

Abstract
Purpose
Infused autologous tumor-infiltrating lymphocytes (TILs) and tumor-targeted chimeric antigen receptor (CAR)-T cells typically surround malignant lesions or penetrate small tumor nodules, but fail to penetrate large solid tumors, significantly compromising their antitumor impact. Strategies to overcome this primary challenge are largely required.
Experimental Design
We tested the effects of IL-12 plus doxorubicin on T cell penetration and efficacy in solid tumors in a murine lung cancer model, a murine breast carcinoma lung metastasis model and two human xenograft tumor models bearing large tumors (>10 mm).
Results
Intriguingly, this simple approach increased the numbers, the distribution, and the depth of penetration of infused CD8+ T cells in these tumors, including both TILs and CAR-T cells. This combined treatment halted tumor progression, and significantly extended survival time. Studies of the underlying mechanism revealed multiple effects. First, the combined treatment maintained the high ratios of immune-stimulatory receptors to immune-inhibitory receptors on infiltrated CD8+ T cells, reduced the accumulation of immune-suppressive regulatory T cells, and enhanced the numbers of T-bet+ effector T cells in the tumors. Second, doxorubicin induced chemokines CXCL9 and CXCL10, which may attract NKG2D+CD8+ T cells to tumors, and this effect was boosted by IL-12–induced IFNγ accumulation in tumors, promoting the penetration of NKG2D+CD8+ T cells.
Conclusions
The deep penetration of infused T cells associated with combined IL-12 plus doxorubicin yielded striking therapeutic effects in murine and human xenograft solid tumors. This approach might broaden the application of T cell therapy to a wider range of solid tumors.
Keywords: intratumoral infiltration, CXCL9, CXCL10, large solid tumor, NKG2D+CD8+ T cells
INTRODUCTION
Many seminal studies have indicated that cytotoxic T cells inhibit the initiation and growth of malignant solid tumors (1,2). Tumor-infiltrating immune effector cells, particularly CD8+ T and natural killer cells, are associated with a promising prognosis and prolonged survival in both the early and late stages of malignancy (3,4). Sato et al. reported that colorectal cancer patients with a high density of infiltrated T cytotoxic and memory cells in their tumor had a much lower tumor recurrence rate and a prolonged survival time (5). The expression of co-stimulatory receptor CD28 on infiltrated T cells reduces the requirement for T cell receptor signaling during T cell activation and rescues exhausted CD8+ T cells (6,7). Tumor-infiltrated NKG2D+CD8+ T cells recognize and eliminate tumor cells that exhibit NKG2D ligands (8,9). At the meanwhile regulatory T cells and immune checkpoint regulators (such as PD-1) in the tumor mass create an immune-suppressive environment that hampers the antitumor response from immune effector cells in advanced stages of cancer (10). Since the balance between immune activation (CD28 and NKG2D) and immune suppression (Tregs and PD1) may represent the effect of immune surveillance, the ratios of immune-stimulatory and immune-inhibitory signals in the tumor microenvironment are better predictors of immunotherapy outcomes (5,11).
The use of T cell immunotherapy has grown rapidly over the past decade; its success has spread from non-solid cancers to melanoma and, gradually, to other solid tumors. Nevertheless, majority of patients with advanced stage of cancers were associated with T cell exhaustion (12), and only a very limited number of patients with a solid tumor experience a response to T cell immunotherapy. These patients have been found to have strong intratumoral infiltration of T cells (13); high levels of cytotoxic T cell effector molecules, such as interferon gamma (IFNγ) (14); and high ratios of CD8/CD4 T cells in tumors (5). These parameters could serve as markers for evaluating the efficacy of T cell immunotherapy.
Infusion of autologous tumor-infiltrating lymphocytes (TILs) has been a remarkable breakthrough in the treatment of patients with refractory melanoma. In practice, however, the response rates are only about 50%, including a 10%-15% complete response rate (15–17). Major challenges in TIL therapy include the reduced tumor-penetration ability of TILs after re-infusion and the immune suppressive tumor microenvironment. In recent clinical trials, patients were infused with 1.5–2×1011 TILs to ensure sufficient tumor-targeting TILs and successful tumor remission (16,17). However, transferring such large numbers of TILs into cancer patients can cause off-target adverse effects. Approaches are needed that enable TILs to be delivered into large solid tumors more efficiently and therefore reduce the number of T cells that must be infused.
The loss of tumor-homing characteristics during ex vivo culture is one critical reason that TILs cannot reach tumor sites; thus, new therapies use T cells that have been engineered with receptors that recognize tumor antigens (such as CD19); this innovation is known as chimeric antigen receptor (CAR)-T cell therapy. CAR-T cell therapy has had substantial success in treating hematologic malignancies, but efficacy in treating large solid tumors is compromised. Because of their heterogeneity, solid tumor cells lack common antigens. Furthermore, host conditioning often prevents T cells from entering the tumor stroma. Caruana et al. made a fundamental breakthrough in CAR-T cell therapy by introducing the enzyme heparanase, which degrades the extracellular matrix to allow T cells to penetrate tumors (18). This discovery opens the door to use CAR-T cell therapy for solid tumors.
In our previous work, we found that the combination of systemic interleukin 12 (IL-12) expression plus low-dose doxorubicin facilitated accumulation of endogenous CD8+ T cells in solid tumors (up to 6mm in diameter) in mice in vivo (9). In this study, we hypothesized that IL-12 plus doxorubicin would also allow ex vivo expanded T cells, TILs, and CAR-T cells to penetrate large solid tumors. We found that this treatment not only boosted NKG2D+CD8+T cell infiltration into large solid tumors but also synergistically upregulated the levels of the T cell–attracting chemokines CXCL9 and CXCL10 production, promoting accumulation of T cells to the tumor microenvironment and enhancing the effector functions of infiltrated T cells by increasing the ratios of stimulation to regulatory factors.
MATERIALS AND METHODS
Animal studies and cells
We purchased 6- to 8-week-old BALB/C, C57BL/6J and NSG mice from Jackson Laboratory (Bar Harbor, ME). The mouse care and handling procedures were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center (Houston, TX).
To create the transplant tumor mouse models, LLC tumor cells (1.5×105 per mouse) were subcutaneously inoculated into C57BL/6 and NSG mice. 4T1 tumor cells (1.5×105 per mouse) were orthotopically inoculated into BABL/C mice via the mammary fat pad. 4T1 primary tumors were surgically removed on day 18 after inoculation. Lungs from mice in all treatment groups were intratracheally injected India ink (15% India Ink, 85% water, 3 drops NH4OH/100 ml), and then washed in Feket’s solution (300 ml 70% EtOH, 30 ml 37% formaldehyde, 5 ml glacial acetic acid) overnight. The tumor cell lines LLC and 4T1 were obtained from ATCC. Mel2549 (tumor-derived human melanoma) cells and autologous TILs, which were expanded and maintained in Dr. Chantale Bernatchez’s laboratory at MD Anderson Cancer Center (19), were inoculated subcutaneously (3×106 per mouse in 30 μL of phosphate-buffered saline solution [PBS]) into NSG mice. These mice also received an intravenous injection of autologous TILs (5×106) the day after each treatment. Nalm6 human lymphoblasts were mixed with Matrigel (1:1 in PBS) and inoculated subcutaneously (5×106 per mouse in 50 μL of solution). CD19–28ζ CAR-T cells were expanded and maintained in Dr. Gianpietro Dotti’s laboratory at University of North Carolina (18). Murine T cells were enriched from splenocytes using an EasySep kit and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. and treated with Mycoplasma Removal Agent from Bio-Rad to ensure mycoplasma negative before inoculation.
All the tumor cell lines were characterized by DNA fingerprinting within 6 months of initiating the experiments at MD Anderson Cancer Center’s Characterized Cell Line Core Facility, and treated with Mycoplasma Removal Agent from Bio-Rad to ensure mycoplasma negative before inoculation.
Tumor-bearing mice were treated with control DNA (10 μg/mouse), control DNA plus doxorubicin (1 mg/kg), mouse or human IL-12–encoding DNA (10 μg/mouse), or mouse or human IL-12–encoding DNA plus doxorubicin, followed by electroporation, as described previously (20). Tumor volume was calculated using the formula V = (π/8)×(a∗b2), where V = tumor volume in cubic centimeters, a = maximum tumor diameter, and b = diameter at 90°.
Sham siRNA or CXCL9 and CXCL10 siRNA (500pmol) was injected intratumorally, followed by electroporation twice weekly. The electroporation was performed using the following parameters: two 50ms pulses of 150 V cm−1 with a 100ms interval between pulses.
mCXCL9 siRNA: GUUUGUAAGCACGAACUUUA[dT][dT]
mCXCL10 siRNA: CCAAUAGUAACAAUUGCUA[dT][dT]
Plasmids and reagents
Our lab generated both mouse and human IL-12 constructs and confirmed by sequence analyses. DNA was prepared by using the endotoxin-free Mega preparation kit from Qiagen, Inc. (Valencia, CA) and following the manufacturer’s instructions. Doxorubicin (Bedford Laboratories, Bedford, OH) was purchased from the pharmacy at MD Anderson.
Immunohistochemical/immunofluorescence analysis
Frozen tumor sections were sequentially fixed with cold acetone, acetone plus chloroform (1:1), and acetone. Tissue sections were blocked with blocking buffer (5% normal horse serum and 1% normal goat serum in PBS) and then incubated with primary antibody overnight at 4°C, secondary antibody for 1 hour at room temperature. For immunohistochemical staining, the secondary antibody was biotin-conjugated and the sections were treated with ABC reagent and the nuclei counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO). Tumor sections were mounted with Cytoseal mounting medium (Life Technologies). For immunofluorescence staining, tumor sections were mounted in antifade with DAPI fluorescence mounting medium. Slides were visualized under a Nikon Eclipse Ti fluorescence microscope (Nikon, Melville, NY).
Flow cytometry
A sample of each tumor from each treatment group was dissociated with liberase TM (Sigma-Aldrich, St. Louis, MO) enzyme cocktail. Cells were sequentially incubated with primary and secondary antibodies for 30 minutes each at 4°C. Stained cells were analyzed on an Attune acoustic focusing cytometer (Applied Biosystems, Foster City, CA). Flow cytometry data were analyzed by the FlowJo software program (BD Biosciences, San Jose, CA).
Antibodies
Alexa Fluor 488–conjugated anti-mouse CD45, anti-human CD45, violet 421–conjugated anti-mouse CD4 and CD8, anti-human CD4 and CD8, and phycoerythrin-conjugated anti-mouse or anti-human CD28, 41BB, NKG2D, CD39, PD-1, LAG3, TIM3, and FOXP3, as well as isotype control antibodies, were purchased from Biolegend (San Diego, CA). Rabbit anti-mouse and human CD3 and rabbit anti-human CD8 antibodies were purchased from Abcam (Cambridge, MA). Biotin anti-mouse NKG2D, mouse anti-human NKG2D, rabbit anti-human FOXP3 and anti-human Tbet antibodies were purchased from R&D Systems (Minneapolis, MN). Anti-mouse FOXP3 antibody was purchased from Thermo Fisher. Horseradish peroxidase–conjugated anti-rabbit IgG was purchased from Cell Signaling Technology (Danvers, MA).
Migration assay
Mel2549 and Nalm6 tumor cells (1×105) were treated with vehicle or doxorubicin (100 nM) in 600 μL of culture medium for 72 hours. The conditioned media were then collected and placed on the bottom chambers of a Boyden transwell chamber system. Human TILs or CAR-T cells (1×104) were labeled with violet tracker (Invitrogen, Carlsbad, CA), re-suspended in 100 μL of chemotaxis buffer (RPMI 1640/0.5% bovine serum albumin), and placed in the upper chambers with 5-μm pores (Corning, Corning, NY) for 1.5 hours at 37°C. Cells that migrated to the bottom chamber were collected, washed once with PBS, and counted by fluorescence-activated cell sorting.
ELISA
Tumor lysates were collected from tumor-bearing mice that underwent the indicated treatments at the indicated time points. The levels of IL-12 and IFNγ were measured by using ELISA Ready-SET-Go ELISA kits (eBioscience, San Diego, CA). The levels of human CXCL9 and CXCL10 in xenograft tumors were measured by using ELISA kits (LSBio, Seattle, WA).
RNA isolation and quantitative PCR
RNA was isolated from tumors with TRIzol reagent (Invitrogen). Residual genomic DNA was removed from total RNA using the TURBO DNA-free kit (Life Technologies).
Two micrograms of RNA were used for cDNA synthesis with the High-Capacity RNA-to-cDNA kit (Life Technologies). The relative gene expression levels were determined by using the RT-qPCR and the SYBR Green labeling method in a StepOnePlus real-time PCR system (Life Technologies). The reaction contained 2 μL of cDNA, 12.5 μL of SYBR Green PCR Master Mix (Life Technologies), and 200 μM primer in a total volume of 25 μL. The PCR cycling conditions were as follows: 40 cycles of 15 s at 95°C and 60 s at 60°C. All samples were run in duplicate. The CT value of each sample was acquired, and the relative level of gene expression was calculated using the delta CT method, which was normalized to the endogenous control, GAPDH. Data are expressed as n-fold relative to the control. The primer sequences for human genes were:
mCXCL9: F: AGCAGTGTGGAGTTCGAGGAA, R: GGTGCTGATGCAGGAGCAT
mCXCL10: F: GACGGTCCGCTGCAACTG, R: GGTGCTGATGCAGGAGCAT
mGAPDH: F: CCAGCCTCGTCCCGTAGAC, R: CGCCCAATACGGCCAAA
hCXCL9: F: AGGTCAGCCAAAAGAAAAAG, R: TGAAGTGGTCTCTTATGTAGTC
hCXCL10: F: AGCTCTACTGAGGTGCTATGT, R: GTACCCTTGGAAGATGGGAAAG
hGAPDH: F: ATGGAAATCCCATCACCATCTT, R: CATCGCCCCACTTGATTTTG
Statistical analysis
The directly measured outcomes were analyzed by using the two-sided Student t-test to compare two treatment groups or one-way analysis of variance to compare more than two treatment groups. The statistical significance of each comparison was determined using GraphPad software (La Jolla, CA). A P value <0.05 indicated statistical significance. * equals P<0.05, ** equals P<0.01, *** equals P<0.005, **** equals P<0.001.
RESULTS
Adoptively transferred murine T cells accumulate in large solid tumors and lung metastatic nodules after treatment with IL-12 plus doxorubicin
Systemic immune therapy in mice has generally been effective only in small tumors (<5 mm in diameter) because the infused T cells do not penetrate into large tumors. Our previous discovery that treatment with a combination of IL-12 plus doxorubicin accumulated endogenous NKG2D+CD8+ T cells into small solid tumors (up to 6 mm in diameter) (9) prompted us to test whether this same treatment enriches exogenous NKG2D+ T cells in large solid tumors. We inoculated C57BL/6 and immune-deficient Nod-SCID-gamma (NSG) mice with Lewis lung carcinoma (LLC) murine lung cancer cells to establish solid tumors, and intentionally waited until the tumors reached 8–10 mm in diameter before beginning treatment with control DNA, mouse IL-12 plasmid DNA, doxorubicin, or mouse IL-12 plasmid DNA plus doxorubicin using a well-established method. This treatment was followed by adoptive intravenous transfer of exogenous T lymphocytes isolated from the tumor-bearing immune-competent C57BL/6 mice (21). The treatments were repeated one week later. Both IL-12 and doxorubicin were administered systematically to treat inaccessible tumors such as metastatic tumors. Tumors were collected 4 days after the second treatment to evaluate T lymphocyte penetration of tumors. Doxorubicin alone greatly increased the numbers of CD4+ T lymphocytes in periphery tumors compared to the control DNA treatment (Fig. 1A, Fig. S1A), whereas IL-12 plus doxorubicin allowed much greater penetration of CD8+ T lymphocytes into tumors (Fig. 1B).
Figure 1. IL-12 plus doxorubicin enhanced the infiltration of infused T cells into murine solid tumors.
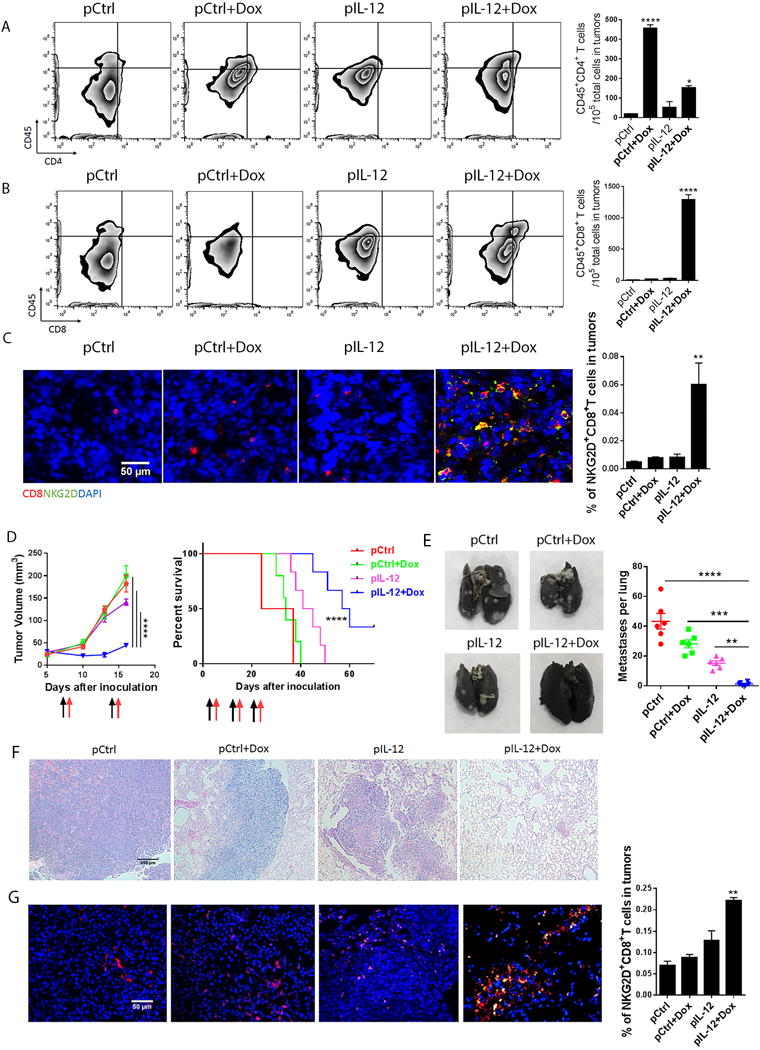
LLC tumor-bearing NSG mice were given one of four standard treatments (control DNA, control DNA plus doxorubicin, mouse IL-12 DNA, or mouse IL-12 DNA plus doxorubicin), followed by an infusion of splenic T cells (5 million cells/mouse) that had been isolated and enriched from tumor-bearing C57BL/6 mice and stimulated with immobilized CD3 and CD28 antibodies for 24 hours. This cycle of treatment followed by T cell infusion was repeated 1 week later. Tumors were collected 4 days after the last T cell infusion. (A, B) The tumor cell suspension was stained with anti-mouse CD45, CD4, and CD8 antibodies and subjected to flow cytometry analyses of CD4+ T (A) and CD8+ T (B) cell infiltration. Bar graphs represent the numbers of the indicated infiltrated lymphocytes, per 105 total cells in tumors, as means ± SEM. (C) Intratumoral T cell infiltration was determined by immunofluorescence staining. Sections from the centers of the tumors were stained with anti-mouse CD8 and NKG2D antibodies. Bar graphs represent the percentage of tumor-infiltrating NKG2D+ CD8+ T cells in five views per slide and three slides per tumor as means ± SEM. (D-G) 4T1 tumor bearing mice were treated with four standard treatments as LLC mice, followed by an infusion of stimulated splenic T cells (5 million cells/mouse) on days 7, 14 and 21 after tumor inoculation. Tumors were surgically removed 4 days after the second administration. (D) Primary tumor sizes and survival time were monitored. Black arrows represent the dates of control or IL-12 plus doxorubicin treatments, and red arrows represent the dates of TIL infusion. (E) Lungs were collected from mice in all treatment groups, and stained with India ink. Macroscopic lung metastatic nodules were observed and counted in all lungs. Bar graphs represent the numbers of metastatic nodules as means ± SEM. (F) H&E staining of lung sections from each treatment group. (G) Sections from lungs were stained with anti-mouse CD8 and NKG2D antibodies. Bar graphs represent the percentage of tumor-infiltrating NKG2D+CD8+ T cells in five views per slide and three slides per tumor as means ± SEM. All results are representative of three repeated experiments.
These results were supported by immunofluorescence staining of tumor sections with CD8 (T cell marker) and NKG2D (activating receptor) showing that large numbers of NKG2D+CD8+ T cells were localized in tumors after treatment with IL-12 plus doxorubicin, whereas only a few NKG2D−CD8+ T cells were found in tumors of mice in the control groups treated with doxorubicin, IL-12 DNA alone, or control DNA (Fig. 1C). Most of the CD8+ T cells recruited into tumors by IL-12 plus doxorubicin treatment were NKG2D+CD8+ T cells (Fig. 1C). These results agree with the reported accumulation of endogenous NGK2D+CD8+ T cells in solid tumors induced by IL-12 plus doxorubicin, suggesting that this treatment also promoted infiltration of the exogenous murine NKG2D+CD8+ T lymphocyte subpopulation in solid tumors.
Next we sought to assess whether our systemic therapeutic approach could delay the metastatic diseases, the primary cause of cancer death. Metastatic mouse breast carcinoma cells 4T1 were orthotopically inoculated into BALB/C mice, and the primary tumors were surgically removed on day 18 after inoculation to establish lung metastases. The tumor bearing mice were treated with the same four groups of treatments as the LLC mice, followed by infusion of T cells that were isolated from 4T1 tumor–bearing BALB/C mice. Treatment with IL-12 plus doxorubicin followed by T cell infusion not only inhibited the primary tumor progression, and significantly prolonged the survival time of 4T1 tumor–bearing mice, with 2/6 long-term survivors (Fig. 1D). Also, this treatment dramatically reduced the numbers and sizes of lung metastases (Fig. 1E). Mice treated with IL-12 plus doxorubicin plus adoptive T cell transfer showed much smaller pulmonary tumor nodules (less than 100μm in diameter) than mice in other treatment groups (Fig. 1F). Intriguingly, a few NKG2D−CD8+ T cells accumulated in lung metastatic tumors after treatment with control DNA or control DNA plus doxorubicin, whereas small numbers of NKG2D+CD8+ T cells infiltrated into the periphery of lung nodules in mice treated with IL-12. Strikingly, IL-12 plus doxorubicin led to influx of NKG2D+CD8+ T cells throughout the tumor nodules, suggesting that this treatment facilitates effector T cell penetration into metastatic tumors (Fig. 1G).
IL-12 plus doxorubicin enhances infiltration of autologous TILs into large tumors in a human melanoma xenograft tumor model
Although infusion of TILs into melanoma patients can result in tumor remission, about 50% of patients experience little response to the treatment. The capacity of TILs to penetrate into a tumor is often correlated with the outcome of TIL transfer. To determine whether the systemic administration of IL-12 plus doxorubicin boosts the penetration of autologous human melanoma-specific TILs as it did in mouse tumors, xenograft tumors were initiated in NSG mice via injection of patient-derived Mel2549 melanoma cells; the tumor-bearing mice were then divided into the same four treatment groups: control DNA, control DNA plus doxorubicin, human IL-12 DNA, human IL-12 DNA plus doxorubicin. Ex vivo–expanded autologous TILs were transferred intravenously as adoptive therapy one day after each treatment. Because TILs often lose their tumor-homing ability after ex vivo expansion and stimulation, most studies infuse large number (1×107) of T cells, but we transferred only 5×106 T cells with the assumption that fewer T cells would be required because of their increased penetration into the solid tumors after IL-12 plus doxorubicin treatment. We detected CD4+ T cells in the periphery tumor areas after control groups (control DNA, control DNA plus doxorubicin and IL-12 DNA) of treatments (Fig. S1B), but rarely any CD4+ T cells in tumors after IL-12 plus doxorubicin treatment, suggesting that CD4+ T cells may not play a critical role in our treatments. While doxorubicin alone slightly increased the TIL infiltration into tumors, the absolute numbers of infiltrated CD8+ TILs did not differ significantly between the group treated with IL-12 DNA and that treated with control DNA. In contrast, IL-12 plus doxorubicin remarkably increased the accumulation of infused CD8+ TILs in the large melanoma tumors (Fig. 2A).
Figure 2. IL-12 plus doxorubicin enhanced the infiltration of infused autologous TILs into human melanoma xenograft solid tumors and delayed tumor development.
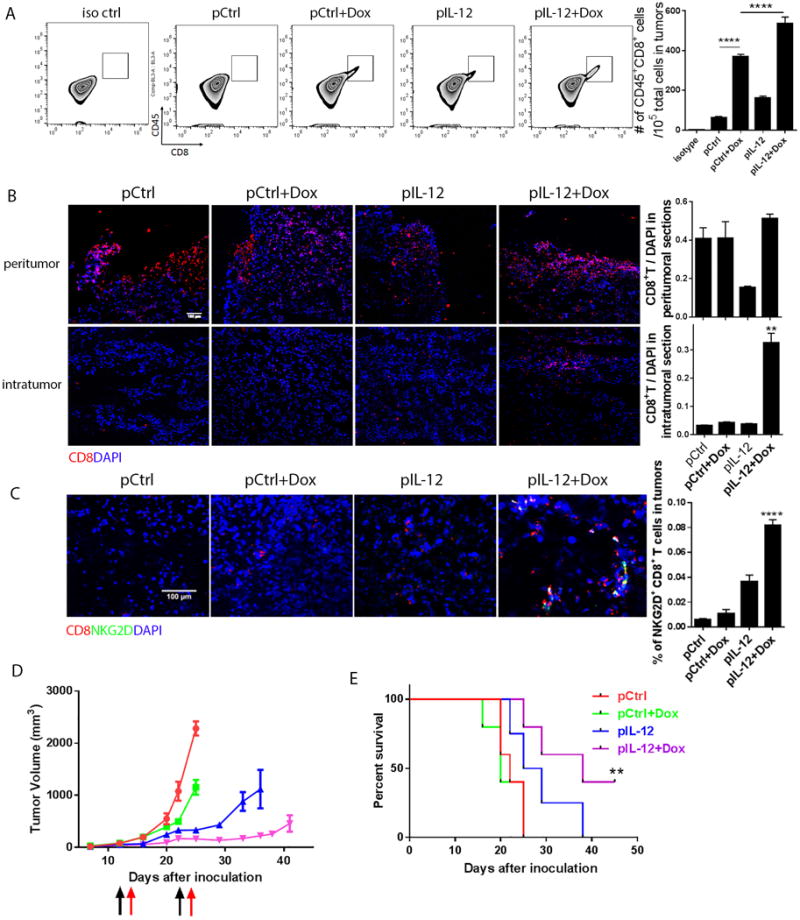
NSG mice were inoculated with Mel2549 tumor cells (3×106 per mouse) to establish a human melanoma xenograft model (N=5). When tumors reached 3–5 mm in diameter, the tumor-bearing mice were subjected to one of four standard treatments: control DNA, control DNA plus doxorubicin, human IL-12 DNA, and human IL-12 DNA plus doxorubicin. After 1 day, the mice received an infusion of 5 million autologous TILs. The mice received a second treatment and TIL infusion 7 days after the first administration. Tumors from each treatment group were collected 4 days after the second T cell infusion. (A) A sample of each tumor was dissociated with liberase enzyme cocktail. Tumor cells in suspension were stained with anti-human CD45 and CD8 antibodies and subjected to flow cytometry analysis to quantify T cell infiltration into tumors. Bar graphs represent the numbers of tumor-infiltrated CD8+ T cells per 105 total cells in tumors as means ± SEM. (B) Intratumoral T cell infiltration was determined by immunofluorescence staining. Sections from both the margins and centers of tumors were stained with anti-human CD8 antibody. Bar graphs represent the numbers of tumor-infiltrating T cells in five views per slide and three slides per tumor as means ± SEM. (C) Sections from the centers of the tumors were stained with anti-human CD8 and NKG2D antibodies. Bar graphs represent the percentage of tumor-infiltrating NKG2D+ CD8+ T cells in five views per slide and three slides per tumor as means ± SEM. (D) Tumor size was measured twice weekly beginning 5 days after inoculation. Black arrows represent the dates of control or IL-12 plus doxorubicin treatments, and red arrows represent the dates of TIL infusion. (E) Survival time was monitored and recorded. All results are representative of three repeated experiments.
To validate the distribution of infused TILs in tumors, we examined tumor sections from the peritumor and intratumor areas. All control groups showed TIL accumulation only in the peritumoral areas. In contrast, IL-12 plus doxorubicin led to 6-fold increase of TIL infiltration into both peritumoral and intratumoral areas (Fig. 2B). Significantly, only treatment with IL-12 plus doxorubicin increased NKG2D+CD8+ TIL cell numbers in the melanoma tumors (Fig. 2C).
Since infused T cells penetrated and distributed into large solid tumors after IL-12 plus doxorubicin treatment, we sought to determine the antitumor efficacy of this combinational therapy. Among the four treatments, only IL-12 plus doxorubicin significantly hampered the progression of melanoma tumors (Fig. 2D). This inhibition of tumor progression also significantly prolonged survival; two of the five mice that received IL-12 plus doxorubicin had particularly long-term survival (Fig. 2E). In contrast, the control groups (control DNA, doxorubicin, or IL-12 DNA) experienced no substantial improvement in tumor volume or survival duration (Fig. 2D, 2E).
CAR-T cells are enriched in tumors after treatment with IL-12 plus doxorubicin
CD19 CAR-T cell therapy is known to be successful in the treatment of hematologic malignancies in which T cells target CD19 antigen on malignant B cells. However, CAR-T cell therapy has so far been largely ineffective in treating large solid tumors. To test the hypothesis that IL-12 plus doxorubicin can boost CAR-T cell penetration into large solid tumors, we inoculated large numbers of CD19+ Nalm6 cells (5×106) mixed with Matrigel subcutaneously into immune-deficient NSG mice to generate solid tumors. CD19 28ζ CAR-T cells were delivered using the same treatment as Mel2549 tumor model and T cell transfer strategy used in the other models until solid CD19+ Nalm6 tumors reached 6–8 mm in diameter. The accumulation of CD4+ T cells in the periphery tumor areas were detected after control groups of treatments, and was substantially reduced in the tumors treated with IL-12 plus doxorubicin (Fig. S1C). The accumulation of CD8+ T cells in large solid tumors was assessed via flow cytometry after two treatment administrations. IL-12 plus doxorubicin (but not the other treatments) resulted in infiltration of a large number of CD8+ T cells into the solid tumors (Fig. 3A).
Figure 3. IL-12 plus doxorubicin enhanced the infiltration of infused CD19 CAR-T cells into human lymphoblast xenograft solid tumors with antitumor therapeutic effects.
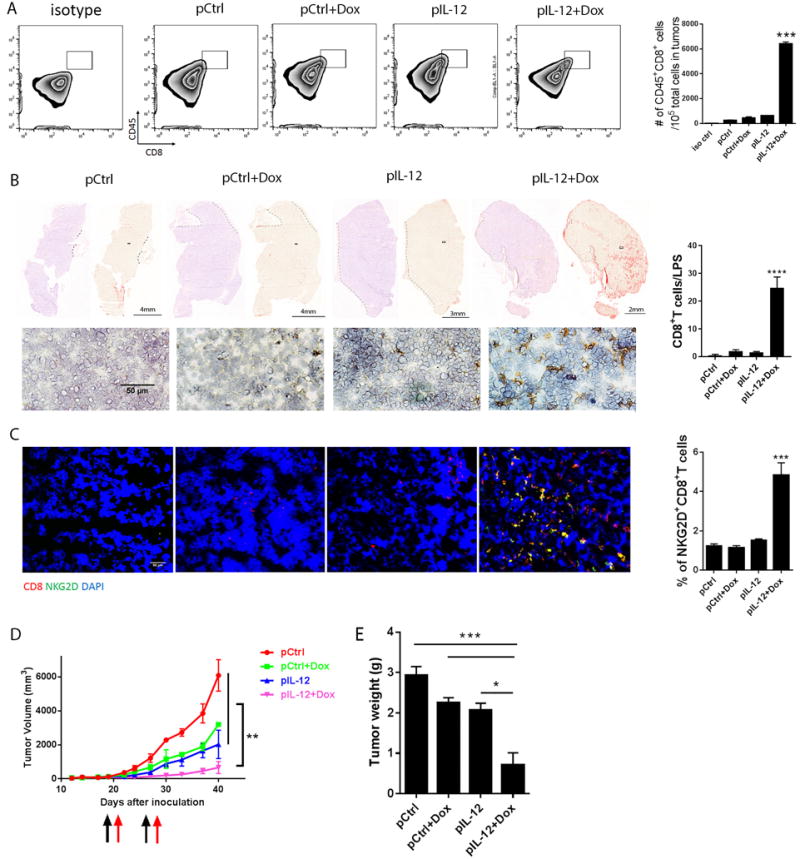
NSG mice were injected with Nalm6 tumor cells (5×106 cells/mouse) to establish a human lymphoblast xenograft model (N=5). When tumors reached 6–8 mm in diameter, the mice were subjected to one of four standard treatments, as shown in Figure 2. The next day, each mouse received 5 million CD19 28ζ CAR-T cells. The mice received a second treatment and CAR-T cell infusion 7 days after the first administration. (A) Tumors were collected from each treatment group 4 days after the second T cell infusion and a sample of each was dissociated with liberase enzyme cocktail. Suspended tumor cells were stained with anti-human CD45 and CD8 antibodies and subjected to flow cytometry analyses to quantify T cell infiltration into tumors. Bar graphs represent the numbers of tumor-infiltrated T cells per 105 total cells in tumors as means ± SEM. (B) Intratumoral CD8+ T cell infiltration was determined by immunochemistry staining. Sections collected >10 mm from the tumor margin were stained with anti-human CD8 antibody. A representative section from each treatment group is shown, including nuclear counterstain on the left, DAB staining alone (red) on the right, and a random view of the core tumor area on the bottom. Bar graphs represent the percentage of tumor-infiltrating CD8+ T cells in five views per slide and three slides per tumor as means ± SEM. (C) Intratumoral T cell infiltration was determined by immunofluorescence staining. Tumor sections were stained with anti-human CD8 and NKG2D antibodies. Bar graphs represent the percentage of tumor-infiltrating NKG2D+ CD8+ T cells in five views per slide and three slides per tumor as means ± SEM. (D) Tumor size was measured twice weekly beginning 5 days after inoculation. Black and red arrows represent treatment dates. (E) Tumor weight was measured at the time of collection. All results are representative of three repeated experiments.
To gain insight into the localization of infiltrated CD8+ CAR-T cells, we dissected the center area of each tumor (>10 mm in diameter) for whole slide scanning. Small numbers of CD8+ CAR-T cells accumulated in the peritumoral areas following all the control treatments. In striking contrast, large numbers (more than 20-fold increase) of CD8+ CAR-T cells accumulated in the central tumor areas of mice receiving systemic IL-12 plus doxorubicin treatment (Fig. 3B). Dual staining by human CD8 and NKG2D on tumor sections (Fig. 3C) confirmed that IL-12 plus doxorubicin allowed significantly greater numbers of NKG2D+CD8+ T cells to penetrate into the tumor centers than the other treatments. This dramatic elevation of intratumoral infiltration by NKG2D+CD8+ CAR-T cells after IL-12 plus doxorubicin treatment reduced tumor volume and tumor weight by more than 5-fold, reflecting significantly enhanced antitumor efficacy (Fig. 3D, 3E).
Inhibition of immunosuppressive factors to enhance the persistence of tumor-infiltrated T cells
The immunosuppressive tumor microenvironment contains a network of factors, including intrinsic inhibitory receptors and extrinsic regulatory immune cells, that may impede the effector functions of antitumor cytotoxic T cells. The expression levels of inhibitory receptors reflect the status of infused T cells and are often altered after infusion in response to cytokines and the tumor microenvironment. The patterns of co-stimulatory and co-inhibitory receptors may characterize the persistence of infiltrated T cells. To test the T cell persistence in mouse LLC tumor model, we primed splenic T cells from LLC tumor-bearing immune-competent mice with plate-bound CD3/CD28 antibodies and IL-2 for 24 hours before infusion and assessed expression of the co-stimulatory receptor CD28 and the exhaustion marker LAG3 and the checkpoint regulator PD-1 (Fig. 4A). These T cells were then infused into LLC tumor-bearing NSG mice following two administrations of control or systemic IL-12 plus doxorubicin treatment. Only IL-12 plus doxorubicin treatment maintained the expression levels of co-stimulatory and co-inhibitory receptors in the infiltrated T cells (Fig. 4B). Along with lower numbers of infiltrated T cells, we observed significant reductions in CD28 expression and substantial upregulation of LAG3 and PD-1 in the control groups, suggesting that the infused T cells became exhausted and lost their effector functions. Intriguingly, the number of infiltrated CD8+ T cells, but not CD4+ T cells, exhibited a strong positive correlation with the ratios of CD28 expression to LAG3 and PD-1 expression (Fig. S2), showing that the infused CD8+ T cells with higher ratios of co-stimulatory/co-inhibitory receptors (S/I) were associated with high persistence states in large solid tumors.
Figure 4. IL-12 plus doxorubicin induced co-stimulatory receptor but suppressed co-inhibitory receptors on infused T cells in murine solid tumors.
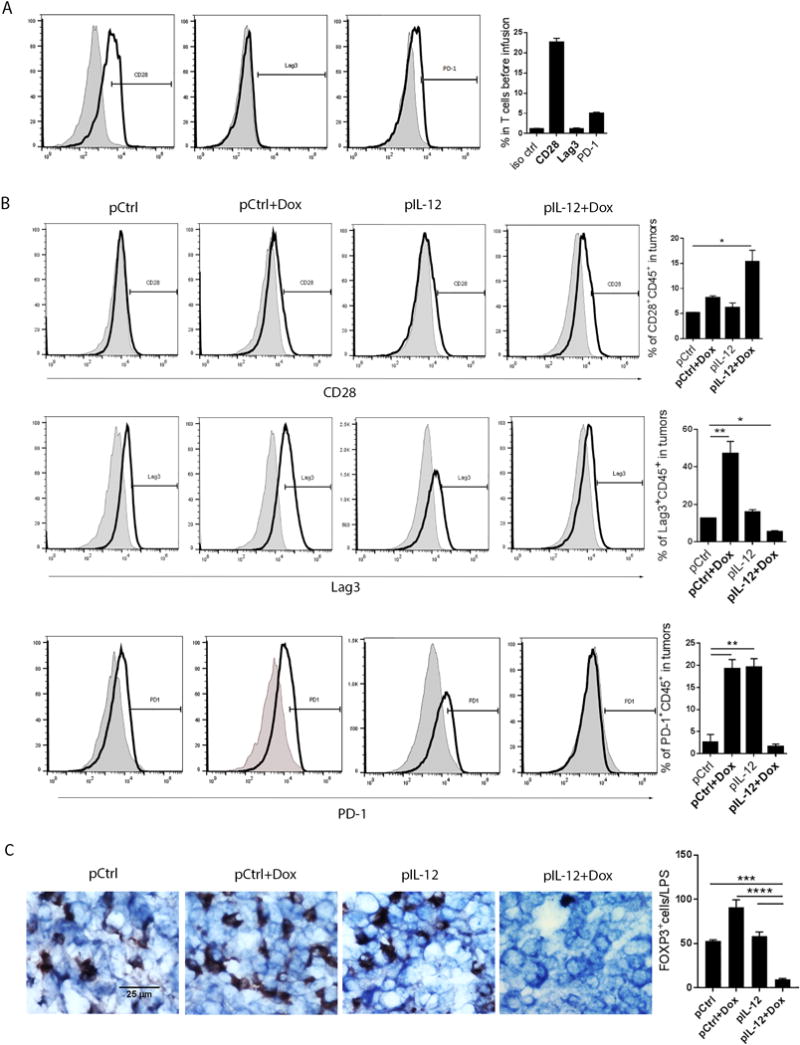
(A) Expression of co-stimulatory and co-inhibitory receptors on murine T cells were measured before infusion. Splenic T cells were isolated and enriched from LLC tumor-bearing C57BL/6 mice, stimulated with immobilized CD3/CD28 antibodies for 24 hours, and stained with anti-mouse CD28, PD-1, and Lag3 antibodies for flow cytometry analyses. (B) Expression of co-stimulatory and co-inhibitory receptors on infused T cells in murine solid tumors. Cells dissociated from tumors from each treatment group were stained with the indicated antibodies to detect CD28, PD-1, and Lag-3 expression on infiltrated T cells by flow cytometry. Shade histogram shows isotype control staining, and black line histogram shows the indicated antibody staining. Bar graphs represent percentages of the indicated cell populations as means ± SEM. (C) Treg infiltration was determined by immunohistochemistry staining. Tumor sections were stained with anti-mouse FOXP3 antibody. Bar graphs represent the percentage of tumor-infiltrating Tregs in five views per slide and three slides per tumor as means ± SEM. All results are representative of three repeated experiments.
Given that the enrichment of regulatory T cells (Tregs) in tumors often compromises the efficacy of T cell adoptive transfer (21–23), we stained LLC tumor sections with forkhead box P3 (FOXP3) antibody to quantify Treg infiltration in tumors (Fig. 4C). Tumors from control groups (control DNA, control DNA plus doxorubicin, or IL-12 DNA alone) showed notable Treg infiltration in clusters. In contrast, treatment with IL-12 plus doxorubicin remarkably reduced the accumulation of Tregs in large solid tumors (Fig. 4C), suggesting that this treatment inhibited the infiltration of suppressive immune cells so as to create an environment “friendly” to effector immune cells.
These encouraging results led us to determine whether the same highly persistent CD8+ T cells accumulated in human xenograft large solid tumors treated with IL-12 plus doxorubicin. CAR expression on CD4+ T and CD8+ T cells was confirmed on CD19 28ζ CAR-T cells before infusion (Fig. 5A). The expression levels of co-stimulatory receptors CD28, 41BB, and NKG2D, as well as inhibitory receptors CD39, PD-1, LAG3, and TIM3, were assessed on expanded CAR-T cells before and after infusions plus the indicated treatments (Fig. 5B, 5C). On the basis of the median fluorescence intensity of each receptor on T cells, we created two heatmaps; these maps showed that after control treatments, infiltrated T cells displayed extremely low levels of co-stimulatory receptors CD28 and NKG2D (Fig. 5B) but increased levels of co-inhibitory receptor LAG3 (Fig. 5C). In striking contrast, IL-12 plus doxorubicin induced sustained expression of CD28 and NKG2D, with some suppression of PD1 and LAG3, in the tumor-infiltrating CD8+ T cells (Fig. 5B, 5C). The levels of these immune cell index markers suggest that IL-12 plus doxorubicin treatment enhanced CD8+ T cell effector function and prevented exhaustion of the infiltrated CD8+ T cells. We further converted the two heatmaps to better assess the S/I ratios on CAR-T cells in vitro and in vivo (Fig. 5D), and found that the control treatments dramatically reduced the S/I ratios on tumor-infiltrated T cells in vivo, whereas IL-12 plus doxorubicin maintained the S/I ratios on tumor-infiltrated CD4+ and CD8+ T cells as high as that on CAR-T cells before infusion.
Figure 5. IL-12 plus doxorubicin maintained high expression levels of CD28 and NKG2D on infused CD8+ CAR-T cells in human lymphoblast solid tumors.
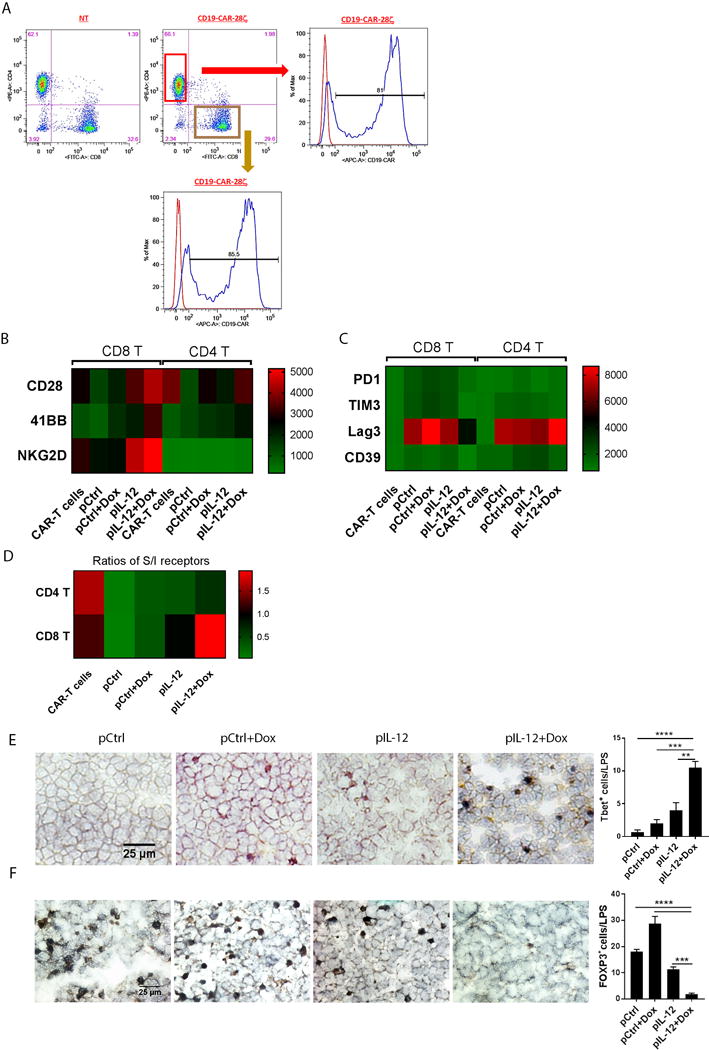
(A) Expression of CAR on CD4+ T and CD8+ T CD19 28ζ CAR-T cells. CAR-T cells were stained with anti-human CAR, CD4, and CD8 antibodies for flow cytometry analyses. (B, C, D) Heatmap based on the expression intensity of co-stimulatory (B), co-inhibitory (C) receptors, and the ratios of co-stimulatory to co-inhibitory receptors (D) on CAR-T cells before and after infusion. CAR-T cells before infusion or from cells dissociated from tumors after infusion were stained with the indicated antibodies, and CD28, 41BB, NKG2D, PD-1, Lag-3, CD39, and TIM3 expression was detected on CD4+ and CD8+ T cells by flow cytometry. (B, C) The heatmap was created on the basis of mean fluorescence intensities. (D) The heatmap was based on the ratios of the co-stimulatory receptor positive to the co-inhibitory receptor positive T cell numbers. (E) Tbet+ effector cell infiltration was determined by immunohistochemistry staining. Tumor sections were stained with anti-human Tbet antibody. Bar graphs represent the percentage of tumor-infiltrating Tbet+ cells in five views per slide and three slides per tumor as means ± SEM. (F) Nalm6 tumor sections were stained with anti-human FOXP3 antibody, and HRP conjugated anti-mouse secondary antibody. Bar graphs represent the density of tumor-infiltrating Tregs in five views per slide and three slides per tumor as means ± SEM. All results are representative of three repeated experiments.
Because it is well accepted that the Tbox transcriptional factor TBX21 (T-bet) plays a very crucial role in T cell development and functions, serving as a marker for effector T cells, we stained tumor sections from indicated groups with anti-T-bet antibody. T-bet+ cells were almost absent in the tumors from the control groups but were present in significantly greater numbers in the tumors from mice treated with IL-12 plus doxorubicin (Fig. 5E). Just as in LLC tumors (Fig. 4C), the infiltration of Tregs in Nalm6 tumors was dramatically reduced by treatment with IL-12 plus doxorubicin (Fig. 5F).Thus, our findings reveal that treatment with IL-12 plus doxorubicin produced the most powerful cytotoxic pattern of infiltrated T cells.
Roles of doxorubicin plus IL-12 in promoting cytotoxic T cell infiltration
Recent reports indicate that a variety of chemotherapeutic agents stimulate tumor cells to release T cell attractants CXCL9, CXC10, and CCL5 (22,23). We therefore first tested whether doxorubicin (100nM) induces these chemokines in tumor cells in vitro. Quantitative PCR analysis showed a remarkable induction of CXCL9 and CXCL10 in LLC cells and Mel2549 treated with doxorubicin (Fig. S3, data not shown). Additionally, the human CAR-T cells we infused into mice expressed high levels of CXCR3, the receptor for CXCL9 and CXCL10 (Fig. 6A).
Figure 6. Roles of doxorubicin and IL-12 in boosting the intratumoral infiltration of infused T cells.
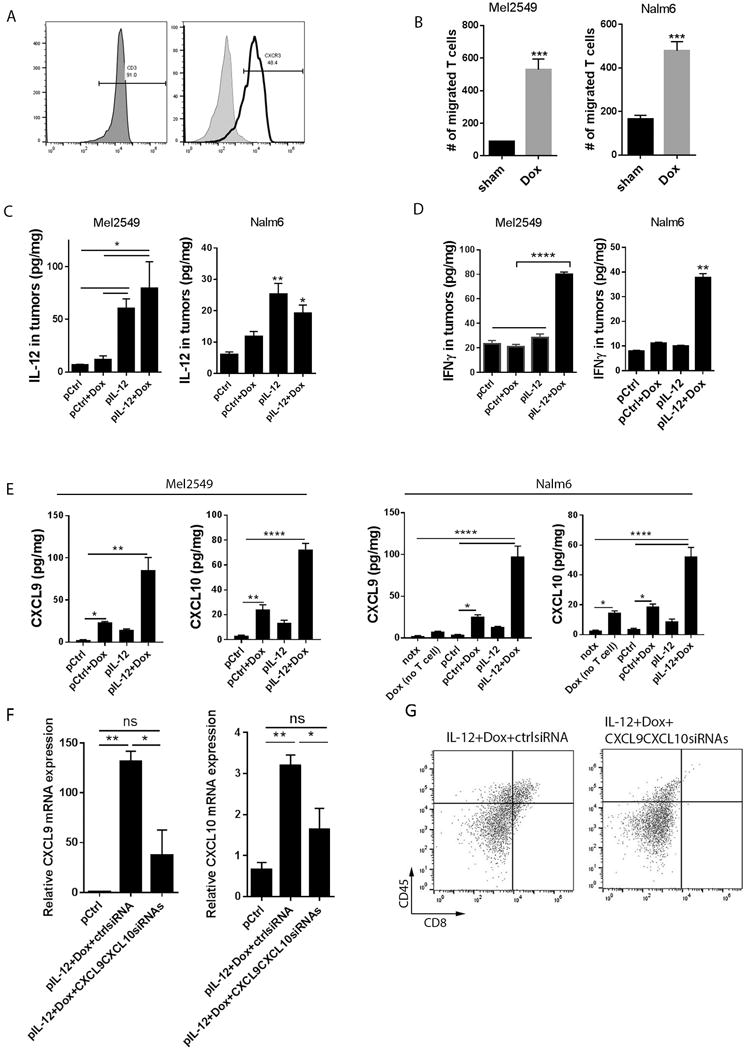
(A) Expression of CXCR3 on human CAR-T cells before infusion. Human CAR-T cells were subjected to CD3 and CXCR3 staining for flow cytometry analyses. (B) Migration assay of T cells and doxorubicin-treated tumor cells in Boyden transwell chambers. Tumor cells were pretreated with 100 nM doxorubicin or vehicle control for 72 hours, and one of the conditioned media was placed in the bottom chamber of each transwell. T cells were labeled with violet cell tracker and placed in the top chamber of each well, which had 5-μm pores. Ninety minutes later, cells in the bottom chambers were counted via flow cytometry analyses. Bar graphs show the numbers of migrated T cells as means ± SEM. (C, D, E) Murine and human xenograft tumors from each treatment group were subjected to lysis in RIPA buffer. The tumor lysates were subjected to ELISA assay to detect IL-12 (C), IFNγ (D) as well as CXCL9 and CXCL10 levels (E). notx, no treatment; Dox (no T cells), doxorubicin treatment without T cell infusions. Bar graphs show the concentration of indicated proteins per milligram of total proteins in the tumor as means ± SEM. (F,G) LLC tumor-bearing mice (N=3) were treated with IL-12 plus doxorubicin as described in Fig. 1 and were then subjected to intratumoral injection of control siRNA or CXCL9 siRNA plus CXCL10 siRNA followed by electroporation delivery twice a week. (F) Quantities of CXCL9 and CXCL10 mRNA (by PCR) in tumors. Bar graphs show the relative mRNA expression normalized to the control DNA-treated group as means ± SEM. (G) Flow cytometry of CD45+CD8+ cells in tumors. All results are representative of three repeated experiments.
In theory, the chemokine-producing tumor cells should be able to promote T cell migration. To test this hypothesis, conditioned medium from Mel2549 or Nalm6 tumor cells treated with doxorubicin was placed at the bottom of Boyden transwell chambers. Violet cell tracker–labeled human TILs or CAR-T cells were cultured in the upper chambers for 1.5 hours. The numbers of T cells that migrated to the lower chambers containing doxorubicin-condition medium were significantly higher (5-fold for Mel2549 and 3-fold for Nalm6) than those that migrated to the lower chambers containing vehicle control medium (Fig. 6B).
Nevertheless, in our study, treatment with low-dose (1mg/kg) systemic doxorubicin alone was unsuccessful in attracting significant numbers of T cells to large solid tumors (Fig. 1–3), which led us to hypothesize that although doxorubicin or IL-12 alone may induce the T cell–attractant chemokines to some extent, only IL-12 plus doxorubicin makes tumor cells produce adequate chemokines to massively attract effector T cells to large solid tumors. Although IL-12 DNA was delivered systemically through intramuscular injection, it induced IL-12 production in tumors (Fig. 6C), which stimulates tumor-infiltrated T cells to secrete the effector molecule IFNγ (Fig. 6D). Treatment with IL-12 plus doxorubicin greatly augmented the intensity of infiltrated T cells, which in turn dramatically increased the expression of IFNγ in tumors (Fig. 6D). IFNγ is a well-known stimulator of chemokines CXCL9, CXCL10, and CXCL11, resulting in the synergistic induction of chemokines after treatment with IL-12 plus doxorubicin. Moreover, IFNγ may initiate positive feedback to stimulate T cell activation. In line with these data, the expression profile of T cell–associated chemokines in mouse and human xenograft tumors after the indicated treatments demonstrated that the levels of CXCL9 and CXCL10 mRNA transcripts were increased in tumors that had been treated with doxorubicin or IL-12 DNA alone and that expression of these chemokines was further boosted in tumors from mice treated with both IL-12 and doxorubicin (Fig. S4). Consistent with induction at the mRNA level, ELISA results showed that the secretion of CXCL9 and CXCL10 in Mel2549 and Nalm6 tumors were substantial induced after treatment with IL-12 plus doxorubicin (Fig. 6E). Strikingly, depletion of these chemokines from tumors impaired the recruitment of CD8+ T cells into tumors from mice treated with both IL-12 plus doxorubicin (Fig. 6F-6G), suggesting that the mechanism regulating CXCL9 and CXCL10 upregulation accounts for the induction of exogenous CD8+ T cell accumulation in large solid tumors by IL-12 plus doxorubicin.
DISCUSSION
We previously reported that IL-12 plus doxorubicin promoted endogenous T cell infiltration into mouse tumors (9), and others found that IL-12 plus cyclophosphamide induced macrophage- and T cell–dependent tumor eradication only in mouse fibrosarcomas, not in mouse melanoma, lung, pancreatic, or colon cancers (24–27). None of these studies showed tumor regression in human tumor models, nor did in-depth characterizations of tumor-infiltrated T cells after treatment. Significantly, our study showed that treatment with IL-12 plus doxorubicin successfully enriched not only murine T cells, but also clinically relevant human T cells (TILs, and CAR-T cells) in large solid tumors, both in peritumoral areas (<3 mm from tumor margin) and in the centers of tumors 10 mm in diameter. Furthermore, this same treatment yielded high ratios of co-stimulatory/co-inhibitory receptors in the tumors and decreased the numbers of Treg cells, creating a tumor microenvironment friendly to infiltrated CD8+ T cells. The infiltration of highly active NKG2D+CD8+ T cells was associated with tumor remission and significantly extended survival in murine and human xenograft tumor models with large tumors (Fig. 1–3).
Although the infiltration of tumors by T lymphocytes, especially CD8+ cytotoxic T cells and memory T cells, has been reported to be associated with positive outcomes in terms of low relapse rates and prolonged overall survival in patients with melanoma (28,29) or lung (30,31), ovarian (4,5), breast (32,33), or colorectal cancer (34,35), the localization of infiltrated CD8+ T cells, which is part of the “immunoscore,” is often neglected. In fact, the localization of infiltrated CD8+ T cells affects the outcomes of T cell therapy in many cancers. Pages et al. observed that the presence of infiltrated CD8+ T cells in both the centers and invasive margins of colorectal tumors served as a marker of a better outcome (35). Sato et al. showed prolonged survival with CD8+ T cells localized in the intraepithelial—instead of the tumor stroma—of ovarian tumors (5). In melanoma metastases, the presence of CD8+ T cells throughout the tumor was correlated with prolonged survival (36). A recent report compared the prognostic outcomes of metastatic melanoma patients with intratumoral versus peritumoral T cell infiltration into the primary tumors and found that the high ratio of intratumoral to peritumoral values was strongly correlated with longer survival duration after dendritic cell therapy (13). Therefore, we studied not only the numbers of infiltrated CD8+ T cells but also their distribution. Treatment with IL-12 or doxorubicin alone attracted CD8+ T cells to the margins of tumors. Only the combination of IL-12 plus doxorubicin recruited CD8+ T cells to accumulate in much deeper areas of the tumors, reaching the center of tumors as large as 10 mm in diameter. Such deep penetration of CD8+ T cells into tumors led to massive tumor cell destruction and delayed tumor progression.
Besides the localization of infused T cells, another key parameter of the immune contexture is the ratio of immune-stimulatory and immune-inhibitory markers, which reflects the functional orientation of infused T cells. T cell therapy can not only augment the infiltration of both cytotoxic and regulatory T cells but also trigger negative feedback, inducing T cell exhaustion. In particular, long-term ex vivo expansion may result in the loss of co-stimulatory receptor CD28 on the CD8+ T cell population, a loss associated with chronic stimulation (37,38). In fact, high numbers of infiltrated T cells are often correlated with high levels of immune-suppressive markers (39,40), which restrains the outcomes of CD8+ T cell infiltration (41). Therefore, it is critical to maintain the effector functions of infiltrated T cells (42).
Our assessment of T cell markers showed elevated numbers of stimulatory CD28+ and NKG2D+ T cells in large solid tumors after treatment with IL-12 plus doxorubicin. CD28 signaling may reinvigorate the antitumor effect of the infiltrated CD8+ T cells. The deep infiltration of NKG2D+CD8+ T cells, besides the tumor specific cytotoxicity, recognize NKG2D ligand expressing tumor cells to induce NKG2D-mediated killing (8,9). Significantly, this same combination treatment (IL-12 plus doxorubicin) is able to restore NKG2D ligand expression in tumors (8), suggesting that this combination therapy could promote engagement between NKG2D+CD8+ T cells and NKG2D ligands on tumor cells to cause tumor rejection. NKG2D co-stimulation also facilitates the formation and rescue of memory CD8+ T cells (43).
Control treatments upregulated exhaustion markers PD1 and LAG3 on infiltrated T cells and increased Treg infiltration, whereas IL-12 plus doxorubicin prevented the exhausted and suppressive T cells present in tumors. It has been reported that CD28 co-stimulation augmented CD8+ T cell proliferation in tumors in response to impairment of PD-1 signaling (7). In the present study, treatment with IL-12 plus doxorubicin not only maintained high levels of CD28 but also suppressed PD-1 expression. Thus, both deep tumor infiltration and cell proliferation may account for the accumulation of great numbers of CD8+ T cells in the large solid tumors. Recent studies indicated that inhibitory receptors suppress the expression of IFNγ by T cells and that these T cells fail to proliferate despite antigen stimulation (44,45). Our treatment with IL-12 resulted in a long duration of IFNγ expression in tumors, which overcame the suppressive effect induced by low levels of inhibitory receptors. As a result, the exogenous source of IFNγ from IL-12 treatment shifted the balance to favor CD8+ T cell antitumor immunity.
Several studies have highlighted the role of chemotherapy in promoting an antitumor immune response. The most frequently proposed mechanism is that chemotherapy, including anthracyclines, causes immunogenic tumor cell death, and the dead cells provide tumor antigens to stimulate immune responses (46–48). Recent studies revealed that chemotherapy also upregulates the expression of chemokines that attract effector T cells to tumors (49). In particular, Ma et al. showed that intratumoral administration of high-dose (2mM) doxorubicin induced expression of T cell–attractant chemokines CXCL9, CXCL10, and CCL5 mRNA in mouse sarcoma tumors in vivo, and in turn recruited CD8+ T and γδ T cells to tumors (22). Hong et al. claimed that temozolomide and cisplatin boosted the production of T cell–attracting chemokines, such as CXCL9, CXCL10, CXCL11, and CCL5, in human xenograft tumors in vitro and in vivo (23). Unlike these previous studies, we administered low-dose doxorubicin (1mg/kg) systemically in our tumor models (20) and showed that such low dose of doxorubicin induced the chemokines CXCL9 and CXCL10 in tumors, which was more or less expected. Surprisingly, doxorubicin in combination with IL-12 significantly boosted this chemokine induction, which became the key mechanism for T cell penetration.
In fact, IFNγ is produced by immune cells (T cells, NK cells, and etc.) upon IL-12 stimulation in tumors. Since there are very few effector immune cells recruited to tumors after IL-12 DNA alone treatment (Fig. 1–3), despite of enhanced IL-12 protein accumulation in tumors (Fig. 6C), it failed to boost IFNγ, a potent inducer of T cell attracting chemokines (Fig. 6D)(21). In contrast, in our combination therapy, doxorubicin allowed effector T cells to penetrate into tumors where in response to accumulated IL-12 (Fig. 6C), IFNγ production was greatly upregulated (Fig. 6D). Subsequently, IFNγ synergized with doxorubicin to enhance chemokine production which in turn recruit more effector T cells to amplify the antitumor immune response.
The correlation between the levels of T cell attracting chemokine, the numbers of infiltrating T cells and antitumor efficacy indicated that only IL-12 plus doxorubicin treatment induced production of CXCL9 and CXCL10 at levels sufficient to recruit the great numbers of CD8+ T cells into large solid tumors needed to obstruct tumor development. However, the source of the chemokines was not limited to tumor cells, because a higher induction of CXCL9 and CXCL10 was observed in tumors from C57BL/6 mice after doxorubicin treatment (data not shown). In fact, T cell–attracting chemokines are also secreted by macrophages, endothelial cells, and other stromal cells (50), and it remains unclear how much other cell types contribute to this induction of chemokines.
Unlike previously published strategies in which T cells had to be engineered to gain a tumor-homing ability, our approach modified the tumor microenvironment to favor T cell accumulation in large solid tumors. Nevertheless, doxorubicin alone was inadequate at inducing the persistent infiltration of effector T cells. However, doxorubicin plus IL-12 promoted the production of IFNγ, which further boosted CXCL9 and CXCL10 expression and stimulated the activation of infiltrated effector NKG2D+CD8+ T cells. These findings suggest that TIL and CAR-T cell therapy may be extended beyond hematological malignancies and melanoma to a broad range of advanced large solid tumors.
Supplementary Material
Translational Relevance.
High density of tumor-infiltrated effector T cells improves the prognosis and prolongs survival of patients with solid tumors. However, the efficacy of T cell immunotherapy in treating large solid tumors remains very limited due to the inadequate effector T cell penetration into large solid tumors. Our discovery of IL-12 DNA plus doxorubicin co-administration prior to TILs or CAR-T cell infusion can promote deep NKG2D+ CD8+ T cell infiltration into large solid tumors in different human xenograft models. This treatment reduced regulatory T cells infiltration, and enhanced the powerful effector patterns of infiltrated T cells in the immune-suppressive tumor microenvironment. Such deep penetration of antitumor effector T cells resulted in massive tumor cell destruction and halted tumor progression. In light of our findings, the use of TILs and CAR-T cell therapy can be extended beyond hematological malignancies and melanoma to a broad range of solid tumors.
Acknowledgments
The authors thank Ms. Kathryn Hale for editing this manuscript. This study was supported by National Institutes of Health grant R01 CA200574 and MD Anderson’s Genetically Engineered Mouse Facility, which is partially supported by the NIH/NCI under award number P30 CA016672.
Footnotes
Author’s contributions
Conception and design: J Hu, S Li
Development of methodology: J Hu, C Sun, C Bernatchez, X Xia, S Li
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J Hu, C Sun, X Xia
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J Hu, C Sun, S Li
Writing, review, and/or revision of the manuscript: J Hu, C Bernatchez, P Hwu, G Dotti, S Li
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C Sun, C Bernatchez, X Xia, P Hwu, G Dotti
Study supervision: S Li
Disclosure of Potential Conflict of interest
No potential conflicts of interest are disclosed.
References
- 1.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556–61. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 3.Giraldo NA, Becht E, Remark R, Damotte D, Sautes-Fridman C, Fridman WH. The immune contexture of primary and metastatic human tumours. Curr Opin Immunol. 2014;27:8–15. doi: 10.1016/j.coi.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 5.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 Costimulation: From Mechanism to Therapy. Immunity. 2016;44:973–88. doi: 10.1016/j.immuni.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355:1423–7. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu J, Bernatchez C, Zhang L, Xia X, Kleinerman ES, Hung MC, et al. Induction of NKG2D Ligands on Solid Tumors Requires Tumor-Specific CD8+ T Cells and Histone Acetyltransferases. Cancer Immunol Res. 2017 doi: 10.1158/2326-6066.CIR-16-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J, Zhu S, Xia X, Zhang L, Kleinerman ES, Li S. CD8+T cell-specific induction of NKG2D receptor by doxorubicin plus interleukin-12 and its contribution to CD8+T cell accumulation in tumors. Mol Cancer. 2014;13:34. doi: 10.1186/1476-4598-13-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–82. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preston CC, Maurer MJ, Oberg AL, Visscher DW, Kalli KR, Hartmann LC, et al. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS One. 2013;8:e80063. doi: 10.1371/journal.pone.0080063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasaturo A, Halilovic A, Bol KF, Verweij DI, Blokx WA, Punt CJ, et al. T-cell Landscape in a Primary Melanoma Predicts the Survival of Patients with Metastatic Disease after Their Treatment with Dendritic Cell Vaccines. Cancer Res. 2016;76:3496–506. doi: 10.1158/0008-5472.CAN-15-3211. [DOI] [PubMed] [Google Scholar]
- 14.Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39:11–26. doi: 10.1016/j.immuni.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2010;16:2646–55. doi: 10.1158/1078-0432.CCR-10-0041. [DOI] [PubMed] [Google Scholar]
- 16.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18:6758–70. doi: 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. 2015;21:524–9. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forget MA, Malu S, Liu H, Toth C, Maiti S, Kale C, et al. Activation and propagation of tumor-infiltrating lymphocytes on clinical-grade designer artificial antigen-presenting cells for adoptive immunotherapy of melanoma. Journal of immunotherapy. 2014;37:448–60. doi: 10.1097/CJI.0000000000000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu S, Waguespack M, Barker SA, Li S. Doxorubicin directs the accumulation of interleukin-12 induced IFN gamma into tumors for enhancing STAT1 dependent antitumor effect. Clin Cancer Res. 2007;13:4252–60. doi: 10.1158/1078-0432.CCR-06-2894. [DOI] [PubMed] [Google Scholar]
- 21.Li S, Zhang X, Xia X. Regression of tumor growth and induction of long-term antitumor memory by interleukin 12 electro-gene therapy. J Natl Cancer Inst. 2002;94:762–8. doi: 10.1093/jnci/94.10.762. [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, et al. Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503. doi: 10.1084/jem.20100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res. 2011;71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466. [DOI] [PubMed] [Google Scholar]
- 24.Le HN, Lee NC, Tsung K, Norton JA. Pre-existing tumor-sensitized T cells are essential for eradication of established tumors by IL-12 and cyclophosphamide plus IL-12. J Immunol. 2001;167:6765–72. doi: 10.4049/jimmunol.167.12.6765. [DOI] [PubMed] [Google Scholar]
- 25.Tsung K, Dolan JP, Tsung YL, Norton JA. Macrophages as effector cells in interleukin 12-induced T cell-dependent tumor rejection. Cancer Res. 2002;62:5069–75. [PubMed] [Google Scholar]
- 26.Guo QY, Yuan M, Peng J, Cui XM, Song G, Sui X, et al. Antitumor activity of mixed heat shock protein/peptide vaccine and cyclophosphamide plus interleukin-12 in mice sarcoma. J Exp Clin Cancer Res. 2011;30:24. doi: 10.1186/1756-9966-30-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malvicini M, Alaniz L, Bayo J, Garcia M, Piccioni F, Fiore E, et al. Single low-dose cyclophosphamide combined with interleukin-12 gene therapy is superior to a metronomic schedule in inducing immunity against colorectal carcinoma in mice. Oncoimmunology. 2012;1:1038–47. doi: 10.4161/onci.20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mackensen A, Ferradini L, Carcelain G, Triebel F, Faure F, Viel S, et al. Evidence for in situ amplification of cytotoxic T-lymphocytes with antitumor activity in a human regressive melanoma. Cancer Res. 1993;53:3569–73. [PubMed] [Google Scholar]
- 29.Clark WH, Jr, Elder DE, Guerry DT, Braitman LE, Trock BJ, Schultz D, et al. Model predicting survival in stage I melanoma based on tumor progression. J Natl Cancer Inst. 1989;81:1893–904. doi: 10.1093/jnci/81.24.1893. [DOI] [PubMed] [Google Scholar]
- 30.Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:4410–7. doi: 10.1200/JCO.2007.15.0284. [DOI] [PubMed] [Google Scholar]
- 31.Al-Shibli KI, Donnem T, Al-Saad S, Persson M, Bremnes RM, Busund LT. Prognostic effect of epithelial and stromal lymphocyte infiltration in non-small cell lung cancer. Clin Cancer Res. 2008;14:5220–7. doi: 10.1158/1078-0432.CCR-08-0133. [DOI] [PubMed] [Google Scholar]
- 32.Alexe G, Dalgin GS, Scanfeld D, Tamayo P, Mesirov JP, DeLisi C, et al. High expression of lymphocyte-associated genes in node-negative HER2+ breast cancers correlates with lower recurrence rates. Cancer Res. 2007;67:10669–76. doi: 10.1158/0008-5472.CAN-07-0539. [DOI] [PubMed] [Google Scholar]
- 33.Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1949–55. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- 34.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 35.Pages F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:5944–51. doi: 10.1200/JCO.2008.19.6147. [DOI] [PubMed] [Google Scholar]
- 36.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012;72:1070–80. doi: 10.1158/0008-5472.CAN-11-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azuma M, Phillips JH, Lanier LL. CD28- T lymphocytes. Antigenic and functional properties. J Immunol. 1993;150:1147–59. [PubMed] [Google Scholar]
- 38.Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30:306–12. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123:2873–92. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denkert C, von Minckwitz G, Brase JC, Sinn BV, Gade S, Kronenwett R, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:983–91. doi: 10.1200/JCO.2014.58.1967. [DOI] [PubMed] [Google Scholar]
- 41.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5:200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–12. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zloza A, Kohlhapp FJ, Lyons GE, Schenkel JM, Moore TV, Lacek AT, et al. NKG2D signaling on CD8(+) T cells represses T-bet and rescues CD4-unhelped CD8(+) T cell memory recall but not effector responses. Nat Med. 2012;18:422–8. doi: 10.1038/nm.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh A, Dey AB, Mohan A, Mitra DK. Programmed death-1 receptor suppresses gamma-IFN producing NKT cells in human tuberculosis. Tuberculosis (Edinb) 2014;94:197–206. doi: 10.1016/j.tube.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 45.Okagawa T, Konnai S, Nishimori A, Ikebuchi R, Mizorogi S, Nagata R, et al. Bovine Immunoinhibitory Receptors Contribute to Suppression of Mycobacterium avium subsp. paratuberculosis-Specific T-Cell Responses. Infect Immun. 2016;84:77–89. doi: 10.1128/IAI.01014-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8:151–60. doi: 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- 47.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 48.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 49.Tan KW, Evrard M, Tham M, Hong M, Huang C, Kato M, et al. Tumor stroma and chemokines control T-cell migration into melanoma following Temozolomide treatment. Oncoimmunology. 2015;4:e978709. doi: 10.4161/2162402X.2014.978709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–22. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.